

Summary
Proteins trafficking through the secretory pathway must first exit the endoplasmic reticulum (ER) through membrane vesicles created and regulated by the COPII coat protein complex. Cranio-lenticulo-sutural dysplasia (CLSD) was recently shown to be caused by a missense mutation in SEC23A, a gene encoding one of two paralogous COPII coat proteins. We now elucidate the molecular mechanism underlying this disease. In vitro assays reveal the mutant form of SEC23A poorly recruits the Sec13-Sec31 complex, inhibiting vesicle formation. Surprisingly, this effect is modulated by the Sar1 GTPase paralog used in the reaction, indicating distinct affinities of the two human Sar1 paralogs for the Sec13-Sec31 complex. Patient cells accumulate numerous tubular cargo-containing ER exit sites devoid of observable membrane coat, likely representing an intermediate step in COPII vesicle formation. Our results indicate the Sar1-Sec23-Sec24 prebudding complex is sufficient to form cargo-containing tubules in vivo, whereas the Sec13-Sec31 complex is required for membrane fission.
Introduction
The eukaryotic secretory pathway is critical for maintaining membrane homeostasis and proper protein localization within cells, and is responsible for secretion of extracellular factors necessary for developmental processes. Entrance into the secretory pathway occurs at the endoplasmic reticulum (ER), where proteins destined for the cell surface or other organelles are incorporated into vesicles that bud from ER membranes. The COPII protein coat is involved in the majority and possibly all of these budding events, by providing the means for membrane deformation as well as incorporation of integral membrane cargo into nascent vesicles (Barlowe et al., 1994; Lee et al., 2004).
COPII coat formation is initiated by the small GTPase Sar1, which is recruited to smooth ER membranes by the GTP/GDP exchange factor Sec12 (Barlowe and Schekman, 1993; Nakano and Muramatsu, 1989). GTP-bound Sar1 then recruits the Sec23-Sec24 protein complex to form a “pre-budding” complex capable of engaging cargo via interactions between Sec24 and the cytosolic tails of transmembrane proteins (Aridor et al., 1998; Kuehn et al., 1998; Miller et al., 2003; Mossessova et al., 2003). Finally, the Sec13-Sec31 complex is recruited to promote coat polymerization, membrane curvature, and membrane fission (Aridor et al., 1998; Fath et al., 2007; Salama et al., 1993; Stagg et al., 2006). Sec23 possesses GTPase-activating protein (GAP) activity, stimulated by Sec13-Sec31, catalyzing hydrolysis of Sar1 bound triphosphate (Antonny et al., 2001; Yoshihisa et al., 1993). The resultant Sar1-GDP has a much reduced affinity for the Sec23-Sec24 complex, leading to coat dissociation some time after vesicle budding.
To date, two human diseases have been found to result from disruption of COPII genes. Chylomicron-retention disease (CMRD), a fat malabsorption disease in which enterocytes fail to secrete large lipoprotein particles into the bloodstream, is caused by missense substitutions in SAR1B, one of two paralogous Sar1 proteins in humans (Jones et al., 2003). Cranio-lenticulo-sutural dysplasia (CLSD) is a syndrome characterized by facial dysmorphisms, skeletal defects, late-closing fontanels, and cataracts (Boyadjiev et al., 2003). This developmental disease was shown to arise from a missense substitution (F382L) in SEC23A, one of two Sec23 paralogs in humans, resulting in disturbed ER-to-Golgi trafficking (Boyadjiev et al., 2006). Not surprisingly, nonsense mutations in Sec23 were also found to have serious developmental consequences in worms and fish (Lang et al., 2006; Roberts et al., 2003). It remains unresolved how a missense mutation in a single COPII paralog – SAR1B in the case of CMRD; SEC23A in the case of CLSD – is sufficient to cause disease.
To determine the effect of the perturbation induced by the CLSD mutation, we have purified recombinant F382L-SEC23A and studied its behavior in vitro. We have also performed morphological studies of primary skin fibroblasts isolated from affected patients (Boyadjiev et al., 2006). In this study, we have characterized the precise nature of the CLSD defect. We have established that the F382L substitution interferes with recruitment of the complete COPII protein coat. Furthermore, taking advantage of differing behavior in vitro between the two Sar1 paralogs, we have identified a putative Sec13-Sec31 binding site at the interface of the Sar1-Sec23 complex. In addition to providing the molecular basis for CLSD, this study sheds light upon the interaction between the Sec13-Sec31 complex and the Sar1-Sec23-Sec24 pre-budding complex (now structurally characterized in an accompanying report (Bi et al., 2007)). Utilizing the COPII defect present in CLSD patient cells, we have been able to distinguish the role of the pre-budding complex from that of the Sec13/Sec31 complex. Indeed, the SEC23A-F382L mutation led to a striking proliferation of tubular ER exit sites devoid of observable coat that are nevertheless enriched with COPII cargo.
Results
F382L-SEC23A loss-of-function is dependent upon SAR1 paralog
In our previous work (Boyadjiev et al., 2006), we demonstrated F382L-SEC23A was incapable of supporting ER-derived vesicle formation in vitro. In that work, we utilized SAR1B as the source of Sar1 in the budding reaction. Surprisingly, the other human paralog, SAR1A, partially rescued the in vitro defect exhibited by F382L-SEC23A (Figure 1A). In this assay, ER membranes prepared by digitonin permeabilization of NIH3T3 cells were incubated with purified recombinant human COPII proteins and a small amount of rat liver cytosol (required to supply an unknown activity). After incubation at 30C, vesicles were separated from the donor membranes by differential centrifugation and analyzed by immunoblot. We assessed the presence of the resident ER protein Ribophorin-I as a negative control, and the cargo receptor ERGIC-53 as a model COPII cargo. Successful incorporation of ERGIC-53 into the vesicle fraction was achieved by adding 4 mg/ml rat liver cytosol, which provided COPII activity at this concentration. Alternatively, the reaction was dependent upon addition of purified COPII proteins if only 1 mg/ml rat liver cytosol was used.
Figure 1. F382L-SEC23A is not competent for budding of cargo-containing vesicles in vitro when paired with SAR1B.
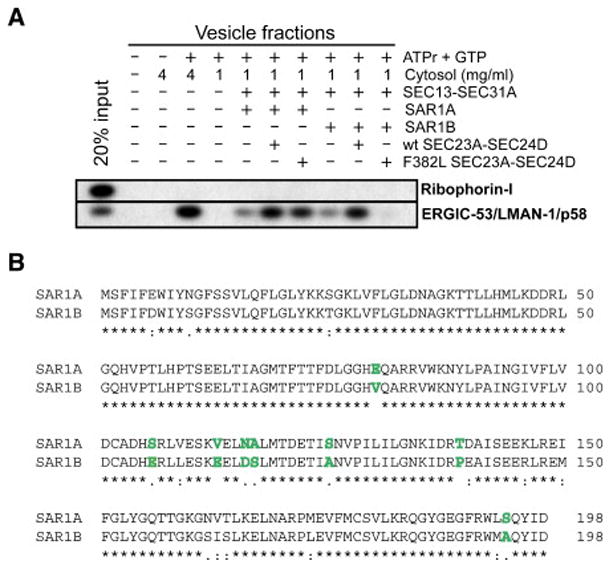
(A) Permeabilized NIH3T3 cells were incubated with an ATP-regenerating system (ATPr), GTP, rat liver cytosol, and purified recombinant human COPII proteins as indicated. Rat liver cytosol served as a source of COPII proteins at 4 mg/ml. At 1 mg/ml cytosol, the reaction was dependent upon the addition of purified human COPII proteins. SAR1A and SAR1B are the two known Sar1 paralogs in humans. Isolated vesicle fractions were subjected to immunoblot analysis to detect the presence of the resident ER protein Ribophorin-I and the COPII cargo protein ERGIC-53/LMAN-1/p58. (B) Protein sequence alignment of human SAR1A and SAR1B. Identical residues are indicated by * and similar residues are indicated by : or . (very similar, or somewhat similar). Those residues differing significantly in charge or character are highlighted in boldface and colored green.
