

Abstract
Macrophages can be both beneficial and detrimental after CNS injury. We previously showed rapid accumulation of macrophages in injured immature brain acutely after ischemia-reperfusion. To determine whether these macrophages are microglia or invading monocytes, we subjected post-natal day 7 (P7) rats to transient 3 h middle cerebral artery (MCA) occlusion and used flow cytometry at 24 and 48 h post-reperfusion to distinguish invading monocytes (CD45high/CD11b+) from microglia (CD45low/medium/CD11b+). Inflammatory cytokines and chemokines were determined in plasma, injured and contralateral tissue 1–24 h post-reperfusion using ELISA-based cytokine multiplex assays. At 24 h, the number of CD45+/CD11b+ cells increased 3-fold in injured compared to uninjured brain tissue and CD45 expression shifted from low to medium with less than 10% of the population expressing CD45high. MCA occlusion induced rapid and transient asynchronous increases in the pro-inflammatory cytokine IL-β and chemokines cytokine-induced neutrophil chemoattractant protein 1 (CINC-1) and monocyte-chemoattractant protein 1 (MCP-1), first in systemic circulation and then in injured brain. Double immunofluorescence with cell-type specific markers showed that multiple cell types in the injured brain produce MCP-1. Our findings show that despite profound increases in MCP-1 in injured regions, monocyte infiltration is low and the majority of macrophages in acutely injured regions are microglia.
Keywords: cytokines, flow cytometry, inflammation, macrophage, microglia, neonatal stroke
Inflammation is a significant contributing factor to neurodegenerative diseases. Depending on injury setting, the relative involvement of systemic and local inflammation, and communication between the two compartments can vary (Carson and Sutcliffe 1999; Baggiolini 2001; Perry 2004). It is well known that stroke in adult triggers a robust inflammatory reaction, largely involving an influx of peripheral leukocytes into the brain parenchyma (for reviews, see Feuerstein et al. 1997; Dirnagl et al. 1999; Han and Yenari 2003) and disruption of the blood–brain barrier (BBB) (Gidday et al. 2005). In the adult, neutrophils are typically the first leukocyte type to infiltrate (Barone et al. 1991; Garcia et al. 1994; Matsuo et al. 1995) and are followed by macrophages and lymphocytes (Barone et al. 1991; Garcia et al. 1994). The damaging role of infiltrating neutrophils in ischemia was shown by studies in which neutrophil depletion (Garcia et al. 1994) or administration of anti-adhesion molecules (Kishimoto and Rothlein 1994; Fassbender et al. 1999) reversed the reduced local tissue perfusion, BBB disruption, and the release of free radicals, proteinases, and other cytotoxins seen after stroke. The key role of macrophages in cerebral ischemic injury has also been firmly established (Feuerstein et al. 1997; Dirnagl et al. 1999; Han and Yenari 2003). Macrophage populations in the injured brain, however, are diverse (Carson et al. 1998; Dalmau et al. 2003) and may consist of infiltrating peripheral monocytes, pericytes, and activated parenchymal microglia. The relative contribution of resident and invading cells may vary depending on injury severity in adult, as is evident from the different dynamics of macrophage accumulation (Morioka et al. 1993; Davies et al. 1998) and increased expression of the common leukocyte antigen CD45 and α2 integrin CD11b (Campanella et al. 2002; Stevens et al. 2002) in the tissue after ischemia of different severity. Activated microglial cells are believed to play a significant role in neuro-inflammation after cerebral ischemia (Liu et al. 2001; Rosenzweig et al. 2004). Distinguishing monocytes from activated microglial cells based on differential antigen expression levels is difficult using conventional immunohistochemical techniques, but can be accomplished using flow cytometry. An understanding of the relative contribution of cells of the monocytic lineage in injury is important for directing the search for novel therapeutic targets.
Compared to the adult, brain susceptibility and the mechanisms of ischemia-related injury are critically affected by immaturity during post-natal brain development (reviewed in Vannucci and Hagberg 2004; Vexler and Ferriero 2006) but less is known regarding the role of the inflammatory component of injury in the neonate. In postnatal day 7 (P7)-P9 rodents, an age when the rat brain is developmentally equivalent to the term human infant brain, up-regulation of structural proteins specific for cells of the monocyte lineage, activated microglia/macrophages, and products of these cells, including cytokines, chemokines and matrix metalloproteinases, have been described following hypoxia-ischemia (McRae et al. 1995; Ivacko et al.1996; Bona et al. 1999; Xu et al. 2001; Cowell et al. 2002; Hedtjarn et al. 2004). Different than in adult status of microglial differentiation (Santambrogio et al. 2001) and activation (Carson et al. 1998) in the early post-natal period, as well as differences in leukocyte trafficking during normal post-natal development (Lorant et al. 1999) and injurious conditions (Anthony et al. 1997, 1998 Hudome et al. 1997) may substantially affect the rapid accumulation of activated microglia/macrophages in injured tissue that we observed following focal transient middle cerebral artery (MCA) occlusion in P7 rats (Derugin et al. 2000; Fox et al. 2005; Dingman et al. 2006).
It is an entirely unanswered question whether macrophages arise from activation of resident microglia or infiltration of peripheral monocytes in neonatal brain post-ischemia. We used a model of transient MCA occlusion in P7 rats in conjunction with flow cytometry of peripheral and brain leukocytes to determine the relative roles of resident microglia and blood-born macrophages in neonatal stroke. ELISA-based multiplex assays of cytokine and chemokine levels in blood and brain were used to investigate whether inflammatory molecules, including chemoattractants for monocytes, are produced in the brain following neonatal stroke. We found that within the first 24 h after ischemia–reperfusion in the neonate, microglia rather than invading monocytes comprise the macrophage population in injured brain and that production of leukocyte chemoattractants is increased in injured tissue.
Materials and methods
Animal preparation
All experimental procedures were performed in accordance with NIH guidelines for humane handling of animals with prior approval from the Committee of Animal Research at UCSF. Female Sprague Dawley rats with 5–6-day-old-litters were obtained from Simonson Laboratories (Gillroy, CA), given food and water ad libitum and housed in a temperature/light controlled animal care facility. P7 rats were subjected to 3 h MCA occlusion, as originally reported (Derugin et al. 1998) with modifications (Derugin et al. 2005). All pups were screened for evidence of injury using diffusion-weighted (DW) MRI 2 h after MCA occlusion and animals with hyperintensity on DW-MRI were selected for experiments (Derugin et al. 2000; Manabat et al. 2003). At 3 h the suture blocking the MCA was removed, allowing reperfusion, and the pups were returned to the dam. Animals were killed 0–48 h after reperfusion.
Isolation of peripheral leukocytes and brain cells
Trunk blood was collected into tubes containing 200 units Heparin (Elkins-Sinn Inc., Cherry Hill, NJ, USA). Following lysis of RBC with NH4Cl PharM Lyse buffer (BD Pharmigen, rt × 15 min), leukocytes were collected by centrifugation (250 g × 5 min), washed in Hanks Balanced Salt Solution (HBSS) containing 1% BSA (flow buffer) and resuspended in 100 µL buffer and placed on ice.
Injured brain tissue, identified based on abnormalities on DW-MRI during occlusion (Derugin et al. 2000; Fox et al. 2005) and anatomically matching tissue from contralateral hemispheres were isolated according to published protocols for adult brain (Carson et al. 1998; Campanella et al. 2002; Stevens et al. 2002) with modifications. All steps were performed at 4°C unless indicated. Animals were perfused intracardially with sterile phosphate-buffered saline (PBS) and euthanized by decapitation. Tissue was placed directly into microfuge tubes containing iced RPMI-1640/25 mM Hepes pre-equilibrated with 5% CO2, homogenized by trituration, and Blendzyme 1 (Roche Applied Sciences, Indianapolis, IN, USA), an endotoxin-free blend of collagenase Types 1 and 2 and the neutral protease Dispase, was added to a final concentration of 45 µg/mL. Gentle isolation with low concentration of Dispase used in our protocol is not associated with degradation of surface markers which was reported using a 100 fold higher Dispase concentration (Ford et al. 1996). Tissue homogenates were incubated (30°C × 20 min) with gentle, discontinuous shaking, placed on ice, and enzyme activity was quenched with Fetal Calf Serum (FCS) (10% final concentration). Large debris was removed by filtration over 70 µm filters and cell suspensions were washed with flow buffer. In pilot experiments, CD45/CD11b expression was compared in samples from the same animals obtained with or without use of Percoll gradient purification in which cells were overlayed onto 1.02/1.10 g/mL Percoll gradients (400 g × 15 min without brake).
