

Abstract
Impaired cognition and memory may be associated with down-regulation of cAMP-response element-binding protein (CREB) in the brain in patients with Alzheimer disease, but the molecular mechanism leading to the down-regulation is not understood. In this study, we found a selective reduction in the levels of the regulatory subunits (RIIα and RIIβ ) and the catalytic subunit (Cβ ) as well as the enzymatic activity of cAMP-dependent protein kinase (PKA), which is the major positive regulator of CREB. We also observed that PKA subunits were proteolyzed by calpain and the levels of PKA subunits correlated negatively with calpain activation in the human brain. These findings led us to propose that in the brain in patients with Alzheimer disease, over-activation of calpain because of calcium dysregulation causes increased degradation and thus decreased activity of PKA, which, in turn, contributes to down-regulation of CREB and impaired cognition and memory.
Keywords: Alzheimer disease, calpain, cAMP-response, element-binding protein, protein kinase
Alzheimer disease (AD) is clinically characterized by slow but progressive disturbance of memory, thinking, and behavior, ending with dementia and death. In the last two decades, many advances have been made concerning the mechanisms of the disease, especially amyloid precursor protein processing, Aβ deposition, and tau abnormalities. However, the mechanisms leading to the impairment of cognition and memory in AD patients are still hardly understood.
The principles of memory storage and processing are very complicated, and little is known to date. Substantial evidence has suggested that the cAMP-response element-binding protein (CREB), a ubiquitous transcription factor, is a key molecule for learning and memory and a core component of the molecular switch that converts short-term to long-term memory (Barco et al. 2003). Disturbance of CREB function has been shown to cause memory deficits in many models. In AD brain and some mouse models of AD, CREB activity is impaired (Yamamoto-Sasaki et al. 1999; Gong et al. 2004; Puzzo et al. 2005; Matsuzaki et al. 2006), but the mechanisms leading to the CREB dysregulation are unknown.
The major regulator of CREB activity is cAMP-dependent protein kinase (PKA). When a signal arrives at the cell surface, it activates the corresponding receptor, which, in turn, leads to the transient elevation of intracellular cAMP and consequently activates PKA by disassociating the regulatory subunits (R subunits) from the catalytic subunits (C subunits). The activated PKA then moves into the cell nucleus, where it activates CREB by phosphorylating it. PKA is a tetrameric holoenzyme consisting of two C subunits and two R subunits in the absence of cAMP. Several isoforms of both C subunit (Cα , Cβ , and Cγ ) and R subunit (RIα , RIβ , RIIα and RIIβ ) have been found in mammalian tissue. The Cα isoform is expressed ubiquitously in most tissue, whereas the Cβ isoform is highly expressed in the brain (Cadd and McKnight 1989). PKA-Cγ is expressed only in testis (Foss et al. 1992). All four isoforms of the R subunits are expressed in human brain (Chang et al. 2003). The R subunit not only controls PKA activity, but also localizes the kinase inside the cell.
To investigate whether deficient CREB function is caused by PKA dysregulation in AD, we studied PKA activity and the levels of various PKA subunits/isoforms in AD brain. We found that PKA is down-regulated in AD brain, and this down-regulation may result from degradation of some PKA isoforms by over-activated calpain.
Methods
Human brain tissue
Medial temporal cortices of the six AD and seven age-matched normal human brains used for this study (Table 1) were obtained from the Sun Health Research Institute Donation Program (Sun City, AZ, USA). All brain samples were confirmed pathologically and stored at − 70° C until used. The use of frozen human brain tissue was in accordance with the National Institutes of Health guidelines and was approved by our institute’s institutional review committee.
Table 1.
Human brain tissue used in this study
| Case | Age at death (years) | Gender | PMI (h) | Braak stagea | Tangle scoreb |
|---|---|---|---|---|---|
| AD 1 | 89 | F | 3 | V | 14.5 |
| AD 2 | 80 | F | 2.25 | VI | 14.5 |
| AD 3 | 78 | F | 1.83 | VI | 15.0 |
| AD 4 | 95 | F | 3.16 | VI | 10.0 |
| AD 5 | 86 | M | 2.25 | VI | 13.5 |
| AD 6 | 91 | F | 3 | V | 8.50 |
| Mean ± SD | 86.5 ± 6.5 | 2.58 ± 0.54 | 12.67 ± 2.73 | ||
| Con 1 | 85 | M | 25 | II | 4.25 |
| Con 2 | 86 | F | 2.5 | III | 5.00 |
| Con 3 | 81 | M | 2.75 | III | 6.41 |
| Con 4 | 88 | F | 3 | II | 2.00 |
| Con 5 | 90 | F | 3 | III | 4.50 |
| Con 6 | 88 | F | 3.5 | III | 2.50 |
| Con 7 | 88 | F | 3 | IV | 4.50 |
| Mean ± SD | 86.6 ± 2.9 | 2.89 ± 0.39 | 4.17 ± 1.50 |
PMI, postmortem interval; AD, Alzheimer disease.
Neurofibrillary pathology was staged according to Braak and Braak (1995).
Tangle score was a density estimate and was designated as none, sparse, moderate, or frequent (0, 1, 2, or 3 for statistics), as defined according to CERAD AD criteria (Mirra et al. 1991). Five areas (frontal, temporal, parietal, hippocampal, and entorhinal) were examined, and the scores were added up for a maximum of 15.
PKA assays
Brain tissue was homogenized in nine volumes of buffer consisting of 50 mmol/L Tris–HCl (pH 7.4), 8.5% sucrose, 10 mmol/L β-mercaptoethanol, 2.0 mmol/L EDTA, 2.0 mmol/L benzamidine, and 2.0 μg/mL each of aprotinin, leupeptin, and pepstatin, followed by centrifugation at 16 000 g at 4° C for 10 min. The supernatants were incubated in an PKA assay mixture (20 μL) containing 50 mmol/L Tris–HCl (pH 7.4), 0.1 mmol/L EGTA, 4.0 μmol/L cyclosporine A, 0.2 μmol/L okadaic acid, 1.0 mmol/L Na3VO4, 10 mmol/L MgCl2, 0.2 mmol/L [γ-32P]ATP (500 cpm/pmol), and 30 μmol/L Malantide. Malantide is a specific peptide substrate for PKA assay (Murray et al. 1990). After incubation at 30° C for 20 min, 10 μL each of 10 mg/mL bovine serum albumin and 40% trichloroacetic acid were added into the assay mixtures to terminate the reaction. The reaction mixture was left in wet ice for 30 min and then centrifuged at 2300 g for 3 min. Aliquots of 10 μL supernatants containing Malantide were spotted onto squares (1 × 1 cm) of P81 chromatography paper (Whatman International Ltd, Maidstone, England). After washing with 75 mmol/L phosphoric acid five times, the chromatography paper squares were dried, and the radioactivities were counted in a scintillation counter to determine the 32P-incorporation into Malantide.
