

Abstract
The PsaF-deficient mutant 3bF of Chlamydomonas reinhardtii was used to modify PsaF by nuclear transformation and site-directed mutagenesis. Four lysine residues in the N-terminal domain of PsaF, which have been postulated to form the positively charged face of a putative amphipathic α-helical structure were altered to K12P, K16Q, K23Q, and K30Q. The interactions between plastocyanin (pc) or cytochrome c6 (cyt c6) and photosystem I (PSI) isolated from wild type and the different mutants were analyzed using crosslinking techniques and flash absorption spectroscopy. The K23Q change drastically affected crosslinking of pc to PSI and electron transfer from pc and cyt c6 to PSI. The corresponding second order rate constants for binding of pc and cyt c6 were reduced by a factor of 13 and 7, respectively. Smaller effects were observed for mutations K16Q and K30Q, whereas in K12P the binding was not changed relative to wild type. None of the mutations affected the half-life of the microsecond electron transfer performed within the intermolecular complex between the donors and PSI. The fact that these single amino acid changes within the N-terminal domain of PsaF have different effects on the electron transfer rate constants and dissociation constants for both electron donors suggests the existence of a rather precise recognition site for pc and cyt c6 that leads to the stabilization of the final electron transfer complex through electrostatic interactions.
Keywords: site-directed mutagenesis, electron transfer, Chlamydomonas
The photosystem I (PSI) complex functions as a light-driven oxidoreductase that transfers electrons from plastocyanin (pc) to ferredoxin in higher plants, most algae, and cyanobacteria. In some cyanobacteria and algae, the type I copper protein pc can be replaced by a class I c-type cytochrome, depending on the relative availability of copper and iron in the culture medium (1–4). The eukaryotic PSI reaction center is a membrane-bound complex consisting of 13–14 polypeptide subunits. The three-dimensional structure of PSI from the cyanobacterium Synechococcus sp. has been determined by x-ray crystallography at a resolution of 4 Å (5).
In Chlamydomonas reinhardtii the PsaF subunit of PSI is essential for efficient electron transfer from pc and cytochrome c6 (cyt c6) to PSI (6), because the in vitro measured electron transfer between both donors and PSI isolated from a PsaF-deficient mutant is drastically reduced compared with wild type. In vivo measurements also revealed that the rate of electron transfer between pc and P700+ is considerably diminished in the PsaF-deficient mutant (7). In contrast, the specific deletion of the psaF gene in cyanobacteria did not affect photoautotrophic growth (8) and the in vivo measured electron transfer rate between cyt c553 and PSI was the same as in wild type (9). In vitro measurements revealed that even at high concentrations of pc or cyt c6 no difference in electron transfer could be measured between the donor proteins and PSI isolated from wild type or the PsaF-deficient Synechocystis mutant (10). In the green alga C. reinhardtii both pc and cyt c6 reduce P700+ in vitro at high concentration of donor proteins with first order kinetics and a half-life of 3 μs (6). This is comparable to the half-life of 4 μs for fast electron transfer between pc and PSI measured in vivo in the unicellular alga Chlorella (11). First order kinetics with a half-life of 12 μs were found for the reaction of pc with PSI in intact thylakoids (12, 13), with digitonin–PSI particles (14, 15), and with PSI–200 particles (16) from higher plants. No corresponding fast phase could be observed in the cyanobacterium Synechocystis (17) and in the thermophilic cyanobacterium Synechococcus elongatus (18). The electron transfer from cyt c6 to P700+ was found to display a first order kinetic component with a half-life of 3, 7, and 4 μs in the green algae C. reinhardtii (6) and Monoraphidium braunii and in the cyanobacterium Anabaena sp. PCC7119 (19), respectively.
In spinach, pc can be crosslinked to the PsaF subunit of PSI (20, 21). The conformation of the crosslinked and authentic pc–PSI complex appears to be similar based on the fast kinetics of reduction of P700+ (21). Lys residues within the N-terminal domain of PsaF from spinach appear to be crosslinked to the conserved acidic amino acids 42–44 and 59–61 of pc as revealed by mass spectroscopic analysis of tryptic peptides of pc and of the crosslinked product of pc and PsaF (10). Because this positively charged N-terminal domain is absent from PsaF of cyanobacteria it was suggested that this motif evolved for binding pc to PSI in a way that leads to the formation of a stable complex, competent for fast electron transfer (10).
Results obtained by site-directed mutagenesis of pc (10, 22–24) suggest that binding to PSI is a “two-step event,” which involves a long-range electrostatic interaction between the positively charged PsaF and the negative patches of pc and a docking mechanism, which brings the flat hydrophobic surface of pc in close contact with PSI and thereby allows efficient electron transfer from copper via His87 to P700+ (23).