Under these conditions, we compared the activity of F382L-SEC23A and wild-type SEC23A (each in complex with SEC24D) together with either SAR1A or SAR1B. The level of ERGIC-53 found in the vesicle fraction of a reaction containing F382L-SEC23A and SAR1B (2% budding efficiency) was lower than without any added SEC23A, confirming our previously reported results. However, when F382L-SEC23A was instead paired with SAR1A, the amount of ERGIC-53 present in the vesicle fraction (17% budding efficiency) was significantly above the background level (9% budding efficiency), although not as much as with wild-type SEC23A (20% budding efficiency). These results suggest that SAR1A can partially rescue the defect associated with F382L-SEC23A. This effect was not dependent upon the concentration of Sar1 used in the reaction (data not shown and Figure 2D), and therefore seemed instead to depend upon the physical properties of each Sar1 paralog. We also employed the three other human paralogs of Sec24 (SEC24A, SEC24B, and SEC24C) paired with wild-type or F382L-SEC23A, and assayed for the presence of additional cargo proteins in vesicle fractions (APP and Sec22b), with similar results (data not shown). SAR1A and SAR1B share 90% amino acid sequence identity (Figure 1B), making the difference in activity when paired with F382L-SEC23A all the more striking.
Figure 2. F382L-SEC23A failure to recruit SEC13-SEC31A to membranes is the cause of the budding defect.
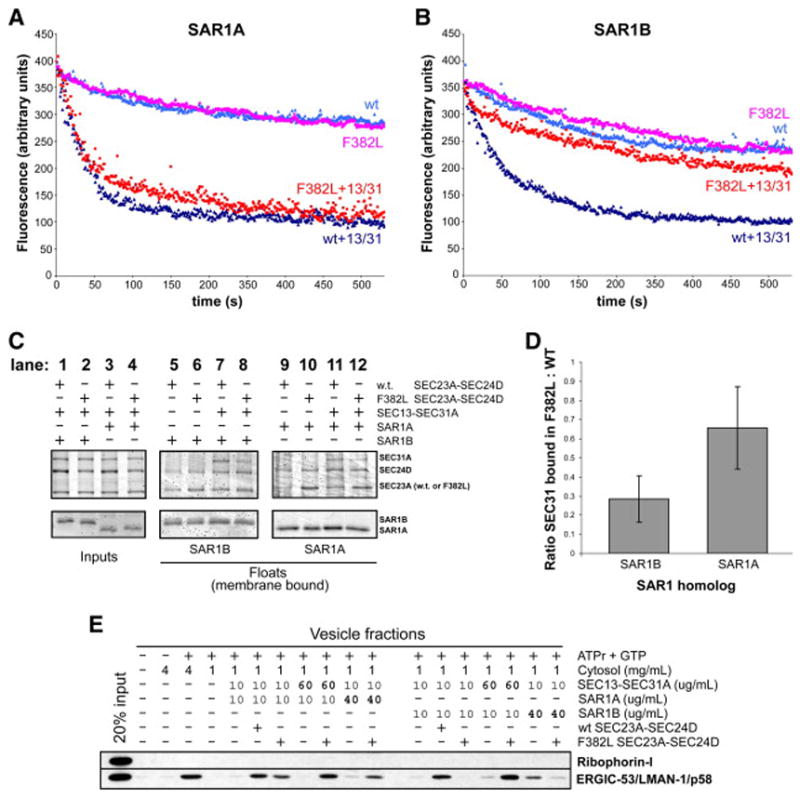
(A,B) GTPase-activity assay using synthetic liposomes, performed at 37C essentially as described (Antonny et al., 2001; Futai et al., 2004). The tryptophan fluorescence signal represents the conformational state of Sar1, owing to GTP or GDP binding. Starting with a sample containing synthetic liposomes and GTP-loaded SAR1A (A) or SAR1B (B), wt or F382L-SEC23A-SEC24D was added and the subsequent decrease in signal over time represents GTP hydrolysis. Where indicated, SEC13-SEC31A was added concurrently with wt or F382L-SEC23A-SEC24D. No cytosol was included in these reactions. (C) Liposome binding assay, performed essentially as described (Kim et al., 2005; Matsuoka et al., 1998). The indicated COPII proteins were incubated with synthetic liposomes at 37C for 20 min and then subjected to flotation on a sucrose density gradient. No cytosol was added to these reactions. Input and float fractions were analyzed by 12% PAGE and stained with SYPRO-Red (Invitrogen). (D) Quantitation of liposome binding experiments, showing the ratio of SEC31A recruited to membranes in the presence of F382L-SEC23A versus wild-type SEC23A, with SAR1A or SAR1B. Error bars are shown for 95% confidence levels calculated from five independent experiments. (D) In vitro vesicle budding assay, similar to Figure 1a. Vesicle fractions from reactions with SAR1A are on the left, and those from reactions with SAR1B are on the right. Amounts of SEC13-SEC31A, SAR1A, and SAR1B added to reactions are indicated.
The F382L substitution interferes with recruitment of Sec13-Sec31 to the membrane
To better understand the nature of the vesicle formation defect associated with the CLSD mutation, we tested the GTPase-activating protein (GAP) activity of F382L-SEC23A compared to wild-type SEC23A using a tryptophan fluorescence-based assay performed with pure proteins (Antonny et al., 2001; Futai et al., 2004). In this assay the nucleotide-bound state of Sar1 is monitored by relative fluorescence measurements, because Sar1-GTP tryptophan fluorescence is higher than that of Sar1-GDP due to distinct conformational states adopted in each case. Starting with a mixture of synthetic liposomes and GTP-loaded SAR1A or SAR1B, addition of wild-type or F382L-SEC23A-SEC24D resulted in similarly modest rates of hydrolysis of SAR1-GTP to SAR1-GDP, as judged by a corresponding decrease in fluorescence (Figures 2A and 2B).
It is known from studies with yeast proteins that the Sec13-Sec31 complex stimulates the GAP activity of Sec23-Sec24 on Sar1 (Antonny et al., 2001). We were curious to see the effect of human SEC13-SEC31A on the GAP activity of F382L-SEC23A. Surprisingly, we saw very different effects depending on which SAR1 paralog was used in the assay. In reactions with SAR1A, addition of SEC13-SEC31A stimulated the GAP activity of wt and F382L-SEC23A-SEC24D equally well (Figure 2A; wt was stimulated from 8.5×10−4 s−1 to 4.5×10−3 s−1; F382L was stimulated from 8.4×10 −4 to 4.8×10−3 s−1). In contrast, in reactions with SAR1B, SEC13-SEC31A failed to stimulate the GAP activity of F382L-SEC23A-SEC24D to the same extent as wild-type (Figure 2B; wt was stimulated from 8.6×10−4 s−1 to 3.0×10−3 s−1; F382L was stimulated from 5.1×10 −4 to 1.1×10−3 s−1). Therefore, the stimulation of SEC23A GAP activity by SEC13-SEC31A is noticeably affected by the F382L substitution when paired with SAR1B.
