

Abstract
There is a need for novel therapies targeting hypoxic cells in tumors. These cells are associated with tumor resistance to therapy and express hypoxia inducible factor-1 (HIF-1), a transcription factor that mediates metabolic adaptation to hypoxia and activates tumor angiogenesis. We previously developed an oncolytic adenovirus (HYPR-Ad) for the specific killing of hypoxic/HIF-active tumor cells, which we now armed with an interleukin-4 gene (HYPR-Ad-IL4). We designed HYPR-Ad-IL4 by cloning the Ad E1A viral replication and IL-4 genes under the regulation of a bidirectional hypoxia/HIF-responsive promoter. The IL-4 cytokine was chosen for its ability to induce a strong host antitumor immune response and its potential antiangiogenic activity. HYPR-Ad-IL4 induced hypoxia-dependent IL-4 expression, viral replication, and conditional cytolysis of hypoxic, but not normoxic cells. The treatment of established human tumor xenografts with HYPR-Ad-IL4 resulted in rapid and maintained tumor regression with the same potency as that of wild-type dl309-Ad. HYPR-Ad-IL4–treated tumors displayed extensive necrosis, fibrosis, and widespread viral replication. Additionally, these tumors contained a distinctive leukocyte infiltrate and prominent hypoxia. The use of an oncolytic Ad that locally delivers IL-4 to tumors is novel, and we expect that HYPR-Ad-IL4 will have broad therapeutic use for all solid tumors that have hypoxia or active HIF, regardless of tissue origin or genetic alterations.
Introduction
Hypoxic/hypoxia inducible factor (HIF)–active cells within solid tumors represent an important clinical problem and have become a critical target for antitumor therapy (reviewed in refs. 1, 2). These tumor cells are a viable and aggressive subpopulation associated with increased patient mortality in a wide variety of human cancers. They contribute to tumor growth, malignancy, and therapeutic resistance to chemo- and radiotherapies. pO2 levels in human tumors are heterogenous, with median levels significantly lower than the surrounding normal tissue (intratumoral hypoxia; ref. 3). Factors that may play a role in the development of intratumoral hypoxia include the presence of a structurally and functionally defective tumor vasculature and the high metabolic rate of tumor cells (4). HIF, a heterodimeric transcription factor composed of α and β subunits, is activated in response to physiologically reduced tissue oxygen levels and plays a central role in mediating the cellular adaptive response to this environmental stress. In addition to oxygen-dependent regulation, the activity of HIF is also increased in tumors by oxygen-independent mechanisms associated with tumor-specific genetic alterations (reviewed in ref. 5). Consistent with this, HIF is overexpressed in numerous cancer types and their metastases, but is not present in the surrounding normal tissue (6, 7). HIF activates more than 70 genes via direct binding to a hypoxia-responsive element (HRE). A large number of HIF target genes function in various aspects of tumor biology, including cellular proliferation, survival/apoptosis, angiogenesis, glucose metabolism, extracellular matrix metabolism, cell adhesion and motility, and drug resistance. Consequently, hypoxic and HIF-expressing cells have become a target for anticancer therapy. There is a clear need to develop new agents that will antagonize and/or eliminate these cells (8, 9).
Oncolytic adenoviruses (Ad) are biological antitumor agents that offer several advantages including an intrinsic ability to kill infected host cells at the completion of the virus replicative cycle and the capacity to deliver therapeutic transgenes (10). Importantly, oncolytic Ads are distinct from classic gene therapy Ads that serve as gene delivery agents and do not replicate. The underlying principle for this developing cancer therapy is that the virus will selectively replicate in tumor cells, resulting in an exponential increase of the virus inoculum and the establishment of repeating cycles of cell infection, virus replication, and tumor cell death. Treatment of cancer patients with oncolytic Ads in clinical trials has shown the overall safety of this therapy and modest antitumor activity (11). The differential activation of HIF/HRE-dependent gene expression in tumors compared with normal tissue provides an opportunity for the design of a targeted oncolytic Ad that would depend on HIF expression for its replication. To develop such a therapeutic agent, we previously designed a novel bidirectional HIF-responsive promoter that can specifically target the expression of two independent genes to hypoxic/HIF-active tumor cells (12). This promoter was then used to drive the expression of the Ad E1A replication gene in the Ad genome, thereby generating a hypoxia/ HIF-dependent replicative oncolytic Ad (HYPR-Ad#1; ref. 13). We showed that HYPR-Ad was able to specifically kill hypoxic/HIF-active tumor cells via its cytolytic replication cycle. This first-generation virus had antitumor activity resulting in tumor growth reduction by up to 5-fold (14), supporting its further development as a novel antitumor therapeutic.
In this report, we describe a second-generation HYPR-Ad that expresses interleukin-4 (HYPR-Ad-IL4), a cytokine with antitumor activity against a wide range of tumor types. IL-4 was chosen for these studies because it exhibits multimodal antitumor activity, including the induction of a host immune response against the tumor and inhibition of tumor angiogenesis (reviewed in ref. 15). IL-4 elicits strong antitumor effects in both immunocompetent and immunocompromised (athymic) animals that are associated with infiltration of the tumor by eosinophils and macrophages (16). Immunocompetent animals also exhibit tumor infiltration by T lymphocytes and the establishment of a long-term antitumor immune response mediated by T-cell–dependent activation of B lymphocytes (17, 18). IL-4 antiangiogenic activity is mediated by binding to its receptor on endothelial cells, which leads to the down-regulation of vascular endothelial growth factor receptor 2 (VEGF-R2) on these cells, resulting in reduced tumor vascularization (19). Interestingly, IL-4 can also directly inhibit the growth of some neoplastic cells in vitro (15). These studies indicate that IL-4 may directly affect tumor cells in addition to modulating host immune and endothelial cells, thus strongly supporting its use as a cancer therapeutic. Finally, local expression of IL-4 in the tumor microenvironment via HYPR-Ad is expected to overcome the short half-life and toxicity associated with systemic recombinant IL-4 delivery (15, 20). This research is innovative because the therapeutic effectiveness of a conditionally replicative Ad that locally expresses IL-4 in the tumor microenvironment has not been designed or tested to date and is expected to have broad therapeutic use for all solid tumors that have hypoxia or active HIF, regardless of tissue origin or genetic alterations.
Materials and Methods
Adenoviruses
AdLacZ is replication deficient, lacks the E1 gene region, and expresses LacZ. dl309-Ad is replication competent and contains a wild-type E1 gene region (21). HYPR-Ad#1 (13) is a conditionally replicative Ad in which expression of the Ad E1A replication gene is regulated by the bidirectional V6R hypoxia/HIF-responsive promoter (12). HYPR-Ad-mIL4 is a second-generation HYPR-Ad in which the bidirectional hypoxia/HIF-responsive promoter coregulates the expression of Ad E1A (right arm of promoter) and mouse IL-4 (left arm of promoter) genes (Fig. 1A). To generate HYPR-Ad-mIL4, the mouse IL-4 coding sequence [American Type Culture Collection (ATCC) IMAGE Consortium clone ID 3376587] was subcloned into the EcoRI/HindIII sites of pcDNA3.1 to generate pcDNA3.1-mIL4 (reverse). A NotI mIL4-fragment of pcDNA3.1-mIL4(rev) was then subcloned into the NotI site within the left arm of the bidirectional hypoxia/ HIF-responsive promoter of pBI-V6R-E1 to generate pBI-V6R-E1+mIL4. A 2,419-bp HpaI fragment of pBI-V6R-E1+mIL4 was ligated with a 9475-bp HpaI fragment of pShuttle-V6R-E1 to generate pShuttle-V6R-E1+mIL4. Recombinant HYPR-Ad-mIL4 was generated from pShuttle-V6R-E1+mIL4 and pAdEasy-1 using the pAdEasy system (22). Purified, high-titer viral stocks were generated at the University of North Carolina Vector Core Facility (Chapel Hill, NC).
