

Abstract
A limited search for hantaviruses in lung and liver tissues of Sorex shrews (family Soricidae, subfamily Soricinae) revealed phylogenetically distinct hantaviruses in the masked shrew (Sorex cinereus) from Minnesota and in the dusky shrew (Sorex monticolus) from New Mexico and Colorado. The discovery of these shrew-borne hantaviruses, named Ash River virus and Jemez Springs virus, respectively, challenges the long-held dogma that rodents are the sole reservoir hosts and forces a re-examination of their co-evolutionary history. Also, studies now underway are aimed at clarifying the epizootiology and pathogenicity of these new members of the genus Hantavirus.
Based on phylogenetic analyses of full-length viral genomic sequences and host mitochondrial DNA (mtDNA) sequences, hantaviruses segregate into clades that parallel the evolution of murinae, arvicolinae, neotominae, and sigmodontinae rodents.1–4 Whether insectivores (or soricomorphs), which are sympatric with rodents, are involved in the evolutionary origins of hantaviruses has not been systematically studied, despite previous reports of hantavirus antigens in tissues of the Eurasian common shrew (Sorex araneus), alpine shrew (Sorex alpinus), Eurasian water shrew (Neomys fodiens), and common mole (Talpa europea) captured in Russia5,6 and the former Yugoslavia.7 Also, the isolation of Thottapalayam virus (TPMV), from the Asian house shrew (Suncus murinus) in India,8–10 would suggest that soricids might serve as legitimate reservoir hosts of hantaviruses.
Armed with the newly acquired full genome of TPMV and emboldened by our recent discovery of genetically distinct hantaviruses in soricine shrews, including Camp Ripley virus (RPLV) in the northern short-tailed shrew (Blarina brevicauda) in the United States11 and Cao Bang virus (CBNV) in the Chinese mole shrew (Anourosorex squamipes) in Vietnam,12 we launched a small-scale search for soricid-borne hantaviruses by accessing the archival tissue collection of Sorex (family Soricidae, subfamily Soricinae), housed in the Museum of Southwestern Biology at the University of New Mexico.
RNA extracts, from 100 mg each of frozen lung or liver tissues from the masked shrew (S. cinereus), dusky or montane shrew (S. monticolus), dwarf shrew (S. nanus), northern water shrew (S. palustris), Trowbridge shrew (S. trowbridgii), tundra shrew (S. tundrensis), and vagrant shrew (S. vagrans), captured in the United States between 1983 and 2005 (Table 1), were analyzed for hantavirus sequences by reverse transcription-polymerase chain reaction (RT-PCR). The remarkably divergent genomes of soricid-borne hantaviruses presented challenges in designing suitable primers, but we eventually succeeded in amplifying regions of the S (outer OSM55: 5′-TAGTAGTAGACTCC-3′ and HTN-S6: 5′-AGCTCIGGATCCATITCATC-3′; inner Cro2R: 5′-AIGAYTGRTARAAIGAIGAYTTYTT-3′ and PHS-5F: 5′-TAGTAGTAGA CTCCTTRAARAGC-3′)13,14 and L (outer HAN-L-F1: 5′-ATGTAYGTBAGTGCWGATGC-3′ and HAN-L-R1: 5′-AACCADTCWGTYCCRTCATC-3′; inner HAN-L-F2: 5′-TGCWGATGCHACIAARTGGTC-3′ and HAN-L-R2: 5′-GCRTCRTCWGARTGRTGDGCA A-3′)14 segments.
TABLE 1.