Western blot analysis
Western blots were carried out by using 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and standard procedure and developed by using ECL kit (Amersham Biosciences, Piscataway, NJ, USA). The primary antibodies used in this study are listed in Table 2.
Table 2.
Primary antibodies used in this study
| Antibody | Type | Specificity | Source/references |
|---|---|---|---|
| Anti-PKA | Polyclonal | PKA-Cα | Abcam, Cambridge, UK |
| PKAβ (C-20) | Polyclonal | PKA-Cβ | Santa Cruz Biotechnology, Santa Cruz, CA, USA |
| PKARI | Monoclonal | PKA-RI | BD, Franklin Lakes, NJ, USA |
| PKARIIα | Monoclonal | PKA-RIIα | BD, Franklin Lakes, NJ, USA |
| PKARIIβ | Monoclonal | PKA-RIIβ | BD, Franklin Lakes, NJ, USA |
| R126d | Polyclonal | Calcineurin | Pei et al. (1998) |
| Anti-calpain I | Polyclonal | Calpain I | Sigma, St Louis, MO, USA |
PKA, cAMP-dependent protein kinase.
In vitro proteolysis of PKA
Forebrain tissue from C57BL/6J mice was homogenized in nine volumes of buffer containing 50 mmol/L Tris–HCl (pH 7.0), 8.5% sucrose, 10 mmol/L β-mercaptoethanol, and 2.0 mmol/L EDTA. After centrifugation of the homogenate at 900 g for 10 min, the resulting supernatant (S1) was centrifuged again at 16 000 g, and the second supernatant (S2) was used as the cytosolic fraction. The first pellet (P1) was resuspended in the homogenizing buffer and centrifuged again at 900 g for 10 min, and this step was repeated twice to remove any residual cytosol contaminants off the pellet. The final pellet was resuspended in the homogenizing buffer, sonicated, and then centrifuged at 16 000 g for 10 min. The resulting extract was used as the nuclear fraction.
For in vitro proteolysis, the cytosolic and nuclear fractions were incubated at 30° C for 10 or 30 min in the presence of 2.5 mmol/L CaCl2 and none or one of the following protease inhibitors: 2.0 μg/mL aprotinin (a serine protease inhibitor; Sigma, St Louis, MO, USA); 2.0 μg/mL pepstatin (an aspartic protease inhibitor, Sigma); 2.0 μg/mL leupeptin (a cysteine and serine protease inhibitor, Sigma); and 1.0 μmol/L calpain inhibitor I (ALLN, Calbiochem, La Jolla, CA, USA). After incubation, the reaction was terminated by adding ¼ volume of concentrated (4× ) sodium dodecyl sulfate–polyacrylamide gel electrophoresis sample buffer into the reaction mixture, and the proteolysis of PKA subunits/isoforms was examined by western blots.
Kainic acid injection of mice
Male FBV mice (25–30 g body weight, 9 weeks old) were housed individually in a 12-h light/dark schedule with free access to food and water. A single dose of kainic acid (20 mg/kg) was injected intraperitoneally. The mice were then killed 2.5, 6, 10, 24, 36, and 48 h after injection, and the forebrains were immediately removed and homogenized in buffer consisting of 50 mmol/L Tris–HCl (pH 7.4), 8.5% sucrose, 10 mmol/L β-mercaptoethanol, and 2.0 mmol/L EDTA. The levels of proteolysis of PKA subunits/isoforms in the brains were examined by western blot analysis of the homogenates.
Correlation analysis
Linear correlation between calpain activation and levels of various PKA subunits/isoforms in human brains was analyzed by using MS Excel. To quantify calpain activation, the active/truncated calpain (76- and 78-kDa bands) and the inactive/full-length calpain (80 kDa) of brain homogenates from six AD and seven control cases were determined by quantitative western blots. Calpain activation was then calculated by using the ratios of the immunore-activities of the active/truncated calpain to the inactive/full-length calpain. Levels of various PKA subunits/isoforms were also determined by quantitative western blots.
Statistic analysis
Comparisons of means between groups were analyzed by student t-test. Correlation analyses between calpain activation and the levels of PKA subunits/isoforms were carried out by linear correlation method using Microsoft Excel 2003.
Results
PKA activity is down-regulated in AD brain
To learn whether PKA is dysregulated in AD brain, we determined PKA activity of the extracts of the medial temporal cortices from postmortem brains of six AD and seven age- and postmortem interval-matched control cases (Table 1). In the basal conditions in vivo, the majority of PKA-C subunits are associated with the R subunits and, therefore, in an inactive state. PKA activity assayed in tissue extracts without the addition of cAMP into the assay mixture represents the active form of PKA (free PKA-C) in situ, whereas the activity assayed in the presence of cAMP added into the assay mixture represents the total activity after full activation (Murray et al. 1990). Therefore, we measured PKA activity in both conditions. We found that both in the absence and in the presence of 5 μmol/L cAMP, PKA activity was decreased by 25–30% in AD brains when compared with controls (Fig. 1a). These results suggest that both active PKA (free PKA-C) and total PKA (free PKA-C plus PKA-C associated with R subunits) are decreased in AD brain, implying that the decreased activity might be due to a decreased level of PKA-C. The ratio of the active to the total PKA activity did not change in AD brain when compared with controls (Fig. 1b).
Fig. 1.
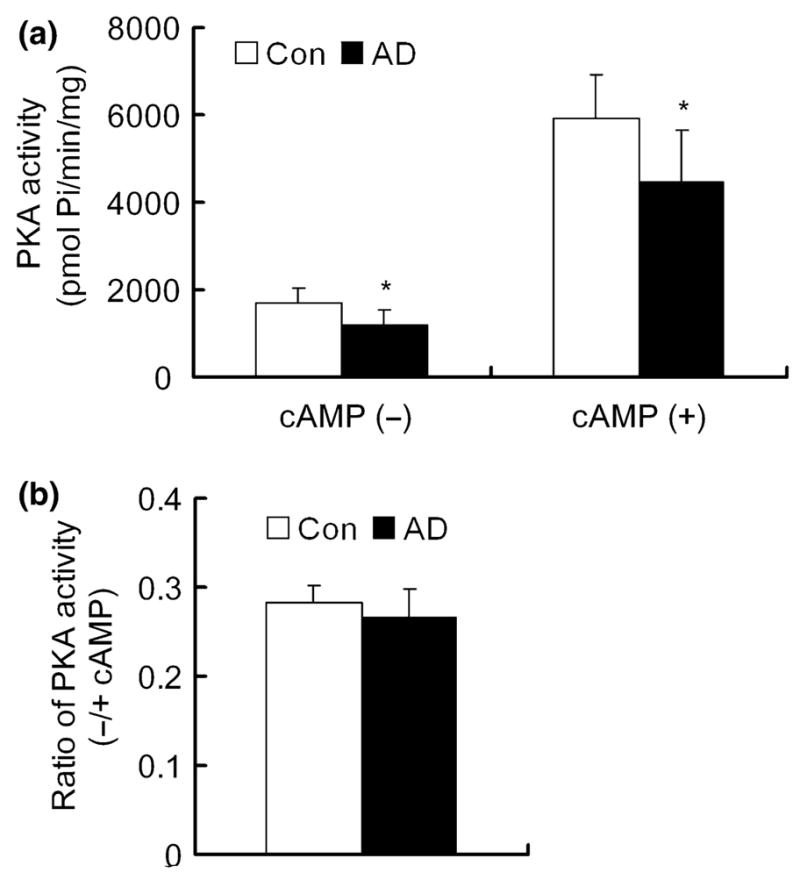
cAMP-dependent protein kinase (PKA) activity in the extracts of Alzheimer disease (AD) and control brains. (a) PKA activity of the 16 000 g extract of the medial temporal cortex from six AD and seven age-matched control brains was assayed in the absence or presence of 5 μmol/L cAMP. (b) Ratio of PKA activity in the absence or in the presence of cAMP. *p < 0.05 as compared with the control group.