Flow cytometry
All steps were performed on ice or at 4°C unless indicated. For both blood and brain samples, cell surface FcR were blocked (gentle rocking × 15 min) with anti-FcgII R antibody followed by double-labeling of cell surface markers with FITC- and PE-conjugated specific and isotype control antibodies. Unlabeled FcgII R antibody (Fc Block), PE- or FITC-conjugated mouse anti-rat CD45, FITC-CD11b, PE-RP-1 and isotype matched controls were from BD Biosciences (San Diego, CA, USA), PE-CD163 and isotype matched control were from Serotec (Oxford, UK). In each experiment, data from equal numbers of events (10 000–100 000) from control and injured animals (blood) or contralateral and injured brain hemispheres were collected on a FACSCalibur flow cytometer running CellQuest Pro software (BD Biosciences) with the following settings for blood, minor adjustments made as needed: Forward Scotter (FSC) V = E^00, gain = 1.2; Side Scatter (SSC) V = 460, gain = 1.05; thresholding on FSC of 52; compensation FL1 = 1–2%, FL2 = 15–17%. For tissue, modifications to the settings were FSC gain = 1.04; secondary thresholding on SSC of 53; compensation FL2 = 15–25%. Results were analyzed with FlowJo software (Tree Star Inc., Ashland, OR, USA) and are presented for CD45-gated populations. For leukocytes, SSC of 150 was used as the threshold for high versus low SSC populations, for tissue SSC of 250 was used since a clear distinction within the cell populations was detected at these values. Using isotype labeled cells in each experiment, a gate was drawn containing at least 95% of isotype labeled cells; the population of cells with staining intensity to the right of this gate was classified as CD45 positive (containing < 5% isotype labeled cells). Assignment of CD45 subpopulations into CD45low, med and high populations was based on the boundaries of five percent contour maps (CD45 vs. SSC plots for leukocytes, CD45 vs. FSC plots in control samples) generated by FlowJo software. The number of experiments performed on samples from individual or pooled animals is indicated in figure legends.
Immunocytochemistry
Brains were flash-frozen, stored at − 80°C, and cut coronally (12 µm) on a cryostat and placed on Superfrost Plus glass slides (Fisher Scientific Publishing, Pittsburg, PA, USA). Slides were thawed overnight, fixed in − 20°C acetone × 10 min, washed with PBS and blocked for 1 h in 10% FCS/0.1% Tx-100/.01% NaN3, as described (Banisadr et al. 2005). Sections were incubated with goat anti-rat MCP-1 (1 : 50, Santa Cruz, CA, USA) together with either mouse anti-GFAP (1 : 200, MP Biomedicals, Aurora, OH, USA) or anti-NeuN (1 : 100, Chemicon, Temecula, CA, USA) antibodies in 5% FCS/0.1% Tx-100/.01% NaN3 overnight at 4°C. Sections were washed with PBS, incubated for 1 h with Cy3-conjugated anti-goat and either Cy2-conjugated anti-mouse or Alexa 488-conjugated isolectin GS-B4 (1 : 100 in 5% FCS/0.1% Tx-100/.01% NaN3), stained with DAPI to label nuclei, washed and coverglassed using Polymount. Images were collected using a Hamamatsu camera and Openlab Software (Improvision, Lexington, MA, USA). Post-acquisition processing of images was performed using identical settings in Adobe Photoshop.
Measurements of cytokine and chemokine levels in plasma and the brain
Concentrations of 14 cytokines, including interleukin-4 (IL-4), IL-1β, IL-1α, IL-6, IL-10, IL-12, IL-18, IL-5, IL-2, interferon gamma (IFNγ), tumor necrosis factor α (TNFα), cytokine induced chemoattractant protein-1 (CINC1/GRO/KC), monocyte chemoattractant protein-1 (MCP-1) and GM-CSF were simultaneously quantified in plasma or brain tissue samples using an ELISA-based bead multiplex assay (Luminex Corp., Austin, TX, USA) as previously reported (Fox et al. 2005). Briefly, injured brain tissue and anatomically matching tissue from contralateral hemispheres as well as tissue from control and sham-operated rat pups was homogenized in a buffer containing 20 mm Tris-HCl (pH 7.5), 150 mm NaCl, 1 mm PMSF, 0.05% Tween-20, and a cocktail of protease inhibitors (Roche), protein concentration was measured using the BCA assay (Pierce, Rockford, IL, USA). Cytokine and chemokine concentrations were determined using a rat cytokine kit (LINCO Research, Inc., Saint Charles, MO, USA) based on the manufacturer’s recommendations. Fluorescent signals originating from cytokine-antibody binding followed by detection with streptavidin-phycoerythrin conjugates were measured with a Luminex100 reader. In each experiment, concentrations for each cytokine in the multiplex assay were calculated from calibration curves using individual recombinant proteins as standards dilued in lysis buffer (for tissue) or matrix (for plasma samples). Plasma samples were diluted (1 : 4) in matrix. StatLIA® software (Brendan Scientific Corp., Calrsbad, CA, USA) with a 5-parameter logistic curve-fitting method was used for data reduction, and tissue sample concentrations were normalized to the amount of protein in each sample. Concentrations of several cytokines, including IL-4, IL-1α, IL-10, IL-12, IL-5, IL-2, TNFα, IFNγ and GM-CSF were either consistently below or at the threshold of detection in more than 50% of the samples and therefore not shown.
Statistical Analysis
In flow cytometry experiments cell numbers were analyzed using a two-tailed student’s t-test (unpaired for blood, paired for tissue). Cytokine concentrations were analyzed by anova with post-hoc testing (Fisher). Differences were considered significant at p < 0.05.
Results
Ischemia-reperfusion triggers activation of peripheral leukocytes
To determine the peripheral leukocyte response to transient MCA occlusion, RBC-depleted leukocytes were analyzed by flow cytometry. In contour plots of CD45 intensity versus SSC, three distinct populations of cells were easily distinguishable in leukocytes from both control and injured animals (Figs 1a and b). Quantitative analysis showed a trend (ns) for greater numbers of CD45+ cells in blood collected from injured animals 24 h post-reperfusion (43 ± 10% events, n = 8) compared to uninjured (34 ± 10% events, n = 8) animals. Although no rightward curve shift in population histograms of CD45 intensity (Fig. 1c) was detected in leukocytes of injured animals, a medium intensity peak (middle horizontal bar) that appeared as a left shoulder in histograms of leukocytes from control animals clearly increased in magnitude and width with injury. Further analysis was restricted to CD45 gated cells, as these cells represent the total leukocyte population in the sample.
Fig. 1.
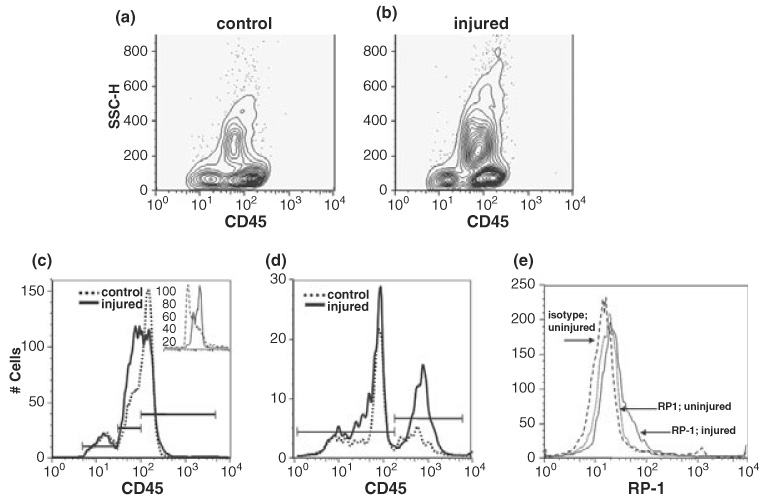
Peripheral leukocytes are activated 24 h after MCA occlusion in P7 rats. (a and b) Three populations of CD45+ cells are detected in CD45 vs. SSC contour plots. (c and d) Increases in CD45med (c) and CD11b (d) expression are detected with injury. In uninjured animals the percentage of CD45+ cells classified as low, med, or high (bars) is, respectively, 14 ± 7.7%, 36 ± 7.8%, and 50 ± 7.1% and in injured animals, these ratios shift to 10 ± 7%, 46 ± 4% and 44 ± 3.7. Statistically significant shifts as a result of injury occur in the CD45medium/CD11b+ positive populations (p = 0.05, n = 4). Histograms in this and all subsequent figures show CD45 gated cells. Bars in CD11b histograms represent negative and positive subpopulations. Inset in (c) shows isotype control antibody (dashed line) compared to CD45 antibody labeling of control (solid) samples. (e) MCA occlusion results in increased RP-1 expression. Quantification of RP-1 positive cells to the right of the isotype matched control antibody (dashed curve) indicates injury results in an increase in RP-1+ cells from 18 ± 9.7% to 22 ± 4.7% (solid curve, control; dotted, injured) of the CD45 population (p > 0.5, n = 3). Shown are data from a representative experiment.
To determine whether ischemia-reperfusion induces expression of the adhesion receptor for ICAM (CD11b/integrin αM) on leukocytes, a receptor needed for leukocyte–endothelium interaction, we colabeled cells with CD45 and CD11b. One major (FI < 10^2, CD11b low) and one minor (FI < 10^3, CD11b high) peak was detected in cells from uninjured animals (Fig. 1d). With injury, the number of cells expressing CD11b increased in both populations, particularly in the population with high signal intensity (at 10^3). In this population, CD11b expression increased significantly from 29 ± 4.8% to 39 ± 6.4% of CD45+ cells (Fig. 1d, p = 0.05, n = 4). CD45 Vs. SSC plots indicated that the population of CD45+ cells with high SSC (higher granularity) increased with injury, from 24 ± 5.3% to 40 ± 8.4% (p = 0.003, n = 6). In attempt to determine whether the observed increased SSC represented granulocytes, colabeling experiments were done with CD45 and RP-1, an antibody that recognizes granulocytes but not lymphocytes and monocytes with low SSC. Although there was considerable overlap in intensities between isotype control and specific RP-1 antibodies on leukocytes from control and injured animals, a shoulder in peak RP-1 intensity (< 10^2) was detected in RP-1 stained cells only from injured animals (Fig. 1e), indicating an increase in RP-1 expression after injury. This increased granularity (SSC) and RP-1 expression suggests activation of peripheral neutrophils. Importantly, alterations in CD45 expression and substantial increases in CD11b expression suggest that focal ischemia-reperfusion injury primes leukocytes to be competent to adhere to the endothelium and transmigrate through the microvasculature within 24 h after injury.
