

Abstract
The incidence of neonatal stroke is high and currently there are no strategies to protect the neonatal brain from stroke or reduce the sequelae. Agents capable of modifying inflammatory processes hold promise. We set out to determine whether delayed administration of one such agent, minocycline, protects the immature brain in a model of transient middle cerebral artery (MCA) occlusion in 7-day-old rat pups. Injury volume in minocycline (45 mg/kg/dose, beginning at 2 h after MCA occlusion) and vehicle-treated pups was determined 24 h and 7 days after onset of reperfusion. Accumulation of activated microglia/macrophages, phosphorylation of mitogen-activated protein kinase (MAPK) p38 in the brain, and concentrations of inflammatory mediators in plasma and brain were determined at 24 h. Minocycline significantly reduced the volume of injury at 24 h but not 7 days after transient MCA occlusion. The beneficial effect of minocycline acutely after reperfusion was not associated with changed ED1 phenotype, nor was the pattern of MAPK p38 phosphorylation altered. Minocycline reduced accumulation of IL-1β and CINC-1 in the systemic circulation but failed to affect the increased levels of IL-1β, IL-18, MCP-1 or CINC-1 in the injured brain tissue. Therefore, minocycline provides early but transient protection, which is largely independent of microglial activation or activation of the MAPK p38 pathway.
Keywords: chemokine, inflammation, MAPK p38, microglia, minocycline, neonatal strokes
Introduction
Neonatal stroke occurs with an incidence of 28 in 100,000 live births (deVeber, 2002) and can result in cognitive deficits, cerebral palsy and epilepsy. Although stroke in the neonatal period is increasingly recognized and studied, much of the understanding of its pathophysiology stems from research performed in adult stroke models. While the underlying cascade of events such as depletion of cellular energy reserves, excitatory amino acid-release and oxygen-free radical production are similar in adult and immature stroke, the neuropathological outcomes (Yager and Thornhill, 1997) and modes of cell death are profoundly influenced by age (Hu et al, 2000). The readily available proapoptotic machinery (Han et al, 2000; Hu et al, 2000), the immature immune system and incomplete myelination make the immature brain prone to a proinflammatory response after an ischemic event (Benjelloun et al, 1999; Bona et al, 1999; Cowell et al, 2002; Derugin et al, 2000; Ivacko et al, 1996; McRae et al, 1995). Therefore, finding an anti-inflammatory drug that is safe and effective in treating neonatal stroke is appealing.
Minocycline is a tetracycline derivative that has generated much interest as a modifier of acute and chronic neuroinflammation. It readily crosses the blood–brain barrier (Brogden et al, 1975), is well tolerated and has pleiotrophic actions (Teng et al, 2004; Tikka et al, 2001a, b; Yrjanheikki et al, 1998, 1999; Zhu et al, 2002) that are separate from its antimicrobial action. Many laboratories have shown minocycline to be a potent therapeutic agent in various models of neurological diseases with chronic inflammatory components, such as models of Parkinson’s (Thomas and Le, 2004, 2003), Huntington’s (Thomas et al, 2003), multiple sclerosis (Popovic et al, 2002), ALS (Zhu et al, 2002) and in models of acute inflammation, such as brain trauma (Teng et al, 2004) and cerebral ischemia (Arvin et al, 2002; Yrjanheikki et al, 1998, 1999; Zhu et al, 2002). At the same time, recent reports show inconsistent and even detrimental effects of minocycline in different models of neurodegeneration, including hypoxia-ischemia (H–I) (Diguet et al, 2004a, b; Tsuji et al, 2004).
The hierarchy among multiple mechanisms of minocycline protection is poorly understood. Minocycline can block the permeability transition pore and the associated release of cytochrome c from mitochondria, resulting in activation of multiple caspases after brain trauma (Teng et al, 2004) and cerebral ischemia (Zhu et al, 2002). It protects neurons against excitotoxic damage directly, independent of microglial activation (Teng et al, 2004; Tikka et al, 2001a, b), as well as against neuronal death amplified by the presence of activated microglial cells (Teng et al, 2004). Minocycline effectively attenuates the delayed phase of injury after focal transient cerebral ischemia (Yrjanheikki et al, 1999) and global brain ischemia (Yrjanheikki et al, 1998) by reducing microglial activation and proliferation (Tikka et al, 2001a, b). Microglial activation, in turn, depends on activation of the mitogen-activated protein (MAP) kinase p38, a stress-activated protein kinase that mediates many cellular responses, including production of inflammatory mediators (Saccani et al, 2002) and direct activation of cell death pathways (Du et al, 2001; Lin et al, 2001). P38 can be inhibited by minocycline (Lin et al, 2001; Teng et al, 2004; Tikka et al, 2001a, b) and is thought to contribute to brain injury reduction (Du et al, 2001; Lin et al, 2001).
Emerging evidence suggests that inflammation plays an important role in neonatal H–I (Cowell et al, 2002, 2003; Hagberg et al, 1996; McRae et al, 1995) and activation of microglial cells is rapid and profound in our neonatal focal stroke model (Derugin et al, 2000).We therefore hypothesized that minocycline would afford protection through the inhibition of microglial activation and signaling through the MAP kinase p38. To test these hypotheses, we determined whether post-treatment of minocycline alters the infarct size during the acute and sub-chronic phases of injury after transient middle cerebral artery (MCA) occlusion in postnatal day 7 (P7) rat (Derugin et al, 1998). To examine the possible mechanisms of protection, we determined if minocycline alters microglial activation, phosphorylation of MAPK p38 and accumulation of proinflammatory molecules acutely after injury. Our results suggest that minocycline confers early but only transient protection, which is largely independent of microglial activation or activation of MAP kinase p38.
Materials and methods
Animals and Surgical Procedures
All animal research was approved by the University of California San Francisco Institutional Animal Care and Use Committee and was performed in accordance with the Guide for the Care and Use of Laboratory Animals (US Department of Health and Human Services, Publication Number 85–23, 1985). Female Sprague–Dawley rats with a 6-day-old litter were purchased and housed in a temperature/light-controlled environment and given food and water ad libitum. On P7, the unsexed pups were subjected to transient MCA occlusion. In preliminary studies where we utilized the previously published procedure (Derugin et al, 1998), animal growth was delayed after this surgical preparation, and baby rats were fed formula to improve weight gain. Mortality and intestinal damage were observed in minocycline-treated, but not in injured PBS-treated pups or minocycline-treated pups with no injury (unpublished observations). We have modified surgical procedure in this study and inserted the coated filament directly through the internal carotid artery (ICA) via the incision made directly over the right common carotid, external carotid and internal carotid bifurcation. These procedures were performed without occluding or coagulating any other cervical vessels. The surgery was performed under 1.5% to 3% isoflurane anesthesia in a mixture of 70% N2O, and 30% O2, using a coated, 6–0 Dermelon suture filament. Throughout the 15 min surgical procedure, the animal temperature was maintained at 38°C by a heating pad and overhead lamp. To select animals with injury that extends throughout the MCA vascular territory, each animal was examined by diffusion weighted (DW) MRI at approximately 1.5 h after occlusion. High-speed spin echo echo-planar images with diffusion sensitizing gradient pulses were employed in eight consecutive 1-mm-thick planes to cover the entire MCA territory. Animals with the desired injury pattern were used in this study, as we have previously shown (Derugin et al, 2000) that evidence of hyperintensity on heavily DW image and the associated decrease of the apparent diffusion coefficient (ADC) during MCA occlusion in P7 rat is predictive of cerebral infarction. After 3 h of occlusion, the suture blocking the MCA was removed, allowing reperfusion, and the pups were then returned to the dam for recovery.
The acute protective effect of minocycline hydrochloride (Sigma, Inc.) was determined at 24 h post-reperfusion in pups treated with minocycline (45 mg/kg) (n = 9) or PBS (n = 7–8 per time point), administered by intraperitoneal injection 2 h after MCA occlusion and again 4 h after reperfusion. The long-term protective effect of minocycline was determined at 7 days after surgery. Since a U-curve in efficacy has been reported with dose escalation (Teng et al, 2004), animals were administered two drug regiments, a single (45 mg/kg, intraperitoneal 2 h after MCA occlusion) or three injections of minocycline (45 mg/kg/dose, intraperitoneal, 2 h MCA occlusion, 4 and 18 h after reperfusion) or PBS. Naïve littermates and treated and nontreated pups were weighed daily to evaluate the effect of minocycline on growth. No adverse effects were seen in all studied groups of pups.