We have previously shown that SEC31A exhibited an altered localization in CLSD patient cells, as observed by immunofluorescence microscopy (Boyadjiev et al., 2006). We therefore considered the possibility that F382L-SEC23A is unable to recruit the Sec13-Sec31 complex necessary for COPII coat polymerization. To test this hypothesis, we performed liposome floatation assays (Kim et al., 2005; Matsuoka et al., 1998) to measure the amount of Sec13-Sec31 complex bound to membranes in the presence of F382L-SEC23A. Purified recombinant human COPII proteins were incubated with synthetic liposomes and the non-hydrolyzable GTP analog GTPγS at 37C for 20 min and applied to the bottom of a sucrose density gradient. After a brief high-speed centrifugation step, floated liposomes were isolated from the top of the sucrose gradient and the COPII proteins present in the liposome fraction were analyzed by SDS-PAGE (Figure 2B). Our previous study demonstrated the F382L-SEC23A-SEC24D complex was competent to bind membranes in the presence of SAR1B (Boyadjiev et al., 2006). Here we monitored SEC13-SEC31A binding to wild-type or F382L-SEC23A-SEC24D in the presence of SAR1A or SAR1B. In the presence of SAR1B, there was a significant decrease in the amount of SEC31A bound in reactions with F382L-SEC23A compared to wild-type (lanes 7 and 8, Figure 2B). This result suggests that the F382L substitution interferes with recruitment of the SEC13-SEC31A complex to the SAR1B-SEC23A-SEC24D membrane-bound complex. A similar effect was seen if SAR1A was included in the reaction instead of SAR1B (lanes 11 and 12, Figure 2B). In addition, the mutant SEC23A-SEC24D complex was sometimes seen to be recruited to membranes more robustly than the wild-type complex.
In order to more clearly examine the differences observed between SAR1A and SAR1B binding reactions, we compared the amounts of SEC31A in the membrane fractions (normalized to the amount of bound SEC23A) as a ratio of the quantity bound to F382L-SEC23A-SEC24D versus wild-type SEC23A-SEC24D (Figure 2C) in five independent experiments. Contrasting this ratio between SAR1A and SAR1B binding reactions, it appeared that SAR1A partially mitigated the SEC13-SEC31A recruitment defect exhibited by F382L-SEC23A with SAR1B. It should be noted that the amount of SEC31A bound in a reaction with SAR1A and F382L-SEC23A was still below that of wild-type SEC23A. These binding data correlate with the results of the in vitro vesicle formation and GAP assays, suggesting inhibition of Sec13-Sec31 binding is responsible for the observed in vitro budding defect, although the difference between SAR1A and SAR1B in the vesicle formation reaction and GAP assays is more dramatic than in the liposome binding assay.
In order to determine if the observed reduction in SEC13-SEC31A recruitment to membrane-bound F382L-SEC23A-SEC24D was the cause of the vesicle budding defect, we returned to the in vitro vesicle formation assay. We reasoned that addition of excess SEC13-SEC31A to the vesicle formation assay might overcome, by mass action, the diminished F382L-SEC23A activity. When 60 μg/ml SEC13-SEC31A (6-fold more than used previously) was added to a reaction containing SAR1B and F382L-SEC23A, the amount of ERGIC-53 incorporated into the vesicle fraction was restored to the level seen in reactions with wild-type SEC23A (Figure 2D, right half). This larger amount of SEC13-SEC31A did not support budding of ERGIC-53-containing vesicles in the absence of SEC23A-SEC24D, indicating the restored activity seen with F382L-SEC23A was due to a mass action effect. Similar results were also observed in reactions utilizing different Sec24 paralogs and detecting other cargo proteins (data not shown). Addition of excess SAR1B to the reaction did not increase the amount of ERGIC-53 found in the vesicle fraction, and together with F382L-SEC23A actually suppressed the background level. This result might be explained as a titration of the small amount of wild-type COPII proteins present in the cytosol added to the reactions.
The same complementation phenomenon was observed with SAR1A (Figure 2D, left half), in which the level of ERGIC-53 found in the vesicle fraction of the F382L-SEC23A reaction with excess SEC13-SEC31A was restored to that seen with wild-type SEC23A. Furthermore, in the GAP activity assay, addition of several-fold excess SEC13-SEC31A over F382L-SEC23A-SEC24D resulted in stimulation of GAP activity to the same extent as with wild-type SEC23A-SEC24D (data not shown). Taken together with the liposome binding and immunocytochemistry results, the results from these functional assays establish that the observed F382L-SEC23A vesicle formation defect arises from a reduced affinity for the Sec13-Sec31 complex.
Calvarial osteoblasts express relatively low levels of SEC23B
We sought to understand why only certain tissues seem to be affected in CLSD patients, despite the essential role of Sec23 in the secretory pathway. Many physiological processes requiring an intact secretory pathway, such as digestion and insulin signaling are seemingly unaffected by the F382L substitution in SEC23A. One explanation is that the reduced activity of the CLSD mutant SEC23A protein may be sufficient for COPII coat assembly in most cell types, but is problematic in tissues with higher secretory burdens. In addition, based on our in vitro data, a high level of SAR1B in comparison to SAR1A might exacerbate the defect in certain cell types. Another possibility is that the paralogous SEC23B protein might provide Sec23 function to complement the defect. Using antibodies we developed to discriminate between SEC23A and SEC23B, and taking advantage of the gel-mobility difference between SAR1A and SAR1B, we examined the relative expression of these proteins in normal primary calvarial osteoblasts, in comparison to other cultured cell types. Calvarial osteoblasts are primarily responsible for ossification of the cranial suture, an event that is almost absent in CLSD patients (Boyadjiev et al., 2003). Strikingly, these cells express very little SEC23B (Figure 3A, lane 6), suggesting that SEC23B is unavailable to functionally compensate for F382L-SEC23A in calvarial osteoblasts, but may do so in phenotypically normal tissues of CLSD patients. In order to address this possibility, we performed immunoblot analysis on several normal human tissues (Figure 3B). Of the five tissues tested, all five express detectable levels of SEC23B. Although this is not an exhaustive analysis, it supports the notion that the tissues affected in CLSD patients are those lacking sufficient SEC23B expression.
Figure 3. Calvarial osteoblasts express little SEC23B.
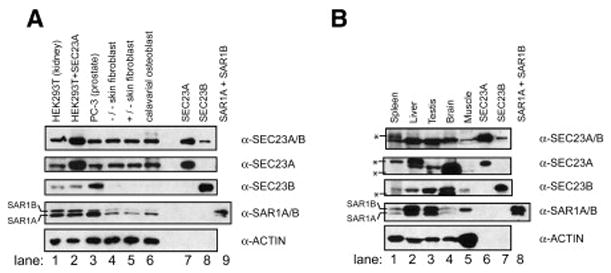
Post-nuclear cultured cell lysates (A) or normal human tissue lysates (B) were analyzed by immunoblot, using antibodies specific for either SEC23A or SEC23B, in addition to an antibody recognizing both human Sec23 paralogs (α-SEC23A/B), an antibody that recognizes both human SAR1 paralogs (α-SAR1A/B), and an antibody for actin as a loading control. (A) Cultured cells used were HEK293T, HEK293T transfected with a plasmid to overexpress SEC23A, PC-3, homozygous CLSD patient skin fibroblasts, heterozygous CLSD patient skin fibroblasts, and primary calavarial osteoblasts. On the right side, purified human proteins (20 ng SEC23A, 20 ng SEC23B, and 400 pg SAR1A + 400 pg SAR1B) were loaded to demonstrate antibody specificity. (B) Similar immunoblot of normal human tissue lysates (35 μg each lane, except for liver, 70 μg), as well as 3 ng SEC23A, 3 ng SEC23B, and 250pg SAR1A + 250 pg SAR1B. Asterisks denote species that we believe to be non-specific cross-reacting proteins, although spleen may contain a slower-migrating modified form of SEC23A (visible in anti-SEC23A/B and anti-SEC23A treatments). As expected, muscle cells have a very high actin:COPII ratio.