Activated microglia, not invading monocytes, are the source of brain macrophages 24 h post-reperfusion
Using immunocytochemical methods we have previously shown increased CD68/ED1 expression in the brain 24 h after neonatal focal stroke (Derugin et al. 2000; Fox et al. 2005; Dingman et al. 2006). To determine the effect of transient MCA occlusion on the pattern of CD45 expression in the brain, flow cytometry was used on cells from contralateral and injured tissue. In pilot experiments with control animals, we found the purification of dissociated cells by Percoll gradient centrifugation, a common component of cell isolation protocols in adult brain used to remove myelin and other debris, to be unnecessary with tissue from neonatal rats because P7 brain is not yet myelinated (Back and Rivkees 2004). Samples isolated either with or without Percoll gradients separated into similar populations in CD45 vs. FSC contour plots (data not shown) and two primary subpopulations, CD45low and CD45med. Between 1 and 3% of the population in each cell preparation was classified as CD45high. In all subsequent experiments cells were prepared without the use of Percoll gradients.
Compared to contralateral hemispheres, total CD45+ expression (before classifying into subpopulations) in injured hemispheres increased 3-fold (p < 0.001, n = 7). Thus, in contrast to peripheral leukocytes, the overall abundance of CD45+ cells increased significantly in the injured brain. A representative example of quadrant analysis of CD45/CD11b double-labeled cells in Fig. 2 shows that while in contralateral hemispheres, 80.9% of the cells were CD45low/ CD11b- (Fig. 2a, lower left quadrant) and only 7.1% were CD45med-high/CD11b+ (Fig. 2a, top right quadrant), 60.3% of the cells acquired CD45med-high/CD11b+ expression (Fig. 2b, top right quadrant) by 24 h following injury.
Fig. 2.
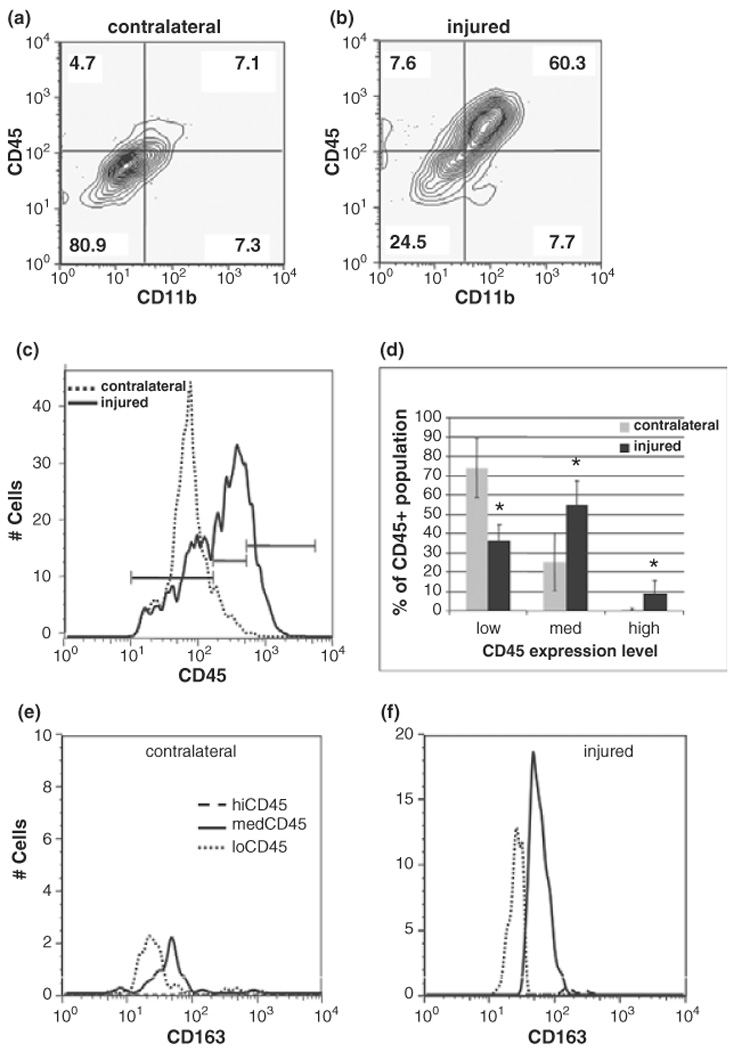
Macrophages in the brain are predominantly activated microglia, not invading monocytes, 24 h post-ischemia reperfusion. (a and b) The number of activated leukocytes (CD45+/CD11+) increases from 7.1% in uninjured (a) to 60.3% in injured tissue (b). Values represent the percent of CD45 gated cells present in that quadrant. Note that in quadrant analysis, CD45+ cells contain both CD45med and CD45high populations. Data are representative of 4 independent experiments. (c) CD45 histograms indicate a shift towards higher intensity and a broadening of the curve with injury. Bars indicate the boundaries of low, medium and high CD45 expression levels. Dotted = contralateral, solid = injured. (d) With injury, there is a shift in the CD45 population from CD45low (36 ± 7.9%) to CD45med (55 ± 12.3%). Less than 10% (8.8 ± 6.7%) of the population in injured tissue is CD45high. Data are expressed as mean ± SD, n = 11. *p < 0.005 compared to contralateral. Grey bars = contralateral, solid = injured. (e and f) Expression of monocytic lineage marker CD163 is rapidly increased on activated microglia 24 h post-reperfusion. In contralateral hemispheres, CD163 expression is minimal (e) whereas in injured tissue CD163 expression is induced in CD45med cells (f). Dotted line, CD45low; solid line, CD45med, dashed line, CD45high. Shown are data from a representative experiment, n = 5.
We then used differential CD45 expression to determine whether the accumulated macrophages are resident or invading cells. A distinction between blood-borne macrophages and parenchymal cells of monocytic lineage (including quiescent or activated resident brain microglia) has been established in adult rodents based on differential CD45 expression levels: high CD45 expression levels in blood-born monocytes in contrast to low CD45 expression in quiescent microglial and a gradual increase in CD45 expression to medium-high levels during microglia activation (Carson et al. 1998; Campanella et al. 2002; Stevens et al. 2002). Contour plots delineated the presence of three peaks; CD45 low, med and high (not shown). Compared to contralateral hemispheres, where one major peak of CD45low (FI < 10^2) was observed (Fig. 2c), in injured tissue, a decrease in the CD45low peak, a clear rightward shift (up to and including FI = 10^3), and the resolution of two peaks, both CD45low and CD45med (10^2 < FI < 10^3) were detected 24 h post-reperfusion. Averaged data (Fig. 2d) indicated that cells from contralateral hemispheres expressed primarily CD45low (74 ± 15%) or CD45med (25 ± 15%) while CD45high macrophages represented less than 1% of the population. With injury, there was a 2-fold decrease in the abundance of cells expressing CD45low and a concomitant 2.2-fold increase in cells expressing CD45med (Fig. 2d), indicating a shift from quiescent to activated microglia. Following injury, the population of CD45high cells comprised less than 10% cells and varied from animal to animal (8.8 ± 6.7%).
We then colabeled CD45+ cells with a specific monocytic lineage marker, CD163, to confirm the lineage of cells in our experiments. CD163, originally identified as the hemoglobin scavenger receptor (Buechler et al. 2000), is constitutively expressed on tissue and perivascular macrophages and is up-regulated on microglia following various injurious conditions (Buechler et al. 2000; Roberts et al. 2004; Fabriek et al. 2005). CD163 expression was low on CD45low cells (Fig. 2e). Compared to contralateral hemispheres, in injured tissue there was an increase in the total number of CD45+/CD163+ double positive cells (49 ± 6.2% vs. 73 ± 8.4%, p = 0.013, n = 5) and a robust (10-fold) increase in CD163 expression within the CD45med population (FI > 10^2) (Fig. 2f). These data demonstrate not only increased CD45+/CD163+ cell numbers after injury, but also a higher cell surface expression of CD163 specifically in the population representing activated microglia.
Injury evolution is associated with further macrophage accumulation
At 48 h post-reperfusion, the distribution profile of CD45 was distinct from that at 24 h post-reperfusion (Fig. 3). The abundance of CD45high cells in injured tissue was substantially increased (reached approximately 30%, p = 0.02, n = 4, Fig. 3b). In addition, CD11b expression increased nearly 5-fold and quadrant analysis of contour plots (defined as for 24 h) from CD45/CD11b double-labeled cells indicated a shift in the CD45med/high/CD11b+ population from ~12% to 70% with injury (data not shown). In contralateral hemispheres, distribution among the three CD45 subpopulations was similar to that at 24 h (compare Fig 2d and Fig 3b). We did not observe accumulation of CD11b-/CD45low lymphocytes (Campanella et al. 2002), a finding consistent with our previous H&E findings (Derugin et al. 2005). In fact, this population decreased 24–48 h post-reperfusion.
Fig. 3.