Histology
For histological studies, pups were anesthetized with pentobarbital and perfused with ice-cold 4% paraformaldehyde (PFA) in 0.1 mol/L PBS through the heart. Brains were removed and post-fixed in 4% PFA at 4°C for 24 h. Fixed brains from pups killed after 24 h were cut into 50 µm sections on a vibratome and stored at 4°C in 0.1 mol/L PBS with 0.01% sodium azide. Owing to the fragility of injured brain tissue 7 days after injury, brains at this time point were placed in 70% ethanol after fixation, paraffin embedded, and cut into 20 µm sections (Biopathology Labs, South San Francisco, CA, USA). Brain sections were stained with cresyl violet for assessment of brain injury.
Immunohistochemistry
Diaminobenzidine peroxidase immunohistochemistry was performed on 50 µm free-floating sections. Endogenous peroxidase was quenched with 1% hydrogen peroxide, and blocked with 5% horse serum/1% Triton-X100/PBS. The tissue was incubated with primary antibody in 1% Triton-X100/PBS according to the manufacturer. Primary antibodies were monoclonal antidually phosphorylated p38 (1:200, Sigma) and ED1, a marker of activated microglia (1:100, Serotec). Sections were washed in 1% Triton-X100/PBS and incubated in biotinylated horse anti-mouse antibody (Vector, 1:200). After washing again with 1% Triton-X100/PBS, sections were incubated in avidin–biotin complex (Vectastain, Vector Laboratories) before visualization with diaminobenzidine (Sigma) and 0.02% H2O2.
Determination of Injury Volume and Cell Density
Injury volumes were determined using systematic random sampling and the Cavalieri principle and guidelines for unbiased stereology for estimating volume (West et al, 1991). Briefly, brains of pups killed 24 h after reperfusion were cut into 50 µm sections and stored serially in a 12-well plate so that each well contained a series of sections obtained at 600 µm intervals. Beginning at the anterior genu of the corpus callosum, a series of sections was selected for staining using a random number generator and all sections from a single well were stained. Volumetric analysis was performed on cresyl violet-stained sections using Neurolucida (Microbrightfield Software Systems) by manually tracing the profiles of the entire ipsilateral hemisphere, contralateral hemisphere, lesion and cystic infarct, then integrating to obtain volumes. Figure 1B demonstrates how we outlined injured and uninjured tissue for a representative coronal section shown in Figure 1A. The same technique was used to determine the injury volumes of paraffin-embedded brain tissues of pups 7 days after surgery, except that the sections were evaluated at 400 µm intervals. As the injury often evolved into a cystic infarct, we used two outcome measures for comparison between minocycline- and PBS-treated pups: the size of the remaining tissue and the morphologically normal (unaffected) tissue in the hemisphere ipsilateral to the occlusion. Data were expressed as a ratio of the size of remaining ipsilateral to contralateral hemisphere. The density of ED1-immunoreactive cells was determined using Stereologer Software to guide systematic random sampling (Microbrightfield Software Systems). The tops of ED1 immunoreactive cells were counted using an × 20 objective. The grid size was chosen using preliminary cell counts and Stereologer software. Sampling sites were generated on a grid 1000 µm apart and placed randomly on each section by the software. Cell density estimates were performed over the entire ipsilateral and contralateral hemispheres in the eight sections comprising the injured region, and densities were calculated from estimated cell population/volume.
Figure 1.

Minocycline treatment preserves tissue in the ipsilateral hemisphere measured 24 h after MCA occlusion. (A) A representative cresyl violet-stained coronal section demonstrates the morphology of the brain infarct 24 h after MCA occlusion in a control rat pup. Minocycline-treated pups show a similar pattern of injury. (B) Contours are drawn around injured (shown in dark gray) and uninjured (shown in light gray) tissue in the hemisphere ipsilateral to MCA occlusion, as well as around the contralateral hemisphere (shown in black). The size of uninjured tissue is calculated and expressed as a percent of the size of the contralateral hemisphere. (C) The volume of uninjured (unaffected) tissue in the ipsilateral hemisphere is larger (P<0.04) in minocycline-treated than in PBS-treated pups. Each dot represents the size of uninjured tissue for an individual pup. (D) Minocycline treatment consistently preserves the tissue in all eight studied sections in the anterior to posterior direction.
Determination of Cytokines and Chemokines in Plasma in the Brain Tissue
Concentrations of IL-4, IL-1β, IL-1α, IL-6, IL-10, IL-12, IL-18, IL-5, IL-2, IFNγ, TNFα, CINC-1/GRO/KC, MCP-1, GM-CSF were simultaneously quantified in a single plasma or brain tissue sample using a LINCOplext™ rat cytokine kit (LINCO Research, Inc., Saint Charles, MO 63304, USA). Briefly, plasma and brain tissue from injured (hyperintense tissue on DW-MRI during occlusion) and anatomically matching tissue from the contralateral hemisphere were collected from control and minocycline- or PBS-treated pups following 24 h of reperfusion. The flashfrozen brain tissue was homogenized in a buffer containing 20 mmol/L Tris-HCl (pH 7.5), 150 mmol/L NaCl, 1 mmol/L PMSF, 0.05% Tween-20, and a cocktail of protease inhibitors (Roche), and protein concentration was measured in each sample. Cytokine and chemokine concentrations were determined using antibodies for each analyte covalently immobilized to a set of microspheres according to protocol developed and validated at LINCO Research, Inc. The analytes on the surface of microspheres were then detected by a cocktail of biotinylated antibodies. Following binding of streptavidin–phycoerythrin conjugate, the reporter fluorescent signal was measured with a Luminex100 reader (Luminex Corp., Austin, TX 78727, USA). Data were calculated using a calibration curve obtained in each experiment using the respective recombinant proteins diluted in kit matrix for plasma samples and lysis buffer for tissue samples. Concentrations of cytokines were calculated using StatLIA® software (Brendan Scientific Corp., Calrsbad, CA 92008, USA) with a five-parameter logistic curve-fitting method, and normalized to the amount of protein in each sample. Concentrations of IL-4, IL-10, IL-12, IL-5, IL-2, IL-1α, IFNγ and GM-CSF were consistently below detectable levels and, therefore, are not shown in the Results section. TNF-α and IL-6 values were below detectable levels in at least 50% of cases, and these data were not summarized. Table 1 and Table 2 show plasma and brain concentrations of interleukin 1β (IL-1β), cytokine-induced chemoattractant protein 1 (CINC-1), interleukin 18 (IL-18) and monocyte chemoattractant protein 1 (MCP-1). Since some of the values for the latter molecules were below detectable level, both the number of values within the detectable range and total number of samples measured for each individual analyte are shown in Table 1 and Table 2.
Table 1.
Plasma concentration of proinflammatory mediators following 3 h MCA occlusion/24 h reperfusion in pups treated with minocycline or PBS
| IL1-β (pg/ml) | CINC-1 (pg/ml) | IL-18 (pg/ml) | MCP-1 (pg/ml) | |
|---|---|---|---|---|
| Control | 261±133 | 221±115 | 3655±1138 | 301±161 |
| n=12/22 | n=17/17 | n=17/17 | n=17/17 | |
| PBS | 725±373**** | 366±170*** | 3429±1411 | 556±288* |
| n=11/17 | n=16/17 | n=9/9 | n=9/9 | |
| Minocycline | 316±290### | 286±131#### | 3909±1644 | 720±321 |
| n=8/11 | n=11/11 | n=11/11 | n=11/11 |
Data are expressed as mean±s.d. For each individual mediator, the number of values within the detectable range of concentrations (included in analysis) and a total number of measured samples are shown below the actual values.
P<0.045 versus Control
P<0.007 versus Control
P<0.0004 versus Control
P<0.007 versus PBS-treated
P<0.0004 versus PBS-treated.
Table 2.