Figure 1.

Hypoxia-dependent E1A and mIL4 protein expression by HYPR-Ad-mIL4–infected cells. A, schematic of HYPR-Ad-mIL4. HYPR-Ad-mIL4 is a novel hypoxia/HIF-dependent oncolytic Ad that is armed with the antitumorigenic mouse interleukin-4 cytokine gene. This virus contains an exogenous bidirectional hypoxia-responsive promoter composed of six tandem copies of the HRE of the human VEGF gene (6× VEGF-HRE) bordered by two independent mini-CMV promoter elements (12). The right arm of this promoter drives the expression of the Ad E1A replication gene, whereas the left arm regulates expression of the mouse IL-4 gene. The E1B/IX gene region was also reconstituted. This modified Ad type 5 E1 gene region was cloned into a replication-deficient Ad5 genome lacking the E1 and E3 genomic regions. B, LN229 human glioblastoma cells were infected at MOI 1 with HYPR-Ad-mIL4, dl309-Ad, HYPR-Ad#1 viruses or mock infected and then maintained under normoxia (N) or hypoxia (H). Total cell lysates were examined by Western blot analysis for Ad E1A and cellular actin (loading control) at days 1 and 3 postinfection. C, LN229 cells were infected at MOI 1 with AdLacZ, dl309-Ad, HYPR-Ad#1, HYPR-Ad-mIL4, or mock infected and then maintained under normoxia (N, white) or hypoxia (Hyp, black). mIL4 in the conditioned media was measured by ELISA at days 3 and 6 postinfection. Columns, mean mIL4 concentration; bars, SD (pg/mL). *, statistically significant (Student’s t test) increase in mIL4 levels under hypoxia.
Cells
Human 293 (CRL-1573, ATCC), normal human foreskin fibroblasts (Hs68, CRL-1635; ATCC), U251MG-T2 human glioma (14), LN229 human glioma (23), U87.ΔEGFR human glioma (24) and D247MG human gliosarcoma (23) cells were maintained in DMEM containing 10% FCS under normoxia (21% O2). Normal human astrocytes (NHA, Cambrex) were maintained under normoxia in astrocyte medium and subcultured according to vendor protocol. Hypoxia (1% O2) was generated as previously described (13, 14).
Western blot analysis
Whole cell extracts were prepared by lysing cells in 50 mmol/L Tris (pH, 7.5), 150 mmol/L NaCl, 0.5% NP40, 50 mmol/L NaF, 1 mmol/L Na3VO4, 1 mmol/L DTT, 1 mmol/L phenylmethylsulfonyl fluoride, and complete mini protease inhibitor cocktail (Roche). Total protein concentrations of cleared supernatants were determined using the Protein Assay Dye (Bio-Rad). For HIF-1α, cells were lysed by resuspension in the Laemmli sample buffer (Bio-Rad) and boiled for 5 min. Electrophoresis and blotting of protein extracts were done using the Bio-Rad Criterion system. Primary Abs were goat anti-actin (Santa Cruz Biotechnology), rat anti-Ad5 E1B 21K (Oncogene), mouse anti-Ad E1B 55K (A. Turnell, University of Birmingham, United Kingdom), mouse anti-Ad Ab-5 (clone M58 + M73, LabVision), and mouse anti–HIF-1α and anti–HIF-1β (BD Biosciences). Secondary Abs (Roche) were goat anti-mouse immunoglobulin G (IgG) F(ab’)2-POD, swine anti-goat IgG-POD, and goat anti-rat IgG-POD. Signal was detected using the Supersignal West Pico chemiluminescent substrate system (Pierce).
mIL4 ELISA and viral replication assays
LN229, D247MG, and NHA (7.5 × 104) cells were seeded in six-well cell culture dishes. The following day, cells were infected with virus (AdLacZ, dl309-Ad, HYPR-Ad#1, HYPR-Ad-mIL4) at multiplicity of infection (MOI) 1 (LN229, NHA), MOI 5 (D247MG) or were mock infected in triplicate for 5 h under normoxia. Subsequently, virus was aspirated, complete media was added, and the cells were incubated under normoxia versus hypoxia. At the indicated time points, conditioned media and cells (by scraping with a rubber policeman) were independently collected. mIL4 levels in the conditioned media were quantified by ELISA (Pierce). For viral replication assays, virus was harvested from the cell lysate samples by three freeze-thaw cycles, followed by microcentrifugation for 5 min at 16,000 × g. Virus in the resulting supernatant and in the conditioned media samples was titered using a protocol similar to the Adeno-X-Rapid Titer assay (BD Biosciences) with the following changes: (a) primary antibody was a mouse anti-Ad monoclonal antibody blend (MAB805; Chemicon); (b) secondary antibody was a horseradish peroxidase-rat anti-mouse IgG (Zymed); and (c) development was done using the immunopure metal enhanced 3,3′-diaminobenzidine (DAB) substrate for 15 min (Pierce). Statistical analysis was done using the Student’s t test, with a P < 0.05 considered a significant difference.
Cytopathic effect and cell viability assays
LN229, D247MG, U251MG-T2, U87MG. ΔEGFR, and Hs68 cells (5 × 104) or NHA (2.5 × 104) were seeded in 12- or 24-well cell culture dishes, respectively. The following day, cells were infected with virus (AdLacZ, dl309-Ad, HYPR-Ad#1, HYPR-Ad-mIL4) at MOI 0.1 (U251MG-T2), MOI 1 (U87MG.δEGFR, NHA), MOI 5 (D247MG), MOI 25 (LN229), MOI 100 (Hs68) or were mock infected for 5 h under normoxia. Subsequently, virus was aspirated, complete media was added, and the cells were incubated under normoxia versus hypoxia. Cells were visually observed for cytopathic effects (CPE; cell lysis/detachment). When >95% CPE was observed in the HYPR-Ad#1 and HYPR-Ad-mIL4–infected cells maintained under hypoxia (6–10 days postinfection), the cells were fixed, stained with crystal violet solution (1% crystal violet, 10% formaldehyde, 20% ethanol) for 15 min, and then destained in water. Photographs were taken at 100× magnification. Cell viability was determined by a standard 3-(4,5-dimethyliazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (in triplicate).
Tumor studies
Three independent tumor growth reduction studies were done. LN229 (7.5 × 106) and U251MG-T2 (7.5 × 106, study 1; 107, study 2) cells were implanted s.c. into the flanks of nu/nu mice (Athymic NCr-nu, National Cancer Institute) individually marked with tattoos (25). Tumors were established to an average size of 100 mm3 (LN229, 28 days after implantation), 100 mm3 (U251MG-T2, study 1, 22 days after implantation) or 350 mm3 (U251MG-T2, study 2, 39 days after implantation). Tumors were injected intratumorally with 2 × 108 infectious forming units (IFU)/day of virus (dl309-Ad, HYPR-Ad#1, HYPR-Ad-mIL4) or PBS (100 μL total injection volume). LN229 tumors were injected with virus on days 33, 35, and 38; U251MG-T2 study 1 tumors were injected with virus on days 27, 29, and 32; and U251MG-T2 study 2 tumors were injected with virus on days 39, 41, and 43. See legend of Fig. 3 and text for the numbers of animals per group for each study. Tumor size was monitored with calipers, and tumor volume was calculated [(length × width2)/2]. Statistical analysis was done using one-way ANOVA, with a P < 0.05 considered a significant difference. Statistical results were independently confirmed using pairwise t test.