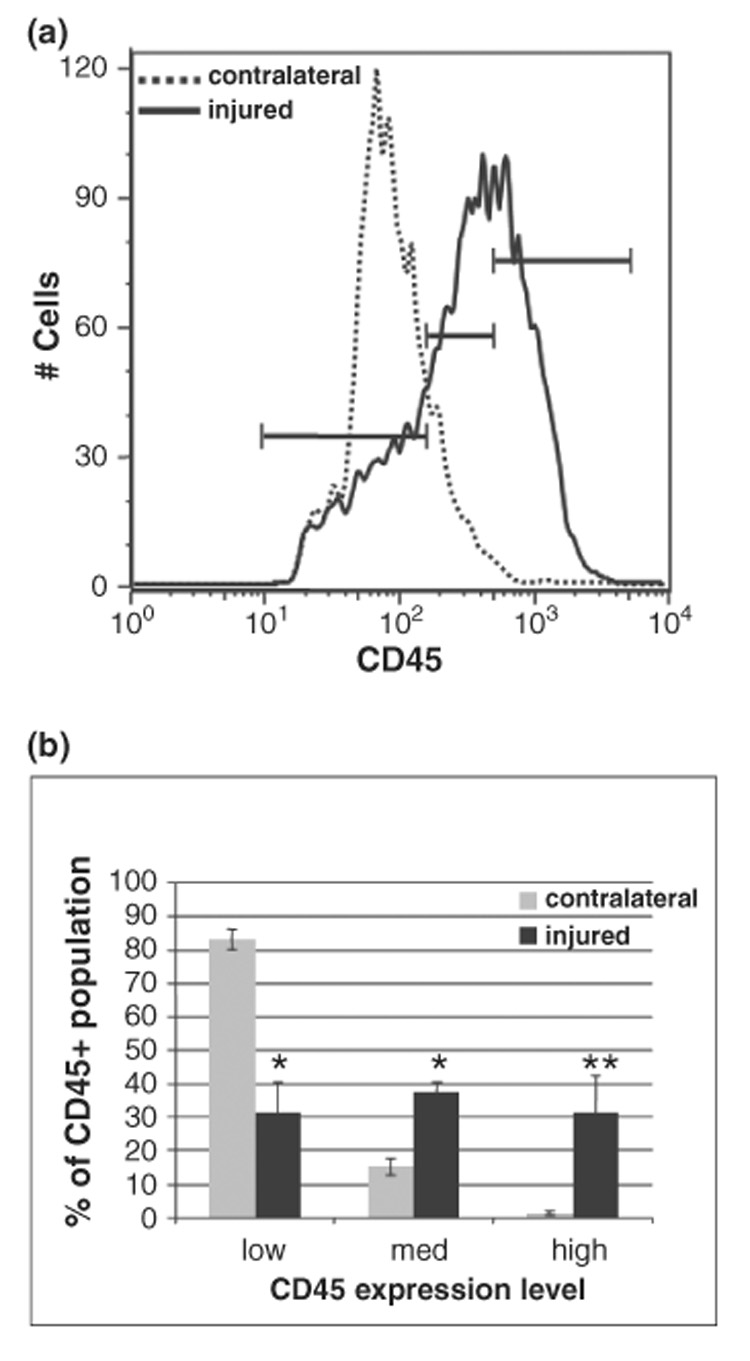
Macrophages continue to accumulate at 48 h post-reperfusion. (a) CD45 histograms show a rightward curve shift with injury evolution. (b) CD45low cells continue to predominate in contralateral hemispheres (85 ± 3.9%, 14 ± 3.4%, 1.2 ± 0.6%, respectively, for low, med and high expression; n = 5), whereas with injury the distribution among the three subpopulations changes dramatically (31 ± 7.3%, 39 ± 3.0% and 30 ± 9%, respectively, for low, med and high; n = 4). Mean ± SD. *p < 0.005 for low, med; **p < 0.02 for high populations compared to contralateral.
Consistent with single labeled CD45 cells, the number of CD45/CD163 expressing cells increased in injured, but not in contralateral hemispheres, predominantly in CD45med activated microglia (not shown). Taken together these data demonstrate that beyond the acute injury phase accumulated macrophages are comprised of both activated microglia and increasing numbers of invading monocytes.
Peripheral and local cytokine levels in injured tissue increase rapidly and robustly after ischemia-reperfusion
Because leukocyte infiltration depends on a gradient of specific chemoattractant proteins, we next determined the effect of transient MCA occlusion on levels of 14 cytokines and chemokines in plasma, and injured and uninjured brain regions. Levels of cytokines that were within detectible levels are shown in Fig. 4. In plasma, the levels of cytokines IL-1β, IL-6 and the chemokine CINC-1 were significantly increased by 3 h MCA occlusion over control levels (2503 ± 1452 Vs. 241 ± 132 pg/mL for IL-1β, p < 0.04; 1998 ± 631 vs. 403 ± 265 pg/mL, p < 0.03 for IL-6; 4350 ± 2698 vs. 366 ± 209 pg/mL, p < 0.015 for CINC-1) (Fig. 4a). The levels of MCP-1 (625 ± 289 vs. 439 ± 148 pg/mL in control, p > 0.05) and IL-18 were not significantly changed at this time point. Upon reperfusion, systemic levels of IL-1β, IL-6, CINC-1, and MCP-1 increased further (Fig. 4a) and reached a maximum at 1 h post-reperfusion for IL-6 (2736 ± 1023 pg/mL, p < 0.0001 vs. control), CINC-1 (8721 ± 5720 pg/mL, p < 0.0001 vs. control) and MCP-1 (1104 ± 363 pg/mL, p < 0.0001 vs. control), and depending on the cytokine, returned to normal levels by 8–24 h post-reperfusion. IL-1β continued to increase over 4 and 8 h post-reperfusion (9384 ± 2610 and 7024 ± 2658 pg/mL, respectively, p < 0.0001 vs. control).
Fig. 4.
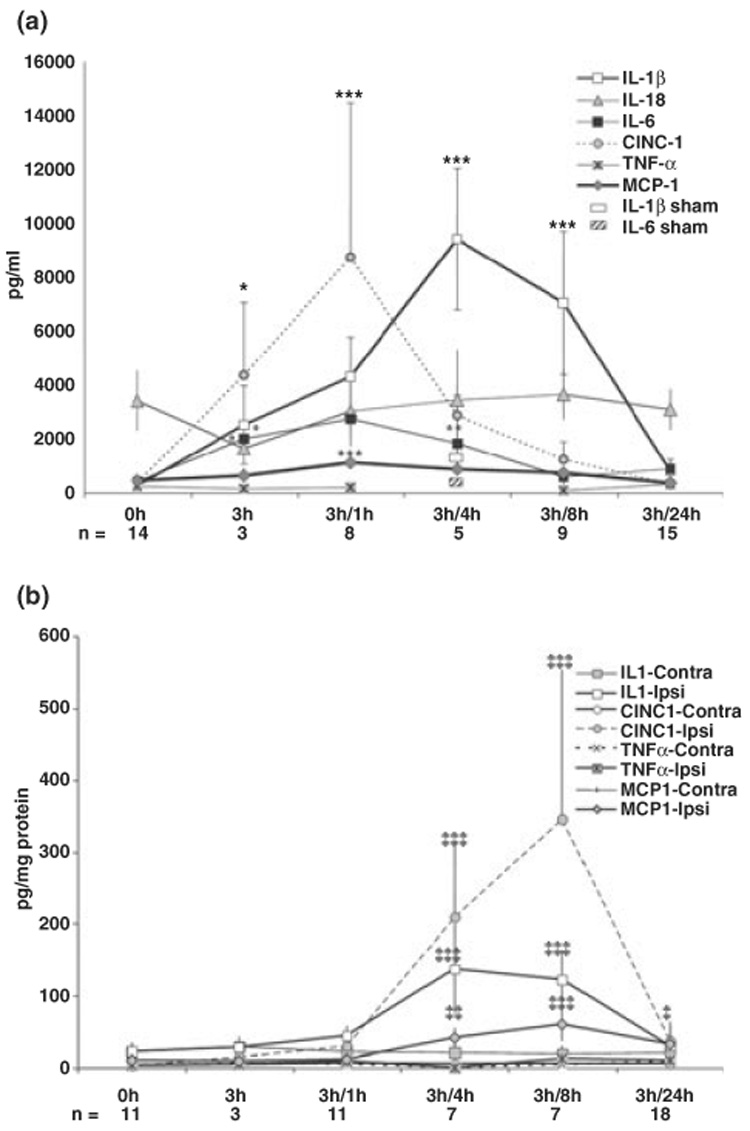
Cytokine and chemokine levels are increased in plasma and injured brain tissue but not in contralateral hemisphere within 24 h after reperfusion. A significant transient increase in IL-1β, IL-6, CINC-1, and MCP-1 occurs first in plasma (a, 0–8 h post-reperfusion) and then in the injured brain (b, 4–8 h post-reperfusion). Measurements were performed using the multiplex cytokine kit (LINCO Research) in animals identified by DW-MRI as injured. Data are expressed as mean ± SD, n = 3–17 per time point, as indicated for individual time points. In a: *p < 0.04 vs. control; **p < 0.015 vs. control; ***p < 0.0001 vs. control. In b: §p < 0.04; §§p < 0.01; §§§p < 0.001 vs. value in the corresponding contralateral hemisphere.