RT-PCR detection of hantavirus sequences in tissues of Sorex shrews
| Genus species | State | County* | Trapping year | Number tested | Number positive |
|---|---|---|---|---|---|
| Sorex cinereus | Alaska | Anchorage | 1998 | 10 | 0 |
| Anchorage | 2005 | 3 | 0 | ||
| Minnesota | Cass | 1983 | 11 | 1 | |
| St. Louis | 1994 | 1 | 1 | ||
| North Carolina | Swain | 1994 | 1 | 0 | |
| Sorex haydeni | New Mexico | Sandoval | 1996 | 1 | 1 |
| Sorex monticolus | Alaska | Anchorage | 1998 | 1 | 0 |
| Colorado | Jackson | 1994 | 1 | 1 | |
| New Mexico | Sandoval | 1996 | 5 | 2 | |
| Sandoval | 1998 | 4 | 1 | ||
| Sandoval | 2000 | 4 | 1 | ||
| Catron | 1995 | 1 | 0 | ||
| Utah | Tooele | 1997 | 1 | 0 | |
| Wasatch | 1994 | 1 | 0 | ||
| Sorex nanus | Colorado | Hinsdale | 1994 | 1 | 0 |
| Sorex palustris | Minnesota | Morrison | 1999 | 1 | 0 |
| New Mexico | Sandoval | 1996 | 1 | 0 | |
| Sorex trowbridgii | Washington | Kitsap | 1996 | 1 | 0 |
| Sorex tundrensis | Alaska | Anchorage | 2005 | 2 | 0 |
| Sorex vagrans | New Mexico | Otero | 1994 | 1 | 0 |
| Santa Fe | 1996 | 2 | 0 |
No counties exist in Alaska, so localities are reported by quadrangle map.
First- and second-round PCR were performed in 20-µL reaction mixtures, containing 250 µM dNTP, 2 mM MgCl2, 1 U of AmpliTaq polymerase (Roche, Basel, Switzerland), and 0.25 µM of each primer. Initial denaturation at 94°C for 2 min was followed by two cycles each of denaturation at 94°C for 30 sec, two-degree step-down annealing from 46°C to 38°C for 40 sec, and elongation at 72°C for 1 min, then 30 cycles of denaturation at 94°C for 30 sec, annealing at 42°C for 40 sec, and elongation at 72°C for 1 min, in a GeneAmp PCR 9700 thermal cycler (Perkin-Elmer, Waltham, MA). PCR products were separated by agarose gel electrophoresis and purified using the Qiaex Gel Extraction Kit (Qiagen, Hilden, Germany). DNA was sequenced directly using an ABI Prism 377XL Genetic Analyzer (Applied Biosystems, Foster City, CA). Sequences were then processed using the Genetyx version 6 software (Genetyx Corporation, Tokyo, Japan) and aligned using Clustal W15 and transAlign.16 For phylogenetic analysis, maximum-likelihood consensus trees were generated by the Bayesian Metropolis–Hastings Markov Chain Monte Carlo (MCMC) tree-sampling methods as implemented by Mr. Bayes using a GTR+I+G model of evolution, as selected by Modeltest v.3.7, partitioned by codon position.17–19
Of the 54 Sorex shrews studied, hantavirus S- and L-segment sequences were detected in S. cinereus, captured near the Ash River Station in Voyageur’s National Park (48°26′N, 92°00′W) in St. Louis County, Minnesota, in August 1994, and in Chippewa National Forest in Cass County, Minnesota, in July 1983, as well as in S. monticolus, captured near Jemez Springs (35°48′N, 106°30′W) in Sandoval County, New Mexico, in September 1996, September 1998 and September 2000, and along Trapline ECJ (40°27′N, 106°00′W) in Jackson County, Colorado, in July 1994 (Table 1). The newly identified hantavirus sequences were designated Ash River virus (ARRV) and Jemez Springs virus (JMSV), respectively. Host identification was verified by morphologic assessment of voucher specimens and by mtDNA sequence analysis of a 756-base pair region of the cytochrome b gene, using previously described universal primers.20
Pairwise alignment and comparison of a 1,083-nucleotide region of the S segment and 347-nucleotide region of the L segment showed low sequence similarities between JMSV and representative rodent-associated hantaviruses, of 59.0–62.1% and 60.2–73.8%, respectively (Table 2). Nucleotide sequence differences in this region of the S and L segments among JMSV strains from Sandoval County in 1996, 1998, and 2000 were 0.4–0.8% and 1.1–6.6%, respectively; amino acid sequences differed by 0.6% and 0.9%, respectively. In the L segment, the JMSV strains from New Mexico and Colorado differed by 11.2–12.1% at the nucleotide level but were identical at the amino acid level.