Here we have taken advantage of the PsaF-deficient mutant 3bF of C. reinhardtii for modifying PsaF using nuclear transformation and site-directed mutagenesis. Our results indicate that (i) the N-terminal domain provides a precise recognition site that is essential for binding and fast electron transfer from pc and cyt c6 to PSI and (ii) although the two alternative donor proteins have different structures and harbor different redox cofactors, their binding to PSI appears to be mediated by the same residues of PsaF. To our knowledge, this is the first report of the substitution of a nonessential nuclear gene with a modified copy of C. reinhardtii and where the effects of the mutations on the corresponding multiprotein complex could be assessed. This study shows that nuclear reverse genetics is possible for nuclear genes of C. reinhardtii involved in photosynthesis or any other dispensible function.
EXPERIMENTAL PROCEDURES
Strains and Media.
C. reinhardtii wild-type and mutant strains were grown as described (25). Tris-acetate phosphate (TAP) medium was solidified with 2% Bacto agar (Difco) and supplemented with 75 μg/ml emetine (Sigma) or 25 μg/ml zeocin (Cayla, Toulouse, France) when required.
Nucleic Acid Techniques.
Procedures for the preparation of recombinant plasmids and DNA sequencing were performed as described (26). Escherichia coli DH5α was used as bacterial host. C. reinhardtii total DNA was isolated as described (27). The site-directed change of K12P, K16Q, K19Q, K19E, K23Q, K27E, and K30Q in PsaF was performed by using the oligonucleotides 5′-TGCTCCGAGAGCCCGGCTTACGCCAAG-3′; 5′-AAGGCTTACGCCCAGCTGGAGAAGAAGG-3′; 5′-TACGCCAAGCTCGAGCAGAAGGAGCTGAAGA-3′; 5′-GCCAAGCTGGAGGAGAAGGAGCTGAAGA-3′; 5′-AAGAAGGAGCTGCAGACCCTGGAGAAGC-3′; 5′-AAGACCCTGGAGGAGCGCCTGAAGC-3′; and 5′-GAGAAGCGCCTGCAGCAGTACGAGG-3′ together with two other oligonucleotides complementary to the 5′ and 3′ ends of the psaF region carrying a SacII and NcoI restriction site, respectively, in a single tube PCR (28). The amplified DNA fragments were gel purified, digested with SacII and NcoI, and cloned into the plasmid pSP731.04 psaF (SacII/NcoI). This plasmid is a derivative of the pSP73 plasmid (Promega), containing the 1.04-kbp BglII/BamHI fragment, from the plasmid p3.8F1 (ref. 7; see also Fig. 2), inserted in its polylinker. The 1.04-kbp BglII/BamHI fragment contains unique SacII and NcoI restriction sites. The amplified mutated DNA was verified by sequencing and inserted into the SacII/NcoI restriction sites of pSP731.04 psaF. The plasmid was digested with BglII and BamHI, and the 1.04-kbp fragment was purified and cloned into the plasmid p2.9F1 (BglII/BamHI). This plasmid is a derivative of the plasmid p3.8F1, from which a 0.88-kbp BamHI/SphI fragment was removed.
Figure 2.

Crosslinking of pc to K23Q–PsaF is strongly impaired. Immunoblot analysis of crosslinked products between pc or cyt c6 and PSI I from wild-type (lanes 1–3, 2 μg Chl loaded on each lane), K23Q transformant (lanes 4–6, 3 μg chl loaded on each lane), K12P transformant (lanes 7–9, 2 μg Chl loaded on each lane), K16Q transformant (lanes 10–12, 2 μg Chl loaded on each lane), and K30Q transformant (lanes 13–15, 2 μg Chl loaded on each lane) fractionated by SDS/PAGE. The blot was probed with anti-PsaF antibodies. Pc or cyt c6 (20 μM) were used for the crosslinking experiments.
Nuclear Transformation and Analysis of Transformants.
Nuclear transformation of the C. reinhardtii PsaF-deficient 3bF cells (7) was performed according to Kindle (29) with the modifications as described (7). Cells were cotransformed with 2 μg DNA of the p2.9F1 plasmid containing the altered psaF gene and 1 μg DNA of the cry1–1 plasmid (30). After transformation the cells were diluted into 10 ml of TAP-N medium, lacking ammonium chloride, and incubated under low light (5 μE/m2 per s) for 3 days. The cells were then concentrated by centrifugation at 2,500 × g, resuspended in 0.5 ml TAP-N, and plated on TAP plates supplemented with 75 μg/ml emetine. The plates were incubated under low light (5 μE/m2 per s) until colonies appeared. Growing colonies were restreaked on fresh TAP/emetine plates and characterized. When cotransformation was done with the pSP108 plasmid (31), cells were diluted after transformation into 10 ml TAP medium and incubated under light intensities of 100 μE/m2 per s for 2 days. The collected cells were resuspended in 0.5 ml TAP and plated on TAP plates supplemented with 25 μg/ml zeocin.