Compared to the response in the blood, the kinetics of the response in the tissue differed (Fig. 4b). IL-1β, IL-18, MCP-1 and CINC-1 in contralateral hemispheres remained unchanged over 24 h post-reperfusion compared to levels in naïve controls. Compared to contralateral tissue, in injured tissue, levels of IL-1β, MCP-1, and CINC-1 were significantly and transiently increased (Fig. 4b). Both local and systemic IL-1β peaked 4–8 h post-reperfusion. However, levels of chemokines MCP-1 and CINC-1 increased later in the brain than in plasma, beginning at 1–4 h after reperfusion, peaking when plasma levels of these chemokines were largely restored, 8 h after reperfusion. The levels of MCP-1 compared to corresponding contralateral levels (in pg/mg) were 8.9 ± 2.7 vs. 9.2 ± 2.8 (ns), 11.3 ± 5.9 vs. 6.3 ± 1.7 (ns), 41.1 ± 17.0 vs. 6.8 ± 2.0 (p < 0.0005), 59.3 ± 22.8 vs. 5.6 ± 3.4 (p < 0.0001), and 31.8 ± 15.3 vs. 7.8 ± 7.8 (p < 0.01) pg/mg protein at 0, 1, 4, 8, and 24 h post-reperfusion, respectively. These data demonstrate the existence of an ischemia-induced chemokine gradient governing leukocyte infiltration from blood to brain, including the major monocyte chemoattractant MCP-1.
MCP-1 expression is elevated in a subset of cells in the injured brain
We then used double-labeling immunofluorescence to determine whether the presence of elevated MCP-1 levels in injured brain was due to local production of MCP-1 in injured tissue. MCP-1 was weakly expressed in contralateral hemispheres (Figs 5a and b) anatomically matched to injured brain regions (Fig. 5c–f). Expression was evident primarily in the vasculature in uninjured hemispheres (Figs 5a and b) and in several cell types within injured tissue, including astrocytes (Figs 5c and e), macrophages (Figs 5d and f) and neurons (data not shown). While the majority of GFAP-immunoreactive astrocytes expressed MCP-1 in the injured region at 8 h post-reperfusion (Fig. 5c, arrows), this cell population was not seen in the core at 24 h (Fig. 5e, arrowheads). However, reactive astrocytes with thickened processes were abundant within the penumbra and a subpopulation of them expressed MCP-1 (Fig. 5e, arrow).
Fig. 5.
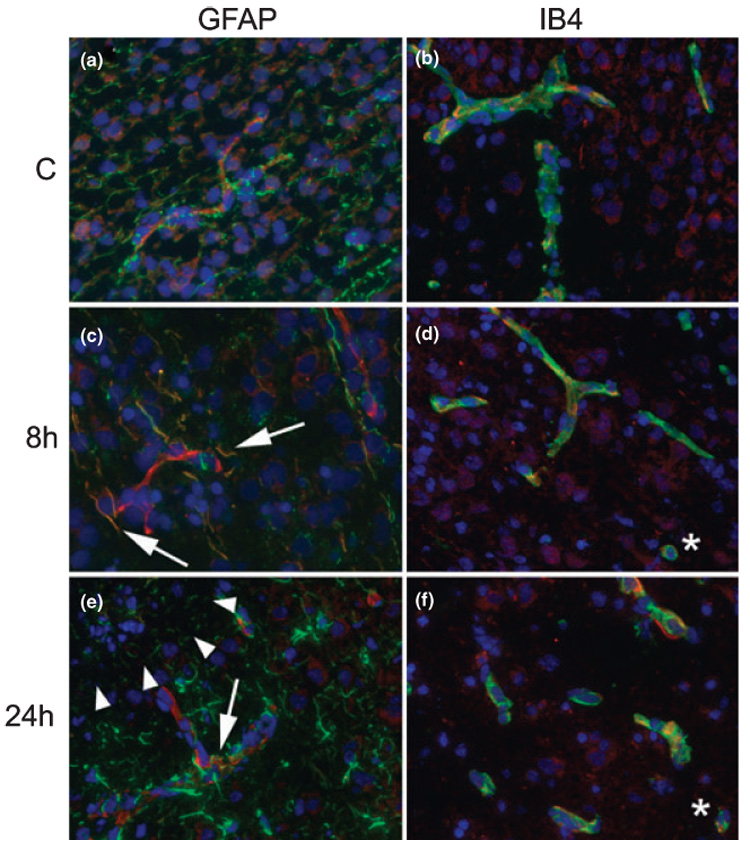
MCP-1 expression is elevated in multiple cell types within injured regions. (a and b) MCP-1 expression is evident in the vasculature in contralateral hemisphere. (c and d) At 8 h post-reperfusion, MCP-1 is expressed in astrocyte processes (c), vessels and macrophages (d) in injured regions. (e and f) At 24 h post-reperfusion, continued expression is detected in the vasculature, astrocyte processes in the penumbra, and macrophages. Arrows in c and e indicate coexpression of MCP-1 with GFAP in astrocyte processes; asterisks in d and f indicate coexpression in cells that likely represent microglia. Arrowheads in e indicate the boundary between injury core and penumbra. All images were captured at 20 ×. In all images red = MCP-1 and blue = nuclei (DAPI), green = GFAP (a, c, e) or IB4 (b, d, f).
Discussion
Findings in this paper demonstrate for the first time that in neonatal focal stroke rapidly accumulated macrophages in ischemic-reperfused brain tissue are microglial cells rather than invading monocytes, a situation different from that reported in similar adult stroke models. Limited monocyte infiltration into acutely ischemic-reperfused neonatal brain does not result from lack of monocyte activation or lack of a monocyte chemoattractant gradient between the blood and injured brain regions, as CD45 and CD11b expression on circulating cells is increased and levels of multiple cytokines and leukocyte chemoattractant proteins, including MCP-1, are elevated in acutely injured neonatal brain.
Despite the traditional view that neonates have greater resistance to CNS injury compared to adults, it has become clear that immaturity in general as well as specific stages of brain development critically affect the susceptibility of the developing brain to hypoxic-ischemic insults and particular modes of neuronal death (reviewed in Vannucci and Hagberg 2004; Vexler and Ferriero 2006). The patterns of inflammation associated with brain injury are also age-dependent (reviewed in Hagberg and Mallard 2005; Vexler 2006). In a focal transient cerebral ischemia model in P7 rat (Derugin et al. 1998; Derugin et al. 2005), it is well documented that CD68-positive macrophages accumulate in injured tissue within hours after reperfusion (Derugin et al. 2000; Fox et al. 2005) and continue to increase over at least a one week period post-injury (Derugin et al. 2005; Dingman et al. 2006). However, the origin of these macrophages, whether invading monocytes or activated resident cells, is not known. This distinction is important for defining the relative contributions of systemic and local inflammatory responses to injury evolution.
Flow cytometry enables determination of cell surface marker expression on subpopulations of leukocytes based on differential expression levels of the common leukocyte antigen CD45; constitutive on peripheral leukocytes and low on resting microglia. Transient or a permanent MCA occlusion in adult animals results in both different extents and temporal patterns of CD45high/CD11b+ cell accumulation in the lesioned brain (Campanella et al. 2002; Stevens et al. 2002). In the present study, using a simplified and accelerated protocol for cell dissociation, we were able to discriminate between microglial cells (CD45low/CD45med) and infilltrated leukocytes (CD45high) within the brain. We found a progressive increase in CD45med cells at 24 and 48 h post-reperfusion, with a concomitant major decline in the number of CD45low cells in injured tissue. The number of CD45high cells remained below 10% at 24 h and reached approximately 30% by 48 h. Colabeling of CD45+ cells with CD163, an antigen selectively expressed on cells of monocyte lineage (Buechler et al. 2000; Roberts et al. 2004; Fabriek et al. 2005), confirmed the presence of activated microglia 24 h post-reperfusion and delineated the presence of a heterogeneous population of CD45high/CD163+ macrophages at 48 h, perhaps comprised of both activated microglia and invading macrophages.
Mechanisms that limit monocyte infiltration into acutely reperfused neonatal brain are yet to be understood. Leukocyte entry into the CNS can occur via multiple routes and is highly regulated under normal conditions (Ransohoff et al. 2003). Transmigration requires coordinated activation of leukocytes and endothelium, and the presence of appropriate chemoattractant gradients between the blood and brain. We found that expression of CD11b on peripheral leukocytes, an antigen required early in the transmigration process for ‘tethering’ and subsequent rolling along the endothelium, is significantly increased on peripheral leukocytes after ischemia-reperfusion. Other aspects of leukocyte activation during early post-natal period are poorly understood.
Maturation of the BBB (Saunders et al. 2000; Engelhardt 2003) can substantially affect leukocyte passage from blood to brain parenchyma. Despite the common belief that the BBB in the neonate is more permissive than in adult, tight junctions are present even during embryonic development (Kniesel et al. 1996) and there are barrier mechanisms in the fetal brain not found in the adult (Saunders et al. 1999). By birth, the BBB is functional with no fenestrations (Engelhardt 2003). Age at time of insult can critically affect leukocyte transmigration, as was evident from a more preserved BBB in P0 and adult rats compared to P21 rats following inflammatory challenge (intrastriatum injections of IL-1β, TNFα or CINC-1) (Anthony et al. 1997; Anthony et al. 1998; Schnell et al. 1999; Blamire et al. 2000).