TABLE 2.
Nucleotide and amino acid sequence similarities (in percent) of the partial S and L segments between JMSV strain MSB89332 and other hantaviruses
| S Segment |
L Segment |
||||
|---|---|---|---|---|---|
| Virus* | Strain | 1083 nt | 356 aa | 347 nt | 115 aa |
| HTN | 76-118 | 62.1 | 60.5 | 72.1 | 78.4 |
| DOB | Greece | 62.0 | 58.0 | 73.8 | 79.3 |
| SEO | HR80-39 | 61.9 | 58.7 | 69.7 | 75.9 |
| SAN | SA14 | 60.7 | 62.0 | 69.7 | 78.4 |
| PUU | Sotkamo | 61.0 | 54.7 | 62.3 | 65.5 |
| TUL | M5302v | 59.9 | 54.9 | 60.2 | 63.8 |
| PH | PH-1 | 59.7 | 53.6 | 67.4 | 67.8 |
| SN | NMH10 | 59.0 | 53.1 | 65.7 | 64.7 |
| AND | Chile 9717869 | 60.4 | 54.5 | 64.3 | 65.5 |
| RPL | MSB89863 | NA | NA | 69.5 | 75.9 |
| CBN | TC-3 | 70.1 | 73.9 | 76.4 | 88.8 |
| ARR | MSB73418 | 71.9 | 77.5 | 75.8 | 94.0 |
| TGN | Tan826 | 63.1 | 64.0 | 68.0 | 76.7 |
| TPM | VRC-66412 | 53.0 | 42.7 | 63.1 | 62.1 |
nt = nucleotide; aa = amino acid; NA = not available.
AND, Andes; ASR, Ash River; CBN, Cao Bang; DOB, Dobrava; HTN, Hantaan; PH, Prospect Hill; PUU, Puumala; RPL, Camp Ripley; SAN, Sangassou; SEO, Seoul; SN, Sin Nombre; TGN, Tanganya; TPM, Thottapalayam; TUL, Tula.
ARRV, from S. cinereus captured in Minnesota, showed a similar degree of sequence divergence in the S and L segments from rodent-borne hantaviruses, with the highest homology to Hantaan virus (HTNV) 76–118 (62.7% and 72.1%) and Dobrava virus (DOBV) Greece (62.2% and 73.5%). When the S- and L-segment sequences were compared with soricid-borne hantaviruses, ARRV was more similar to JMSV (71.9% and 75.8%) and CBNV (68.8% and 78.1%), than to TPMV (54.4% and 64.0%) and Tanganya virus (TGNV) (64.4% and 73.8%), a hantavirus detected recently in the Therese shrew (Crocidura theresae) in Guinea.21 At the amino acid level, ARRV and JMSV were 77.5% and 94.0% identical in the partial S and L segments, respectively (Table 2).
Hantavirus S- and L-segment nucleotide sequences, detected in a shrew, which was initially identified as S. cinereus, were only 77.5% and 79.5% similar to ARRV, respectively, but 99.1–99.6% and 93.7–99.1% similar to JMSV. Amino acid sequence similarities were 99.1–99.6% and 99.2–100%. This shrew was captured in Sandoval County, New Mexico, at the identical site and time as S. monticolus in which JMSV was detected. On mtDNA analysis, this shrew was shown to be S. haydeni (not S. cinereus). Isolation of JMSV from S. monticolus and subsequent plaque-reduction neutralization tests will be necessary to definitively prove that S. monticolus is the true reservoir host of JMSV.