Total DNA from the transformants was used for PCR amplification of a 330-bp fragment by using PCR primers complementary to the NcoI site and to a sequence located 80 bp downstream from the SacII site of the psaF gene. To confirm the presence of the desired mutations, the PCR amplified fragments were digested with appropriate restriction enzymes such as BanII for K12P, PvuII for K16Q, and PstI for K23Q and K30Q. These unique restriction sites were introduced with the site-directed mutation. These sites are not present in the wild-type gene.
Isolation of pc and cyt c6.
The isolation of pc and cyt c6 followed published procedures (4, 32), with modifications as described (6). The concentrations of pc and cyt c6 were determined spectroscopically by using an extinction coefficient of 4.9 mM−1 cm−1 at 597 nm for the oxidized form of pc (33) and 20 mM−1 cm−1 at 552 nm for the reduced form of cyt c6 (1).
Growth Rate Measurements.
Doubling times of the mutants were determined from four independent measurements.
Isolation of Thylakoid Membranes and of the PSI Complex.
The isolation of PSI particles and thylakoid membranes purified by centrifugation through a sucrose step gradient were as described (6, 34). Chlorophyll (Chl) concentrations were determined according to Porra et al. (35).
SDS/PAGE and Western Blot Analysis.
SDS/PAGE (15.5% T/2.66% C) was carried out according to Laemmli (36). After the electrophoretic fractionation the proteins were electroblotted onto nitrocellulose and incubated with antibodies as described (21). Immunodetection was carried out according to Hippler et al. (6).
Crosslinking Procedures.
Crosslinking was performed as described (6), except that crosslinking with PSI particles from the 3bF transformants was done in the presence of 10 mM MgCl2.
Flash Absorption Spectroscopy.
Kinetics of flash-induced absorbance changes at 817 nm were measured essentially as described (16). The measuring light was provided by a luminescence diode [Hitachi HE8404SG, 40 mW, full-width at half-maximum (FWHM) 30 nm] supplied with a stabilized battery-driven current source. The light was filtered through a 817 nm interference filter (FWHM 9 nm) and passed through a cuvette containing 200 μl of the sample with an optical pathlength of 1 cm.
RESULTS
Expression of PsaF After Nuclear Transformation of 3bF.
The proposed α-helix motif close to the N-terminal end of the PsaF subunit (Fig. 1), found to be involved in the crosslink of pc to PSI from spinach, is also conserved in the PsaF protein of C. reinhardtii (37). In particular, Lys12, Lys16, Lys19, or Lys23 within this α-helix motif of PsaF were suggested to interact with the conserved acidic patch 42–44 of pc (10). To test this model, we have changed Lys16, Lys19, Lys23, and Lys30 to Gln, Lys12 to Pro, and Lys19 and Lys27 to Glu. A direct selection for the expression of PsaF is not possible because the PSI complex is stable without PsaF and the 3bF cells grow photoautotrophically. Nuclear cotransformation of the 3bF mutant was performed with a plasmid containing the cry1 [which confers cryptopleurine and emetine resistance (30)] or the ble gene [which confers phleomycin and zeocin resistance (31)] and a plasmid containing the mutated psaF gene (Fig. 1). Forty transformants were checked by PCR for the presence of the altered psaF gene. A cotransformation rate of about 60% was observed. Because of this high cotransformation rate, expression of psaF was tested directly by immunoblot analysis. The frequency of expression of PsaF was variable among the transformants. The psaF constructs K12P, K16Q, K23Q, K30Q, and the wild-type psaF were found to be expressed after transformation of 3bF. From these constructs a total of 67 transformants were obtained after selection of resistence to emetine, and 11 of these expressed PsaF. For constructs K19Q, K19E, and K27E, between 34 and 48 transformants were tested by immunoblot analysis in each case, but no expression of PsaF was found.
Figure 1.

Restriction map of the genomic clone p3.8Fcos1 containing the psaF gene. psaF exons are shaded and introns are hatched. The strategy used for the site-directed mutagenesis is shown. (Inset) Helical wheel structure of the N terminus of PsaF. The positive and hydrophobic sites are indicated by solid and open curves.
The doubling times of the mutant strains K12P, K16Q, K23Q, and K30Q in high salt medium at a light intensity of 60 μE/m2 per s were 7.8 ± 2.1, 5.8 ± 0.9, 7.8 ± 2.1, and 7.3 ± 3.8 h, respectively, and comparable to the value of 5.8 ± 1.0 found for the 3bF strain rescued with the wild-type psaF gene. This is consistent with the growth properties of the PsaF-deficient strain, which is comparable to those of wild type (7).