An insufficient gradient of chemoattractant molecules in the brain and/or a limited engagement of G-protein coupled chemokine receptors on monocytes with the appropriate ligands may also adversely affect leukocyte extravasation. MCP-1 and its receptor, CCR2 (Charo et al. 1994), play a central role in monocyte transmigration into the brain, as demonstrated by profound deficits in the recruitment of monocytes to sites of inflammation and injury in CCR2 deleted mice (Gosling et al. 1999; Gerard and Rollins 2001; Huang et al. 2001). A variety of cells have been shown to express MCP-1 after focal ischemia (Che et al. 2001) and trauma (Glabinski et al. 1996) in both adult and newborn rodent brain following hypoxia-ischemia (Xu et al. 2001) and excitotoxic insult (Galasso et al. 2000b). A pathophysiologic role of MCP-1 in neonatal brain injury was demonstrated by protection of the neonatal brain following functional inactivation of MCP-1 post-insult (Galasso et al. 2000b). Our data show that MCP-1 brain levels are profoundly elevated, up to 11 fold, and that MCP-1 is produced by several brain cell types, including endothelial cells and astrocytes.
Altered temporal-spatial expression of cytokines is an important modulator of trafficking of subsets of leukocytes after injury. In adult stroke, the most studied cytokines, IL-1β and TNFα, have been found to exacerbate brain damage by both inducing neuronal injury directly and via induction of glial and endothelial cells, with a consequent production of additional cytokines/chemokines and up-regulation of adhesion molecules (reviewed in (Allan et al. 2005; Lovering and Zhang 2005)). In adult rats, IL-1β injection preferentially triggers recruitment of neutrophils and propagates cytokine expression, whereas TNFα injection induces infiltration of monocytes, not neutrophils, and does not induce cytokine production (Blond et al. 2002). In P7 rats, increases in IL-1β were both substantial and sustained after MCA occlusion but were not associated with neutrophil extravasation or increased BBB permeability acutely after transient MCA occlusion (Dingman et al. 2004). Importantly, although substantial increases in local levels of IL-1β, MCP-1 and CINC-1 and lack of detectible levels of the anti-inflammatory cytokine IL-10 are consistent with a robust pro-inflammatory response, the specific lack of TNFα induction may contribute to limited/delayed monocyte infiltration in ischemia-reperfusion.
Biological functions of cytokines are not limited to regulation of leukocyte trafficking but affect a broad-range of processes after injury. In hypoxia-ischemia in neonatal rodents, for example, pharmacological inhibition of cytokines, either by IL-ra (Hagberg et al. 1996) or neutralizing anti-MCP-1 antibody (Galasso et al. 2000a), or deficiency of inflammatory cytokines, such as IL-18, IL-6, or of the FAS receptor (Hagberg et al. 1996; Hedtjarn et al. 2002; Graham et al. 2004), reduce injury and diminish microglial activation. In the immature brain, activated microglia/macrophages contribute to hypoxic-ischemic, ischemia-reperfusion and excitotoxic injury via several mechanisms (for a review see Vexler et al. 2006). The production of toxic products by microglia, such as inflammatory cytokines and chemokines (Szaflarski et al. 1995; Hagberg et al. 1996; Cowell et al. 2002; Hedtjarn et al. 2002; Cowell et al. 2003; Campbell et al. 2005) and high levels of NO (Tsuji et al. 2000), up-regulation of FAS receptor (Northington et al. 2001; Graham et al. 2004), complement receptor (Cowell et al. 2003; Ten et al. 2003, 2004) and iNOS (Dingman et al. 2006) can, in turn, further activate microglia, potentially propagating tissue injury. Furthermore, reduction of injury when macrophage accumulation is diminished (Arvin et al. 2002; Dommergues et al. 2003) as opposed to unaffected injury severity when microglial activation is unchanged (Fox et al. 2005; Dingman et al. 2006) supports the notion that microglial activation is important in ischemic and excitotoxic injury in the immature brain. Taken together, our data on the relatively preserved BBB acutely after neonatal stroke (Dingman et al. 2004) and findings in this study that microglia cells rather than invading monocytes comprise the early macrophage population, and that local inflammation is profound suggest that the search for therapeutic agents designed to intervene with inflammatory processes early after neonatal stroke should be focused on molecules capable of crossing the BBB and accessing the brain parenchyma to target microglial cells.
Acknowledgements
This study was supported by NIH NS44025 and UCP R766-04 to Z.S.V. The authors thank Dr Scott Zamvil for critical reading of the manuscript and Joel Faustino for technical assistance.
Abbreviations used
- BBB
blood–brain barrier
- CINC′ 1
cytokine-induced neutrophil chemoattractant protein-1
- CNS
central nervous system
- DW
diffusion-weighted
- IL-1b
interlukin 1b
- FI
fluorescence intensity
- MCA
occlusion, middle cerebral artery occlusion
- MCP-1
monocyte chemoattractant protein-1
- P7
post-anatal day 7
- PBS
phosphate-buffered saline
- TNF-α
tumor necrosis factor
References
- Allan SM, Tyrrell PJ, Rothwell NJ. Interleukin-1 and neuronal injury. Nat Rev. Immunol. 2005;5:629–640. doi: 10.1038/nri1664. [DOI] [PubMed] [Google Scholar]
- Anthony DC, Bolton SJ, Fearn S, Perry VH. Age-related effects of interleukin-1 beta on polymorphonuclear neutrophil-dependent increases in blood–brain barrier permeability in rats. Brain. 1997;120:435–444. doi: 10.1093/brain/120.3.435. [DOI] [PubMed] [Google Scholar]
- Anthony D, Dempster R, Fearn S, Clements J, Wells G, Perry VH, Walker K. CXC chemokines generate age-related increases in neutrophil-mediated brain inflammation and blood–brain barrier breakdown. Curr. Biol. 1998;8:923–926. doi: 10.1016/s0960-9822(07)00373-9. [DOI] [PubMed] [Google Scholar]
- Arvin KL, Du Han BHY, Lin SZ, Paul SM, Holtzman DM. Minocycline markedly protects the neonatal brain against hypoxic-ischemic injury. Ann. Neurol. 2002;52:54–61. doi: 10.1002/ana.10242. [DOI] [PubMed] [Google Scholar]
- Back SA, Rivkees SA. Emerging concepts in periventricular white matter injury. Semin. Perinatol. 2004;28:405–414. doi: 10.1053/j.semperi.2004.10.010. [DOI] [PubMed] [Google Scholar]
- Baggiolini M. Chemokines in pathology and medicine. J. Intern. Med. 2001;250:91–104. doi: 10.1046/j.1365-2796.2001.00867.x. [DOI] [PubMed] [Google Scholar]
- Banisadr G, Gosselin RD, Mechighel P, Rostene W, Kitabgi P, Melik Parsadaniantz S. Constitutive neuronal expression of CCR2 chemokine receptor and its colocalization with neurotransmitters in normal rat brain: functional effect of MCP-1/CCL2 on calcium mobilization in primary cultured neurons. J. Comp. Neurol. 2005;492:178–192. doi: 10.1002/cne.20729. [DOI] [PubMed] [Google Scholar]
- Barone FC, Hillegass LM, Price WJ, White RF, Lee EV, Feuerstein GZ, Sarau HM, Clark RK, Griswold DE. Polymorphonuclear leukocyte infiltration into cerebral focal ischemic tissue: myeloperoxidase activity assay and histologic verification. J. Neurosci. Res. 1991;29:336–345. doi: 10.1002/jnr.490290309. [DOI] [PubMed] [Google Scholar]
- Blamire AM, Anthony DC, Rajagopalan B, Sibson NR, Perry VH, Styles P. Interleukin-1beta-induced changes in blood–brain barrier permeability, apparent diffusion coefficient, and cerebral blood Volume in the rat brain: a magnetic resonance study. J. Neurosci. 2000;20:8153–8159. doi: 10.1523/JNEUROSCI.20-21-08153.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blond D, Campbell SJ, Butchart AG, Perry VH, Anthony DC. Differential induction of interleukin-1beta and tumour necrosis factor-alpha may account for specific patterns of leukocyte recruitment in the brain. Brain Res. 2002;958:89–99. doi: 10.1016/s0006-8993(02)03473-x. [DOI] [PubMed] [Google Scholar]
- Bona E, Andersson AL, Blomgren K, Gilland E, Puka-Sundvall M, Gustafson K, Hagberg H. Chemokine and inflammatory cell response to hypoxia-ischemia in immature rats. Pediatr. Res. 1999;45:500–509. doi: 10.1203/00006450-199904010-00008. [DOI] [PubMed] [Google Scholar]
- Buechler C, Ritter M, Orso E, Langmann T, Klucken J, Schmitz G. Regulation of scavenger receptor CD163 expression in human monocytes and macrophages by pro- and antiinflammatory stimuli. J. Leukoc. Biol. 2000;67:97–103. [PubMed] [Google Scholar]