Figure 3.
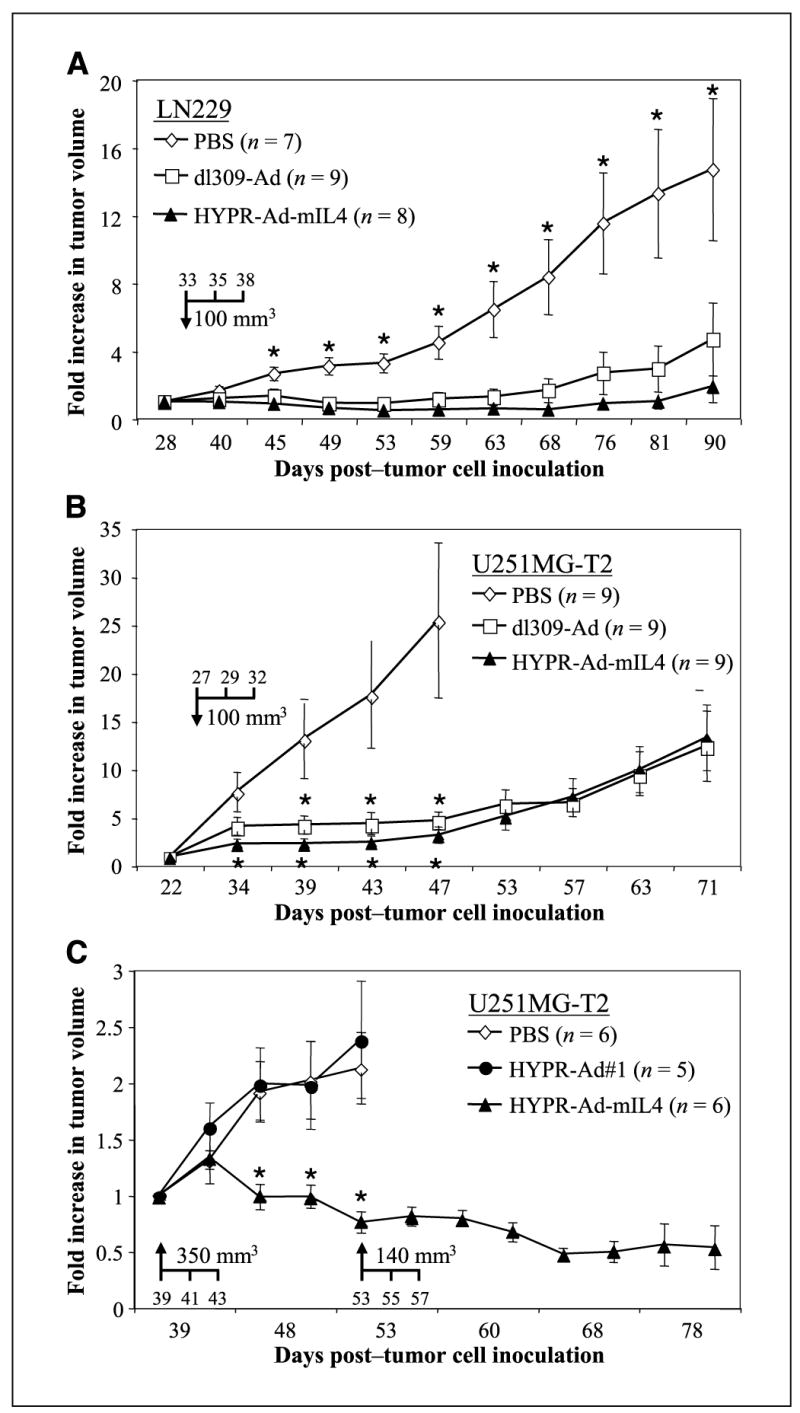
HYPR-Ad-mIL4 therapy leads to regression of established human tumor xenografts. A, established s.c. LN229 tumors (average size of 100 mm3) were injected intratumorally (arrow, days 33, 35, and 38) with PBS (n = 7), dl309-Ad (n = 9), or mIL4-HYPR-Ad (n = 8), and tumor growth was monitored by caliper measurement. B, established s.c. U251MG-T2 tumors (average size of 100 mm3) were injected intratumorally (arrow, days 27, 29, and 32) with PBS (n = 9), dl309-Ad (n = 9), or HYPR-Ad-mIL4 (n = 9), and tumor growth was monitored. C, established s.c. U251MG-T2 tumors (average size of 350 mm3) were injected intratumorally (arrow, days 39, 41, and 43) with PBS (n = 6), HYPR-Ad#1 (n = 5), or HYPR-Ad-mIL4 (n = 6), and tumor growth was monitored. Points, mean; bars, SE. *, statistically significant difference (see Materials and Methods, P < 0.05) in tumor size in the PBS versus virus treatment groups (see text for details).
Histologic and immunohistochemical analysis
Mice containing s.c. LN229 tumors treated with PBS, HYPR-Ad-mIL4, or dl309-Ad (LN229 tumor study described above) were injected i.p. with 60 mg/kg pimonidazole hydrochloride, a 2-nitroimidazole hypoxia marker (Chemicon) on day 15 from the start of virus injection. Ninety minutes later, the tumors were harvested, stored in 10% buffered formalin overnight at room temperature, and transferred to 70% ethanol at 4°C. The tumors were embedded in paraffin and sectioned. Deparaffinized sections were stained with H&E for tumor histology and detection of infiltrating polymorphonuclear (PMN) leukocytes and with Masson’s trichrome to detect collagen. Immunostaining was done for pimonidazole adducts (Hypoxyprobe-1 MAb-1, Chemicon), Ad hexon protein (MAB805, Chemicon), von Willebrandt factor (vWF), and CD45 leukocyte common antigen (LCA, Ly-5, clone 30-F11, BD Biosciences, 1:50 dilution, a pan-lymphocyte marker) as described in ref. (14). vWF-positive blood vessels in two independent tumors per treatment group were quantified by counting the average number of blood vessels in five view fields at 200× magnification. For detection of LCA-positive cells, tissue sections were treated with 1× Target Retrieval Solution (DAKO) for 5 min in a pressure cooker and then immunostained using Vectastain Elite ABC and DAB peroxidase substrate kits (Vector Laboratories). Immunostained sections were counterstained with hematoxylin.
Results
Generation of HYPR-Ad-mIL4, a dual oncolytic and gene therapy Ad
HYPR-Ad-mIL4 is a second-generation hypoxia/HIF-dependent oncolytic HYPR-Ad modified to selectively express the antitumorigenic IL-4 cytokine in the tumor microenvironment (Fig. 1A). HYPR-Ad-mIL4 contains a hypoxia/HIF-responsive promoter composed of six tandem copies of the human VEGF HRE flanked by two CMVmini promoters that bidirectionally regulate the expression of the Ad E1A replication (right arm of promoter) and mIL4 (left arm of promoter) genes. The E1B/IX gene region was restored in HYPR-Ad-mIL4 with its endogenous regulatory elements.