Crosslinking of pc or cyt c6 to PsaF Is Diminished in PSI from the K23Q Transformant.
The interactions between the altered PsaF subunit and pc or cyt c6 were examined by crosslinking studies by using purified PSI particles from the K12P, K16Q, K23Q, and K30Q transformants and from wild type (Fig. 2). The crosslinked products were fractionated by SDS/PAGE and identified by immunoblotting by using PsaF antibodies (Fig. 2). From the intensities of the PsaF signal it can be estimated that PsaF accumulates between 50–100% of wild-type levels in the four different PSI mutant complexes. The crosslinking products between pc or cyt c6 and PsaF were found at 29 kDa and 28.5 kDa, respectively. The change of K12P, K16Q, K23Q, or K30Q within the N-terminal domain of PsaF did not totally impair its ability to crosslink with both donor proteins. cyt c6 was less efficiently crosslinked to PSI from the K12P, K16Q, and K23Q transformants than to PSI from wild type. The crosslinking between pc and PSI from the K16Q transformant was also less efficient compared with the wild-type PSI. The largest effect was observed with the mutant K23Q, where the crosslinking between pc and PsaF was strongly diminished (Fig. 2). The same results were obtained when the PSI complex from a second independent K23Q transformant was isolated and crosslinked to pc and cyt c6. It is thus unlikely that another mutation induced by the transformation is responsible for the lowered affinity of pc and cyt c6 to the altered PsaF subunit of the K23Q mutant.
Single Amino Acid Changes Within the N-Terminal Domain of PsaF Decrease the Efficiency of the Electron Transfer Reaction Between pc or cyt c6 and P700.
The electron transfer from pc or cyt c6 to the altered PSI was investigated further using excitation by single turnover flashes. Fig. 3 shows the absorbance transients at 820 nm induced by a laser flash for PSI particles in the presence of 300 μM cyt c6 (Left) or 300 μM pc (Right). In all cases, the time course of the P700+ reduction can be deconvoluted into three kinetic components. The fast component with a constant half-life of 3–4 μs for both donor proteins and a variable amplitude A(1) reflects a first order electron transfer, the rate of which is independent from the concentration of the donor proteins. This phase can be explained by an electron transfer reaction within a preformed complex between pc or cyt c6 and PSI. This fast phase, which was identified in the kinetics of P700+ reduction for wild type (ref. 6 and data not shown) and all four mutants (Fig. 3) appears to be unchanged. The intermediate component with an amplitude A(2) shows a half-life that decreases with increasing concentration of reduced donor protein as known for second order reactions between soluble reactants (see also Fig. 5). The amplitude A(1) increases with increasing concentration of reduced donor protein at the expense of A(2) (Fig. 4). The third very slow component with an amplitude of about 35, 16, 54, and 20% of the total signal for PSI isolated from mutants K12P, K16Q, K23Q, and K30Q, respectively, has an electron transfer rate constant in the range of 2–5 × 105 M−1 s−1 for pc or cyt c6 (Fig. 3), which is comparable to the values found for electron transfer between both donors and PSI from the PsaF-deficient mutant. Thus, it is most likely that the third very slow component can be attributed to PSI without PsaF (6). The amplitude of the very slow phase is always higher with pc than with cyt c6. Because the oxidation of pc during the two faster phases gives rise to a positive absorption change at 820 nm, its subsequent rereduction by ascorbate contributes to the very slow kinetics in the millisecond time range. We will consider mainly the two kinetic components, A(1) and A(2), which are attributed to PSI with functional PsaF. Drepper et al. (16) introduced a kinetic model for the binding and the electron transfer between pc and PSI that also takes into account the redox equilibrium of the electron transfer. In this model, a simple dissociation equilibrium of the complex between the reduced donor protein [D] and PSI was used to describe the concentration dependence of the amplitude A(1). The dissociation constant, KD, was determined by using the following equation
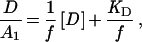 |
1 |
where f represents an empirical factor (f < 1), which relates the amplitude A(1) observed after the flash to the fraction of PSI in a complex with the reduced donor before the flash and corresponds to the maximum of A(1) at infinite [D] (16). Eq. 1 indicates that a plot of the concentration [D] over the relative amplitude A(1) as a function of the donor protein concentration [D] should yield a linear relationship with a slope 1/f and −KD as the intercept of the abscissa.
Figure 3.