The antibody used to recognize SAR1A and SAR1B displays much higher avidity to SAR1B, judging by the signals obtained when equal amounts of recombinant SAR1A and SAR1B proteins were probed simultaneously (Figure 3, lane 9). Therefore, the level of SAR1A is much higher than SAR1B in calvarial osteoblasts (Figure 3, lane 6). Taken together, these results suggest that low SAR1A expression in calvarial osteoblasts is not the source of the cranial suture phenotype in CLSD patients, but instead the cause is likely the low level of SEC23B expressed in this cell type. It remains possible that low SAR1A expression in certain tissues may contribute to other developmental problems associated with CLSD.
CLSD patient fibroblasts accumulate tubular profiles lacking an observable coat
In order to analyze the cellular phenotype caused by the F382L-SEC23A substitution, we examined CLSD patient skin fibroblasts by thin section electron microscopy, and discovered a surprising abundance of tubular extensions projecting from distended peripheral ER compartments (Figure 4A), which were virtually absent in micrographs of heterozygote control cells. In some homozygous mutant cells we observed a very large number of tubulo-vesicular elements within a small area of the ER (Figure S1), an occurrence never observed in heterozygote or wild-type control cells. In comparison to peripheral budding profiles in wild-type (data not shown) and phenotypically normal heterozygote cells (Figures 4B, 4C, and 4D), the tubular extensions observed in homozygote mutant cells were virtually devoid of an observable coat on the cytosolic membrane surface (Figure 4E and Table 1). The same phenomenon was also observed at transitional ER sites proximal to the cis-Golgi complex (Figure S2 and Table 1). In rare instances, a cytosolic coat could be observed on ER budding profiles in homozygote mutant cells. In such cases, the coat was observed at the tip of tubules connected to the ER (Figure 4F) or on simple budding profiles (Figure 4G).
Figure 4. Peripheral ER exit sites are numerous in CLSD patient cells.

(A) Thin section electron micrograph of a homozygote mutant cell in the region of ER. Note the many tubular protrusions (arrows) projecting from swollen ER compartments. (B–D) ER exit sites in heterozygous (unaffected) cells showing the COPII coat on the cytosolic membrane surface of tubules. Budding profiles assumed a variety of shapes, including constricted tubules. Note also the thin morphology of the ER compartments from which the tubules project. (E) Typical ER exit site in a homozygous mutant (affected) cell, in which the tubules exhibited no obvious membrane coat. In ~10% of cases, coated surfaces were observed on tubular ER exit sites in homozygous mutant cells (see Table 1), but then only at the tip of a tubule (F) or on simple budding profiles (G).
Table 1.
Quantitation of coated budding profiles in thin-section electron micrographs.
| Budding profiles in peripheral ER exit sites | |||
|---|---|---|---|
| Cell type | Total budding profiles examined | Percentage of coated profiles | Images analyzed |
| Homozygous mutant (affected) | 477 | 10 % | 40 |
| Heterozygous (unaffected) | 22 | 100 % | 20 |
| Unrelated wild-type | 23 | 100 % | 20 |
| Budding profiles in ER exit sites near the cis-Golgi (transitional elements) | |||
| Cell type | Total budding profiles examined | Percentage of coated profiles | Transitional elements analyzed |
| Homozygous mutant (affected) | 72 | 11 % | 30 |
| Heterozygous (unaffected) | 44 | 88 % | 30 |
| Unrelated wild-type | 37 | 100 % | 30 |
We performed quantitative immunocytochemistry on primary skin fibroblasts isolated from homozygous (F382L/F382L) CLSD patients, to determine if a reduction of Sec13/Sec31 binding to ER exit sites (ERES) could be observed in vivo. We treated thin sections with anti-SEC31A antibody and gold-labeled secondary antibody, and counted the number of gold particles found per tubular profile. In the mutant cells there was a significant reduction in the number of SEC31A-positive tubules observed compared to the heterozygote and wild-type controls (Figure S3). Therefore, the F382L substitution appears to impair Sec13-Sec31 recruitment to ER membranes in vivo as well as in vitro. The lack of an observable coat in mutant cells is consistent with the reduced SEC31A immunogold labeling of ER exit sites in these cells. In contrast, when we performed immunogold analysis with anti-Sar1 antibody, we observed a marked increase in Sar1 labeling at ERES in patient cells compared to unaffected heterozygotes (Figure 5). The F382L substitution therefore results in accumulation of Sar1 at ERES in vivo.
Figure 5. Sar1 is enriched at ER exit sites in CLSD patient cells.

Thin sections were analyzed by immuno-electron microscopy using anti-Sar1 antibody and gold labeled secondary antibody. The average number of gold particles per tubular-vesicular profile is given with standard errors. The number of images evaluated is in parentheses. * p<0.001 for homozygote versus heterozygote and unrelated wild type (Student t-Test).
Sorting of COPII cargo into ER exit sites is unaffected in CLSD patient fibroblasts
We were curious to observe the steady state localization of COPII cargo in patient cells, given the marked proliferation of uncoated ER exit sites. We therefore examined cells stained with a monoclonal antibody for ERGIC-53/LMAN-1/p58, a secretory cargo receptor that cycles between the ER and cis-Golgi compartments (Nichols et al., 1998; Saraste and Svensson, 1991; Schindler et al., 1993). ERGIC-53 staining is thought to represent ER exit sites, the ERGIC, the cis-Golgi, and vesicular-tubular clusters (VTCs) transiting to the cis-Golgi (Ben-Tekaya et al., 2005; Presley et al., 1997; Scales et al., 1997). We noticed a significant accumulation in homozygous mutant cells of ERGIC-53-positive punctae in the peri-nuclear region and periphery of cells (Figures 6A and 6C). These punctae were more numerous and more intensely stained than the more typical ERGIC-53-containing structures found in control cells (Figures 6B and 6D). Co-staining with an antibody for the lumenal ER marker PDI revealed ERGIC-53 punctae localized proximally to PDI-filled ER distensions (Figures 6E and S4). Closer examination revealed that ERGIC-53 is present in dot-like structures adjacent to, but distinct from, swollen ER cisternae containing PDI (Figures 6F, 6G, 6H, and S4). These structures very likely correspond to the numerous tubular ER exit sites we observed by thin-section electron microscopy (Figure 4).
Figure 6. Sorting of the COPII cargo ERGIC-53 into ER exit sites is unaffected in CLSD patient cells.