Levels of various subunits/isoforms of PKA are selectively decreased in AD brain
To investigate whether a decrease in protein levels of any components of the PKA complex might underlie the decreased kinase activity in AD brain, we studied the protein levels of each major brain subunit of PKA by western blots. We found that Cα and RIIβ of PKA displayed as a single band at 42 and 53 kDa, respectively, in human brain samples (Fig. 2a), which is consistent with previous observations (Chang et al. 2003). Cβ showed two bands at the apparent molecular weights of 53 and 42 kDa, and the 42-kDa Cβ was the predominant form in human brain. RI displayed as a predominant 48-kDa band and a weak 51-kDa band, representing RIα and RIβ isoforms, respectively. RIIα displayed as close duplets at the apparent molecular weight of 51 kDa. When the PKA subunits/isoforms between AD and control cases were compared, we found a 50–60% reduction of the 53-kDa Cβ , the 51-kDa RIIα , and the 53-kDa RIIβ in AD brain (Fig. 2a and b). There was also an ~15% reduction of PKA-Cα and the 42-kDa PKA-Cβ in AD brain, but these decreases did not reach statistical significance. In the cases of RIIα (51 kDa) and RIIβ (53 kDa), the reduction of the levels was accompanied by the appearance of lower molecular weight bands (47 and 50 kDa, respectively; Fig. 2a), suggesting that the decrease of full-length RIIα and RIIβ may be due to their selective proteolysis in AD brain.
Fig. 2.
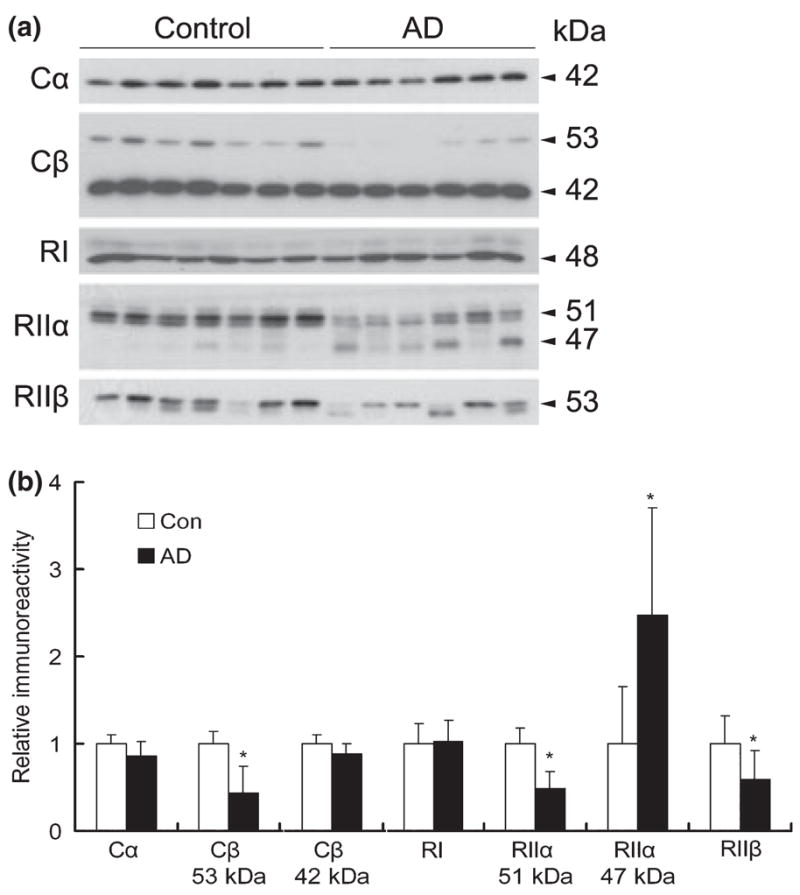
Levels of cAMP-dependent protein kinase (PKA) subunits/isoforms in Alzheimer disease (AD) and control brains. (a) Western blots of the medial temporal cortical homogenate from six AD and seven control brains, which were probed with antibodies against various subunits/isoforms of PKA, as indicated on the left of the blots. (b) Densitometric quantitation of the blots shown in panel (a). Mean ± SD of the relative immunoreactivities are shown. *p < 0.05 as compared with the control groups.
PKA subunits are degraded by calpain in the mouse brain
To elucidate by which protease(s) PKA subunits might have been proteolyzed in AD brain, we first studied proteases responsible for PKA degradation in mouse brain. Because PKA is distributed in both the cytoplasm and nucleus, the cytosol and the nuclear fractions from mouse brain homogenate were prepared and incubated at 30° C for 10 or 30 min in the presence of calcium and specific protease inhibitors. We found that incubation of brain cytosol and, to a lesser extent, nuclear fraction with calcium-induced proteolysis of RIIα from 51 to 47 kDa polypeptide (Fig. 3), which were similar to the partial truncation of RIIα observed in AD brain (compare Fig. 3 with Fig 2a). The RIIα proteolysis was not inhibited by aprotinin or pepstatin, the most commonly used potent inhibitors of serine protease and aspartic protease, respectively, but was almost completely inhibited by leupeptin (a cysteine protease) and calpain inhibitor I (Fig. 3). These results indicate that PKA-RIIα is proteolyzed by calpain, but not by serine or aspartic proteases, from 51 into 47 kDa, and suggest that calpain-mediated RIIα proteolysis could occur in AD brain.
Fig. 3.