Phylogenetic analysis based on partial S- and L-segment sequences, generated by the maximum-likelihood method using Bayesian tree-sampling, placed JMSV and ARRV in a distinct group that included CBNV (Figure 1). This clade was most closely related to that of the murinae rodent-borne hantaviruses. By contrast, the relationship of JMSV and ARRV to TPMV and TGNV, two hantaviruses harbored by crocidurine shrews, varied according to the S- and L-segment data (Figure 1). Similar topologies, supported by bootstrap analysis, were obtained using the neighbor-joining method. Also, amino acid sequence phylogenies, constructed using Tree Puzzle22 and the BLOSUM 62 model of evolution with a gamma rate-heterogeneity parameter, yielded nearly identical topologies (data not shown).
FIGURE 1.
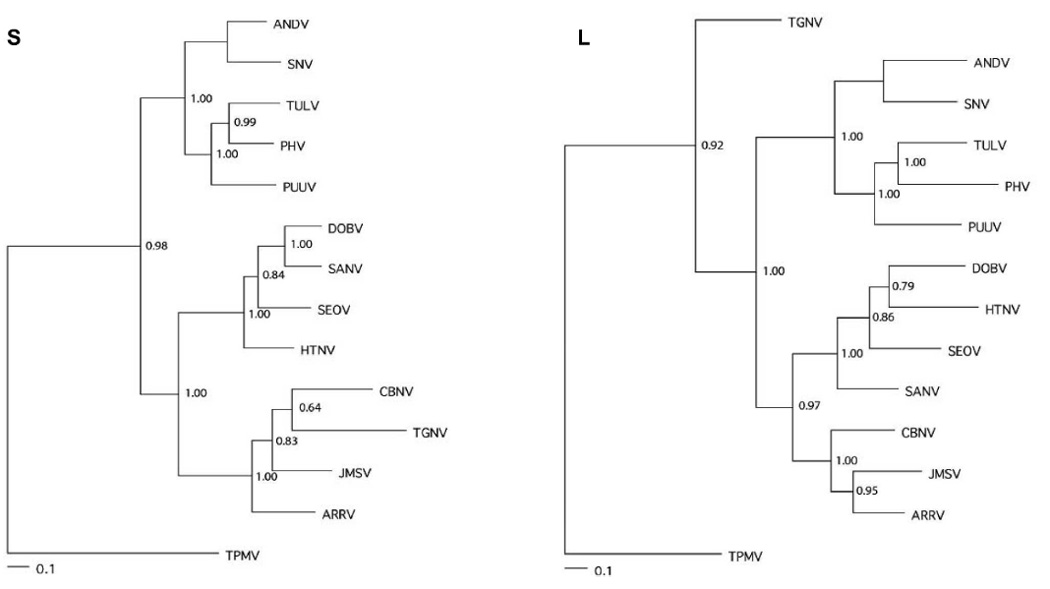
Maximum-likelihood phylogenetic consensus trees, generated by Bayesian Metropolis–Hastings Markov Chain Monte Carlo (MCMC) tree-sampling methods using a GTR+I+G model of evolution partitioned by codon position, based on the alignment of the partial 1048-nucleotide S- and 347-nucleotide L-genomic segments of Jemez Springs virus (JMSV MSB89332, EF619960, EF619962) from the dusky shrew (Sorex monticolus) and Ash River virus (ARRV MSB73418, EF619961, EF650086) from the masked shrew (Sorex cinereus), as well as representative murinae rodent-borne hantaviruses, including Hantaan virus (HTNV 76-118, NC_005219, NC_005222), Sangassou virus (SANV SA14, DQ268651, DQ268652), Dobrava virus (DOBV Greece, NC_005234, NC_005235), and Seoul virus (SEOV 80 39, NC_005237, NC_005238); arvicolinae rodent-borne hantaviruses, including Tula virus (TULV M5302v, NC_005228, NC_005226), Prospect Hill virus (PHV PH-1, X55128, EF646763) and Puumala virus (PUUV Sotkamo, NC_005223, NC_005225); and sigmodontinae and neotominae rodent-borne hantaviruses, including Andes virus (ANDV Chile 9717869, NC_003467, NC_003468) and Sin Nombre virus (SNV NMH10, NC_005215, NC_005217). Cao Bang virus (CBNV TC-3, EF543524, EF543525) from the Chinese mole shrew (Anourosorex squamipes), Tanganya virus (TGNV Tan826, EF050454, EF050455) from the Therese shrew (Crocidura theresae), and Thottapalayam virus (TPMV VRC-66412, AY526097, EU001330) from the Asian house shrew (Suncus murinus) are also shown. Numbers at each node are posterior node probabilities based on 30,000 trees: two replicate MCMC runs consisting of four chains of 2 million generations each sampled every 100 generations with a burn-in of 5000 (25%). Sufficiency of chain length was determined based on convergence of likelihood values, giving effective sample sizes well over 400 (implemented in MrBayes v. 3). The scale bar indicates 0.1 nucleotide substitutions per site. Alternate phylogenetic methods gave rise to essentially identical topologies, with only minor, unsupported differences. Host identifications of S. monticolus and S. cinereus were confirmed by mitochondrial DNA sequencing (data not shown).