- Campanella M, Sciorati C, Tarozzo G, Beltramo M. Flow cytometric analysis of inflammatory cells in ischemic rat brain. Stroke. 2002;33:586–592. doi: 10.1161/hs0202.103399. [DOI] [PubMed] [Google Scholar]
- Campbell SJ, Perry VH, Pitossi FJ, Butchart AG, Chertoff M, Waters S, Dempster R, Anthony DC. Central nervous system injury triggers hepatic CC and CXC chemokine expression that is associated with leukocyte mobilization and recruitment to both the central nervous system and the liver. Am. J. Pathol. 2005;166:1487–1497. doi: 10.1016/S0002-9440(10)62365-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson MJ, Reilly CR, Sutcliffe JG, Lo D. Mature microglia resemble immature antigen-presenting cells. Glia. 1998;22:72–85. doi: 10.1002/(sici)1098-1136(199801)22:1<72::aid-glia7>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Carson MJ, Sutcliffe JG. Balancing function vs. self defense: the CNS as an active regulator of immune responses. J. Neurosci. Res. 1999;55:1–8. doi: 10.1002/(SICI)1097-4547(19990101)55:1<1::AID-JNR1>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Charo IF, Myers SJ, Herman A, Franci C, Connolly AJ, Coughlin SR. Molecular cloning and functional expression of two monocyte chemoattractant protein 1 receptors reveals alternative splicing of the carboxyl-terminal tails. Proc. Natl Acad. Sci. USA. 1994;91:2752–2756. doi: 10.1073/pnas.91.7.2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Che X, Ye W, Panga L, Wu DC, Yang GY. Monocyte chemoattractant protein-1 expressed in neurons and astrocytes during focal ischemia in mice. Brain Res. 2001;902:171–177. doi: 10.1016/s0006-8993(01)02328-9. [DOI] [PubMed] [Google Scholar]
- Cowell RM, Plane JM, Silverstein FS. Complement activation contributes to hypoxic-ischemic brain injury in neonatal rats. J. Neurosci. 2003;23:9459–9468. doi: 10.1523/JNEUROSCI.23-28-09459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowell RM, Xu H, Galasso JM, Silverstein FS. Hypoxic-ischemic injury induces macrophage inflammatory protein-1alpha expression in immature rat brain. Stroke. 2002;33:795–801. doi: 10.1161/hs0302.103740. [DOI] [PubMed] [Google Scholar]
- Dalmau I, Vela JM, Gonzalez B, Finsen B, Castellano B. Dynamics of microglia in the developing rat brain. J. Comp. Neurol. 2003;458:144–157. doi: 10.1002/cne.10572. [DOI] [PubMed] [Google Scholar]
- Davies CA, Loddick SA, Stroemer RP, Hunt J, Rothwell NJ. An integrated analysis of the progression of cell responses induced by permanent focal middle cerebral artery occlusion in the rat. Exp. Neurol. 1998;154:199–212. doi: 10.1006/exnr.1998.6891. [DOI] [PubMed] [Google Scholar]
- Derugin N, Dingman A, Wendland M, Fox C, Vexler ZS. Magnetic Resonance Imaging as a Surrogate Measure for Histological Sub-Chronic Endpoint in a Neonatal Rat Stroke Model. Brain Res. 2005;1066:49–56. doi: 10.1016/j.brainres.2005.10.043. [DOI] [PubMed] [Google Scholar]
- Derugin N, Ferriero DM, Vexler ZS. Neonatal reversible focal cerebral ischemia: a new model. Neurosci. Res. 1998;32:349–353. doi: 10.1016/s0168-0102(98)00096-0. [DOI] [PubMed] [Google Scholar]
- Derugin N, Wendland M, Muramatsu K, Roberts T, Gregory G, Ferriero D, Vexler Z. Evolution of brain injury after transient middle cerebral artery occlusion in neonatal rat. Stroke. 2000;31:1752–1761. doi: 10.1161/01.str.31.7.1752. [DOI] [PubMed] [Google Scholar]
- Dingman A, Derugin N, Ji S, Wendland M, Bollen A, Vexler ZS. Society for Neuroscience. San Diego: 2004. Increased levels of cytokine-induced neutrophil chemoattractant protein1 (CINC-1) acutely after neonatal focal ischemia-reperfusion are not associated with neutrophil accumulation. [Google Scholar]
- Dingman A, Lee SY, Derugin N, Wendland MF, Vexler ZS. Aminoguanidine inhibits caspase-3 and calpain activation without affecting microglial activation following neonatal transient ischemia. J. Neurochem. 2006;96:1467–1479. doi: 10.1111/j.1471-4159.2006.03672.x. [DOI] [PubMed] [Google Scholar]
- Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- Dommergues MA, Plaisant F, Verney C, Gressens P. Early microglial activation following neonatal excitotoxic brain damage in mice: a potential target for neuroprotection. Neuroscience. 2003;121:619–628. doi: 10.1016/s0306-4522(03)00558-x. [DOI] [PubMed] [Google Scholar]
- Engelhardt B. Development of the blood–brain barrier. Cell Tissue Res. 2003;314:119–129. doi: 10.1007/s00441-003-0751-z. [DOI] [PubMed] [Google Scholar]
- Fabriek BO, Van Haastert ES, Galea I, et al. CD163-positive perivascular macrophages in the human CNS express molecules for antigen recognition and presentation. Glia. 2005;51:297–305. doi: 10.1002/glia.20208. [DOI] [PubMed] [Google Scholar]
- Fassbender K, Bertsch T, Mielke O, Muhlhauser F, Hennerici M. Adhesion molecules in cerebrovascular diseases. Evidence for an inflammatory endothelial activation in cerebral large- and small-vessel disease. Stroke. 1999;30:1647–1650. doi: 10.1161/01.str.30.8.1647. [DOI] [PubMed] [Google Scholar]
- Feuerstein GZ, Wang X, Barone FC. Inflammatory gene expression in cerebral ischemia and trauma. Potential new therapeutic targets. Ann. N Y Acad. Sci. 1997;825:179–193. doi: 10.1111/j.1749-6632.1997.tb48428.x. [DOI] [PubMed] [Google Scholar]
- Ford AL, Foulcher E, Goodsall AL, Sedgwick JD. Tissue digestion with dispase substantially reduces lymphocyte and macrophage cell-surface antigen expression. J. Immunol. Meth. 1996;194:71–75. doi: 10.1016/0022-1759(96)00067-1. [DOI] [PubMed] [Google Scholar]
- Fox C, Dingman A, Derugin N, Wendland MF, Manabat C, Ji S, Ferriero DM, Vexler ZS. Minocycline confers early but transient protection in the immature brain following focal cerebral ischemia-reperfusion. J. Cereb. Blood Flow. Metab. 2005;25:1138–1149. doi: 10.1038/sj.jcbfm.9600121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galasso JM, Liu Y, Szaflarski J, Warren JS, Silverstein FS. Monocyte chemoattractant protein-1 is a mediator of acute excitotoxic injury in neonatal rat brain. Neuroscience. 2000a;101:737–744. doi: 10.1016/s0306-4522(00)00399-7. [DOI] [PubMed] [Google Scholar]
- Galasso JM, Miller MJ, Cowell RM, Harrison JK, Warren JS, Silverstein FS. Acute excitotoxic injury induces expression of monocyte chemoattractant protein-1 and its receptor, CCR2, in neonatal rat brain. Exp. Neurol. 2000b;165:295–305. doi: 10.1006/exnr.2000.7466. [DOI] [PubMed] [Google Scholar]
- Garcia JH, Liu KF, Yoshida Y, Lian J, Chen S, del Zoppo GJ. Influx of leukocytes and platelets in an evolving brain infarct (Wistar rat) Am. J. Pathol. 1994;144:188–199. [PMC free article] [PubMed] [Google Scholar]
- Gerard C, Rollins BJ. Chemokines and disease. Nat. Immunol. 2001;2:108–115. doi: 10.1038/84209. [DOI] [PubMed] [Google Scholar]
- Gidday JM, Gasche YG, Copin JC, Shah AR, Perez RS, Shapiro SD, Chan PH, Park TS. Leukocyte-derived matrix metalloproteinase-9 mediates blood–brain barrier breakdown and is proinflammatory after transient focal cerebral ischemia. Am. J. Physiol. Heart Circ. Physiol. 2005;289:H558–H568. doi: 10.1152/ajpheart.01275.2004. [DOI] [PubMed] [Google Scholar]
- Glabinski AR, Balasingam V, Tani M, Kunkel SL, Strieter RM, Yong VW, Ransohoff RM. Chemokine monocyte chemoattractant protein-1 is expressed by astrocytes after mechanical injury to the brain. J. Immunol. 1996;156:4363–4368. [PubMed] [Google Scholar]
- Gosling J, Slaymaker S, Gu L, Tseng S, Zlot CH, Young SG, Rollins BJ, Charo IF. MCP-1 deficiency reduces susceptibility to atherosclerosis in mice that overexpress human apolipoprotein. B. J. Clin. Invest. 1999;103:773–778. doi: 10.1172/JCI5624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham EM, Sheldon RA, Flock DL, Ferriero DM, Martin LJ, O’Riordan DP, Northington FJ. Neonatal mice lacking functional Fas death receptors are resistant to hypoxic-ischemic brain injury. Neurobiol. Dis. 2004;17:89–98. doi: 10.1016/j.nbd.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Hagberg H, Gilland E, Bona E, Hanson LA, Hahin-Zoric M, Blennow M, Holst M, McRae A, Soder O. Enhanced expression of interleukin (IL)-1 and IL-6 messenger RNA and bioactive protein after hypoxia-ischemia in neonatal rats. Pediatr. Res. 1996;40:603–609. doi: 10.1203/00006450-199610000-00015. [DOI] [PubMed] [Google Scholar]