Hypoxia-dependent expression of E1A and mIL4 by HYPR-Ad-mIL4
The activity and specificity of the bidirectional hypoxia/ HIF-responsive promoter within HYPR-Ad-mIL4 was evaluated by measuring E1A and mIL4 protein expression by infected cells grown under normoxia (21% O2) versus hypoxia (1% O2) on days 1 and 3 postinfection. As controls, dl309-Ad–, HYPR-Ad#1–, or mock-infected cells were used. HYPR-Ad-mIL4– and parental HYPR-Ad#1–infected cells, displayed strong hypoxia-dependent E1A expression on days 1 and 3 (Fig. 1B). HYPR-Ad-mIL4 showed tighter hypoxia regulation than parental HYPR-Ad#1 as low levels of E1A appeared for HYPR-Ad#1 under normoxia on day 3 (Fig. 1B). E1A expression by these viruses correlated with the hypoxia-dependent expression of cellular HIF-1α (Supplementary Fig. S1A). In contrast, dl309-Ad–infected cells expressed similar levels of E1A under normoxia and hypoxia throughout the 3-day study. We also confirmed the expression of the Ad E1B 21K and 55K proteins in these infected cells, as the corresponding E1B gene region was restored in HYPR-Ad-mIL4 with its own promoter (Supplementary Fig. S1B). Because E1A activates E1B gene expression, hypoxic up-regulation of E1B was also observed. These results show that HYPR-Ad-mIL4–infected cells conditionally express E1A under hypoxia.
To verify that the IL-4 therapeutic gene was expressed in HYPR-Ad-mIL4–infected cells, we measured mIL4 protein levels by ELISA in the conditioned media of infected LN229, NHA, and D247MG cells postinfection (Fig. 1C and Supplementary Fig. S2B). A significant increase in mIL4 levels was detectable in the conditioned media of infected LN229 cells maintained under hypoxia versus normoxia on day 3 (28-fold increase; P = 0.002, Fig. 1C, left) and day 6 (16-fold increase; P = 0.000, Fig. 1C, right). Importantly, the levels of mIL4 detectable under normoxia were comparable to those obtained with control-infected cells (AdLacZ, dl309-Ad, HYPR-Ad#1, or mock). Similar findings were obtained in NHA and D247MG cells (Supplementary Fig. S2). These results show that the bidirectional hypoxia/HIF-dependent promoter maintains its proper regulation in the context of the Ad genome, and that we created a recombinant Ad able to conditionally express mIL4 and E1A under hypoxia.
Hypoxia-dependent replication of HYPR-Ad-mIL4
The ability of HYPR-Ad-mIL4 to conditionally replicate under hypoxia versus normoxia was examined in LN229, D247MG, and NHA cells (Table 1). On the indicated days postinfection, the amount of virus was titered in collected cell lysates and conditioned media as described in Materials and Methods. A significant increase in HYPR-Ad-mIL4 production under hypoxia was detectable in LN229 cell lysates on day 3 (50-fold increase; P = 0.02) and day 6 (23-fold increase; P = 0.0001, Table 1). This translated into a 28-fold increase in the conditioned media on day 6 derived from these cells (Table 1, P = 0.0007). Under hypoxia, HYPR-Ad-mIL4 production was comparable to that obtained with the parental HYPR-Ad#1 (P > 0.25). In contrast, the production of viral particles in normoxic versus hypoxic cells infected with the control dl309-Ad was similar at day 3 (P > 0.06) and showed a decrease under hypoxia at day 6 (P < 0.002; Table 1). As expected, AdLacZ did not replicate in these cells (Table 1) and, therefore, served as a negative control and a visual marker that the hypoxic and viral infection conditions are not cytotoxic (evidenced by the lack of CPE; data not shown). Similar findings were obtained in NHA and D247MG cells (Table 1). These results show that the replication of HYPR-Ad-mIL4 is hypoxia dependent in culture.
Table 1.
Virus replication in LN229, D247MG, and NHA cells under normoxia (N) versus hypoxia (Hyp) following infection at MOI 1, 5, and 1, respectively
| Cell lysate
|
Conditioned media
|
|||||
|---|---|---|---|---|---|---|
| N (×105) | Hyp (×105) | Fold change | N (×103) | Hyp (×103) | Fold change | |
| LN229 day 3 | ||||||
| AdLacZ | 0 ± 0 | 0 ± 0 | 0 | 0 ± 0 | 0 ± 0 | 0 |
| dl309-Ad | 376 ± 77 | 324 ± 66 | 1 | 513 ± 46 | 728 ± 133 | 1 |
| HYPR-Ad#1 | 4 ± 1 | 25 ± 6 | 6* | 2 ± 1 | 7 ± 3 | 4 |
| HYPR-Ad-mIL4 | 0.4 ± 0.1 | 20 ± 4 | 50* | 1 ± 0.7 | 2 ± 1 | 2 |
| LN229 day 6 | ||||||
| AdLacZ | 0 ± 0 | 0 ± 0 | 0 | 0 ± 0 | 0 ± 0 | 0 |
| dl309-Ad | 3,192 ± 793 | 1,060 ± 310 | −3* | 141,181 ± 12,546 | 79,743 ± 9,406 | −2* |
| HYPR-Ad#1 | 10 ± 1 | 31 ± 6 | 3* | 6 ± 3 | 95 ± 30 | 16* |
| HYPR-Ad-mIL4 | 1 ± 0.3 | 23 ± 5 | 23* | 3 ± 3 | 83 ± 14 | 28* |
| NHA day 3 | ||||||
| AdLacZ | 0 ± 0 | 0 ± 0 | 0 | 0 ± 0 | 0 ± 0 | 0 |
| dl309-Ad | 1,392 ± 175 | 1,538 ± 266 | 1 | 859 ± 159 | 696 ± 144 | 0.8 |
| HYPR-Ad#1 | 57 ± 5 | 282 ± 44 | 5* | 1.4 ± 1.0 | 9.5 ± 1.6 | 6.7* |
| HYPR-Ad-mIL4 | 18 ± 3 | 319 ± 47 | 23* | 0.0 ± 0.4 | 10.8 ± 3.9 | 12.0* |
| D247MG day 3 | ||||||
| AdLacZ | 0 ± 0 | 0 ± 0 | 0 | 0 ± 0 | 0 ± 0 | 0 |
| dl309-Ad | 228 ± 73 | 452 ± 101 | 2* | 69 ± 17 | 132 ± 22 | 1.9 |
| HYPR-Ad#1 | 10 ± 4 | 102 ± 15 | 10* | 2 ± 0.3 | 9 ± 0.4 | 4.5* |
| HYPR-Ad-mIL4 | 5 ± 2 | 101 ± 16 | 20* | 2 ± 0.5 | 13 ± 0.4 | 6.5* |
NOTE: The data represent the mean IFU/mL ± SD.
Statistically significant (Student’s t test) increase or decrease in virus production under hypoxia (P < 0.0118).
HYPR-Ad-mIL4 specifically lyses hypoxic cells
The ability of HYPR-Ad-mIL4 to conditionally induce cytolysis of infected cells under hypoxia and its potential cytotoxicity under normoxia was examined in four histologically, genetically, and biologically diverse human brain tumor cell lines (LN229: Fig. 2A, U251MG-T2; Fig. 2B, U87. ΔEGFR; Supplementary Fig. S3A, D247MG; Supplementary Fig. S3B) and two normal human cells (primary astrocytes; Fig. 2C, Hs68 foreskin fibroblasts; Supplementary Fig. S3C). HYPR-Ad-mIL4 elicited a hypoxia-dependent CPE response in all of the cells tested, with >95% of infected cells displaying CPE under hypoxia but not normoxia. The CPE-inducing ability of HYPR-Ad-mIL4 was similar to that of the parental HYPR-Ad#1, with both viruses inducing comparable CPE under hypoxia at the same MOI and point in time in the different cell lines tested. As expected, cells infected with dl309-Ad underwent >95% CPE under both normoxia and hypoxia, whereas AdLacZ-infected cells showed no cell toxicity. Viral mediated cytotoxicity under normoxia versus hypoxia was independently confirmed and quantified by MTT assay in LN229, U251MG-T2, and D247MG cells (Supplementary Fig. S4). These data show that HYPR-Ad-mIL4 is able to selectively induce cytolysis under hypoxia in a panel of tumor and normal cells with no apparent toxicity to infected cells maintained under normoxia.