Kinetics of the electron transfer from pc or cyt c6 to P700+. Absorbance transients monitored as ΔA817 induced by a laser flash in PSI particles isolated from the four mutants, K12P, K16Q, K23Q, and K30Q (from top to bottom) in the presence of 300 μM cyt c6 (Left) or 300 μM pc (Right). The cuvette contained PSI particles at a concentration of 90 μg Chl per ml, 0.05% (wt/vol) β-dodecyl maltoside, 10 mM MgCl2, 30 mM 3-(N-morpholino)propanesulfonic acid (pH 7.0), 1 mM sodium ascorbate, 0.1 mM methylviologen, and 0.2 mM diaminodurene. The absorbance transients measured in the absence of donor proteins are shown as control by dotted traces. Vertical broken lines separate regions recorded with a different time base. The half-lives of the kinetic components attributed to the donor proteins are as follows: in the presence of 300 μM cyt: c6 K12P, 4 μs and 95 μs; K16Q, 4 μs and 178 μs; K23Q, 4 μs and 393 μs; K30Q, 4 μs and 169 μs, and in the presence of 300 μM pc: K12P, 4 μs and 45 μs; K16Q, 4 μs and 128 μs; K23Q, 4 μs and 239 μs; K30Q, 4 μs and 86 μs.
Figure 5.

Estimation of the rate constant k2. Half-life of the slow component as a function of the reciprocal value of the concentration of pc (A) and cyt c6 (B) concentration. PSI particles were isolated from wild-type (•), K12P transformant (□), K16Q transformant (Δ), K23Q transformant (◊), and the K30Q transformant (○). Measurements were performed in the presence of 10 mM MgCl2, except for wild-type PSI with pc (3 mM MgCl2).
Figure 4.

Estimation of dissociation constants. Amplitude of the fast kinetic component of P700+ reduction as a function of the concentration of donor proteins pc (A) and cyt c6 (B). PSI particles were isolated from wild type (•), K12P transformant (□), K16Q transformant (Δ), K23Q transformant (◊), and K30Q transformant (○). MgCl2 concentration was 10 mM, except for wild-type PSI with pc (3 mM).
The amplitude of the fast phases of P700+ reduction for kinetic experiments performed with various concentrations of pc and cyt c6 is displayed in Fig. 4 according to Eq. 1. For wild type and all four mutants the data points follow a straight line. The dissociation constants and the values of f for pc and cyt c6 are summarized in Table 1. For the K12P transformant, the dissociation constants for the binding of pc or cyt c6 to PSI are comparable to those for wild type. The dissociation constants for the complex between both donors and PSI from the K16Q and K30Q transformants are 3- and 2-fold higher than for wild type, respectively. For K23Q the dissociation constant is ≥500 μM for pc and ≥600 μM for cyt c6.
Table 1.
Properties of electron transfer from pc and cyt c6 to PSI from wild type and from 3bF transformants with an altered PsaF subunit
| PSI | pc
|
cyt c6
|
||||
|---|---|---|---|---|---|---|
| k2 × 107/M−1s−1 | KD/μM* | f | k2 × 107/M−1s−1 | KD/μM | f | |
| Wild type | 9.2 | 83 | 0.67 | 3.4 | 116 | 0.62 |
| K12P | 6.9 | 83 | 0.67 | 2.9 | 82 | 0.66 |
| K16Q | 1.61 | 241 | 0.41 | 1 | 303 | 0.5 |
| K23Q | 0.7 | >500 | 0.21 | 0.46 | >600 | 0.36 |
| K30Q | 3.4 | 154 | 0.48 | 1.8 | 210 | 0.52 |
KD value for the transient complex allowing fast electron transfer (t½ = 3–4 μs) to P700+.
The kinetic component A(2) follows an exponential time course because the concentration of pc and cyt c6 exceeds that of P700 by more than one order of magnitude. The plots of the half-life of A(2) versus the reciprocal donor concentration yield straight lines (Fig. 5). The second order rate constant k2 = ln2/(t1/2 × [D]) can be determined from the slope of the curves in Fig. 5.
For wild-type PSI, k2 values of 3.4 × 107 M−1 s−1 and 9.2 × 107 M−1 s−1 for cyt c6 and pc, respectively, are determined. The bimolecular reaction between cyt c6 and PSI from the transformants K12P, K30Q, K16Q, and K23Q was slower by a factor of 1.2, 1.9, 3.4, and 7.4, relative to the binding to wild-type PSI, respectively. Similar effects (Table 1) because of the changes to K12P, K30Q, K16Q, and K23Q are seen on the binding of pc, where the second order rate constants decrease 1.3-, 2.7-, 5.7-, and 13.1-fold, respectively, compared with wild type. It can be seen that the changes of K16Q and K23Q in PsaF have the strongest influence on binding and electron transfer between pc or cyt c6 and P700. Interestingly, both changes affect the rate constant for binding of pc twice as much as that for cyt c6.