The masked shrew is the most widely distributed shrew in North America, extending from Alaska and Canada, across Washington, Idaho, south-central Utah, north-central New Mexico, and Nebraska, and throughout the Appalachians in the east. Also among the most common shrews in North America, the dusky shrew is found in Alaska and western Canada, extending southerly through Washington, Idaho, Montana, Utah, Colorado, New Mexico, and Arizona to Mexico. Both species (subfamily Soricinae) occupy a variety of moist habitats, as well as grassy areas near streams or rivers; meadows; thickets of willow and alder; spruce-fir forests; and alpine tundra.
Dusky shrews occasionally co-exist with as many as four other soricid species, including masked shrews. As such, sharing of nesting materials, as well as inter- and intra-specific fighting and wounding, might result in hantavirus spillover. The high degree of sequence similarity between hantaviruses detected in S. monticolus and S. haydeni in Sandoval County, New Mexico, may be instructive. Based on its dominant position in this community, the natural host of JMSV was assigned to S. monticolus.
Using parsimony and maximum likelihood methods, phylogenetic analyses based on the mtDNA cytochrome b gene have revealed significant molecular variation among S. monticolus and eight related species (S. bairdi, S. bendirii, S. neomexicanus, S. ornatus, S. pacificus, S. palustris, S. sonomae and S. vagrans).23 Poorly resolved internal nodes within topologies suggest rapid diversification within this group. S. monticolus was not monophyletic under current taxonomic nomenclature, but formed two distinct clades: a continental clade and a coastal clade. The high degree of sequence similarity of the hantaviruses detected in S. monticolus from New Mexico and Colorado are consistent with their grouping into the (southern) continental clade.
In analyzing the cytochrome b and nicotinamide adenine dinucleotide dehydrogenase 4 mtDNA genes, eight members of the S. cinereus group (S. camtschatica, S. cinereus, S. haydeni, S. jacksoni, S. portenkoi, S. preblei, S. pribilofensis, and S. ugyunak) and S. longirostris were placed into northern and southern clades, with S. cinereus and S. longirostris in the latter.24 A recent analysis estimates the divergence time at approximately 10 million years before present.25
The newly identified hantaviruses in the masked shrew and dusky shrew in the United States, when considered within the context of the recently identified hantavirus in the northern short-tailed shrew,11 heralds the discovery of additional hantaviruses in shrews within North America and beyond. Studies, now underway, will examine the epizootiology and molecular phylogeny of JMSV and ARRV throughout the respective geographic ranges of S. monticolus and S. cinereus. Investigations will also focus on the possibility of genetic re-assortment among hantaviruses harbored by sympatric Sorex species. Finally, the isolation of these novel hantaviruses in cell culture will facilitate investigations aimed at determining their definitive host associations, as well as their pathogenicity for humans.