Figure 2.

HYPR-Ad-mIL4 selectively lyses hypoxic cells. LN229 (A), U251MG-T2 (B), and NHA (C) cells were infected at MOI 25, 0.1, or 1, respectively, with AdLacZ, HYPR-Ad#1, HYPR-Ad-mIL4, dl309-Ad, or mock infected and then maintained under normoxia (N) and hypoxia (Hyp). Cells were visually monitored for CPE. Shown are photographs (magnification, 100×) of crystal violet–stained cells taken 6 to 10 d postinfection.
HYPR-Ad-mIL4 has potent antitumor activity resulting in the regression of established tumors
The antitumor activity of HYPR-Ad-mIL4 against established tumors was examined in three independent studies in nu/nu mice using two aggressive s.c. xenograft tumor models (LN229, U251MG-T2; refs. 14, 23). Established s.c. LN229 glioma tumor xenografts (average size of 100 mm3, seven to nine animals per group) were treated intratumorally with 6 × 108 IFU of dl309-Ad, HYPR-Ad-mIL4, or PBS (Fig. 3A). Tumor growth was monitored for 57 days when animals had to be sacrificed due to the large size of the PBS-treated tumors. A significant reduction in tumor growth was detectable in the HYPR-Ad-mIL4 (P = 0.004) and dl309-Ad (P = 0.046) versus PBS treatment groups 12 days from the start of treatment (day 45) through the end of the study (day 90). Importantly, the antitumor activity of HYPR-Ad-mIL4 was as potent as the wild-type dl309-Ad throughout the study. Regressions in tumor size were observed in a large number of the HYPR-Ad-mIL4 and dl309-Ad–treated tumors from day 45 until day 90 (Table 2). At the end of the study, HYPR-Ad-mIL4 and dl309-Ad treatment had significantly reduced tumor growth by 7.7-fold (P = 0.022) and 3.1-fold (P = 0.039), respectively, compared with PBS treatment. These data show that HYPR-Ad-mIL4 has potent antitumor activity that leads to regression of established LN229 tumor xenografts.
Table 2.
Regression in tumor size in the LN229 s.c. tumor xenograft study
| Day | Treatment group
|
||
|---|---|---|---|
| PBS | dl309-Ad | HYPR-Ad-mIL4 | |
| 45 | 0/7 (0%) | 4/9 (44%) | 5/8 (63%) |
| 53 | 0/7 (0%) | 5/9 (56%) | 8/8 (100%) |
| 90 | 1/7 (14%) | 5/9 (56%) | 5/8 (63%) |
NOTE: Listed is the number of tumors showing a regression in size/ total number of tumors in treatment group. Listed in parentheses is the percentage of tumors in group showing a regression in size.
To verify that the antitumor activity of HYPR-Ad-mIL4 was not restricted to a single cell line, we extended these studies to U251MG-T2 glioma cells. In the first study (Fig. 3B), established s.c. xenografts (average size of 100 mm3, nine animals per group) were treated intratumorally as described above. A significant reduction in tumor growth was evidenced in the HYPR-Ad-mIL4 (P = 0.03) and dl309-Ad (P = 0.043) versus PBS treatment groups 7 and 12 days from the start of treatment, respectively, and this significant difference continued through 20 days posttreatment (day 47). At that time, HYPR-Ad-mIL4 and dl309-Ad treatment had reduced tumor growth 7.8- and 5.4-fold, respectively, compared with PBS treatment. Animals within the PBS-treated group were then sacrificed due to the large size of the tumors. The growth of HYPR-Ad-mIL4 and dl309-Ad–treated tumors began to increase after day 47. By 44 days posttreatment (day 71), the growth of HYPR-Ad-mIL4 and dl309-Ad tumors was still reduced 2-fold compared with PBS-treated tumors on day 47. Importantly, the antitumor activity of HYPR-Ad-mIL4 was as potent as the wild-type dl309-Ad throughout the study (P > 0.085). These data independently confirm that HYPR-Ad-mIL4 has strong and sustained antitumor activity.
To examine viral antitumor potency in larger tumors and compare its efficacy versus parental HYPR-Ad#1, we did a second study (Fig. 3C). Established s.c. xenografts (average size of 350 mm3) were treated intratumorally as above. In this more challenging setting, tumors in the PBS and HYPR-Ad#1 groups showed no therapeutic response and continued to exponentially grow until they reached a size mandating animal sacrifice. In contrast, the growth of HYPR-Ad-mIL4–treated tumors was significantly reduced from 9 (day 48) to 14 (day 53) days posttreatment (P < 0.029). At day 53, 5/6 HYPR-Ad-mIL4–treated tumors even showed a regression in size by 5% to 44%. To overcome the tumor regrowth observed in the first study beyond initial viral efficacy, tumors in the HYPR-Ad-mIL4 group received a second round of virus injection starting on day 53 (average size of 140 mm3). We found that a second round of HYPR-Ad-mIL4 treatment was well tolerated by the animals, and there was a continued regression of tumor size that lasted up to 43 days posttreatment (day 82) when the experiment was terminated. Altogether, these results show that HYPR-Ad-mIL4 has potent antitumor activity, which is similar to wild-type dl309-Ad and significantly greater than the parental HYPR-Ad#1. Moreover, there were no virus treatment–related deaths or observable toxicity (activity or weight change, Supplementary Fig. S5) in these three animal tumor studies.
Mechanism of HYPR-Ad-mIL4–induced tumor regression
To better understand the mechanisms underlying the rapid regressions in tumor size following HYPR-Ad-mIL4 treatment, a histologic and immunohistochemical analysis was done on tumors from the LN229 tumor study described above at 15 days from the start of viral treatment. Analysis of a PBS-treated tumor revealed a solid high-grade tumor growing in fascicles with numerous mitotically active tumor cells with no evidence of necrosis or fibrosis (Fig. 4A). The HYPR-Ad-mIL4– and dl309-Ad–treated tumors also presented as high-grade malignant tumor, but were histologically distinct from the PBS-treated tumor. HYPR-Ad-mIL4– and dl309-Ad–treated tumors showed extensive sheetlike necrosis, numerous bands of fibrosis traversing viable tumor (highlighted by Masson’s trichrome stains) and viral CPEs in tumor cells seen as nuclear enlargement with eosinophilic inclusions (owl eyes; Fig. 4B and C and data not shown). Consistent with this, widespread Ad hexon coat protein expression, a marker of newly synthesized virus (26), was detected throughout the HYPR-Ad-mIL4– and dl309-ad–treated tumors (Fig. 4B and C). A unique characteristic of HYPR-Ad-mIL4–treated tumor was extensive hypoxia throughout the tumor mass that was most pronounced around areas of necrosis (Fig. 4B). In contrast, the PBS-treated tumor had isolated pockets of hypoxic cells throughout the tumor mass (Fig. 4A). Another distinguishing feature of HYPR-Ad-mIL4–treated tumors was a prominent infiltration by lymphocytes and PMN leukocytes in a perivascular distribution into the tumor stroma and around necrosis. Staining for CD45 leukocyte common antigen showed cells with either lymphocyte or PMN morphology (Fig. 4B). Only scattered eosinophils were noted. The dl309-Ad–treated tumor also contained leukocytes, but the degree of infiltrate was less compared with the HYPR-Ad-mIL4–treated tumor (Fig. 4C). In contrast, the PBS-treated tumor only occasionally contained leukocytes within the tumor mass (Fig. 4A) and had a more modest leukocyte infiltration within the tumor capsule (data not shown). Because IL-4 modulates angiogenesis, the blood vessel density in the treated tumors was quantified by vWF immunostaining. The average number of vessels observed per field was similar in the dl309-Ad– (26.4 ± 7.7) and HYPR-Ad-mIL4– (29.1 ± 7.1) treated tumors (P > 0.5). These blood vessel density values were 2.2- to 2.5-fold greater than those found in the PBS-treated tumors (11.8 ± 3.3, P < 0.001). In summary, these data are consistent with a model in which HYPR-Ad-mIL4 reduces tumor growth by directly killing hypoxic tumor cells via the virus cytolytic replicative cycle and by the induction of a host immune response.