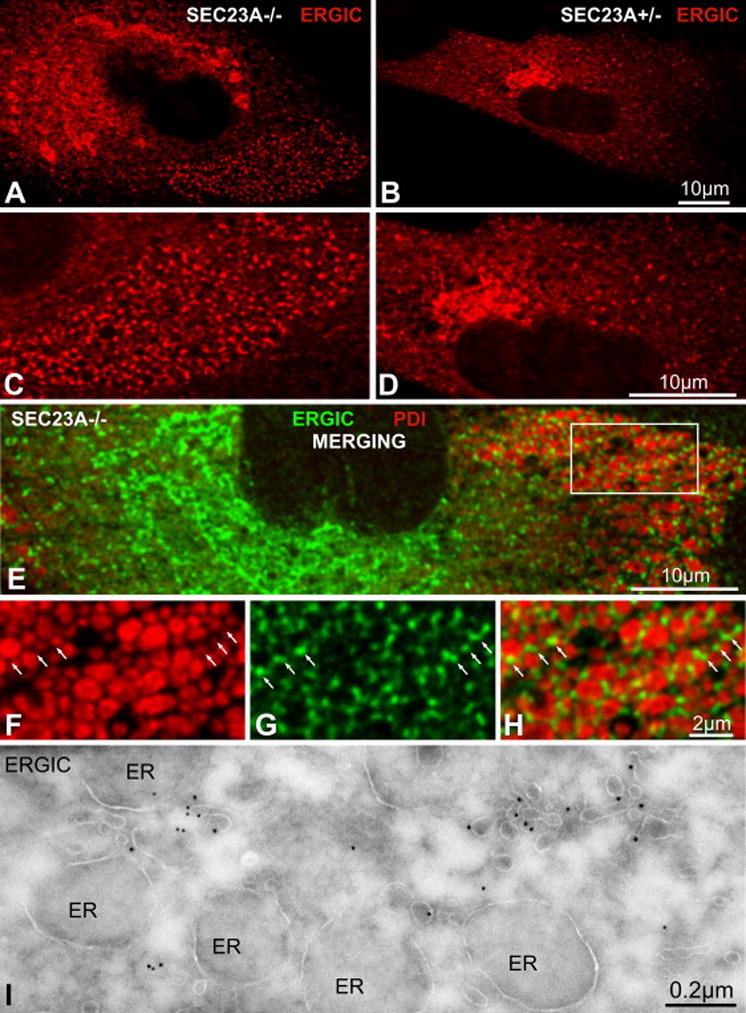
Confocal immunofluorescence images of anti-ERGIC-53 staining in primary skin fibroblasts from affected homozygotes (A, C) and unaffected heterozygotes (B, D). C and D are magnifications of A and B, respectively. (E) Confocal immunofluorescence image of anti-ERGIC-53 (green) and anti-PDI (red) staining of an affected homozygote cell. (F–H) Magnification of the framed area in E. The PDI-positive structures are distended ER compartments. The ERGIC-53-positive structures are located proximally to, but not co-localized with, the ER distensions, and correspond to the tubular ER exit sites seen in Figure 4. (I) Thin sections were analyzed by immuno-electron microscopy using an anti-ERGIC-53 antibody and gold-coupled secondary antibody. Gold particles concentrate in tubular ER exit sites and are virtually absent in distended ER cisternae.
Seeking a higher resolution view, we next performed anti-ERGIC-53 immunocytochemistry on fixed thin sections of CLSD patient skin fibroblasts. Tubular ER protrusions were specifically decorated with gold particles, and labeling was virtually absent from dilated ER cisternae, indicating enrichment of ERGIC-53 at ER-exit sites (Figure 6I). This result confirms the peripheral ERGIC-53 punctae we observed by immunofluorescence correspond to ER exit sites. We have therefore established that CLSD patient cells accumulate COPII cargo at steady state, and membrane protein cargo is specifically localized to numerous tubular ER exit sites virtually devoid of observable cytoplasmic coat. These structures may represent an intermediate step in COPII carrier formation, occurring after membrane protein cargo capture but prior to completion of coat assembly.
Discussion
The mechanisms behind genetic diseases are usually quite complex, involving misfunction or dysregulation of several genes. Even for those diseases in which only a single gene is thought to be involved, such as CLSD, understanding how genotype leads to phenotype can be problematic when the gene involved is poorly understood. Here we have been able to ascertain the molecular basis for CLSD, based on in vitro analysis of the effects of the F382L substitution and morphological examination of cultured primary cells from patients. Furthermore, our work has uncovered a remarkable cellular phenotype shedding light on the role of the pre-budding complex in the formation of ER exit sites.
Basis of the CLSD cellular phenotype
In order to understand the root cause of the ER export defect observed in CLSD patient cells, we compared F382L-SEC23A to wild-type SEC23A in several in vitro assays. F382L-SEC23A is able to bind to synthetic membranes at a level equivalent to wild-type and retains intrinsic GAP activity similar to wild-type protein (Figure 2). However, F382L-SEC23A shows reduced recruitment of the Sec13-Sec31 outer coat complex both in vitro and in vivo (Figures 2 and S3), especially when paired with SAR1B (Figures 1A, 2B, and 2D). We found that the in vitro budding defect observed with F382L-SEC23A can be ameliorated by addition of more SEC13-SEC31A complex. Taken together, these results establish that weak Sec13-Sec31 binding to F382L-SEC23A is the cause of the budding defect and, by extension, of the cellular CLSD phenotype.
CLSD is characterized by open calvarial sutures with large, late-closing fontanels (the “soft-spot” on the skull found on newborns), sutural cataracts, facial dysmorphisms, and skeletal defects. To begin to address why certain tissues are affected in CLSD patients, we looked at the levels of SEC23A and SEC23B in primary calvarial osteoblasts (Figure 3). These cells, found on the skullcap, are responsible for membraneous ossification of the fontanels. We observed a very low level of SEC23B in calvarial osteoblast lysates, suggesting these cells are specifically affected because of insufficient SEC23B protein available to provide normal Sec23 function. Conversely, unaffected tissues in CLSD patients might retain normal function through expression of sufficient SEC23B to complement the loss of SEC23A function. Analysis of several normal human tissues showed detectable levels of SEC23B, providing evidence in support of this idea. The primary skin fibroblasts from CLSD patients we examined also expressed low levels of SEC23B, explaining the morphological ER defects we observe in these cells. It should be noted that there is no obvious skin defect associated with CLSD, so it seems skin fibroblasts remain functional despite an altered ER morphology in CLSD patients.
COPII function at ER exit sites
One might have expected to observe a complete absence of ER exit sites in CLSD patient cells, because of impaired COPII function stemming from mutant SEC23A. Our biochemical analysis revealed that the main effect of this mutation is to limit the recruitment of the Sec13-Sec31 complex by the Sar1-Sec23-Sec24 pre-budding complex, allowing us to separate the roles of these two protein complexes in the formation of ER exit sites. We have observed a proliferation of Sar1-enriched tubular ER exit sites protruding from distended ER compartments (Figures 4, 5, and S1), and the COPII cargo ERGIC-53 is being properly sorted to these ER exit sites (Figures 6 and S4). These results suggest that the pre-budding complex is sufficient to generate tubular extensions at ER exit sites and to sort COPII cargo. Accordingly, the role of the Sec13-Sec31 complex is to fragment these tubular extensions into transport vesicles.
This interpretation is compatible with previous reports that Sar1 is able to tubulate ER membranes and to sort cargo in the absence of COPII function. This mechanism was proposed as the result of a study utilizing a permeabilized cell system, in which addition of a GTP-locked mutant (H79G) form of Sar1 led to the formation of extended ER tubules enriched in COPII cargo and visible by light microscopy (Aridor et al., 2001). These structures formed without addition of other COPII proteins, but were capable of recruiting Sec23 from cytosol added subsequently. To our knowledge, no similar structures (produced by Sar1 without contribution from other COPII proteins) have been observed in vivo. In contrast, based upon our biochemical data demonstrating that F382L-SEC23A is efficiently recruited to membranes by Sar1, it is likely that the tubules we observe in CLSD patient cells are decorated by Sar1-F382L-SEC23A-Sec24 pre-budding complexes. This would be expected given the role of the Sec13-Sec31 complex in stimulating GTP hydrolysis of Sar1-GTP, a prerequisite for coat dissociation from the membrane (Antonny et al., 2001). Our observation of increased Sar1 labeling at ERES in patient cells (Figure 5) is consistent with this notion. It is possible that these complexes are not large enough or are too dispersed to be observed as a coat by electron microscopy (Figures 4 and S2). In either case, the failure to recruit Sec13-Sec31 (Figure S3) likely accounts for the lack of observable membrane coat. Our results therefore provide insight into the mechanisms underlying ER tubulation and vesiculation in vivo.