Acknowledgements
The authors thank the staff of the Greenwood Molecular Biology Facility, Pacific Biosciences Research Center, for excellent technical assistance with DNA sequencing. This work was supported in part by grants P20RR018727 (Centers of Biomedical Research Excellence) and G12RR003061 (Research Centers in Minority Institutions) from the National Center for Research Resources, National Institutes of Health. Shrew samples from Alaska were collected through support of grants DEB0196095 and DEB9981915 from the National Science Foundation.
REFERENCES
- 1.Plyusnin A, Vapalahti O, Vaheri A. Hantaviruses: genome structure, expression and evolution. J Gen Virol. 1996;77:2677–2687. doi: 10.1099/0022-1317-77-11-2677. [DOI] [PubMed] [Google Scholar]
- 2.Heiske A, Anheier B, Pilaski J, Volchkov VE, Feldmann H. A new Clethrionomys-derived hantavirus from Germany: evidence for distinct genetic sublineages of Puumala viruses in Western Europe. Virus Res. 1999;61:101–112. doi: 10.1016/s0168-1702(99)00024-6. [DOI] [PubMed] [Google Scholar]
- 3.Hughes AL, Friedman R. Evolutionary diversification of protein-coding genes of hantaviruses. Mol Biol Evol. 2000;17:1558–1568. doi: 10.1093/oxfordjournals.molbev.a026254. [DOI] [PubMed] [Google Scholar]
- 4.Vapalahti O, Mustonen J, Lundkvist A, Henttonen H, Plyusnin A, Vaheri A. Hantavirus infections in Europe. Lancet Infect Dis. 2003;3:653–661. doi: 10.1016/s1473-3099(03)00774-6. [DOI] [PubMed] [Google Scholar]
- 5.Gavrilovskaya IN, Apekina NS, Myasnikov Yu A, Bernshtein AD, Ryltseva EV, Gorbachkova EA, Chumakov MP. Features of circulation of hemorrhagic fever with renal syndrome (HFRS) virus among small mammals in the European U.S.S.R. Arch Virol. 1983;75:313–316. doi: 10.1007/BF01314898. [DOI] [PubMed] [Google Scholar]
- 6.Tkachenko EA, Ivanov AP, Donets MA, Myasnikov YA, Ryltseva EV, Gaponova LK, Bashkirtsev VN, Okulova NM, Drozdov SG, Slonova RA, Somov GP. Potential reservoir and vectors of haemorrhagic fever with renal syndrome (HFRS) in the U.S.S.R. Ann Soc Belg Med Trop. 1983;63:267–269. [PubMed] [Google Scholar]
- 7.Gligic A, Stojanovic R, Obradovic M, Hlaca D, Dimkovic N, Diglisic G, Lukac V, Ler Z, Bogdanovic R, Antonijevic B, Ropac D, Avsic T, LeDuc JW, Ksiazek T, Yanagihara R, Gajdusek DC. Hemorrhagic fever with renal syndrome in Yugoslavia: epidemiologic and epizootiologic features of a nationwide outbreak in 1989. Eur J Epidemiol. 1992;8:816–825. doi: 10.1007/BF00145326. [DOI] [PubMed] [Google Scholar]
- 8.Carey DE, Reuben R, Panicker KN, Shope RE, Myers RM. Thottapalayam virus: a presumptive arbovirus isolated from a shrew in India. Indian J Med Res. 1971;59:1758–1760. [PubMed] [Google Scholar]
- 9.Zeller HG, Karabatsos N, Calisher CH, Digoutte J-P, Cropp CB, Murphy FA, Shope RE. Electron microscopic and antigenic studies of uncharacterized viruses. II. Evidence suggesting the placement of viruses in the family Bunyaviridae. Arch Virol. 1989;108:211–227. doi: 10.1007/BF01310935. [DOI] [PubMed] [Google Scholar]