Figure 4.

Histology and immunohistochemistry of HYPR-Ad-mIL4–treated tumors. LN229 tumors were intratumorally treated with PBS (A), HYPR-Ad-mIL4 (B), or dl309-Ad (C) as described in the legend of Fig. 3. Fifteen days from the start of treatment, animals were injected with pimonidazole hydrochloride (a 2-nitroimidazole hypoxia marker) and sacrificed, and the tumors were harvested. Deparaffinized tumor sections were stained with H&E for tumor histology and detection of infiltrating PMN leukocytes and with Masson’s trichrome to detect collagen (blue). Immunostaining was done for Ad hexon protein, pimonidazole (hypoxia) adduct, and CD45 leukocyte common antigen, a pan-lymphocyte marker. Magnifications: A, C, and D, 50×; B, 100×; and E, 400×. Representative sections are shown.
Discussion
Hypoxia, a prevalent and distinguishing feature of tumors, is associated with poor patient survival and resistance to conventional radio- and chemotherapies. Hypoxia increases malignant progression by activating signaling cascades that promote angiogenesis, cell survival, metabolic adaptation to anaerobic metabolism, and cellular invasion (1). Hypoxia also generates selective pressure for cells to acquire genetic alterations, such as TP53 mutations, that will circumvent hypoxia-induced apoptosis (27). The HIF transcription factor is the primary mediator of the cellular response to hypoxia (1). HIF is induced in response to hypoxia and deregulation of numerous oncogenic and tumor suppressor signaling pathways. The development of new therapeutic strategies that counteract the hypoxic/HIF response and/or specifically target hypoxic/HIF-active tumor cells are needed. For this purpose, we developed the HYPR-Ad series of oncolytic viruses whose activation is dependent on the presence of HIF (13, 14). To increase the potency of HYPR-Ad beyond the direct killing of infected hypoxic/HIF-active tumor cells, we armed it with IL-4, thereby creating a dual oncolytic and IL-4 gene therapy delivery Ad (HYPR-Ad-IL4). We show that HYPR-Ad-IL4 exhibits hypoxia-dependent IL-4 expression and viral replication with subsequent cytolysis of infected hypoxic cells. Treatment of established human tumor xenografts with HYPR-Ad-IL4 led to a strong and sustained antitumor response characterized by extensive necrosis and fibrosis, widespread viral replication, and a profuse leukocyte infiltrate. This is the first study demonstrating that expression of IL-4 by an oncolytic Ad in the tumor microenvironment leads to a potent antitumor response.
To more thoroughly evaluate the potential therapeutic benefit of HYPR-Ad-IL4, we compared it in parallel to the first-generation HYPR-Ad#1 and the wild-type dl309-Ad. HYPR-Ad-IL4 and HYPR-Ad#1 contain an identical bidirectional hypoxia/HIF-responsive promoter with the right arm of this promoter being used to regulate E1A gene expression. The left arm of the promoter was not used in HYPR-Ad#1, whereas in HYPR-Ad-IL4, it regulates the IL-4 gene. We found that under hypoxia, these viruses had similar levels of E1A expression and virus production, whereas under normoxia, HYPR-Ad-IL4 had more stringent regulation of these events compared with HYPR-Ad#1 (see Figs. 1B and 2A). These data suggest a tighter regulation of the bidirectional hypoxia/HIF-responsive promoter in HYPR-Ad-IL4 relative to HYPR-Ad#1. Although we have not formally investigated the reasons for this, we had designed the virus with the gene therapy expression cassette in opposing direction with the intent of blocking any potential read-through transcription from surrounding viral promoter elements. We also found that the antitumor activity of HYPR-Ad-IL4 is superior to that of the parental HYPR-Ad#1. HYPR-Ad#1 treatment results predominantly in a slower tumor growth rate evidenced by a reduction in tumor volume by up to 5-fold without evidence of tumor regression (14). In contrast, 61% (14/23) of tumors treated with HYPR-Ad-IL4 underwent a regression in size. When compared concurrently using large tumor xenografts (average size of 350 mm3), HYPR-Ad#1 treatment led to a negligible therapeutic response, whereas HYPR-Ad-IL4 treatment resulted in 5/6 tumors showing a regression in size by 5% to 44%. The therapeutic effect of HYPR-Ad-IL4 is visible immediately in the days following tumor injection. Macroscopically, tumors develop a white appearance, and regression is apparent within a week. In contrast, the effects of HYPR-Ad#1 are only evidenced after several weeks and are most efficacious in combination with chemotherapy (14). These results are consistent with the potent antitumor activities of IL-4 (16) and show that HYPR-Ad-IL4 represents a significant advancement compared with the first-generation HYPR-Ad#1.
dl309-Ad is a replication-competent Ad that contains a wild-type E1 gene region and, therefore, represents an important benchmark in our studies. dl309-Ad is not tumor selective, and therefore, this virus could not be used in a patient due to systemic toxicity to normal cells/tissues. The goal of armed oncolytic Ads should be to have tumor-selective action, yet display antitumor potency similar or better than that of wild-type Ad without increasing toxicity. We show that E1A expression by HYPR-Ad-IL4 under hypoxia was similar to or greater than dl309-Ad (Fig. 1B), demonstrating the strength of the hypoxia/HIF-responsive promoter. E1B expression was also similar for both viruses (Supplementary Fig. S1). The replicative capacity of HYPR-Ad-IL4 in vitro was attenuated 10- to 100-fold compared with dl309-Ad depending on the cell line tested (Table 1). The reasons for this are currently unknown but may in part relate to differences in the E3 gene region between the two viruses (11). Despite this, HYPR-Ad-IL4 was able to effectively induce cytolysis in vitro, and most importantly, its in vivo antitumor activity was equivalent to dl309-Ad. The reduced toxicity of HYPR-Ad-mIL4 compared with dl309-Ad is most evident in vitro against infected normoxic cells. Collectively, the data show that the antitumor efficacy of the first-generation HYPR-Ad can be augmented by its ability to deliver adjuvant therapeutic genes. Although these results are promising, the necessary use of a human tumor xenograft in nu/nu mice for our in vivo animal studies restricts our ability to evaluate the full potential of HYPR-Ad-IL4–mediated antitumor effects. This limitation in animal tumor models is based on the severe attenuation of human Ad replication in a majority of rodent cells. It is certainly possible that HYPR-Ad-mIL4 will exhibit improved antitumor potency compared with dl309-Ad in an immunocompetent animal tumor model or metastatic cancer model where IL-4 antitumor activity mediated by an induced T- and B-cell response can be evaluated. Recently, several rodent tumor cell lines that support human Ad replication have been identified (28–31), including a syngeneic metastatic breast cancer model (32). Now that this paper has shown proof of principle, a next useful study would be to evaluate the efficacy of HYPR-Ad-mIL4 in these new models. Alternatively, the strategy of a hypoxia/HIF-dependent oncolytic virus can be reproduced in a virus such as herpes simplex virus-1 (HSV-1). Because HSV-1 can replicate in rodent cells, the use of syngeneic rodent tumor models would then be possible.