Brain concentrations of proinflammatory mediators following 3 h MCA occlusion/24 h reperfusion in minocycline and PBS-treated pups
| IL1-β (pg/mg protein) | CINC-1, (pg/mg protein) | IL-18 (pg/mg protein) | MCP-1 (pg/mg protein) | |
|---|---|---|---|---|
| Control | 9.6±4.4 | 3.2±2.2 | 155.0±87.5 | 5.6±6.1 |
| n=13/17 | n=17/17 | n=17/17 | n=17/17 | |
| PBS | ||||
| Ipsilateral | 19.4±8.4** | 24.5±15.7**** | 397.6±92.3**** | 42.7±23.1**** |
| n=7/7 | n=7/7 | n=7/7 | n=7/7 | |
| Contralateral | 14.4±10.5 | 4.1±3.0§§§ | 193.6±80.1§§ | 9.4±5.2§§§ |
| n=6/7 | n=7/7 | n=7/7 | n=4/7 | |
| Minocycline | ||||
| Ipsilateral | 19.7±13.4** | 29.7±12.6**** | 420.5±239.3**** | 42.1±11.3**** |
| n=8 | n=9 | n=9 | n=9 | |
| Contralateral | 11.7±4.6 | 3.5±1.9§§§ | 207.5±96.3§§§ | 6.6±2.1§§§ |
| n=8/9 | n=8/9 | n=9/9 | n=5/9 |
Data are expressed as mean±s.d. For each individual mediator, the number of values within the detectable range of concentrations (included in analysis) and number of measured samples are shown below actual values.
P<0.017 versus Control
P<0.0001 versus Control
P<0.017 value in the ipsilateral hemisphere
P<0.001 value in the ipsilateral hemisphere.
Statistical Analysis
The volume of brain tissue and density of ED1 immunoreactive cells in treated and nontreated groups were analyzed by ANOVA with post hoc testing. For systematic random sampling of injury, the Gunderson coefficient of error (Gundersen et al, 1999) was used to estimate sampling error in analysis of volume of brain tissue and density of ED1 immunoreactive cells. Counting parameters were chosen for a Gunderson CE<0.1. All differences were considered significant at P<0.05. Results are represented as mean±s.d.
Results
Minocycline Treatment Reduces Injury Size 24 h Post-Reperfusion
All pups that underwent MCA occlusion were subjected to a DW-MRI session at 1.5 h after MCA occlusion. Animals with no apparent intensity changes within the MCA territory on DW-MRI were excluded from further studies. Consistent with our previous findings (Manabat et al, 2003), injury was extensive by 24 h after reperfusion (Figure 1A). The volume of uninjured tissue in the hemisphere ipsilateral to the occlusion was significantly larger in minocycline- pups, 76±15mm³ (n=9), compared to that in PBS-treated controls, 60±11mm³ (n=8) (P<0.04) The volume of uninjured tissue for individual minocycline- and PBS-treated pups is shown in Figure 1C. The volumes of the whole ipsilateral hemisphere and contralateral hemisphere were 112±9 and 109±8mm³ in minocycline group and 114±7 and 111±8mm³ in PBS -treated group respectively, and were not significantly different within and between groups. The reduction of injury size in the minocycline-treated pups was consistent throughout the anterior to posterior distribution, as shown in Figure 1D.
Minocycline Treatment Does not Alter Microglial Response 24 h After Transient MCA Occlusion
To determine whether minocycline protects the neonatal brain via attenuation of microglial activation, we used acquisition of ED1 immunoreactivity as the indicator. Activation of microglial cells can be evaluated using several outcomes, including acquisition of an activated phenotype when microglial cells retract their processes and enlarge their cell body, a process known as transformation from ramified to ameboid phenotype (Raivich et al, 1999). Sections adjacent to Nissl-stained were immunostained for ED1. In naïve brain (data not shown) and in the contralateral hemisphere (Figure 2A), ED1 immunoreactivity was localized almost exclusively to the corpus callosum. No ED1 immunoreactive cells were observed 1 to 8 h after reperfusion in the injured tissue (not shown). At 24 h after reperfusion, ED1 immunoreactive cells were consistently observed throughout the injured tissue (Figure 2B), but in greater numbers at the edge of the injury (Figure 2C versus 2D). No such cells were apparent in non-injured cortex of the ipsilateral hemisphere. Owing to difficulty in delineating the exact border of injured versus noninjured, the number of ED1 immunoreactive cells was measured in the entire ischemic hemisphere rather than only in the injured tissue. The density of ED1 immunoreactive cells in the ischemic hemisphere more than doubled compared to contralateral hemisphere, from 2123±508 to 5217±2163 cells/mm³ (P<0.0003; n=6/group), demonstrating a robust accumulation of activated microglia/macrophages in the injured hemisphere after ischemia–reperfusion in the neonatal brain. Minocycline treatment did not alter the density of ED1 immunoreactive cells in the corpus collosum of the contralateral hemisphere, 2123±453 cells/mm³ in minocycline-treated animals and 2124±634 cells/mm³ in PBS-treated animals. In the ischemic hemisphere, the density of ED1 immunoreactive cells was 4959±2457 cells/mm³ (n=7) and was not significantly different from that seen in PBS-treated group, 5579±1881 cells/mm³ (n=5, P=0.65) (Figure 2D), suggesting that minocycline administration does not affect acquisition of the activated microglial phenotype.
Figure 2.
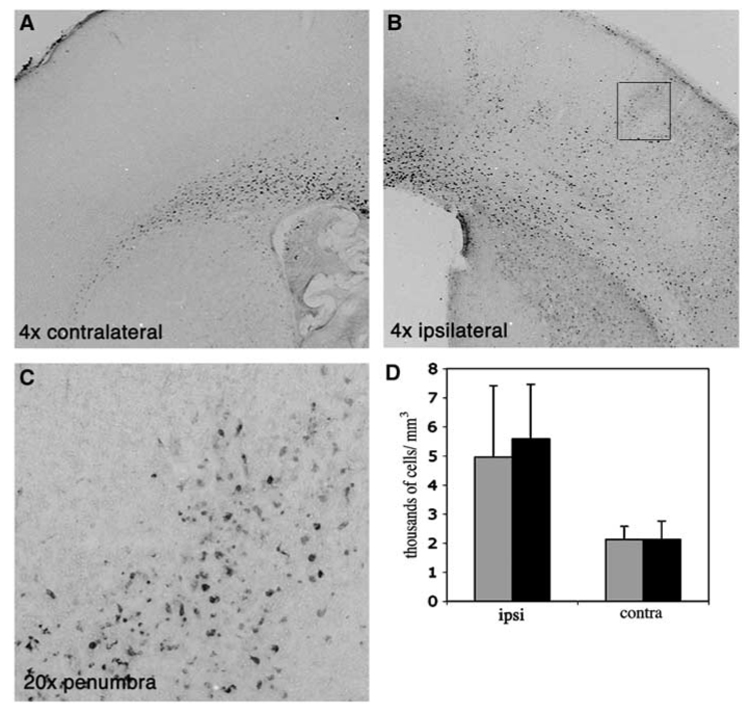
Accumulation of ED1 immunoreactive cells in the injured hemisphere is not affected by minocycline treatment 24 h after MCA occlusion. Representative examples of ED1 immunostained sections contralateral (A) and ipsilateral (B, C) to MCA occlusion. (C) shows a higher magnification picture (20 ×) of the frame shown in (B). While in the uninjured hemisphere the presence of ED1-positive cells is limited to corpus collosum (A), rapid acquisition of ED1 phenotype is evident in the lesioned tissue (B, C). (D) Density of ED1 immunoreactive cells in minocycline and PBS-treated pups 24 h after reperfusion. Black—PBS-treated, Gray—minocycline-treated pups.