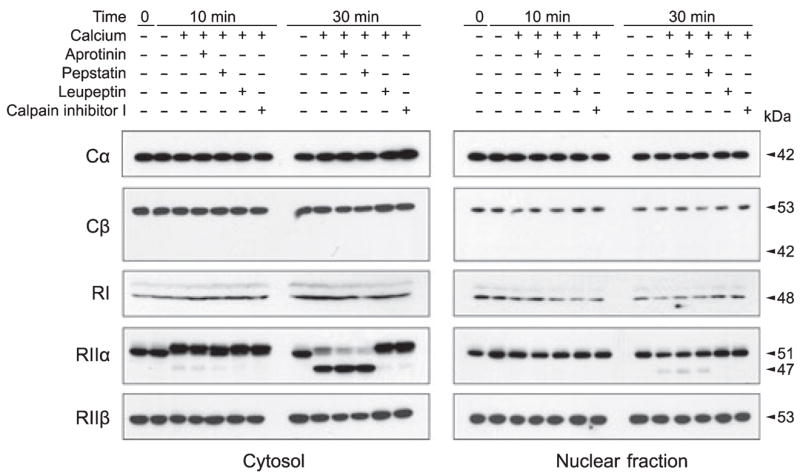
In vitro proteolysis of cAMP-dependent protein kinase (PKA) subunits/isoforms. The cytosolic and nuclear fractions of mouse brain homogenate were incubated at 30° C for 10 or 30 min in the presence of calcium and selective protease inhibitors, as indicated above the blots. The levels and proteolysis of various PKA subunits/isoforms of the samples were then analyzed by western blots.
A similar but much less marked proteolysis of PKA-RIIβ was also seen after 30 min incubation of mouse brain cytosol (Fig. 3). Like that of RIIα , the RIIβ proteolysis was not inhibited by aprotinin or pepstatin but by leupeptin and calpain inhibitor I, indicating that RIIβ is also proteolyzed by calpain. The RIIβ degradation of the mouse brain cytosol was not accompanied by the appearance of a lower molecular weight band, as seen in AD brain (compare Fig. 3 with Fig. 2a). We did not observe any readily detectable degradation of Cα , Cβ , or RI after incubation of mouse brain cytosol or nuclear fraction with calcium (Fig. 3).
Unlike the human brain, in which the 42-kDa Cβ is the predominant form of Cβ (Fig. 2a), mouse brain did not have the 42-kDa form but only had the 53-kDa form (Fig. 3), which is consistent with what was observed in rat brain previously (Nagakura et al. 2002). It is interesting to note that RIIα showed a small upward gel mobility shift of its apparent molecular weight after incubation of mouse brain cytosol but not nuclear fraction with calcium (Fig. 3). It is possible that this gel mobility shift might have resulted from phosphorylation of RIIα by a calcium-activated protein kinase and residual ATP that were present in the cytosol but not active in the nuclear fraction that was sonicated.
To investigate whether the calpain-mediated PKA-R proteolysis that we observed above in the brain cytosol also occurs in vivo, we studied PKA proteolysis of mouse brain after the mice were injected with kainic acid intraperitoneally. Kainic acid injection is well known to increase intraneuronal calcium and, thus, activate calcium-activated enzymes including calpain (Wu et al. 2004). As expected, 24 h after injection, we observed activation of calpain activity, as evidenced by the cleavage of calcineurin from 60 kDa into 57 and 48 kDa (Fig. 4). Calcineurin is known to be cleaved by activated calpain from 60 kDa into 57 and 48 kDa both in vitro and in vivo (Liu et al. 2005a). Similar to calcineurin cleavage, we also observed slightly decreased levels of all subunits/isoforms of PKA examined, including Cα , Cβ , RI, RIIα , and RIIβ , 24–48 h after injection of kainic acid (Fig. 4). In the cases of Cβ and RIIα , we also observed a proteolyzed product of Cβ (42 kDa band) and of RIIα (47 kDa band). The 42-kDa band derived from the 53-kDa Cβ might also be present in AD brain, but the small level of the truncated band might not be visible because there was a heavy 42-kDa band of Cβ in human brain (Fig. 2a). The 47-kDa truncated RIIα seen in the kainic acid-injected mouse brains had the same apparent molecular weight as the truncated RIIα of AD brain (compare Fig. 4 with Fig. 2a). These results suggest that PKA subunits can be proteolyzed in the brain by activated calpain.
Fig. 4.
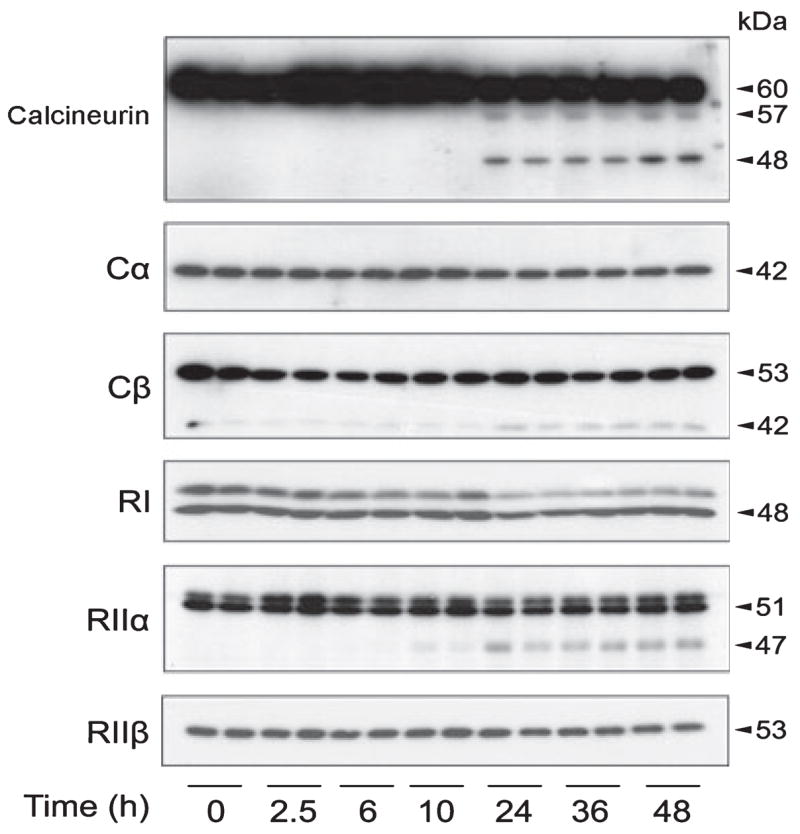
cAMP-dependent protein kinase (PKA) degradation in mouse brain. Western blots of calcineurin and various isoforms of PKA subunits in the brain homogenate of mice after a single intraperitoneal injection of kainic acid at the indicated time points before killing the animals.