There are a large number of therapeutic genes with antitumor activity that could potentially be introduced into HYPR-Ad such as prodrug-converting enzymes, cell cycle inhibitors, growth and tumor suppressors, immune stimulatory factors, or angiogenesis inhibitors. It is currently unknown which adjuvant therapeutic gene or category of genes will offer the greatest therapeutic benefit with the least toxicity. There is also no a priori guarantee that expression of a particularly attractive candidate therapeutic gene by an oncolytic Ad will lead to a measurable antitumor effect (33). In addition, the potential impact of adjuvant therapeutic gene expression on virus replication needs to be carefully evaluated. This is particularly important in the case of prodrug-converting enzymes, which generate products that inhibit both cellular and viral DNA replication (34, 35). In our opinion, genes that mediate bystander effects on adjacent noninfected cells without restricting viral replication and spread throughout the tumor will be most efficacious. As proof of principle, we chose the IL-4 cytokine for our studies. In numerous preclinical studies, IL-4 has shown potent multimodal antitumor activity associated with pleiotropic effects on various immune effector cells (such as B, T, and natural killer cells, macrophages) and inhibition of angiogenesis and cell growth (15). Currently, the clinical use of IL-4 for antitumor therapy is hindered by its short half-life and dose-limiting toxicity when administered systemically as a recombinant protein (20). IL-4 gene delivery has also been attempted using classic gene therapy with a replication-deficient retroviral vector (36), although over time, expression is lost in these systems, and they are limited by initial gene transduction efficacy of dividing cells. The development of replication-competent viruses, such as HYPR-Ad-IL4, addresses these issues. In support of this, HSV (37) and vesicular stomatitis virus (38) that express IL-4 and selectively replicate in cells containing activating Ras mutations or IFN pathway defects, respectively, have shown potent oncolytic activity. We show that local expression of IL-4 by HYPR-Ad-IL4 in the tumor microenvironment leads to a strong antitumor effect. This effect was associated with immune infiltrates but no changes in tumor vascularization as compared with dl309 control adenovirus. This may reflect the complexity of IL-4 action on angiogenesis, being stimulatory or inhibitory under different experimental conditions (39, 40). The increased activity of HYPR-Ad-IL4 over the parental HYPR-Ad virus may, therefore, mostly relate to immune-mediated functions of IL-4. These antitumor effects are observed in genetically and biologically diverse tumor cells because HYPR-Ad replication is not restricted to a tumor with a particular genetic defect. Furthermore, to reduce toxicity associated with high levels of IL-4, we placed its expression under the regulation of a promoter that is preferentially active in tumor cells, rather than a strong constitutively active promoter such as the cytomegalovirus (CMV). We have not detected a significant change in animal weight or activity following direct injection of HYPR-Ad-mIL4 into s.c. tumor xenografts, suggesting that the local levels of IL-4 generated were sufficient to mediate a potent antitumor effect without any obvious signs of general toxicity to the host. Alternative approaches to using IL-4 as a cancer therapeutic are being actively pursued by others, including targeting Pseudomonas exotoxin (PE) to tumor cells expressing IL-4 receptors using a chimeric IL4-PE protein (41) and a vaccine strategy using autologous tumor cells admixed with IL-4 gene–transfected fibroblasts (42). Both of these strategies have shown strong antitumor activity preclinically and have entered clinical trial testing. Now that we have established the therapeutic efficacy of HYPR-Ad-IL4, it will be important to conduct detailed studies evaluating the safety and toxicity of this virus toward clinical translation. These future preclinical studies will need to carefully examine the histology of normal tissues and cells to detect any microscopic signs of pathology and unwanted viral replication, which may not manifest clinically as a change in animal weight or behavior.
In conclusion, we designed a dual oncolytic and IL-4 gene therapy Ad. This virus contains a novel bidirectional tumor-restrictive hypoxia/HIF promoter to drive viral E1A gene expression and subsequent viral replication resulting in the targeted death of hypoxic/HIF-active tumor cells. The same promoter controls IL-4 expression. Our preclinical in vitro and in vivo studies show that HYPR-Ad-IL4 selectively replicates in hypoxic/HIF active cells thereby inducing cytolysis and has strong antitumor activity resulting in tumor regression. The therapeutic potential of this virus is applicable to all tumors that develop hypoxia/HIF activation.
Supplementary Material
Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Acknowledgments
Grant support: NIH grants NS49300 (D.E. Post) and CA87830, CA86335, and NS41403 (E.G. Van Meir).
We thank Saroja N. Devi and Zhaobin Zhang for lab operation support, Misako Hwang for technical assistance, Carol Tucker-Burden, Anita Bellail, and Azita Djalilvand for helpful suggestions, and A. Turnell (University of Birmingham, United Kingdom) for the anti-E1B 55K antibody.
References
- 1.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–32. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 2.Brown JM, Wilson WR. Exploiting tumour hypoxia in cancer treatment. Nat Rev Cancer. 2004;4:437–47. doi: 10.1038/nrc1367. [DOI] [PubMed] [Google Scholar]
- 3.Evans SM, Judy KD, Dunphy I, et al. Comparative measurements of hypoxia in human brain tumors using needle electrodes and EF5 binding. Cancer Res. 2004;64:1886–92. doi: 10.1158/0008-5472.can-03-2424. [DOI] [PubMed] [Google Scholar]
- 4.Brat DJ, Van Meir EG. Vaso-occlusive and prothrombotic mechanisms associated with tumor hypoxia, necrosis, and accelerated growth in glioblastoma. Lab Invest. 2004;84:397–405. doi: 10.1038/labinvest.3700070. [DOI] [PubMed] [Google Scholar]
- 5.Bardos JI, Ashcroft M. Negative and positive regulation of HIF-1: a complex network. Biochim Biophys Acta. 2005;1755:107–20. doi: 10.1016/j.bbcan.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 6.Zagzag D, Zhong H, Scalzitti JM, Laughner E, Simons JW, Semenza GL. Expression of hypoxia-inducible factor 1α in brain tumors: association with angiogenesis, invasion, and progression. Cancer. 2000;88:2606–18. [PubMed] [Google Scholar]
- 7.Talks KL, Turley H, Gatter KC, et al. The expression and distribution of the hypoxia-inducible factors HIF-1α and HIF-2α in normal human tissues, cancers, and tumor-associated macrophages. Am J Pathol. 2000;157:411–21. doi: 10.1016/s0002-9440(10)64554-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Powis G, Kirkpatrick L. Hypoxia inducible factor-1α as a cancer drug target. Mol Cancer Ther. 2004;3:647–54. [PubMed] [Google Scholar]