Minocycline Treatment Does not Alter the Pattern of p38 Phosphorylation Acutely After Neonatal Focal Ischemia
To determine whether the protective effect of minocycline depends on activation of the MAP kinase p38, we identified the spatial pattern of p38 phosphorylation following minocycline and PBS treatment. In naïve pups, diffuse p38 phosphorylation was observed throughout the cortex in a laminar pattern in the superficial cortical layers. Staining in neocortex was predominantly in neurons in the upper layers (II/III–IV) in a cellular pattern, with the most intense staining occurring in cell bodies and the apical dendrite of layer IV neurons (Figure 3A). Intense immunostaining was also observed in the piriform cortex (Figure 3B), similar to that previously reported in adult rat brain (Mielke et al, 1999). At the boundaries of neocortex, the cingulum medially and entorhinal cortex laterally, staining involved all cell layers. We then determined the spatial pattern of p38 phosphorylation at 8 h post-reperfusion when cell death is already apparent (Manabat et al, 2003) and microglial activation is at an early stage (not shown), and at 24 h after ischemia when cell death is profound and microglial cells exhibit an activated phenotype (Manabat et al, 2003). In PBS-treated pups, the pattern of p38 phosphorylation changed by 8 h, outlining the injured territory (Figure 3C), while remaining the same in the contralateral hemisphere (Figure 3D). In regions perifocal to ischemia, there was patchy loss of p38 immunoreactivity, and the spatial pattern of reduced p38 phosphorylation matched injured MCA vascular territory (Figures 3C and 3G). Within the infarct zone, the diffuse laminar pattern was lost and only scattered intensely stained cells were seen throughout the cortex and basal ganglia (Figure 3H). The overall loss of p38 phosphorylation did not result from tissue loss, because cresyl violet staining of adjacent sections showed intact tissue (not shown). At 24 h, p38 phosphorylation was seen in both hemispheres. Comparison of the patterns of p38 phosphorylation in brain sections of minocycline- and PBS-treated pups at 8 h post-reperfusion showed a similar pattern in the contralateral hemisphere and similar spatial distribution of loss of p38 phosphorylation in the injured territory (Figure 3E versus 3C), suggesting that the early effects of minocycline are largely independent of p38 activation.
Figure 3.
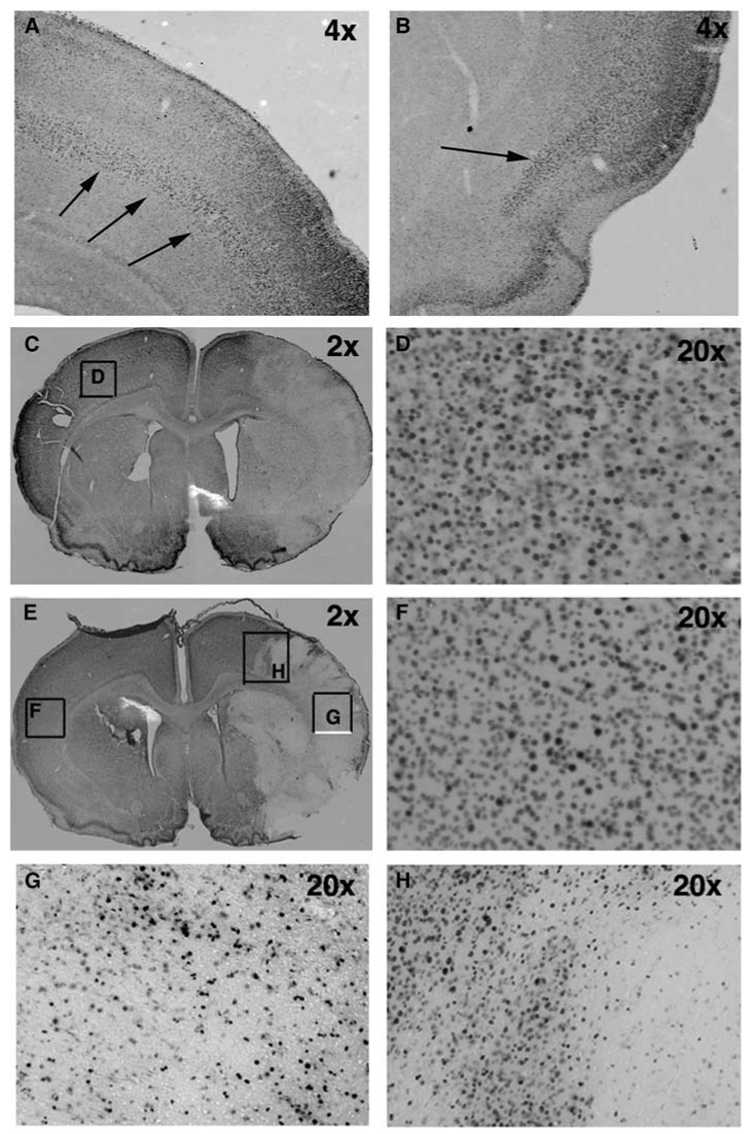
The pattern of the expression of phosphorylated form of MAP kinase p38 is not affected by minocycline treatment at 8 h after reperfusion. (A, B) Expression of p-p38 in a naïve brain of P7 rat. Arrows demonstrate the laminar pattern of p38 phosphorylation. (C) The pattern of p38 phosphorylation following PBS treatment (2 ×). A unilateral decrease of p38 phosphorylation is observed in injured but not in unaffected brain tissue. (D) A higher magnification picture obtained from the box in panel C. (E) The pattern of p38 phosphorylation following minocycline treatment (2 ×). (F) A higher magnification picture obtained from the box in panel E. (G) A higher magnification picture (20 ×, box in panel E) shows that p38 phosphorylation is seen only in a subpopulation of cells in the ischemic core. (H) A higher magnification picture (20 ×, box in panel E) of p38 phosphorylation. A heterogeneous pattern of p38 phosphorylation is observed in the ischemic penumbra 8 h post-reperfusion.
Minocycline Attenuates Early Accumulation of Pro-Inflammatory Molecules in Plasma but not in the Injured Brain Tissue at 24 h
To determine whether minocycline attenuates accumulation of inflammatory molecules, we simultaneously measured concentrations of multiple cytokines and chemokines in plasma and brain tissue samples from control and injured pups treated with minocycline or PBS. Data for plasma concentrations of IL-β, IL-18, MCP-1 and CINC-1 are shown in Table 1. Transient MCA occlusion resulted in a significant increase in plasma concentration of cytokine IL-1β (approximately 2.8-fold, P<0.0004) and chemokines MCP-1 (1.9-fold, P<0.045) and CINC-1 (1.7-fold, P<0.007), while IL-18 concentration remained the same (Table 1). Minocycline treatment significantly diminished levels of IL-1β (P<0.007) and CINC-1 (P<0.0004) in plasma of injured animals, consistent with the known anti-inflammatory action of this drug. It did not change the elevated levels of MCP-1 (P>0.1). There was a significant increase of IL-1β (~2-fold, P<0.017), CINC-1 (7.7-fold, P<0.0001), MCP-1 (7.6-fold, P<0.0001) and IL-18 (2.6-fold, P<0.0001) concentrations in the injured tissue of PBS-treated animals compared to control animals (Table 2). The concentrations of these inflammatory mediators in the injured tissue of PBS-treated animals were also significantly higher compared to the contralateral hemisphere, a 6-fold increase for CINC-1 (P<0.001), 4.5-fold for MCP-1 (P<0.001) and 2-fold for IL-18 (2.6-fold, P<0.017). Unlike in plasma, minocycline did not alter accumulation of IL-1β, IL-18, MCP-1 or CINC-1 in injured brain tissue (Table 2) at 24 h postreperfusion, demonstrating that the anti-inflammatory action of minocycline is limited to its effects in the systemic circulation.
Minocycline Treatment has no Effect on Brain Injury 7 Days Post-Reperfusion
To determine whether minocycline provides sustained protection, animals survived for 7 days after reperfusion and tissue loss and the size of morphologically normal tissue were measured. We used two different administration protocols, a single injection at 2 h after occlusion or three injections (45 mg/kg each) over an 18-hour period beginning at 2 hours after occlusion, because published reports show that timing and cumulative dose of the drug can affect outcome (Teng et al, 2004). In all the three groups, pups grew at a similar rate and gained 12.6±1.2 g in the PBS-treated group, 15.4±3.1 g following a single minocycline injection and 12.6±0.9 g following triple injection of the drug (P>0.05). By 7 days, the infarct evolved into a loss of tissue and scar formation in many, but not all, cases in the hemisphere ipsilateral to the occlusion, making the size of injury more variable compared to that at 24 h after reperfusion (Figure 4A). The volumetric sizes of the remaining tissue in the injured hemisphere were 55%±11%, 49%±13% and 54%±16% of the contralateral hemisphere in pups following PBS, single or triple injection of minocycline, respectively (Figure 4A). In addition to evaluating tissue loss, we determined whether administration of minocycline reduced the size of the injury itself. The volume of preserved tissue, tissue that appeared morphologically normal on the same Nissl-stained sections, was also unaffected by the treatment. The volumes of the preserved tissue were not significantly different, comprising 47%±79%, 40%±12% and 45%±15% of the contralateral hemisphere following PBS, single or triple injection of minocycline, respectively (Figure 4B). Administration of minocycline also did not affect the anterior-to-posterior injury distribution (not shown). Taken together, these results demonstrate that minocycline did not provide lasting protection against transient ischemia in neonatal brain.