The second order rate constant of P700+ reduction by pc and cyt c6 with PSI particles from wild type and the PsaF-deficient mutant revealed a strong dependence on the MgCl2 concentration. For wild-type PSI particles a maximal reaction rate was observed at low MgCl2 concentrations for both donors, whereas the maximal rate with PSI particles from 3bF was found at MgCl2 concentrations of 1 M (6). The rate constants for the interaction of pc or cyt c6 with PSI particles from two independent K23Q transformants were measured at different MgCl2 concentrations. The maximum reaction rates were found between 1–10 mM MgCl2 and between 10–100 mM MgCl2 for pc and cyt c6, respectively (data not shown). The reaction rate decreased with increasing salt concentrations to values close to those found for the PsaF-deficient mutant.
DISCUSSION
In a previous study (6) we investigated the function of the PsaF subunit of PSI by analyzing the interactions between pc or cyt c6 and PSI isolated from wild type and a PsaF-deficient mutant of C. reinhardtii. We showed that efficient electron transfer from both pc and cyt c6 to PSI depends on PsaF. In this study we have taken advantage of a PsaF-deficient mutant to modify PsaF using nuclear transformation and site-directed mutagenesis. PSI particles from different mutant strains containing a specific amino acid change in the PsaF subunit were isolated together with PSI particles from wild type and used with purified pc and cyt c6 to characterize the electron transfer from these donors to P700+in vitro.
As reported earlier we found that nuclear cotransformation occurs with high frequency in C. reinhardtii (29). Selection for resistance to emetine or zeocin allowed us to screen for the expression of the cotransformed psaF gene. On average, only one of four transformants containing the mutant K12P, K16Q, K23Q, and K30Q or the wild-type psaF gene accumulated its protein, which was stably incorporated into the PSI complex (Fig. 2). However, in spite of screening numerous K19Q, K19E, and K27E transformants, the corresponding PsaF protein was not detectable. It is possible that these mutations affect the stability of the PsaF protein, its targeting to the thylakoid lumen, or its proper insertion into PSI.
It had previously been proposed that Lys12, Lys16, and Lys23 of PsaF, together with Lys19, could form potential crosslinking sites with the acidic patch 42–44 of pc (10). In this work, we have shown that K16Q and K23Q affect significantly the formation of the electron transfer complex between pc or cyt c6 and the corresponding mutant PSI complex. In contrast, the K12P change has almost no effect. The bimolecular rate for the interaction of pc and PSI from the K23Q mutant is reduced 13-fold (see Table 1). In a previous study, changes of acidic to neutral (10) or basic (24) residues at the southern acidic patch of pc were found to result in a comparable decrease of the second order rate constant. Taken together, these observations suggest that Lys23 of PsaF is essential for the productive interaction with the southern acidic patch of pc. The observation that crosslinking between pc and PsaF from the K23Q mutant is strongly reduced strengthens this view. However, Lys16 and Lys30 of PsaF also appear to play an important role in the binding of pc, because the electron transfer rate constants are diminished 6- and 3-fold compared with wild type. Lys30 has been proposed to be involved in the crosslinking of the northern acidic patch, although it was argued that it would not contribute significantly to the formation of the electron transfer complex between pc and PSI (10). The observation that pc can be crosslinked to PSI from the K30Q transformant and that the dissociation constant is only increased by a factor of two indicates that Lys30 is not essential for the formation of the complex with pc. However, Lys 30 does contribute to the stabilization of this complex. In contrast, the change of Lys16 to Gln has a more drastic effect on the formation of the electron transfer complex between the mutant PSI and pc, suggesting an interaction of Lys16 with the southern acidic patch of pc.
The changes of K16Q, K23Q, and K30Q affect more strongly the binding of pc than that of cyt c6 to the corresponding PSI complex. The rate of electron transfer was also reduced twice as much for pc than for cyt c6 in the PsaF-deficient mutant (6). The strongest reduction of the electron transfer rate constant from cyt c6 to PSI is observed with the K23Q mutant. Crosslinking between cyt c6 and PSI from the K23Q mutant is also affected, although not as strongly as with pc. Comparable to the results obtained with pc, the electron transfer rate constants for cyt c6 and the PSI complexes K16Q and K30Q are 2- or 4-fold higher than with the K23Q mutant PSI complex. It is noticeable that crosslinking between cyt c6 and K16Q–PsaF is also diminished. The x-ray crystal structure of cyt c6 has revealed that it has a northern and southern negative patch similar to those of pc (32). Because the electrostatic interactions between cyt c6 and PsaF are needed for the efficient electron transfer and because cyt c6 reduces P700+ with first order kinetics and a half-life of 3 μs (6), it is possible that Lys16 and Lys23 of PsaF interact with the southern negative patch of cyt c6 to allow stable complex formation.