Implications for COPII coat protein assembly
The F382L-substitution, although occurring at a conserved position in primary sequence, is subtle, maintaining the hydrophobic character of the original residue. The human F382L-SEC23A protein retains many aspects of wild-type function, including Sec24 binding, membrane binding, and intrinsic GAP activity (Figure 2). We found that the equivalent substitution in yeast Sec23p (F380L) had no obvious functional consequence (supplementary results and Figure S5), underlining the subtle nature of this change. Accordingly, the structural perturbation induced by the F382L substitution in human SEC23A appears to be quite local in nature, as the only functional consequences relate to Sec13-Sec31 binding. This suggests that F382 should occupy, or be very near to, a Sec13-Sec31 binding site on Sec23. Furthermore, the effect of the F382L substitution on Sec13-Sec31 binding is modulated by the identity of the Sar1 paralog (Figures 1 and 2), presumably because of higher affinity of SEC13-SEC31A for SAR1A than for SAR1B. Thus, Sec13-Sec31 may bind to Sar1 at a site comprising at least one residue that is distinct between SAR1A and SAR1B. A new crystal structure of the complex between yeast Sar1p, Sec23p, and a fragment of Sec31p (Bi et al., 2007) corroborates both of these predictions arising from our biochemical data. F382 is not itself surface exposed, but lies buried just beneath the surface of Sec23 as part of a surface-loop. The Sec31 fragment makes direct contact with this loop, and additional contacts are made between Sec31 and Sar1 in the vicinity of several residues whose identity differs between SAR1A and SAR1B, specifically residues equivalent to positions 80, 113, 116, and 117 in human SAR1 (Figure 7).
Figure 7. The Sec31 binding site is proximal to the CLSD mutation and residues of Sar1 that differ between homologs.
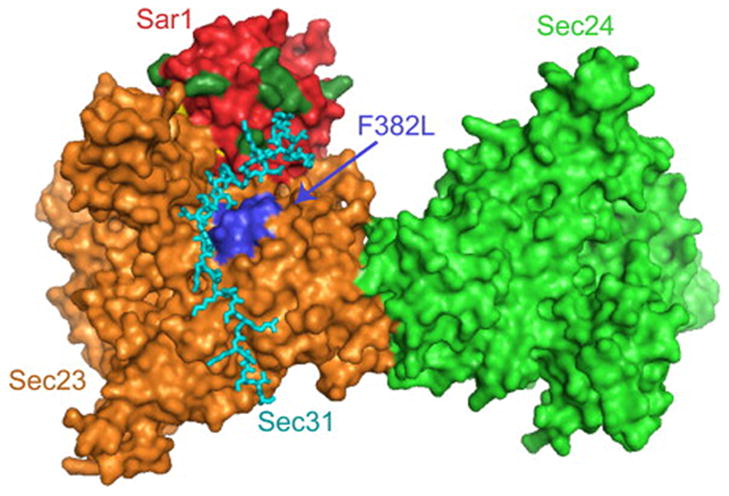
Atomic structure of a fragment of yeast Sec31 (cyan) in complex with Sar1p (red) and Sec23p (orange) (Bi et al., 2007), with Sec24p (light green) included based on the structure of the Sar1p-Sec23p-Sec24p pre-budding complex (Bi et al., 2002). The membrane binding surface of this complex is thought to lie underneath this perspective. The position of the yeast residue equivalent to human F382 (F380) is indicated by blue coloring of several residues surrounding position 380 in primary sequence, and by the label “F382L”. The eight residues of human Sar1 that are significantly different in chemical composition between SAR1A and SAR1B (as shown in Figure 1B) are highlighted in dark green. All eight of these residues lie on the globular surface of Sar1, and six of them are on the same face of the complex as is F380. In particular, residues corresponding to positions 80, 113, 116, and 117 in human Sar1 appear capable of making direct contact with Sec31.
Our suggestion that SEC13-SEC31A has a higher affinity for SAR1A than for SAR1B has further implications for COPII vesicle formation. Within the cellular milieu, the repertoire of COPII paralogs available for coat polymerization should dictate the nature of the vesicles budding from the endoplasmic reticulum. It has been assumed that in different tissues, differential expression of COPII paralogs would confer distinct properties to vesicles according to the requirements of ER export, most simply in the case of distinct cargos selected by Sec24 paralogs (Miller et al., 2002; Tang et al., 1999; Wendeler et al., 2007). Here we have provided the first evidence for distinct affinities between COPII subunit paralogs, suggesting that variation of Sar1 paralog expression is another means of regulation.
Human genetic evidence has implicated SAR1B in formation of ER-derived carriers larger than a typical 60–70 nm COPII vesicle (Jones et al., 2003). Accordingly, we are tempted to speculate on a possible functional role for the weaker affinity of SAR1B for SEC13-SEC31A in the formation of larger COPII vesicles. Perhaps the Sec13-Sec31 cage (Fath et al., 2007; Stagg et al., 2006) is less constrained when in complex with SAR1B. This added flexibility might allow Sec13-Sec31 to assemble into larger caged structures consistent with larger vesicles.
Experimental Procedures
Antibodies and cell culture
Anti-Ribophorin I serum was provided by Peter Walter. Anti-SEC23A/B was provided by Jean-Pierre Paccaud (Pagano et al., 1999). Anti-Sec22b was provided by Jesse Hay (Hay et al., 1996). Rabbit anti-SEC31 was provided by A. Hubbard (Johns Hopkins University). Monoclonal anti-PDI was a gift from S. Fuller (EMBL-Heidelberg). Anti-ERGIC-53/LMAN-1/p58 antiserum used for immunoblots was raised in rabbits injected with a purified GST fusion to residues 49–484 of rat ERGIC-53/LMAN-1/p58 produced in E. coli. Monoclonal anti-ERGIC-53/LMAN-1/p58 antibody was purchased from Alexis Biochemicals. Anti-SAR1A/B antiserum was raised in rabbits injected with a mixture of purified human SAR1A and SAR1B. Anti-SEC23A antiserum was raised in rabbits injected with the peptide (CG)-SKVPLTQATRGPQVQQPPPSNR conjugated to rabbit serum albumin. Anti-SEC23B antiserum was raised in rabbits injected with the peptide (CG)- TKPAMPMQQARPAQPQEHPFASSR conjugated to rabbit serum albumin. Both of these peptide-derived antisera were purified using peptide-conjugated Sulfolink beads (Pierce).
Cell lines were cultured and transfected using standard procedures. Patient fibroblasts were cultured as described (Boyadjiev et al., 2006). The calvarial osteoblast cell lysate was obtained from ScienCell Research Laboratories. The tissue lysates were purchased from Clontech (“Protein Medleys”).
Immunofluorescence and electron microscopy
Fibroblasts plated on glass coverslips were fixed with 4% PAF for 30 min and permeabilized with methanol at −20°C for 3 min. Primary antibodies used were polyclonal anti-ERGIC-53 (diluted 1:400), polyclonal anti-SEC31 (diluted 1:200), monoclonal anti-ERGIC-53 (diluted 1:4000) and monoclonal anti-PDI (diluted 1:20). Secondary antibodies used were Alexa Fluor 488 goat anti-rabbit IgG, and Alexa Fluor 546 goat anti-mouse IgG (diluted 1:400, Molecular Probes/Invitrogen, Eugene, OR). After staining cells with the appropriate primary and secondary antibodies, images were visualized and captured with a Zeiss LSM 510 confocal microscope (Carl Zeiss, Feldbach, Switzerland).