- 10.Song J-W, Baek LJ, Schmaljohn CS, Yanagihara R. Thottapalayam virus: a prototype shrewborne hantavirus. Emerg Infect Dis. 2007;13:980–985. doi: 10.3201/eid1307.070031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arai S, Song J-W, Sumibcay L, Bennett SN, Nerurkar VR, Parmenter C, Cook JA, Yates TL, Yanagihara R. Hantavirus in northern short-tailed shrew, United States. Emerg Infect Dis. 2007;13:1420–1423. doi: 10.3201/eid1309.070484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Song J-W, Kang HJ, Song KJ, Truong TT, Bennett SN, Arai S, Truong NU, Yanagihara R. Newfound hantavirus in Chinese mole shrew, Vietnam. Emerg Infect Dis. 2007;13:1784–1787. doi: 10.3201/eid1311.070492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arthur RR, Lofts RS, Gomez J, Glass GE, LeDuc JW, Childs JE. Grouping of hantaviruses by small (S) genome segment polymerase chain reaction and amplification of viral RNA from wild-caught rats. Am J Trop Med Hyg. 1992;47:210–224. doi: 10.4269/ajtmh.1992.47.210. [DOI] [PubMed] [Google Scholar]
- 14.Klempa B, Fichet-Calvet E, Lecompte E, Auste B, Aniskin V, Meisel H, Denys C, Koivogui L, ter Meulen J, Krüger DH. Hantavirus in African wood mouse, Guinea. Emerg Infect Dis. 2006;12:838–840. doi: 10.3201/eid1205.051487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bininda-Emonds ORP. transAlign: using amino acids to facilitate the multiple alignment of protein-coding DNA sequences. BMC Bioinformatics. 2005;6:156. doi: 10.1186/1471-2105-6-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huelsenbeck JP, Ronquist F, Nielsen R, Bollback JP. Bayesian inference of phylogeny and its impact on evolutionary biology. Science. 2001;294:231–234. doi: 10.1126/science.1065889. [DOI] [PubMed] [Google Scholar]
- 18.Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- 19.Posada D, Crandall KA. MODELTEST: testing the model of DNA substitution. Bioinformatics. 1998;14:817–818. doi: 10.1093/bioinformatics/14.9.817. [DOI] [PubMed] [Google Scholar]
- 20.Smith MF, Patton JL. Variation in mitochondrial cytochrome b sequence in natural populations of South American Akodontine rodents (Murinae: Sigmodontinae) Mol Biol Evol. 1991;8:85–103. doi: 10.1093/oxfordjournals.molbev.a040638. [DOI] [PubMed] [Google Scholar]
- 21.Klempa B, Fichet-Calvet E, Lecompte E, Auste B, Aniskin V, Meisel H, Barrier P, Koivogui L, ter Meulen J, Krüger DH. Novel hantavirus sequences in shrew, Guinea. Emerg Infect Dis. 2007;13:520–552. doi: 10.3201/eid1303.061198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schmidt HA, Strimmer K, Vingron M, von Haeseler A. TREE-PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics. 2002;18:502–504. doi: 10.1093/bioinformatics/18.3.502. [DOI] [PubMed] [Google Scholar]
- 23.Demboski JR, Cook JA. Phylogeography of the dusky shrew, Sorex monticolus (Insectivora, Soricidae): insight into deep and shallow history in northwestern North America. Mol Ecol. 2001;10:1227–1240. doi: 10.1046/j.1365-294x.2001.01260.x. [DOI] [PubMed] [Google Scholar]
- 24.Demboski JR, Cook JA. Phylogenetic diversification within the Sorex cinereus group (Soricidae) J Mammal. 2003;84:144–158. [Google Scholar]
- 25.Dubey S, Salamin N, Ohdachi SD, Barriére P, Vogel P. Molecular phylogenetics of shrews (Mammalia: Soricidae) reveals timing of transcontinental colonizations. Mol Phylogenet Evol. 2007;44:126–137. doi: 10.1016/j.ympev.2006.12.002. [DOI] [PubMed] [Google Scholar]