Decreases in PKA subunits/isoforms are correlated to calpain activation in human brain
It is well known that calpain is over-activated in AD brain (reviewed by Raynaud and Marcilhac 2006). To investigate whether the degradation of PKA in AD brain is caused by calpain over-activation, we carried out correlation analyses between calpain activation and the levels of PKA subunits/isoforms. Calpain I is the major calpain isoform in the brain (Mattson and Chan 2003). The full-length, inactive calpain I (80 kDa) is activated after calcium-stimulated autoproteolysis into 78- and 76-kDa truncated forms (Saito et al. 1993; Veeranna et al. 2004). In normal control human brains, calpain I displayed as two major bands (80 and 76 kDa) and a very minor band (78 kDa), indicating that both inactivated and activated calpain I are present in normal brain (Fig. 5a). In AD brain, most calpain I was truncated into the 76-kDa active form, which is consistent with previous observations (Saito et al. 1993; Liu et al. 2005a). For correlation analysis, we used the ratio of the active calpain (78 and 76 kDa) over the inactive calpain (80 kDa) to represent calpain activation in the brain samples. We found that, except for RI, levels of Cα , Cβ (53 kDa), Cβ (42 kDa), RIIα (51 kDa), and RIIβ all negatively correlated to calpain activation in human brain (Fig. 5b–h). The negative correlation was more remarkable for Cβ (r = − 0.81), the 51-kDa RIIα (r = − 0.86), and RIIβ (r = − 0.77). On the other hand, level of truncated 47-kDa RIIα correlated positively to calpain activation (Fig. 5g). Taken together with the in vitro and in vivo studies presented above, these results suggest that the decreased levels of PKA components in AD brain are most likely caused by calpain over-activation.
Fig. 5.

Correlation analysis between the levels of cAMP-dependent protein kinase (PKA) subunits/isoforms and calpain activation in human brain. (a) Western blots of calpain I of medial temporal cortical homogenate from six Alzheimer disease (AD) and seven control cases. The apparent molecular weights of the full length and the truncated calpain are marked on the right of the blot. (b–h) Linear correlation analyses between the levels of each PKA subunit/isoform and calpain activation in 13 human brains (six AD and seven controls).
Discussion
Impaired cognition and memory in AD are believed to be associated with down-regulation of CREB activity (Yamamoto-Sasaki et al. 1999; Gong et al. 2004; Puzzo et al. 2005; Matsuzaki et al. 2006), but the molecular mechanism leading to CREB down-regulation has not been understood. CREB activity is regulated mainly by the cAMP–PKA–CREB pathway. In this study, we used a cohort of human brain tissue with very short postmortem delays (average: 2.5–3 h) and found decreased PKA activity in AD brain. These findings are consistent with a previous report showing decreased PKA activity in AD brain (Kim et al. 2001). We further demonstrated the decreased levels of Cβ and RII subunits of PKA and increased calpain-mediated PKA degradation in AD brain. These findings provide, for the first time, a molecular mechanism of PKA down-regulation through its increased degradation by over-activated calpain. Our conclusion is consistent with several gene expression microarray studies that did not find significant alteration of expression of PKA subunits/isoforms (reviewed by Reddy and McWeeney 2006).
Under non-stimulated conditions, PKA is present as an inactive heterotetramer consisting of two C subunits and two R subunits. Association of the R subunits to the C subunits has several biological roles. First, it keeps the catalytic C subunits inactive and dynamically regulates the activity in response to alteration of intracellular cAMP level. Second, it protects the C subunits from degradation (Burton et al. 1997). Upon stimulation, increased cAMP binds to the R subunits and dissociates them from the C subunits. The free C subunits catalyze phosphorylation of the substrate proteins and then become vulnerable to degradation. A loss of PKA-C subunit and PKA activity has been observed in PKA-R knockout mice (Burton et al. 1997). Finally, the R subunits, especially RII, guide the C subunits to specific subcellular localizations and substrates via binding to various PKA anchoring proteins (reviewed by Wong and Scott 2004). In the present study, we demonstrate that the RIIα and RIIβ of PKA were selectively degraded by calpain both in vitro and in vivo, and this degradation was correlated to calpain activation in human brain. On the basis of these observations, we postulate the following mechanism leading to down-regulation of PKA and CREB in AD brain (Fig. 6). First, over-activation of calpain because of calcium dysregulation causes increased degradation and, thus, decreased levels of PKA-RII in AD brain. Because there is less PKA-RII available at the basal conditions to protect PKA-C from degradation, the C subunits, especially Cβ , of PKA are also decreased in AD brain. This analysis is consistent with our finding that Cβ is more affected than Cα in AD brain, because it has much less affinity to bind to the R subunits (Gamm et al. 1996) and, thus, is more vulnerable to degradation caused by the lack of protection from sufficient PKA-R. The consequent decrease in both PKA-C and PKA-RII leads to down-regulation of PKA activity in AD brain. This is supported by our observation of decreased activity of PKA both in the absence (representing the constitutive free PKA-C) and the presence (representing the total PKA-C) of external cAMP. Finally, the down-regulation of PKA results in down-regulation of CREB, both at basal and stimulated conditions and, consequently, less expression of the CREB-regulating genes that are vital to cognition and memory.
Fig. 6.