Figure 4.
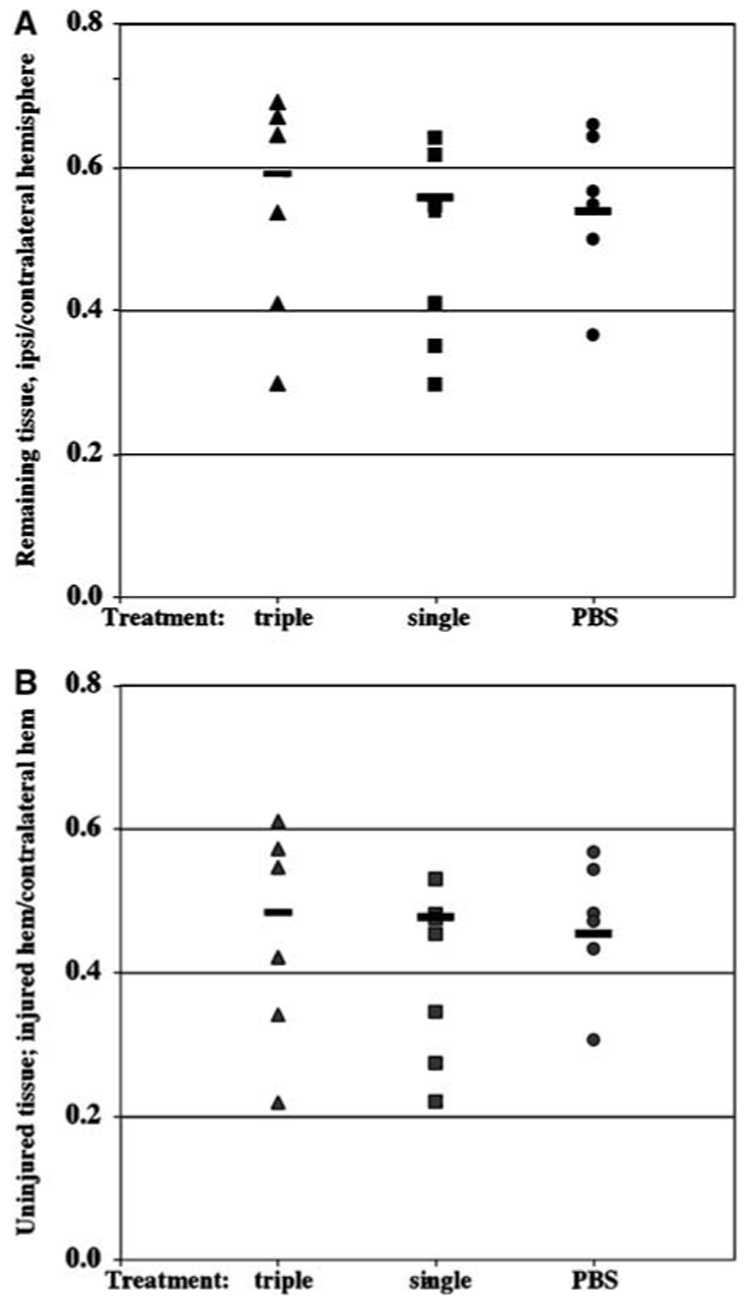
Minocycline treatment does not change the volume of the remaining and uninjured tissue of lesioned hemisphere 7 days after transient MCA occlusion. Volumes of the contralateral hemisphere, remaining ipsilateral and morphologically preserved ipsilateral hemisphere were determined in eight consecutive 15-µm-thick paraffin-embedded cresyl violet-stained coronal sections. Sections were obtained at 400-µm intervals. Volumes of the remaining ipsilateral (A) and morphologically normal/unaffected (B) tissues were normalized to the volume of contralateral hemisphere in serial sections. Pups treated with a single or triple minocycline injections show no difference in the volume of the remaining (A) or preserved (B) tissue in the hemisphere ipsilateral to the occlusion.
Discussion
These results show that delayed administration of minocycline after transient MCA occlusion in P7 rats reduced injury at 24 h following ischemia and reperfusion but the effect was transient and protection was not sustained. Our data also show that early protection against ischemia–reperfusion by minocycline is largely independent of microglial activation or activation of MAP kinase p38.
Many previous studies conducted to determine the protective effects of minocycline in experimental models of neurodegeneration have shown promising results (Arvin et al, 2002; Popovic et al, 2002; Teng et al, 2004; Thomas and Le, 2004; Thomas et al, 2003; Yrjanheikki et al, 1998, 1999; Zhu et al, 2002), while other studies show no protection (Diguet et al, 2004a, b) or even injury deterioration (Tsuji et al, 2004).
In the developing brain, activated microglia/macrophages rapidly accumulate in the injured tissue in our focal ischemia model (Derugin et al, 2000) as well as following H–I (Bona et al, 1999; Cowell et al, 2002; Ivacko et al, 1996; McRae et al, 1995). Microglia can play a dual role in the brain by protecting the CNS by phagocyting tissue debris or amplifying the effects of potentially harmful inflammation. Activation of these cells is a complex and multi-step process that results in changes in morphologic phenotype, acquisition of the ED1 phenotype, release of inflammatory mediators, increased chemotactic activity, phagocytic activity and the ability to serve as antigen-presenting cells (Bohatschek et al, 2001; Carson et al, 1998; Raivich et al, 1999). We chose to use acquisition of ED1 immuoreactive cells as an indicator of activation. In naïve brain the presence of ED1-immunoreactive cells was limited to corpus collosum, consistent with previous studies (Giulian and Baker, 1986; Graeber et al, 1998; Raivich et al, 1999). Acquisition of the ED1 phenotype was limited to lesioned tissue and occurred rapidly after ischemia–reperfusion in P7 pups (Derugin et al, (2000) and Figure 2), while in adult rodents it became apparent later. Unbiased stereological analysis showed a significantly higher density of ED1-positive cells in the injured hemisphere than in the hemisphere contralateral to the occlusion. The analysis also showed that minocycline administration did not change the density of ED1 immunoreactive cells in the injured hemisphere. This finding is somewhat surprising considering the body of literature showing that minocycline diminishes injury by attenuating microglial activation. While it is possible that difference in the ED1 estimates between treated and nontreated pups in the entire ipsilateral hemisphere can be diluted by the presence of regions constitutively expressing ED1 cells, the finding that minocycline did not reduce local inflammation in the injured tissue is consistent with unaffected microglial activation.
Since release of inflammatory molecules both substantially depends on and enhances microglial activation, we measured proinflammatory mediators in plasma and brains of injured minocycline- and PBS-treated pups at 24 h after reperfusion. After cerebral ischemia, there was a significant increase in plasma levels of inflammatory cytokine IL-1β, a monocyte chemoattractant protein MCP-1 and a chemoattractant protein for neutrophils CINC-1, and minocycline significantly decreased the levels of IL-1β and CINC-1. In the lesioned tissue, concentrations of IL-1β, IL-18, MCP-1 and CINC-1 were significantly elevated but not affected by minocycline. Since minocycline easily crosses the intact blood–brain barrier when given orally, intravenously or intraperitoneally (Brogden et al, 1975), the lack of effect in the brain is unlikely to be due to inadequate penetration although changes may occur at earlier or later times after injury.
These data show that minocycline is bioactive and exerts its anti-inflammatory effect systemically rather than intraparenchymally. These data also show the presence of local inflammation acutely after neonatal ischemia–reperfusion and suggest that IL-1β, IL-18, MCP-1 and CINC-1 are being produced locally in the brain. While these inflammatory molecules are not necessarily produced by microglial cells, they are persistently elevated in the injured tissue, indicating the presence of ongoing local inflammation despite treatment. Studies in the H–I model in P7 rats also show rapid accumulation of multiple cytokines and chemokines, including MCP-1 (Galasso et al, 2000a, b; Xu et al, 2001), MIP1 (Cowell et al, 2002) and complement activation (Cowell et al, 2003; Ten et al, 2003, 2004). In addition to exacerbating local inflammation via astrocyte and microglia activation, elevated levels of IL-1β (Barone and Feuerstein, 1999), IL-18 (Kanno et al, 2004), MCP-1 (Galasso et al, 2000a) and CINC-1 (Popivanova et al, 2003) can be neurotoxic. MCP-1 and CINC-1, in turn, are major chemoattractant molecules for circulating monocytes and neutrophils (Gerard and Rollins, 2001), cells that are largely responsible for production of reactive oxygen species and inflammatory mediators. These factors can ultimately contribute to the evolution of ischemic injury in the neonatal brain in ways that may be both dependent and independent on microglia.