In contrast to the effects observed on the rate constants, the differences between the dissociation constants for pc and cyt c6 for the interaction with PSI from wild type and the K16Q, K23Q, and K30Q mutants are minor. However, the f value for the interaction of both donors with PSI from the transformants K16Q and K23Q is significantly lower for pc than for cyt c6. The f value corresponds to the maximal fraction of the total amplitude of the productive intermolecular electron transfer complex. This can be limited by competition between productive and unproductive binding, by the rate-limiting formation of an intermediate (38), or by the altered redox equilibrium within the electron transfer complex. Because it is very unlikely that the change of single amino acids within the N-terminal domain of PsaF affects significantly the redox midpoint potential of P700 or of the two electron donors, it is therefore more likely that the mutations of PsaF interfere with the formation of a productive electron transfer complex.
Because the change of Lys to Gln is not known to change the α-helical structure (39), predicted in the region between the residues A9 and Y32 (40, 41), it is likely that the results obtained with the changes K16Q, K23Q, and K30Q reflect a local charge effect rather than a conformational change within the N-terminal region of PsaF. That the 3–4 μs first order electron transfer occurs with the mutant PSI complexes provides strong evidence that the overall conformation of PsaF is unchanged because the rate of electron transfer is very sensitive to changes in distance between electron transfer partners (38). We observe a clear hierarchy in the effects on the electron transfer rate constants and dissociation constants for both electron donors caused by the single amino acid changes within the N-terminal domain of PsaF with the order K23Q > K16Q > K30Q > K12P. We can conclude (i) that although pc and cyt c6 have different primary structures and carry distinct redox cofactors, they both bind to a similar recognition site within the N-terminal domain of PsaF and (ii) that this recognition site is formed mostly by residues between K23 and K16. These residues appear to stabilize the final electron transfer complex through electrostatic interactions with negatively charged amino acids of the southern acidic patch of pc and, most likely, of cyt c6, but do not alter the microsecond electron transfer within the intermolecular electron transfer complex.
Acknowledgments
We thank N. Roggli for drawings and photography. This work was supported by a grant from the Human Frontier Science Program and by Grant 31.34014.92 from the Swiss National Fund to J.-D.R. and by Grant SFB388/A1 from the Deutsche Forschungsgemeinschaft to W.H. M.H. gratefully acknowledges a long-term fellowship from the Human Frontier Science Program.
ABBREVIATIONS
- Chl
chlorophyll
- cyt c6
cytochrome c6
- pc
plastocyanin
- PSI
photosystem I
- TAP
tris-acetate phosphate
References
- 1.Wood P M. Eur J Biochem. 1978;87:9–19. doi: 10.1111/j.1432-1033.1978.tb12346.x. [DOI] [PubMed] [Google Scholar]
- 2.Ho K K, Krogmann D W. Biochim Biophys Acta. 1984;766:310–316. [Google Scholar]
- 3.Sandmann G. Arch Microbiol. 1986;21:6366–6375. [Google Scholar]
- 4.Merchant S, Bogorad L. Mol Cell Biol. 1986;6:462–469. doi: 10.1128/mcb.6.2.462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schubert W, Klukas O, Saenger W, Fromme P, Witt H T. J Mol Biol. 1997;272:741–769. doi: 10.1006/jmbi.1997.1269. [DOI] [PubMed] [Google Scholar]
- 6.Hippler M, Drepper F, Farah J, Rochaix J-D. Biochemistry. 1997;36:6343–6349. doi: 10.1021/bi970082c. [DOI] [PubMed] [Google Scholar]
- 7.Farah J, Rappaport F, Choquet Y, Joliot P, Rochaix J-D. EMBO J. 1995;14:4976–4984. doi: 10.1002/j.1460-2075.1995.tb00180.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chitnis P R, Purvis D, Nelson N. J Biol Chem. 1991;266:20146–20151. [PubMed] [Google Scholar]
- 9.Xu Q, Xu L, Chitnis V, Chitnis P. J Biol Chem. 1994;269:3205–3211. [PubMed] [Google Scholar]