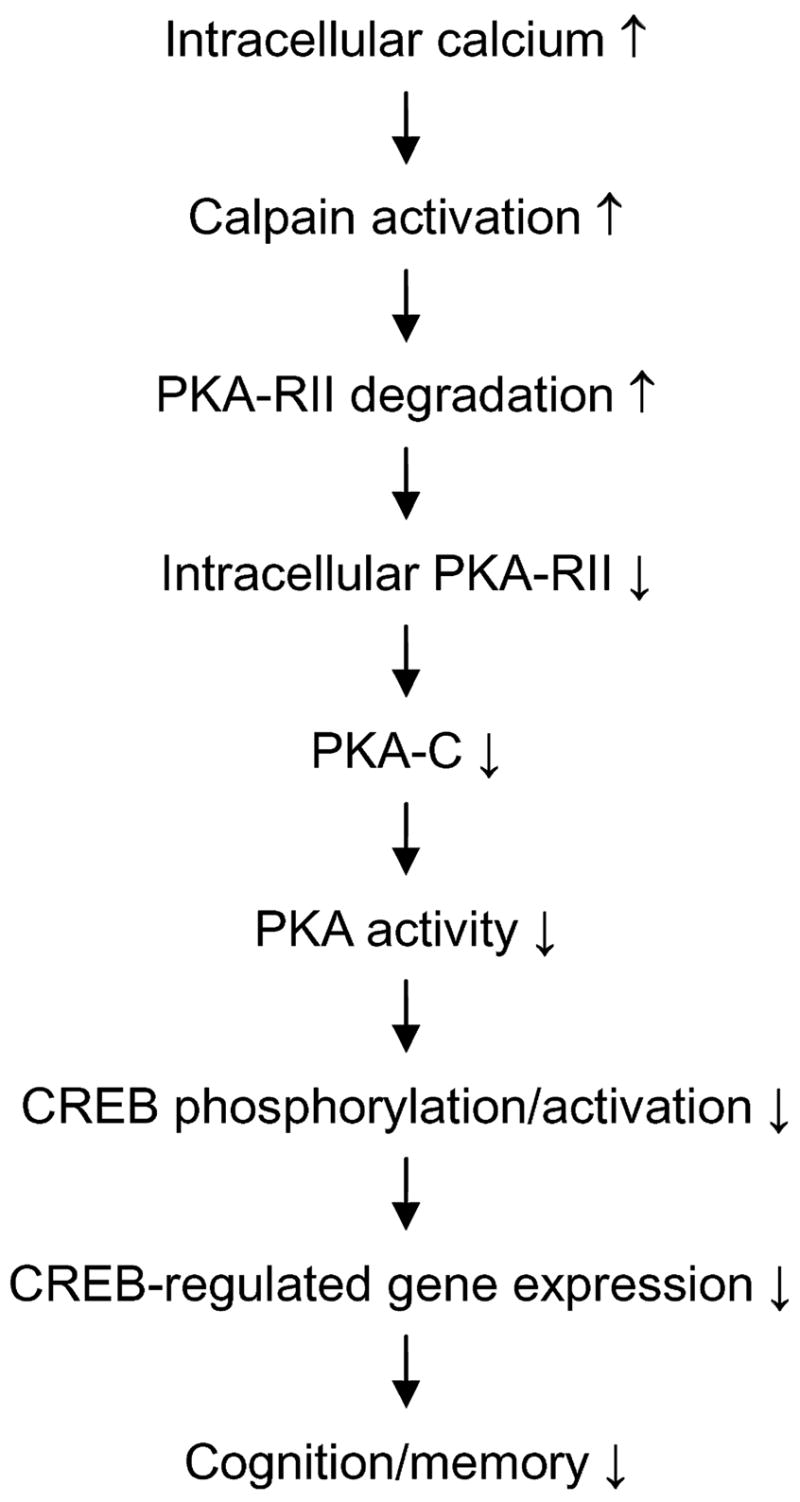
A proposed scheme of calpain–cAMP-dependent protein kinase (PKA)–cAMP-response element-binding protein (CREB) dysregulation in Alzheimer disease (AD) brain.
To confirm the above-outlined hypothesis of calpain–PKA–CREB dysregulation in AD brain, we studied CREB by western blots and found a dramatic decrease in both the level and the phosphorylation of CREB in AD brain when compared with controls (unpublished observations). Further studies indicated that CREB is also a substrate of calpain and that over-activated calpain degraded CREB in AD brain (unpublished observations). Thus, calpain over-activation could down-regulate CREB activity by degrading it directly and by inhibiting its activity indirectly via down-regulation of PKA.
The PKA–CREB pathway has been shown to be vital to synaptic plasticity and cognition (reviewed by Tully et al. 2003; Josselyn and Nguyen 2005). Animals deficient in either PKA-Cβ or CREB show dramatic changes in synaptic plasticity and long-term potentiation (Qi et al. 1996; Pittenger et al. 2006). CREB is activated via phosphorylation by PKA and is inactivated via dephosphorylation by protein phosphatase 1 (PP1). We recently found that calpain over-activation also truncates and activates calcineurin (also called PP2B) in AD brain, resulting in calcineurin’s phosphatase activity being twofold higher in AD than in controls (Liu et al. 2005a). Activated calcineurin can dephosphorylate and inactivate inhibitor 1 of PP1 (Cohen 1989), resulting in increased PP1 activity and dephosphorylation of CREB. Therefore, the abnormal calpain over-activation in AD brain can also cause down-regulation of CREB by up-regulating calcineurin via the calcineurin–inhibitor 1–PP1–CREB pathway.
It has been shown previously that PKA-R can be degraded by the ubiquitin-proteosome system (Hegde et al. 1993). In rat alveolar epithelial type II cells, PKA is proteolyzed by calpain also (Zimmerman et al. 1996). To our knowledge, the present study is the first to demonstrate the proteolysis of PKA-RII by calpain in the brain and a marked increase of PKA-RII proteolysis in AD brain. This increased proteolysis leads to decreased levels of PKA-Cβ and decreased PKA activity. It remains to be elucidated where the exact proteolysis site of RIIα by calpain is which produces the 47-kDa truncated form and whether the truncated form can still bind to the C subunits of PKA.
The down-regulation of PKA in AD brain might be involved in several mechanisms in addition to impaired CREB functioning. In the brain, PKA can phosphorylate multiple protein substrates and regulate several important pathways. For example, PKA may play an important role in maturation of β-amyloid precursor protein and in β-amyloid peptide production (Su et al. 2003). PKA also phosphorylates tau and primes tau for further phosphorylation by another important tau kinase, glycogen synthase kinase-3β (Jicha et al. 1999; Liu et al. 2004, 2006; Wang et al. 2007). The most favorable sites for phosphorylation of tau by PKA are Ser214 and Ser409 (Liu et al. 2006). Probably because of the decreased activity of the major tau phosphatase, PP2A (Gong et al. 1993, 1995; Liu et al. 2005b), tau is abnormally hyperphosphorylated in AD brain. In contrast to the hyper-phosphorylation of tau at multiple phosphorylation sites, tau phosphorylation at Ser214 and Ser409 was not markedly increased in AD brain (unpublished observations). These observations are interesting and could be explained by the down-regulation of PKA, which might have neutralized the increased tau phosphorylation at these two sites caused by the phosphatase down-regulation.
We found that PKA-RII, but not RI, was degraded by calpain and was decreased in AD brain, where calpain is over-activated. These findings are interesting because they may imply an impaired PKA substrate specificity in AD brain. RI is known to be diffused in the cytoplasm of the cell, whereas RII has much higher affinity to PKA-anchoring proteins for targeting PKA-C to specific subcellular localizations and substrates (Wong and Scott 2004). Thus, some selective PKA-regulated pathways might be more affected than others in AD brain.
Calpain activation could proteolyze many other proteins, including other protein kinases, and thus modulate their activities/functions. For example, calpain cleaves p35 and p39 into p25 and p29, respectively, thereby activating cyclin-dependent protein kinase 5 (Patzke and Tsai 2002). Calpain also cleaves protein kinase C during differentiation of embryonic myoblasts and leads to the promotion of tumor cell adhesion in human breast carcinoma cells (Aragon et al. 2002; Kennett et al. 2004). Therefore, PKA down-regulation could just be one of the consequences of calpain over-activation that occurs in AD brain.
In conclusion, we have demonstrated that the RII subunits of PKA are proteolyzed both in vitro and in the brain by calpain, and the proteolysis leads to decreased PKA-C level and PKA activity in AD brain because of over-activation of calpain. Because PKA is the major regulator of CREB, our findings provide a molecular mechanism leading to deficient CREB function and impaired cognition and memory in AD. Correcting calpain over-activation might inhibit cognitive impairment in AD and related disorders.