For electron microscopy analysis, we fixed confluent fibroblast cell cultures with 2% glutaraldehyde in 0.1M phosphate buffer and either gently scraped before postfixation with osmium tetroxide, dehydration and embedding, or processed in situ. En bloc staining was performed with uranyl acetate (Tandler, 1990). For immunoelectron microscopy, we fixed cells with 2.5% glutaraldehyde and processed for cryo-ultramicrotomy as described (Volchuk et al., 2000). Ultrathin frozen sections were prepared and incubated for immunolabeling as described (Liou et al., 1996).
Protein expression and purification
Human SAR1A and SAR1B proteins were overexpressed in E. coli and purified as cleaved GST-fusions as described for hamster Sar1a (Kim et al., 2005). SEC23A-hisSEC24D, F382L-SEC23A-hisSEC24D, and SEC13-hisSEC31A were purified using immobilized metal affinity chromatography from baculovirus-infected insect cells as described (Boyadjiev et al., 2006; Kim et al., 2005).
In vitro vesicle formation assay
The in vitro vesicle budding assay was performed essentially as described (Kim et al., 2005) in B880 buffer (composition) using semi-intact cells permeabilized with digitonin (Plutner et al., 1992; Wilson et al., 1995) and washed with B880 buffer containing 1M KOAc as a donor ER membrane source.
GTPase activity assay
The tryptophan fluorescence GTPase activity assay was performed at 37C as described for yeast COPII proteins (Antonny et al., 2001; Futai et al., 2004), using major-minor mix synthetic liposomes containing 10% cholesterol, in a stirred-cell cuvette. To 0.3 mM liposomes in HKM buffer (20 mM HEPES pH 7.2, 150 mM KOAc, 1 mM Mg(OAc)2), we added SAR1A or SAR1B to a final concentration of 1.33 μM. One min later, GTP was added to 30 μM. After spontaneous exchange of GDP for GTP was complete (another 10 to 15 min), wild-type or F382L-SEC23A-SEC24D complex was added to 250 nM. Where indicated, SEC13-SEC31A complex was added to 400 nM simultaneously with wild-type or F382L-SEC23A-SEC24D.
Liposome binding assay
The liposome binding assay was performed as described for yeast COPII proteins (Kim et al., 2005; Matsuoka et al., 1998) using 10% cholesterol major-minor mix liposomes. Following a 20 min incubation at 37C, the protein-liposome mixture was separated by flotation through a sucrose density step gradient (in HKM buffer), achieved by centrifugation at 391,000 g for 25 min at 22C.
Supplementary Material
Acknowledgments
We thank Jonathan Goldberg and Xiping Bi for sharing their data prior to publication. We are grateful to Jesse Hay, Hans-Peter Hauri, Jean-Pierre Paccaud, Jaakko Saraste, and Peter Walter for generous gifts of reagents. We acknowledge the important contributions of Jinoh Kim and Bertrand Kleizen in development of the in vitro vesicle formation assay. We thank Chris Nauta and Ann Fischer for assistance with cell culture, and Robyn Barfield, Trevor Starr, and Chao-Wen Wang for valuable advice with yeast assays. We are grateful to Ariane Widmer and Ebrahim Malek for technical assistance and to Nadine Dupont for image processing. The Pole Facultaire de Microscopie Ultrastructurale at the University of Geneva Medical School kindly provided access to electron microscopy equipment. We thank Adam Schindler for helpful discussions.
J.C.F. was supported by a fellowship from the Miller Institute for Basic Research. S.A.B. was partially funded through a Children’s Miracle Network Endowed Chair and through grants K23 DE00462, R03 DE016342, and R01 DE016886 from NIDCD/NIH and M01-RR00052 from NCRR/NIH. L.O. and P.C. were supported by the Swiss National Science Foundation. R.S. is supported by the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Antonny B, Madden D, Hamamoto S, Orci L, Schekman R. Dynamics of the COPII coat with GTP and stable analogues. Nat Cell Biol. 2001;3:531–537. doi: 10.1038/35078500. [DOI] [PubMed] [Google Scholar]
- Aridor M, Fish KN, Bannykh S, Weissman J, Roberts TH, Lippincott-Schwartz J, Balch WE. The Sar1 GTPase coordinates biosynthetic cargo selection with endoplasmic reticulum export site assembly. J Cell Biol. 2001;152:213–229. doi: 10.1083/jcb.152.1.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aridor M, Weissman J, Bannykh S, Nuoffer C, Balch WE. Cargo selection by the COPII budding machinery during export from the ER. J Cell Biol. 1998;141:61–70. doi: 10.1083/jcb.141.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlowe C, Orci L, Yeung T, Hosobuchi M, Hamamoto S, Salama N, Rexach MF, Ravazzola M, Amherdt M, Schekman R. COPII: a membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell. 1994;77:895–907. doi: 10.1016/0092-8674(94)90138-4. [DOI] [PubMed] [Google Scholar]
- Barlowe C, Schekman R. SEC12 encodes a guanine-nucleotide-exchange factor essential for transport vesicle budding from the ER. Nature. 1993;365:347–349. doi: 10.1038/365347a0. [DOI] [PubMed] [Google Scholar]
- Ben-Tekaya H, Miura K, Pepperkok R, Hauri HP. Live imaging of bidirectional traffic from the ERGIC. J Cell Sci. 2005;118:357–367. doi: 10.1242/jcs.01615. [DOI] [PubMed] [Google Scholar]
- Bi X, Corpina RA, Goldberg J. Structure of the Sec23/24-Sar1 pre-budding complex of the COPII vesicle coat. Nature. 2002;419:271–277. doi: 10.1038/nature01040. [DOI] [PubMed] [Google Scholar]
- Bi X, Mancias JD, Goldberg J. Insights into COPII coat nucleation from the structure of Sec23-Sar1 complexed with the active fragment of Sec31. 2007 doi: 10.1016/j.devcel.2007.10.006. co-submission with this manuscript. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyadjiev SA, Fromme JC, Ben J, Chong SS, Nauta C, Hur DJ, Zhang G, Hamamoto S, Schekman R, Ravazzola M, et al. Cranio-lenticulo-sutural dysplasia is caused by a SEC23A mutation leading to abnormal endoplasmic-reticulum-to-Golgi trafficking. Nat Genet. 2006;38:1192–1197. doi: 10.1038/ng1876. [DOI] [PubMed] [Google Scholar]
- Boyadjiev SA, Justice CM, Eyaid W, McKusick VA, Lachman RS, Chowdry AB, Jabak M, Zwaan J, Wilson AF, Jabs EW. A novel dysmorphic syndrome with open calvarial sutures and sutural cataracts maps to chromosome 14q13-q21. Hum Genet. 2003;113:1–9. doi: 10.1007/s00439-003-0932-6. [DOI] [PubMed] [Google Scholar]
- Fath S, Mancias JD, Bi X, Goldberg J. Structure and organization of coat proteins in the COPII cage. Cell. 2007;129:1325–1336. doi: 10.1016/j.cell.2007.05.036. [DOI] [PubMed] [Google Scholar]
- Futai E, Hamamoto S, Orci L, Schekman R. GTP/GDP exchange by Sec12p enables COPII vesicle bud formation on synthetic liposomes. EMBO J. 2004;23:4146–4155. doi: 10.1038/sj.emboj.7600428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay JC, Hirling H, Scheller RH. Mammalian vesicle trafficking proteins of the endoplasmic reticulum and Golgi apparatus. J Biol Chem. 1996;271:5671–5679. doi: 10.1074/jbc.271.10.5671. [DOI] [PubMed] [Google Scholar]