Several studies have suggested that MAP kinase p38 may play a role in ischemic and excitotoxic injury in adult (Barone et al, 2001; Kummer et al, 1997; Lee et al, 2000; Legos et al, 2001; Mielke et al, 1999; Takagi et al, 2000) and immature brain (Hee Han et al, 2002) via its activation in microglial and astroglial cells (Bhat et al, 1998; Da Silva et al, 1997; Kaur et al, 2003; Kummer et al, 1997; Lee et al, 2000). Both in vivo and in vitro data have linked the protective effects of minocycline to its ability to attenuate p38 activation (Du et al, 2001; Lin et al, 2001; Suk, 2004; Tikka et al, 2001a; Zhu et al, 2002). In this study, we observed an overall profound decrease in p38 phosphorylation in the injured tissue at 8 h post-reperfusion, with the exception of a small population of scattered intensely stained cells, while the pattern of p38 phosphorylation remained unchanged in the contralateral hemisphere and in uninjured regions of the ipsilateral hemisphere. Since only patches of dead cells are apparent at this time point by Nissl stain, it is unlikely that cell death accounts for the diminished p38 phosphorylation. Decrease in p38 phosphorylation has been described by others after CNS injury, including exposure of adult rats to kainic acid (Mielke et al, 1999), neonatal rats to H–I (Han et al, 2002), and in cultured neurons to insulin (Heidenreich and Kummer, 1996). The fact that minocycline did not affect the overall spatial distribution of p38 phosphorylation suggests that its acute protection is largely p38-independent in this immature model.
According to recent studies (Zhu et al, 2002; Wang et al, 2003; Teng et al, 2004), mitochondria are an important target for minocycline and minocycline indirectly inhibits caspase-3 activation via the permeability-transition pore-mediated release of cytochrome c from mitochondria to the cytosol. This cascade was shown to be affected in cortical neurons exposed to NMDA (Zhu et al, 2002), in hSODG93A(ALS) mice (Zhu et al, 2002), and after spinal cord injury and cerebral ischemia (Teng et al, 2004). The protective effect we observed 24 h post-reperfusion may be due to reduction of caspase-3 activation, since we have shown an 18–20-fold increase in caspase-3 activity at this time point (Manabat et al, 2003).
Importantly, while minocycline appeared to provide protection acutely after injury, the protective effect did not last. For 7-day survival, we used two different administration protocols to eliminate the possibility that a high cumulative dose adversely affects outcome, as previous reports show a U-curve in efficacy (Teng et al, 2004). However, neither regimen resulted in lasting neuroprotection despite evidence that minocycline attenuated systemic accumulation of proinflammatory molecules. Although we cannot eliminate the possibility that administration of minocycline over days could have provided better protection, the timing of our administration is consistent with those reported by others and does produce a reduction in circulating inflammatory mediators. Also, pharmacokinetic data show that intraperitoneal injection of minocycline results in a delayed but sustained plasma concentration (Fagan et al, 2004). The inability of minocycline to affect concentrations of inflammatory mediators in the injured brain or microglial activation supports the finding of inadequate long-term protection.
These results add to the growing controversy surrounding the effects of minocycline in the setting of neurodegeneration (Diguet et al, 2004a, b), including neonatal cerebral ischemic injury (Tsuji et al, 2004). While Arvin et al (2002) reported that minocycline substantially preserved brain tissue and prevented the formation of cleaved caspase-3 one week after H–I insult in rat pups, Tsuji et al (2004) using a neuropathologic injury score as a measurement of injury, reported that minocycline treatment provided only mild protection from H–I in rat pups, and worsened injury in mouse pups after each of the five treatment regimens used. Species-specific differences in the response to minocycline, severity of ischemic injury or differences in stroke models may contribute to varying results.
In summary, in a model of transient neonatal cerebral ischemia, minocycline protection was short lived and did not appear to be mediated by a reduction in the accumulation of activated microglia/macrophages or by inhibition of p38 phosphorylation. Our study suggests that although minocycline has generated excitement as a modifier of brain injury, due to possible toxicity and lack of long-term efficacy, therapeutic trials in newborns should be approached with caution.
Acknowledgements
The authors thank Patrick McQuillen for helpful discussions concerning 3-D volumetric measurements and cell counting using systematic random sampling techniques, and Ann Sheldon for technical support.
This study is supported by UCP R-738-02 (ZV), NIH NS44025 (ZV), NIH NS35902 (DMF, ZV), HHMI and Genentech scholarships (CF)
References
- Arvin KL, Han BH, Du Y, Lin SZ, Paul SM, Holtzman DM. Minocycline markedly protects the neonatal brain against hypoxic–ischemic injury. Ann Neurol. 2002;52:54–61. doi: 10.1002/ana.10242. [DOI] [PubMed] [Google Scholar]
- Barone FC, Feuerstein GZ. Inflammatory mediators and stroke: new opportunities for novel therapeutics. J Cereb Blood Flow Metab. 1999;19:819–834. doi: 10.1097/00004647-199908000-00001. [DOI] [PubMed] [Google Scholar]
- Barone FC, Irving EA, Ray AM, Lee JC, Kassis S, Kumar S, Badger AM, White RF, McVey MJ, Legos JJ, Erhardt JA, Nelson AH, Ohlstein EH, Hunter AJ, Ward K, Smith BR, Adams JL, Parsons AA. SB 239063, a second-generation p38 mitogen-activated protein kinase inhibitor, reduces brain injury and neurological deficits in cerebral focal ischemia. J Pharmacol Exp Ther. 2001;296:312–321. [PubMed] [Google Scholar]
- Benjelloun N, Renolleau S, Represa A, Ben-Ari Y, Charriaut-Marlangue C. Inflammatory responses in the cerebral cortex after ischemia in the P7 neonatal rat. Stroke. 1999;30:1916–1923. doi: 10.1161/01.str.30.9.1916. discussion 1923–1914. [DOI] [PubMed] [Google Scholar]
- Bhat NR, Zhang P, Lee JC, Hogan EL. Extracellular signal-regulated kinase and p38 subgroups of mitogen-activated protein kinases regulate inducible nitric oxide synthase and tumor necrosis factor-alpha gene expression in endotoxin-stimulated primary glial cultures. J Neurosci. 1998;18:1633–1641. doi: 10.1523/JNEUROSCI.18-05-01633.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohatschek M, Kloss CU, Kalla R, Raivich G. In vitro model of microglial deramification: ramified microglia transform into amoeboid phagocytes following addition of brain cell membranes to microglia-astrocyte cocultures. J Neurosci Res. 2001;64:508–522. doi: 10.1002/jnr.1103. [DOI] [PubMed] [Google Scholar]
- Bona E, Andersson AL, Blomgren K, Gilland E, Puka-Sundvall M, Gustafson K, Hagberg H. Chemokine and inflammatory cell response to hypoxia-ischemia in immature rats. Pediatr Res. 1999;45:500–509. doi: 10.1203/00006450-199904010-00008. [DOI] [PubMed] [Google Scholar]