- Hagberg H, Mallard C. Effect of inflammation on central nervous system development and vulnerability. Curr. Opin. Neurol. 2005;18:117–123. doi: 10.1097/01.wco.0000162851.44897.8f. [DOI] [PubMed] [Google Scholar]
- Han HS, Yenari MA. Cellular targets of brain inflammation in stroke. Curr. Opin. Invest. Drugs. 2003;4:522–529. [PubMed] [Google Scholar]
- Hedtjarn M, Leverin AL, Eriksson K, Blomgren K, Mallard C, Hagberg H. Interleukin-18 involvement in hypoxic-ischemic brain injury. J. Neurosci. 2002;22:5910–5919. doi: 10.1523/JNEUROSCI.22-14-05910.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedtjarn M, Mallard C, Hagberg H. Inflammatory gene profiling in the developing mouse brain after hypoxia-ischemia. J. Cereb. Blood Flow Metab. 2004;24:1333–1351. doi: 10.1097/01.WCB.0000141559.17620.36. [DOI] [PubMed] [Google Scholar]
- Huang DR, Wang J, Kivisakk P, Rollins BJ, Ransohoff RM. Absence of monocyte chemoattractant protein 1 in mice leads to decreased local macrophage recruitment and antigen-specific T helper cell type 1 immune response in experimental autoimmune encephalomyelitis. J. Exp. Med. 2001;193:713–726. doi: 10.1084/jem.193.6.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudome S, Palmer C, Roberts RL, Mauger D, Housman C, Towfighi J. The role of neutrophils in the production of hypoxic-ischemic brain injury in the neonatal rat. Pediatr. Res. 1997;41:607–616. doi: 10.1203/00006450-199705000-00002. [DOI] [PubMed] [Google Scholar]
- Ivacko JA, Sun R, Silverstein FS. Hypoxic-ischemic brain injury induces an acute microglial reaction in perinatal rats. Pediatr Res. 1996;39:39–47. doi: 10.1203/00006450-199601000-00006. [DOI] [PubMed] [Google Scholar]
- Kishimoto TK, Rothlein R. Integrins, ICAMs, and selectins: role and regulation of adhesion molecules in neutrophil recruitment to inflammatory sites. Adv. Pharmacol. 1994;25:117–169. doi: 10.1016/s1054-3589(08)60431-7. [DOI] [PubMed] [Google Scholar]
- Kniesel U, Risau W, Wolburg H. Development of blood–brain barrier tight junctions in the rat cortex. Brain Res. Dev. Brain Res. 1996;96:229–240. doi: 10.1016/0165-3806(96)00117-4. [DOI] [PubMed] [Google Scholar]
- Liu J, Bartels M, Lu A, Sharp FR. Microglia/macrophages proliferate in striatum and neocortex but not in hippocampus after brief global ischemia that produces ischemic tolerance in gerbil brain. J. Cereb. Blood Flow. Metab. 2001;21:361–373. doi: 10.1097/00004647-200104000-00005. [DOI] [PubMed] [Google Scholar]
- Lorant DE, Li W, Tabatabaei N, Garver MK, Albertine KH. P-selectin expression by endothelial cells is decreased in neonatal rats and human premature infants. Blood. 1999;94:600–609. [PubMed] [Google Scholar]
- Lovering F, Zhang Y. Therapeutic potential of TACE inhibitors in stroke. Curr. Drug Targets CNS Neurol. Disord. 2005;4:161–168. doi: 10.2174/1568007053544147. [DOI] [PubMed] [Google Scholar]
- Manabat C, Han BH, Wendland M, Derugin N, Fox CK, Choi J, Holtzman DM, Ferriero DM, Vexler ZS. Reperfusion differentially induces caspase-3 activation in ischemic core and penumbra after stroke in immature brain. Stroke. 2003;34:207–213. doi: 10.1161/01.STR.0000047101.87575.3C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo Y, Kihara T, Ikeda M, Ninomiya M, Onodera H, Kogure K. Role of neutrophils in radical production during ischemia and reperfusion of the rat brain: effect of neutrophil depletion on extracellular ascorbyl radical formation. J. Cereb. Blood Flow Metab. 1995;15:941–947. doi: 10.1038/jcbfm.1995.119. [DOI] [PubMed] [Google Scholar]
- McRae A, Gilland E, Bona E, Hagberg H. Microglia activation after neonatal hypoxic-ischemia. Brain Res. Dev. Brain Res. 1995;84:245–252. doi: 10.1016/0165-3806(94)00177-2. [DOI] [PubMed] [Google Scholar]
- Morioka T, Kalehua AN, Streit WJ. Characterization of microglial reaction after middle cerebral artery occlusion in rat brain. J. Comp. Neurol. 1993;327:123–132. doi: 10.1002/cne.903270110. [DOI] [PubMed] [Google Scholar]
- Northington FJ, Ferriero DM, Flock DL, Martin LJ. Delayed neurodegeneration in neonatal rat thalamus after hypoxia-ischemia is apoptosis. J. Neurosci. 2001;21:1931–1938. doi: 10.1523/JNEUROSCI.21-06-01931.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry VH. The influence of systemic inflammation on inflammation in the brain: implications for chronic neurodegenerative disease. Brain Behav. Immun. 2004;18:407–413. doi: 10.1016/j.bbi.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, Kivisakk P, Kidd G. Three or more routes for leukocyte migration into the central nervous system. Nat. Rev. Immunol. 2003;3:569–581. doi: 10.1038/nri1130. [DOI] [PubMed] [Google Scholar]
- Roberts ES, Masliah E, Fox HS. CD163 identifies a unique population of ramified microglia in HIV encephalitis (HIVE) J. Neuropathol. Exp. Neurol. 2004;63:1255–1264. doi: 10.1093/jnen/63.12.1255. [DOI] [PubMed] [Google Scholar]
- Rosenzweig HL, Lessov NS, Henshall DC, Minami M, Simon RP, Stenzel-Poore MP. Endotoxin preconditioning prevents cellular inflammatory response during ischemic neuroprotection in mice. Stroke. 2004;35:2576–2581. doi: 10.1161/01.STR.0000143450.04438.ae. [DOI] [PubMed] [Google Scholar]
- Santambrogio L, Belyanskaya SL, Fischer FR, Cipriani B, Brosnan CF, Ricciardi-Castagnoli P, Stern LJ, Strominger JL, Riese R. Developmental plasticity of CNS microglia. Proc. Natl Acad. Sci. USA. 2001;98:6295–6300. doi: 10.1073/pnas.111152498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders NR, Habgood MD, Dziegielewska KM. Barrier mechanisms in the brain, II. Immature brain. Clin. Exp. Pharmacol. Physiol. 1999;26:85–91. doi: 10.1046/j.1440-1681.1999.02987.x. [DOI] [PubMed] [Google Scholar]
- Saunders NR, Knott GW, Dziegielewska KM. Barriers in the immature brain. Cell Mol. Neurobiol. 2000;20:29–40. doi: 10.1023/A:1006991809927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell L, Fearn S, Schwab ME, Perry VH, Anthony DC. Cytokine-induced acute inflammation in the brain and spinal cord. J. Neuropathol. Exp. Neurol. 1999;58:245–254. doi: 10.1097/00005072-199903000-00004. [DOI] [PubMed] [Google Scholar]
- Stevens SL, Bao J, Hollis J, Lessov NS, Clark WM, Stenzel-Poore MP. The use of flow cytometry to evaluate temporal changes in inflammatory cells following focal cerebral ischemia in mice. Brain Res. 2002;932:110–119. doi: 10.1016/s0006-8993(02)02292-8. [DOI] [PubMed] [Google Scholar]
- Szaflarski J, Burtrum D, Silverstein FS. Cerebral hypoxia-ischemia stimulates cytokine gene expression in perinatal rats. Stroke. 1995;26:1093–1100. doi: 10.1161/01.str.26.6.1093. [DOI] [PubMed] [Google Scholar]
- Ten VS, Bradley-Moore M, Gingrich JA, Stark RI, Pinsky DJ. Brain injury and neurofunctional deficit in neonatal mice with hypoxic-ischemic encephalopathy. Behav. Brain Res. 2003;145:209–219. doi: 10.1016/s0166-4328(03)00146-3. [DOI] [PubMed] [Google Scholar]
- Ten VS, Wu EX, Tang H, Bradley-Moore M, Fedarau MV, Ratner VI, Stark RI, Gingrich JA, Pinsky DJ. Late measures of brain injury after neonatal hypoxia-ischemia in mice. Stroke. 2004;35:2183–2188. doi: 10.1161/01.STR.0000137768.25203.df. [DOI] [PubMed] [Google Scholar]
- Tsuji M, Higuchi Y, Shiraishi K, Kume T, Akaike A, Hattori H. Protective effect of aminoguanidine on hypoxic-ischemic brain damage and temporal profile of brain nitric oxide in neonatal rat. Pediatr. Res. 2000;47:79–83. doi: 10.1203/00006450-200001000-00015. (in press) [DOI] [PubMed] [Google Scholar]
- Vannucci SJ, Hagberg H. Hypoxia-ischemia in the immature brain. J. Exp. Biol. 2004;207:3149–3154. doi: 10.1242/jeb.01064. [DOI] [PubMed] [Google Scholar]
- Vexler ZS. Inflammation and ischemia in the developing brain. In: Yenari MA, Giffard RG, editors. Glia and Inflammation in Neurodegenerative Disease. Nova Science Publishers, Inc.; 2006. [Google Scholar]
- Vexler ZS, Ferriero DM. Mechanisms of Ischemic Cell Death in the Developing Brain. In: Chan P, editor. Handbook of Neurochemistry. New York, NY, USA: Kluwer Academic Publishers; 2006. Volume in Press. [Google Scholar]
- Xu H, Barks JD, Schielke GP, Silverstein FS. Attenuation of hypoxia-ischemia-induced monocyte chemoattractant protein-1 expression in brain of neonatal mice deficient in interleukin-1 converting enzyme. Brain Res. Mol. Brain Res. 2001;90:57–67. doi: 10.1016/s0169-328x(01)00087-0. [DOI] [PubMed] [Google Scholar]