Acknowledgments
This work was supported in part by funds from the New York State Office of Mental Retardation and Developmental Disabilities; NIH grants AG027429 and AG019158; a US Alzheimer’s Association grant (IIRG-05-13095); and a fellowship from the Li Foundation, Inc., New York. We thank Ms Janet Murphy for secretarial assistance, and Ms Maureen Marlow for editorial suggestions. We are also grateful to the Sun Health Research Institute Brain Donation Program of Sun City, Arizona, USA, for the provision of postmortem human brain tissue. The Brain Donation Program is partially supported by a National Institute on Aging grant (P30 AG19610, Arizona Alzheimer’s Disease Core Center).
Abbreviations used
- AD
Alzheimer disease
- CREB
cAMP-response element-binding protein
- PKA
cAMP-dependent protein kinase
- PP
protein phosphatase
References
- Aragon B, Poussard S, Dulong S, Touyarot K, Dargelos E, Brustis JJ, Levieux D, Ducastaing A, Cottin P. Protein kinase Calpha is a calpain target in cultured embryonic muscle cells. Mol Cell Biochem. 2002;231:97–106. doi: 10.1023/a:1014460730664. [DOI] [PubMed] [Google Scholar]
- Barco A, Pittenger C, Kandel ER. CREB, memory enhancement and the treatment of memory disorders: promises, pitfalls and prospects. Expert Opin Ther Targets. 2003;7:101–114. doi: 10.1517/14728222.7.1.101. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol Aging. 1995;16:271–284. doi: 10.1016/0197-4580(95)00021-6. [DOI] [PubMed] [Google Scholar]
- Burton KA, Johnson BD, Hausken ZE, Westenbroek RE, Idzerda RL, Scheuer T, Scott JD, Catterall WA, McKnight GS. Type II regulatory subunits are not required for the anchoring-dependent modulation of Ca2+ channel activity by cAMP-dependent protein kinase. Proc Natl Acad Sci USA. 1997;94:11067–11072. doi: 10.1073/pnas.94.20.11067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadd G, McKnight GS. Distinct patterns of cAMP-dependent protein kinase gene expression in mouse brain. Neuron. 1989;3:71–79. doi: 10.1016/0896-6273(89)90116-5. [DOI] [PubMed] [Google Scholar]
- Chang A, Li PP, Warsh JJ. Altered cAMP-dependent protein kinase subunit immunolabeling in post-mortem brain from patients with bipolar affective disorder. J Neurochem. 2003;84:781–791. doi: 10.1046/j.1471-4159.2003.01605.x. [DOI] [PubMed] [Google Scholar]
- Cohen P. The structure and regulation of protein phosphatases. Annu Rev Biochem. 1989;58:453–508. doi: 10.1146/annurev.bi.58.070189.002321. [DOI] [PubMed] [Google Scholar]
- Foss KB, Simard J, Bérubé D, Beebe SJ, Sandberg M, Grzeschik KH, Gagné R, Hansson V, Jahnsen T. Localization of the catalytic subunit C gamma of the cAMP-dependent protein kinase gene (PRKACG) to human chromosome region 9q13. Cytogenet Cell Genet. 1992;60:22–25. doi: 10.1159/000133286. [DOI] [PubMed] [Google Scholar]
- Gamm DM, Baude EJ, Uhler MD. The major catalytic subunit isoforms of cAMP-dependent protein kinase have distinct biochemical properties in vitro and in vivo. J Biol Chem. 1996;271:15736–15742. doi: 10.1074/jbc.271.26.15736. [DOI] [PubMed] [Google Scholar]
- Gong C-C-X, Singh TJ, Grundke-Iqbal I, Iqbal K. Phosphoprotein phosphatase activities in Alzheimer disease brain. J Neurochem. 1993;3:921–927. doi: 10.1111/j.1471-4159.1993.tb03603.x. [DOI] [PubMed] [Google Scholar]
- Gong C-X, Shaikh S, Wang J-Z, Zaidi T, Grundke-Iqbal I, Iqbal K. Phosphatase activity toward abnormally phosphorylated tau: decrease in Alzheimer disease brain. J Neurochem. 1995;2:732–738. doi: 10.1046/j.1471-4159.1995.65020732.x. [DOI] [PubMed] [Google Scholar]
- Gong B, Vitolo OV, Trinchese F, Liu S, Shelanski M, Arancio O. Persistent improvement in synaptic and cognitive functions in an Alzheimer mouse model after rolipram treatment. J Clin Invest. 2004;114:1624–1634. doi: 10.1172/JCI22831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde AN, Goldberg AL, Schwartz JH. Regulatory subunits of cAMP-dependent protein kinases are degraded after conjugation to ubiquitin: a molecular mechanism underlying long-term synaptic plasticity. Proc Natl Acad Sci USA. 1993;90:7436–7440. doi: 10.1073/pnas.90.16.7436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jicha GA, Weaver C, Lane E, Vianna C, Kress Y, Rockwood J, Davies P. cAMP-dependent protein kinase phosphorylations on tau in Alzheimer’s disease. J Neurosci. 1999;19:7486–7494. doi: 10.1523/JNEUROSCI.19-17-07486.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josselyn SA, Nguyen PV. CREB, synapses and memory disorders: past progress and future challenges. Curr Drug Targets CNS Neurol Disord. 2005;4:481–497. doi: 10.2174/156800705774322058. [DOI] [PubMed] [Google Scholar]
- Kennett SB, Roberts JD, Olden K. Requirement of protein kinase C micro activation and calpain-mediated proteolysis for arachidonic acid-stimulated adhesion of MDA-MB-435 human mammary carcinoma cells to collagen type IV. J Biol Chem. 2004;279:3300–3307. doi: 10.1074/jbc.M305734200. [DOI] [PubMed] [Google Scholar]
- Kim SH, Nairn AC, Cairns N, Lubec G. Decreased levels of ARPP-19 and PKA in brains of Down syndrome and Alzheimer’s disease. J Neural Transm Suppl. 2001;61:263–272. doi: 10.1007/978-3-7091-6262-0_21. [DOI] [PubMed] [Google Scholar]
- Liu SJ, Zhang JY, Li HL, Fang ZY, Wang Q, Deng HM, Gong CX, Grundke-Iqbal I, Iqbal K, Wang JZ. Tau becomes a more favorable substrate for GSK-3 when it is prephosphorylated by PKA in rat brain. J Biol Chem. 2004;279:50078–50088. doi: 10.1074/jbc.M406109200. [DOI] [PubMed] [Google Scholar]
- Liu F, Grundke-Iqbal I, Iqbal K, Oda Y, Tomizawa K, Gong CX. Truncation and activation of calcineurin A by calpain I in Alzheimer disease brain. J Biol Chem. 2005a;280:37755–37762. doi: 10.1074/jbc.M507475200. [DOI] [PubMed] [Google Scholar]