- Brogden RN, Speight TM, Avery GS. Minocycline: a review of its antibacterial and pharmacokinetic properties and therapeutic use. Drugs. 1975;9:251–291. doi: 10.2165/00003495-197509040-00005. [DOI] [PubMed] [Google Scholar]
- Carson MJ, Reilly CR, Sutcliffe JG, Lo D. Mature microglia resemble immature antigen-presenting cells. Glia. 1998;22:72–85. doi: 10.1002/(sici)1098-1136(199801)22:1<72::aid-glia7>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Cowell RM, Plane JM, Silverstein FS. Complement activation contributes to hypoxic-ischemic brain injury in neonatal rats. J Neurosci. 2003;23:9459–9468. doi: 10.1523/JNEUROSCI.23-28-09459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowell RM, Xu H, Galasso JM, Silverstein FS. Hypoxic-ischemic injury induces macrophage inflammatory protein-1 alpha expression in immature rat brain. Stroke. 2002;33:795–801. doi: 10.1161/hs0302.103740. [DOI] [PubMed] [Google Scholar]
- Da Silva J, Pierrat B, Mary JL, Lesslauer W. Blockade of p38 mitogen-activated protein kinase pathway inhibits inducible nitric-oxide synthase expression in mouse astrocytes. J Biol Chem. 1997;272:28373–28380. doi: 10.1074/jbc.272.45.28373. [DOI] [PubMed] [Google Scholar]
- Derugin N, Ferriero DM, Vexler ZS. Neonatal reversible focal cerebral ischemia: a new model. Neurosci Res. 1998;32:349–353. doi: 10.1016/s0168-0102(98)00096-0. [DOI] [PubMed] [Google Scholar]
- Derugin N, Wendland M, Muramatsu K, Roberts T, Gregory G, Ferriero D, Vexler Z. Evolution of brain injury after transient middle cerebral artery occlusion in neonatal rat. Stroke. 2000;31:1752–1761. doi: 10.1161/01.str.31.7.1752. [DOI] [PubMed] [Google Scholar]
- deVeber G. Stroke and the child’s brain: an overview of epidemiology, syndromes and risk factors. Curr Opin Neurol. 2002;15:133–138. doi: 10.1097/00019052-200204000-00002. [DOI] [PubMed] [Google Scholar]
- Diguet E, Fernagut PO, Wei X, Du Y, Rouland R, Gross C, Bezard E, Tison F. Deleterious effects of minocycline in animal models of Parkinson’s disease and Huntington’s disease. Eur J Neurosci. 2004a;19:3266–3276. doi: 10.1111/j.0953-816X.2004.03372.x. [DOI] [PubMed] [Google Scholar]
- Diguet E, Gross CE, Tison F, Bezard E. Rise and fall of minocycline in neuroprotection: need to promote publication of negative results. Exp Neurol. 2004b;189:1–4. doi: 10.1016/j.expneurol.2004.05.016. [DOI] [PubMed] [Google Scholar]
- Du Y, Ma Z, Lin S, Dodel RC, Gao F, Bales KR, Triarhou LC, Chernet E, Perry KW, Nelson DL, Luecke S, Phebus LA, Bymaster FP, Paul SM. Minocycline prevents nigrostriatal dopaminergic neurodegeneration in the MPTP model of Parkinson’s disease. Proc Natl Acad Sci USA. 2001;98:14669–14674. doi: 10.1073/pnas.251341998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan SC, Edwards DJ, Borlongan CV, Xu L, Arora A, Feuerstein G, Hess DC. Optimal delivery of minocycline to the brain: implication for human studies of acute neuroprotection. Exp Neurol. 2004;186:248–251. doi: 10.1016/j.expneurol.2003.12.006. [DOI] [PubMed] [Google Scholar]
- Galasso JM, Liu Y, Szaflarski J, Warren JS, Silverstein FS. Monocyte chemoattractant protein-1 is a mediator of acute excitotoxic injury in neonatal rat brain. Neuroscience. 2000a;101:737–744. doi: 10.1016/s0306-4522(00)00399-7. [DOI] [PubMed] [Google Scholar]
- Galasso JM, Miller MJ, Cowell RM, Harrison JK, Warren JS, Silverstein FS. Acute excitotoxic injury induces expression of monocyte chemoattractant protein-1 and its receptor, CCR2, in neonatal rat brain. Exp Neurol. 2000b;165:295–305. doi: 10.1006/exnr.2000.7466. [DOI] [PubMed] [Google Scholar]
- Gerard C, Rollins BJ. Chemokines and disease. Nat Immunol. 2001;2:108–115. doi: 10.1038/84209. [DOI] [PubMed] [Google Scholar]
- Giulian D, Baker TJ. Characterization of ameboid microglia isolated from developing mammalian brain. J Neurosci. 1986;6:2163–2178. doi: 10.1523/JNEUROSCI.06-08-02163.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graeber MB, Lopez-Redondo F, Ikoma E, Ishikawa M, Imai Y, Nakajima K, Kreutzberg GW, Kohsaka S. The microglia/macrophage response in the neonatal rat facial nucleus following axotomy. Brain Res. 1998;813:241–253. doi: 10.1016/s0006-8993(98)00859-2. [DOI] [PubMed] [Google Scholar]
- Gundersen HJ, Jensen EB, Kieu K, Nielsen J. The efficiency of systematic sampling in stereology—reconsidered. J Microsc. 1999;193:199–211. doi: 10.1046/j.1365-2818.1999.00457.x. [DOI] [PubMed] [Google Scholar]
- Hagberg H, Gilland E, Bona E, Hanson LA, Hahin-Zoric M, Blennow M, Holst M, McRae A, Soder O. Enhanced expression of interleukin (IL)-1 and IL-6 messenger RNA and bioactive protein after hypoxia-ischemia in neonatal rats. Pediatr Res. 1996;40:603–609. doi: 10.1203/00006450-199610000-00015. [DOI] [PubMed] [Google Scholar]
- Han BH, D’Costa A, Back SA, Parsadanian M, Patel S, Shah AR, Gidday JM, Srinivasan A, Deshmukh M, Holtzman DM. BDNF blocks caspase-3 activation in neonatal hypoxia-ischemia. Neurobiol Dis. 2000;7:38–53. doi: 10.1006/nbdi.1999.0275. [DOI] [PubMed] [Google Scholar]
- Han BH, Xu D, Choi J, Han Y, Xanthoudakis S, Roy S, Tam J, Vaillancourt J, Colucci J, Siman R, Giroux A, Robertson GS, Zamboni R, Nicholson DW, Holtzman DM. Selective, reversible caspase-3 inhibitor is neuroprotective and reveals distinct pathways of cell death following neonatal hypoxic-ischemic brain injury. J Biol Chem. 2002;10:10. doi: 10.1074/jbc.M202931200. [DOI] [PubMed] [Google Scholar]
- Hee Han B, Choi J, Holtzman DM. Evidence that p38 mitogen-activated protein kinase contributes to neonatal hypoxic-ischemic brain injury. Dev Neurosci. 2002;24:405–410. doi: 10.1159/000069046. [DOI] [PubMed] [Google Scholar]
- Heidenreich KA, Kummer JL. Inhibition of p38 mitogen-activated protein kinase by insulin in cultured fetal neurons. J Biol Chem. 1996;271:9891–9894. doi: 10.1074/jbc.271.17.9891. [DOI] [PubMed] [Google Scholar]
- Hu BR, Liu CL, Ouyang Y, Blomgren K, Siesjo BK. Involvement of caspase-3 in cell death after hypoxia-ischemia declines during brain maturation. J Cereb Blood Flow Metab. 2000;20:1294–1300. doi: 10.1097/00004647-200009000-00003. [DOI] [PubMed] [Google Scholar]
- Ivacko JA, Sun R, Silverstein FS. Hypoxic-ischemic brain injury induces an acute microglial reaction in perinatal rats. Pediatr Res. 1996;39:39–47. doi: 10.1203/00006450-199601000-00006. [DOI] [PubMed] [Google Scholar]
- Kanno T, Nagata T, Yamamoto S, Okamura H, Nishizaki T. Interleukin-18 stimulates synaptically released glutamate and enhances postsynaptic AMPA receptor responses in the CA1 region of mouse hippocampal slices. Brain Res. 2004;1012:190–193. doi: 10.1016/j.brainres.2004.03.065. [DOI] [PubMed] [Google Scholar]
- Kaur J, Woodman RC, Kubes P. P38 MAPK: critical molecule in thrombin-induced NF-kappa B-dependent leukocyte recruitment. Am J Physiol Heart Circ Physiol. 2003;284:H1095–H1103. doi: 10.1152/ajpheart.00016.2002. [DOI] [PubMed] [Google Scholar]
- Kummer JL, Rao PK, Heidenreich KA. Apoptosis induced by withdrawal of trophic factors is mediated by p38 mitogen-activated protein kinase. J Biol Chem. 1997;272:20490–20494. doi: 10.1074/jbc.272.33.20490. [DOI] [PubMed] [Google Scholar]