- Jones B, Jones EL, Bonney SA, Patel HN, Mensenkamp AR, Eichenbaum-Voline S, Rudling M, Myrdal U, Annesi G, Naik S, et al. Mutations in a Sar1 GTPase of COPII vesicles are associated with lipid absorption disorders. Nat Genet. 2003;34:29–31. doi: 10.1038/ng1145. [DOI] [PubMed] [Google Scholar]
- Kim J, Hamamoto S, Ravazzola M, Orci L, Schekman R. Uncoupled packaging of amyloid precursor protein and presenilin 1 into coat protein complex II vesicles. J Biol Chem. 2005;280:7758–7768. doi: 10.1074/jbc.M411091200. [DOI] [PubMed] [Google Scholar]
- Kuehn MJ, Herrmann JM, Schekman R. COPII-cargo interactions direct protein sorting into ER-derived transport vesicles. Nature. 1998;391:187–190. doi: 10.1038/34438. [DOI] [PubMed] [Google Scholar]
- Lang MR, Lapierre LA, Frotscher M, Goldenring JR, Knapik EW. Secretory COPII coat component Sec23a is essential for craniofacial chondrocyte maturation. Nat Genet. 2006;38:1198–1203. doi: 10.1038/ng1880. [DOI] [PubMed] [Google Scholar]
- Lee MC, Miller EA, Goldberg J, Orci L, Schekman R. Bi-directional protein transport between the ER and Golgi. Annu Rev Cell Dev Biol. 2004;20:87–123. doi: 10.1146/annurev.cellbio.20.010403.105307. [DOI] [PubMed] [Google Scholar]
- Liou W, Geuze HJ, Slot JW. Improving structural integrity of cryosections for immunogold labeling. Histochem Cell Biol. 1996;106:41–58. doi: 10.1007/BF02473201. [DOI] [PubMed] [Google Scholar]
- Matsuoka K, Orci L, Amherdt M, Bednarek SY, Hamamoto S, Schekman R, Yeung T. COPII-coated vesicle formation reconstituted with purified coat proteins and chemically defined liposomes. Cell. 1998;93:263–275. doi: 10.1016/s0092-8674(00)81577-9. [DOI] [PubMed] [Google Scholar]
- Miller E, Antonny B, Hamamoto S, Schekman R. Cargo selection into COPII vesicles is driven by the Sec24p subunit. EMBO J. 2002;21:6105–6113. doi: 10.1093/emboj/cdf605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller EA, Beilharz TH, Malkus PN, Lee MC, Hamamoto S, Orci L, Schekman R. Multiple cargo binding sites on the COPII subunit Sec24p ensure capture of diverse membrane proteins into transport vesicles. Cell. 2003;114:497–509. doi: 10.1016/s0092-8674(03)00609-3. [DOI] [PubMed] [Google Scholar]
- Mossessova E, Bickford LC, Goldberg J. SNARE selectivity of the COPII coat. Cell. 2003;114:483–495. doi: 10.1016/s0092-8674(03)00608-1. [DOI] [PubMed] [Google Scholar]
- Nakano A, Muramatsu M. A novel GTP-binding protein, Sar1p, is involved in transport from the endoplasmic reticulum to the Golgi apparatus. J Cell Biol. 1989;109:2677–2691. doi: 10.1083/jcb.109.6.2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols WC, Seligsohn U, Zivelin A, Terry VH, Hertel CE, Wheatley MA, Moussalli MJ, Hauri HP, Ciavarella N, Kaufman RJ, Ginsburg D. Mutations in the ER-Golgi intermediate compartment protein ERGIC-53 cause combined deficiency of coagulation factors V and VIII. Cell. 1998;93:61–70. doi: 10.1016/s0092-8674(00)81146-0. [DOI] [PubMed] [Google Scholar]
- Pagano A, Letourneur F, Garcia-Estefania D, Carpentier JL, Orci L, Paccaud JP. Sec24 proteins and sorting at the endoplasmic reticulum. J Biol Chem. 1999;274:7833–7840. doi: 10.1074/jbc.274.12.7833. [DOI] [PubMed] [Google Scholar]
- Plutner H, Davidson HW, Saraste J, Balch WE. Morphological analysis of protein transport from the ER to Golgi membranes in digitonin-permeabilized cells: role of the P58 containing compartment. J Cell Biol. 1992;119:1097–1116. doi: 10.1083/jcb.119.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presley JF, Cole NB, Schroer TA, Hirschberg K, Zaal KJ, Lippincott-Schwartz J. ER-to-Golgi transport visualized in living cells. Nature. 1997;389:81–85. doi: 10.1038/38001. [DOI] [PubMed] [Google Scholar]
- Roberts B, Clucas C, Johnstone IL. Loss of SEC-23 in Caenorhabditis elegans causes defects in oogenesis, morphogenesis, and extracellular matrix secretion. Mol Biol Cell. 2003;14:4414–4426. doi: 10.1091/mbc.E03-03-0162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salama NR, Yeung T, Schekman RW. The Sec13p complex and reconstitution of vesicle budding from the ER with purified cytosolic proteins. EMBO J. 1993;12:4073–4082. doi: 10.1002/j.1460-2075.1993.tb06091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraste J, Svensson K. Distribution of the intermediate elements operating in ER to Golgi transport. J Cell Sci. 1991;100(Pt 3):415–430. doi: 10.1242/jcs.100.3.415. [DOI] [PubMed] [Google Scholar]
- Scales SJ, Pepperkok R, Kreis TE. Visualization of ER-to-Golgi transport in living cells reveals a sequential mode of action for COPII and COPI. Cell. 1997;90:1137–1148. doi: 10.1016/s0092-8674(00)80379-7. [DOI] [PubMed] [Google Scholar]
- Schindler R, Itin C, Zerial M, Lottspeich F, Hauri HP. ERGIC-53, a membrane protein of the ER-Golgi intermediate compartment, carries an ER retention motif. Eur J Cell Biol. 1993;61:1–9. [PubMed] [Google Scholar]
- Stagg SM, Gurkan C, Fowler DM, LaPointe P, Foss TR, Potter CS, Carragher B, Balch WE. Structure of the Sec13/31 COPII coat cage. Nature. 2006;439:234–238. doi: 10.1038/nature04339. [DOI] [PubMed] [Google Scholar]
- Tandler B. Improved uranyl acetate staining for electron microscopy. J Electron Microsc Tech. 1990;16:81–82. doi: 10.1002/jemt.1060160110. [DOI] [PubMed] [Google Scholar]
- Tang BL, Kausalya J, Low DY, Lock ML, Hong W. A family of mammalian proteins homologous to yeast Sec24p. Biochem Biophys Res Commun. 1999;258:679–684. doi: 10.1006/bbrc.1999.0574. [DOI] [PubMed] [Google Scholar]
- Volchuk A, Amherdt M, Ravazzola M, Brugger B, Rivera VM, Clackson T, Perrelet A, Sollner TH, Rothman JE, Orci L. Megavesicles implicated in the rapid transport of intracisternal aggregates across the Golgi stack. Cell. 2000;102:335–348. doi: 10.1016/s0092-8674(00)00039-8. [DOI] [PubMed] [Google Scholar]
- Wendeler MW, Paccaud JP, Hauri HP. Role of Sec24 isoforms in selective export of membrane proteins from the endoplasmic reticulum. EMBO Rep. 2007;8:258–264. doi: 10.1038/sj.embor.7400893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson R, Allen AJ, Oliver J, Brookman JL, High S, Bulleid NJ. The translocation, folding, assembly and redox-dependent degradation of secretory and membrane proteins in semi-permeabilized mammalian cells. Biochem J. 1995;307(Pt 3):679–687. doi: 10.1042/bj3070679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihisa T, Barlowe C, Schekman R. Requirement for a GTPase-activating protein in vesicle budding from the endoplasmic reticulum. Science (New York, NY) 1993;259:1466–1468. doi: 10.1126/science.8451644. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.