- Liu F, Grundke-Iqbal I, Iqbal K, Gong CX. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur J Neurosci. 2005b;8:1942–1950. doi: 10.1111/j.1460-9568.2005.04391.x. [DOI] [PubMed] [Google Scholar]
- Liu F, Liang Z, Shi J, Yin D, El-Akkad E, Grundke-Iqbal I, Iqbal K, Gong CX. PKA modulates GSK-3beta- and cdk5-catalyzed phosphorylation of tau in site- and kinase-specific manners. FEBS Lett. 2006;580:6269–6274. doi: 10.1016/j.febslet.2006.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki K, Yamakuni T, Hashimoto M, Haque AM, Shido O, Mimaki Y, Sashida Y, Ohizumi Y. Nobiletin restoring beta-amyloid-impaired CREB phosphorylation rescues memory deterioration in Alzheimer’s disease model rats. Neurosci Lett. 2006;400:230–234. doi: 10.1016/j.neulet.2006.02.077. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Chan SL. Neuronal and glial calcium signaling in Alzheimer’s disease. Cell Calcium. 2003;34:385–397. doi: 10.1016/s0143-4160(03)00128-3. [DOI] [PubMed] [Google Scholar]
- Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- Murray KJ, England PJ, Lynham JA, Mills D, Schmitz-Peiffer C, Reeves ML. Use of a synthetic dodecapeptide (malantide) to measure the cyclic AMP-dependent protein kinase activity ratio in a variety of tissues. Biochem J. 1990;267:703–708. doi: 10.1042/bj2670703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagakura A, Niimura M, Takeo S. Effects of a phosphodiesterase IV inhibitor rolipram on microsphere embolism-induced defects in memory function and cerebral cyclic AMP signal transduction system in rats. Br J Pharmacol. 2002;135:1783–1793. doi: 10.1038/sj.bjp.0704629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patzke H, Tsai LH. Calpain-mediated cleavage of the cyclin-dependent kinase-5 activator p39 to p29. J Biol Chem. 2002;277:8054–8060. doi: 10.1074/jbc.M109645200. [DOI] [PubMed] [Google Scholar]
- Pei JJ, Gong CX, Iqbal K, Grundke-Iqbal I, Wu QL, Winblad B, Cowburn RF. Subcellular distribution of protein phosphates and abnormally phosphorylated tau in the temporal cortex from Alzheimer’s disease and control brains. J Neural Transm. 1998;105:69–83. doi: 10.1007/s007020050039. [DOI] [PubMed] [Google Scholar]
- Pittenger C, Fasano S, Mazzocchi-Jones D, Dunnett SB, Kandel ER, Brambilla R. Impaired bidirectional synaptic plasticity and procedural memory formation in striatum-specific cAMP response element-binding protein-deficient mice. J Neurosci. 2006;26:2808–2813. doi: 10.1523/JNEUROSCI.5406-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puzzo D, Vitolo O, Trinchese F, Jacob JP, Palmeri A, Arancio O. Amyloid-beta peptide inhibits activation of the nitric oxide/cGMP/cAMP-responsive element-binding protein pathway during hippocampal synaptic plasticity. J Neurosci. 2005;25:6887–6897. doi: 10.1523/JNEUROSCI.5291-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi M, Zhuo M, Skalhegg BS, Brandon EP, Kandel ER, McKnight GS, Idzerda RL. Impaired hippocampal plasticity in mice lacking the Cbeta1 catalytic subunit of cAMP-dependent protein kinase. Proc Natl Acad Sci USA. 1996;93:1571–1576. doi: 10.1073/pnas.93.4.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raynaud F, Marcilhac A. Implication of calpain in neuronal apoptosis. A possible regulation of Alzheimer’s disease. FEBS J. 2006;273:3437–3443. doi: 10.1111/j.1742-4658.2006.05352.x. [DOI] [PubMed] [Google Scholar]
- Reddy PH, McWeeney S. Mapping cellular transcriptosomes in autopsied Alzheimer’s disease subjects and relevant animal models. Neurobiol Aging. 2006;27:1060–1077. doi: 10.1016/j.neurobiolaging.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Saito K, Elce JS, Hamos JE, Nixon RA. Widespread activation of calcium-activated neutral proteinase (calpain) in the brain in Alzheimer disease: a potential molecular basis for neuronal degeneration. Proc Natl Acad Sci USA. 1993;90:2628–2632. doi: 10.1073/pnas.90.7.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y, Ryder J, Ni B. Inhibition of Abeta production and APP maturation by a specific PKA inhibitor. FEBS Lett. 2003;546:407–410. doi: 10.1016/s0014-5793(03)00645-8. [DOI] [PubMed] [Google Scholar]
- Tully T, Bourtchouladze R, Scott R, Tallman J. Targeting the CREB pathway for memory enhancers. Nat Rev Drug Discov. 2003;2:267–277. doi: 10.1038/nrd1061. [DOI] [PubMed] [Google Scholar]
- Veeranna Kaji T, Boland B, et al. Calpain mediates calcium-induced activation of the erk1,2 MAPK pathway and cytoskeletal phosphorylation in neurons: relevance to Alzheimer’s disease. Am J Pathol. 2004;165:795–805. doi: 10.1016/S0002-9440(10)63342-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JZ, Grundke-Iqbal I, Iqbal K. Kinases and phosphatases and tau sites involved in Alzheimer neurofibrillary degeneration. Eur J Neurosci. 2007;25:59–68. doi: 10.1111/j.1460-9568.2006.05226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong W, Scott JD. AKAP signalling complexes: focal points in space and time. Nat Rev Mol Cell Biol. 2004;5:959–970. doi: 10.1038/nrm1527. [DOI] [PubMed] [Google Scholar]
- Wu HY, Tomizawa K, Oda Y, Wei FY, Lu YF, Matsushita M, Li ST, Moriwaki A, Matsui H. Critical role of calpain-mediated cleavage of calcineurin in excitotoxic neurodegeneration. J Biol Chem. 2004;279:4929–4940. doi: 10.1074/jbc.M309767200. [DOI] [PubMed] [Google Scholar]
- Yamamoto-Sasaki M, Ozawa H, Saito T, Rösler M, Riederer P. Impaired phosphorylation of cyclic AMP response element binding protein in the hippocampus of dementia of the Alzheimer type. Brain Res. 1999;824:300–303. doi: 10.1016/s0006-8993(99)01220-2. [DOI] [PubMed] [Google Scholar]
- Zimmerman UJ, Wang M, Nelson JB, Ekwunife FS, Liu L. Secretagogue-induced proteolysis of cAMP-dependent protein kinase in intact rat alveolar epithelial type II cells. Biochim Biophys Acta. 1996;1311:117–123. doi: 10.1016/0167-4889(95)00181-6. [DOI] [PubMed] [Google Scholar]