- Lee YB, Schrader JW, Kim SU. p38 map kinase regulates TNF-alpha production in human astrocytes and microglia by multiple mechanisms. Cytokine. 2000;12:874–880. doi: 10.1006/cyto.2000.0688. [DOI] [PubMed] [Google Scholar]
- Legos JJ, Erhardt JA, White RF, Lenhard SC, Chandra S, Parsons AA, Tuma RF, Barone FC. SB 239063, a novel p38 inhibitor, attenuates early neuronal injury followingischemia. Brain Res. 2001;892:70–77. doi: 10.1016/s0006-8993(00)03228-5. [DOI] [PubMed] [Google Scholar]
- Lin S, Zhang Y, Dodel R, Farlow MR, Paul SM, Du Y. Minocycline blocks nitric oxide-induced neurotoxicity by inhibition p38 MAP kinase in rat cerebellar granule neurons. Neurosci Lett. 2001;315:61–64. doi: 10.1016/s0304-3940(01)02324-2. [DOI] [PubMed] [Google Scholar]
- Manabat C, Han BH, Wendland M, Derugin N, Fox CK, Choi J, Holtzman DM, Ferriero DM, Vexler ZS. Reperfusion differentially induces caspase-3 activation in ischemic core and penumbra after stroke in immature brain. Stroke. 2003;34:207–213. doi: 10.1161/01.STR.0000047101.87575.3C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McRae A, Gilland E, Bona E, Hagberg H. Microglia activation after neonatal hypoxic-ischemia. Brain Res Dev Brain Res. 1995;84:245–252. doi: 10.1016/0165-3806(94)00177-2. [DOI] [PubMed] [Google Scholar]
- Mielke K, Brecht S, Dorst A, Herdegen T. Activity and expression of JNK1, p38 and ERK kinases, c-Jun N-terminal phosphorylation, and c-jun promoter binding in the adult rat brain followingkainate-induced seizures. Neuroscience. 1999;91:471–483. doi: 10.1016/s0306-4522(98)00667-8. [DOI] [PubMed] [Google Scholar]
- Popivanova BK, Koike K, Tonchev AB, Ishida Y, Kondo T, Ogawa S, Mukaida N, Inoue M, Yamashima T. Accumulation of microglial cells expressing ELR motif-positive CXC chemokines and their receptor CXCR2 in monkey hippocampus after ischemia-reperfusion. Brain Res. 2003;970:195–204. doi: 10.1016/s0006-8993(03)02343-6. [DOI] [PubMed] [Google Scholar]
- Popovic N, Schubart A, Goetz BD, Zhang SC, Linington C, Duncan ID. Inhibition of autoimmune encephalomyelitis by a tetracycline. Ann Neurol. 2002;51:215–223. doi: 10.1002/ana.10092. [DOI] [PubMed] [Google Scholar]
- Raivich G, Bohatschek M, Kloss CU, Werner A, Jones LL, Kreutzberg GW. Neuroglial activation repertoire in the injured brain: graded response, molecular mechanisms and cues to physiological function. Brain Res Brain Res Rev. 1999;30:77–105. doi: 10.1016/s0165-0173(99)00007-7. [DOI] [PubMed] [Google Scholar]
- Saccani S, Pantano S, Natoli G. p38-dependent marking of inflammatory genes for increased NF-kappa B recruitment. Nat Immunol. 2002;3:69–75. doi: 10.1038/ni748. [DOI] [PubMed] [Google Scholar]
- Suk K. Minocycline suppresses hypoxic activation of rodent microglia in culture. Neurosci Lett. 2004;366:167–171. doi: 10.1016/j.neulet.2004.05.038. [DOI] [PubMed] [Google Scholar]
- Takagi Y, Nozaki K, Sugino T, Hattori T, Hashimoto N. Phosphorylation of c-Jun NH(2)-terminal kinase and p38 mitogen-activated protein kinase after transient forebrain ischemia in mice (in process citation) Neurosci Lett. 2000;294:117–120. doi: 10.1016/s0304-3940(00)01552-4. [DOI] [PubMed] [Google Scholar]
- Ten VS, Bradley-Moore M, Gingrich JA, Stark RI, Pinsky DJ. Brain injury and neurofunctional deficit in neonatal mice with hypoxic-ischemic encephalopathy. Behav Brain Res. 2003;145:209–219. doi: 10.1016/s0166-4328(03)00146-3. [DOI] [PubMed] [Google Scholar]
- Ten VS, Wu EX, Tang H, Bradley-Moore M, Fedarau MV, Ratner VI, Stark RI, Gingrich JA, Pinsky DJ. Late measures of brain injury after neonatal hypoxia-ischemia in mice. Stroke. 2004;35:2183–2188. doi: 10.1161/01.STR.0000137768.25203.df. [DOI] [PubMed] [Google Scholar]
- Teng YD, Choi H, Onario RC, Zhu S, Desilets FC, Lan S, Woodard EJ, Snyder EY, Eichler ME, Friedlander RM. Minocycline inhibits contusion-triggered mitochondrial cytochrome c release and mitigates functional deficits after spinal cord injury. Proc Natl Acad Sci USA. 2004;101:3071–3076. doi: 10.1073/pnas.0306239101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas M, Le WD. Minocycline: neuroprotective mechanisms in Parkinson’s disease. Curr Pharm Des. 2004;10:679–686. doi: 10.2174/1381612043453162. [DOI] [PubMed] [Google Scholar]
- Thomas M, Le WD, Jankovic J. Minocycline and other tetracycline derivatives: a neuroprotective strategy in Parkinson’s disease and Huntington’s disease. Clin Neuropharmacol. 2003;26:18–23. doi: 10.1097/00002826-200301000-00005. [DOI] [PubMed] [Google Scholar]
- Tikka T, Fiebich BL, Goldsteins G, Keinanen R, Koistinaho J. Minocycline, a tetracycline derivative, is neuroprotective against excitotoxicity by inhibiting activation and proliferation of microglia. J Neurosci. 2001a;21:2580–2588. doi: 10.1523/JNEUROSCI.21-08-02580.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tikka T, Usenius T, Tenhunen M, Keinanen R, Koistinaho J. Tetracycline derivatives and ceftriaxone, a cephalosporin antibiotic, protect neurons against apoptosis induced by ionizing radiation. J Neurochem. 2001b;78:1409–1414. doi: 10.1046/j.1471-4159.2001.00543.x. [DOI] [PubMed] [Google Scholar]
- Tsuji M, Wilson MA, Lange MS, Johnston MV. Minocycline worsens hypoxic-ischemic brain injury in a neonatal mouse model. Exp Neurol. 2004;189:58–65. doi: 10.1016/j.expneurol.2004.01.011. [DOI] [PubMed] [Google Scholar]
- Wang CX, Yang T, Shuaib A. Effects of minocycline alone and in combination with mild hypothermia in embolic stroke. Brain Res. 2003;963:327–329. doi: 10.1016/s0006-8993(02)04045-3. [DOI] [PubMed] [Google Scholar]
- West MJ, Slomianka L, Gundersen HJ. Unbiased stereological estimation of the total number of neurons in the subdivisions of the rat hippocampus using the optical fractionator. Anat Rec. 1991;231:482–497. doi: 10.1002/ar.1092310411. [DOI] [PubMed] [Google Scholar]
- Xu H, Barks JD, Schielke GP, Silverstein FS. Attenuation of hypoxia-ischemia-induced monocyte chemoattractant protein-1 expression in brain of neonatal mice deficient in interleukin-1 converting enzyme. Brain Res Mol Brain Res. 2001;90:57–67. doi: 10.1016/s0169-328x(01)00087-0. [DOI] [PubMed] [Google Scholar]
- Yager JY, Thornhill JA. The effect of age on susceptibility to hypoxic-ischemic brain damage. Neurosci Biobehav Rev. 1997;21:167–174. doi: 10.1016/s0149-7634(96)00006-1. [DOI] [PubMed] [Google Scholar]
- Yrjanheikki J, Keinanen R, Pellikka M, Hokfelt T, Koistinaho J. Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. Proc Natl Acad Sci USA. 1998;95:15769–15774. doi: 10.1073/pnas.95.26.15769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yrjanheikki J, Tikka T, Keinanen R, Goldsteins G, Chan PH, Koistinaho J. A tetracycline derivative, minocycline, reduces inflammation and protects against focal cerebral ischemia with a wide therapeutic window. Proc Natl Acad Sci USA. 1999;96:13496–13500. doi: 10.1073/pnas.96.23.13496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, Stavrovskaya IG, Drozda M, Kim BY, Ona V, Li M, Sarang S, Liu AS, Hartley DM, Wu du C, Gullans S, Ferrante RJ, Przedborski S, Kristal BS, Friedlander RM. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature. 2002;417:74–78. doi: 10.1038/417074a. [DOI] [PubMed] [Google Scholar]