- 10.Hippler M, Reichert J, Sutter M, Zak E, Altschmied L, Schröer U, Herrmann R G, Haehnel W. EMBO J. 1996;15:6374–6384. [PMC free article] [PubMed] [Google Scholar]
- 11.Delosme R. Photosynth Res. 1991;29:45–54. doi: 10.1007/BF00035205. [DOI] [PubMed] [Google Scholar]
- 12.Haehnel W, Döring G, Witt H T. Z Naturforsch B. 1971;26:1171–1174. doi: 10.1515/znb-1971-1118. [DOI] [PubMed] [Google Scholar]
- 13.Haehnel W, Ratajczak R, Robenek H. J Cell Biol. 1989;108:1397–1405. doi: 10.1083/jcb.108.4.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bottin H, Mathis P. Biochemistry. 1985;24:6453–6460. [Google Scholar]
- 15.Bottin H, Mathis P. Biochim Biophys Acta. 1987;892:91–98. [Google Scholar]
- 16.Drepper F, Hippler M, Nitschke W, Haehnel W. Biochemistry. 1996;35:1282–1295. doi: 10.1021/bi951471e. [DOI] [PubMed] [Google Scholar]
- 17.Hervás M, Ortega J M, Navarro J A, De la Rosa M A, Bottin H. Biochim Biophys Acta. 1994;1184:235–241. [Google Scholar]
- 18.Hatakana H, Sonoike K, Hirano M, Katoh S. Biochim Biophys Acta. 1993;1141:45–51. doi: 10.1016/0005-2728(93)90187-k. [DOI] [PubMed] [Google Scholar]
- 19.Hervás M, Navarro J A, Díaz A, Bottin H, De la Rosa M A. Biochemistry. 1995;34:11321–11326. doi: 10.1021/bi00036a004. [DOI] [PubMed] [Google Scholar]
- 20.Wynn R M, Malkin R. Biochemistry. 1988;27:5863–5869. doi: 10.1021/bi00416a007. [DOI] [PubMed] [Google Scholar]
- 21.Hippler M, Ratajczak R, Haehnel W. FEBS Lett. 1989;250:280–284. [Google Scholar]
- 22.Nordling M, Sigfridsson K, Young S, Lundberg L G, Hansson O. FEBS Lett. 1991;291:327–330. doi: 10.1016/0014-5793(91)81313-w. [DOI] [PubMed] [Google Scholar]
- 23.Haehnel W, Jansen T, Gause K, Klösgen R B, Stahl B, Michl D, Huvermann B, Karas M, Herrmann R G. EMBO J. 1994;13:1028–1038. doi: 10.1002/j.1460-2075.1994.tb06351.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee B H, Hibino T, Takabe T, Weisbeek P J, Takabe T. J Biochem. 1995;117:1209–1217. doi: 10.1093/oxfordjournals.jbchem.a124846. [DOI] [PubMed] [Google Scholar]
- 25.Harris E H. The Chlamydomonas Sourcebook. San Diego: Academic; 1989. [Google Scholar]
- 26.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 27.Rochaix J-D, Mayfield S, Goldschmidt-Clermont M, Erickson J. In: Plant Molecular Biology: A Practical Approach. Shaw C H, editor. Oxford: IRL; 1988. pp. 253–275. [Google Scholar]
- 28.Picard V, Ersdal-Badju E, Aiqin L, Bock S C. Nucleic Acids Res. 1994;22:2587–2591. doi: 10.1093/nar/22.13.2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kindle K L. Proc Natl Acad Sci USA. 1990;87:1228–1232. doi: 10.1073/pnas.87.3.1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nelson J E, Savereide P B, Lefebvre P A. Mol Cell Biol. 1994;14:4011–4019. doi: 10.1128/mcb.14.6.4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stevens D, Rochaix J D, Purton S. Mol Gen Genet. 1996;251:23–30. doi: 10.1007/BF02174340. [DOI] [PubMed] [Google Scholar]
- 32.Kerfeld C A, Anwar H A, Interrante R, Merchant S, Yeates O T. J Mol Biol. 1995;250:627–647. doi: 10.1006/jmbi.1995.0404. [DOI] [PubMed] [Google Scholar]
- 33.Katoh S, Shiratori I, Takamiya A. J Biochem. 1962;51:32–40. doi: 10.1093/oxfordjournals.jbchem.a127497. [DOI] [PubMed] [Google Scholar]
- 34.Chua N-H, Bennoun P. Proc Natl Acad Sci USA. 1975;72:2175–2179. doi: 10.1073/pnas.72.6.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Porra R J, Thompson W A, Kriedemann P E. Biochim Biophys Acta. 1989;975:384–394. [Google Scholar]
- 36.Laemmli U K. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 37.Franzén L G, Frank G, Zuber H, Rochaix J-D. Plant Mol Biol. 1989;12:463–474. doi: 10.1007/BF00017585. [DOI] [PubMed] [Google Scholar]
- 38.Marcus R A, Sutin N. Biochim Biophys Acta. 1985;811:265–322. [Google Scholar]
- 39.Chou P Y, Fasman G D. Biochemistry. 1974;13:222–245. doi: 10.1021/bi00699a002. [DOI] [PubMed] [Google Scholar]
- 40.Rost B, Sander C. J Mol Biol. 1993;232:584–599. doi: 10.1006/jmbi.1993.1413. [DOI] [PubMed] [Google Scholar]
- 41.Rost B, Sander C. Proteins. 1994;20:216–226. doi: 10.1002/prot.340200303. [DOI] [PubMed] [Google Scholar]