- 9.Belozerov VE, Van Meir EG. Hypoxia inducible factor-1: a novel target for cancer therapy. Anticancer Drugs. 2005;16:901–9. doi: 10.1097/01.cad.0000180116.85912.69. [DOI] [PubMed] [Google Scholar]
- 10.Chu RL, Post DE, Khuri FR, Van Meir EG. Use of replicating oncolytic adenoviruses in combination therapy for cancer. Clin Cancer Res. 2004;10:5299–312. doi: 10.1158/1078-0432.CCR-0349-03. [DOI] [PubMed] [Google Scholar]
- 11.Post DE, Shim H, Toussaint-Smith E, Van Meir EG. Cancer scene investigation: how a cold virus became a tumor killer. Future Oncol. 2005;1:247–58. doi: 10.1517/14796694.1.2.247. [DOI] [PubMed] [Google Scholar]
- 12.Post DE, Van Meir EG. Generation of bidirectional hypoxia/HIF-responsive expression vectors to target gene expression to hypoxic cells. Gene Ther. 2001;8:1801–7. doi: 10.1038/sj.gt.3301605. [DOI] [PubMed] [Google Scholar]
- 13.Post DE, Van Meir EG. A novel hypoxia-inducible factor (HIF) activated oncolytic adenovirus for cancer therapy. Oncogene. 2003;22:2065–72. doi: 10.1038/sj.onc.1206464. [DOI] [PubMed] [Google Scholar]
- 14.Post DE, Devi NS, Li Z, et al. Cancer therapy with a replicating oncolytic adenovirus targeting the hypoxic microenvironment of tumors. Clin Cancer Res. 2004;10:8603–12. doi: 10.1158/1078-0432.CCR-04-1432. [DOI] [PubMed] [Google Scholar]
- 15.Okada H, Kuwashima N. Gene therapy and biologic therapy with interleukin-4. Curr Gene Ther. 2002;2:437–50. doi: 10.2174/1566523023347625. [DOI] [PubMed] [Google Scholar]
- 16.Tepper RI, Pattengale PK, Leder P. Murine interleukin-4 displays potent anti-tumor activity in vivo. Cell. 1989;57:503–12. doi: 10.1016/0092-8674(89)90925-2. [DOI] [PubMed] [Google Scholar]
- 17.Golumbek PT, Lazenby AJ, Levitsky HI, et al. Treatment of established renal cancer by tumor cells engineered to secrete interleukin-4. Science. 1991;254:713–6. doi: 10.1126/science.1948050. [DOI] [PubMed] [Google Scholar]
- 18.Pericle F, Giovarelli M, Colombo MP, et al. An efficient Th2-type memory follows CD8+ lymphocyte-driven and eosinophil-mediated rejection of a spontaneous mouse mammary adenocarcinoma engineered to release IL-4. J Immunol. 1994;153:5659–73. [PubMed] [Google Scholar]
- 19.Saleh M, Davis ID, Wilks AF. The paracrine role of tumour-derived mIL-4 on tumour-associated endothelium. Int J Cancer. 1997;72:664–72. doi: 10.1002/(sici)1097-0215(19970807)72:4<664::aid-ijc19>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 20.Leach MW, Rybak ME, Rosenblum IY. Safety evaluation of recombinant human interleukin-4. II. Clinical studies. Clin Immunol Immunopathol. 1997;83:12–4. doi: 10.1006/clin.1997.4304. [DOI] [PubMed] [Google Scholar]
- 21.Bett AJ, Krougliak V, Graham FL. DNA sequence of the deletion/insertion in early region 3 of Ad5 dl309. Virus Res. 1995;39:75–82. [PubMed] [Google Scholar]
- 22.He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci U S A. 1998;95:2509–14. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ishii N, Maier D, Merlo A, et al. Frequent co-alterations of TP53, p16/CDKN2A, p14ARF, PTEN tumor suppressor genes in human glioma cell lines. Brain Pathol. 1999;9:469–79. doi: 10.1111/j.1750-3639.1999.tb00536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nishikawa R, Ji XD, Harmon RC, et al. A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc Natl Acad Sci U S A. 1994;91:7727–31. doi: 10.1073/pnas.91.16.7727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van Meir EG. Identification of nude mice in tumorigenicity assays. Int J Cancer. 1997;71:310. doi: 10.1002/(sici)1097-0215(19970410)71:2<310::aid-ijc29>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 26.Bischoff JR, Kirn DH, Williams A, et al. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science. 1996;274:373–6. doi: 10.1126/science.274.5286.373. [DOI] [PubMed] [Google Scholar]
- 27.Graeber TG, Osmanian C, Jacks T, et al. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature. 1996;379:88–91. doi: 10.1038/379088a0. [DOI] [PubMed] [Google Scholar]
- 28.Wang Y, Hallden G, Hill R, et al. E3 gene manipulations affect oncolytic adenovirus activity in immunocompetent tumor models. Nat Biotechnol. 2003;21:1328–35. doi: 10.1038/nbt887. [DOI] [PubMed] [Google Scholar]
- 29.Toth K, Spencer JF, Tollefson AE, et al. Cotton rat tumor model for the evaluation of oncolytic adenoviruses. Hum Gene Ther. 2005;16:139–46. doi: 10.1089/hum.2005.16.139. [DOI] [PubMed] [Google Scholar]
- 30.Hallden G, Hill R, Wang Y, et al. Novel immunocompetent murine tumor models for the assessment of replication-competent oncolytic adenovirus efficacy. Mol Ther. 2003;8:412–24. doi: 10.1016/s1525-0016(03)00199-0. [DOI] [PubMed] [Google Scholar]
- 31.Nagayama Y, Nakao K, Mizuguchi H, Hayakawa T, Niwa M. Enhanced antitumor effect of combined replicative adenovirus and nonreplicative adenovirus expressing interleukin-12 in an immunocompetent mouse model. Gene Ther. 2003;10:1400–3. doi: 10.1038/sj.gt.3302001. [DOI] [PubMed] [Google Scholar]
- 32.Guo W, Zhu H, Zhang L, et al. Combination effect of oncolytic adenovirotherapy and TRAIL gene therapy in syngeneic murine breast cancer models. Cancer Gene Ther. 2006;13:82–90. doi: 10.1038/sj.cgt.7700863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lamfers ML, Gianni D, Tung CH, et al. Tissue inhibitor of metalloproteinase-3 expression from an oncolytic adenovirus inhibits matrix metalloproteinase activity in vivo without affecting antitumor efficacy in malignant glioma. Cancer Res. 2005;65:9398–405. doi: 10.1158/0008-5472.CAN-04-4264. [DOI] [PubMed] [Google Scholar]
- 34.Wildner O, Morris JC. The role of the E1B 55 kDa gene product in oncolytic adenoviral vectors expressing herpes simplex virus-tk: assessment of antitumor efficacy and toxicity. Cancer Res. 2000;60:4167–74. [PubMed] [Google Scholar]
- 35.Wildner O, Morris JC. Therapy of peritoneal carcinomatosis from colon cancer with oncolytic adenoviruses. J Gene Med. 2000;2:353–60. doi: 10.1002/1521-2254(200009/10)2:5<353::AID-JGM130>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 36.Benedetti S, Bruzzone MG, Pollo B, et al. Eradication of rat malignant gliomas by retroviral-mediated, in vivo delivery of the interleukin 4 gene. Cancer Res. 1999;59:645–52. [PubMed] [Google Scholar]
- 37.Andreansky S, He B, van Cott J, et al. Treatment of intracranial gliomas in immunocompetent mice using herpes simplex viruses that express murine interleukins. Gene Ther. 1998;5:121–30. doi: 10.1038/sj.gt.3300550. [DOI] [PubMed] [Google Scholar]
- 38.Fernandez M, Porosnicu M, Markovic D, Barber GN. Genetically engineered vesicular stomatitis virus in gene therapy: application for treatment of malignant disease. J Virol. 2002;76:895–904. doi: 10.1128/JVI.76.2.895-904.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Volpert OV, Fong T, Koch AE, et al. Inhibition of angiogenesis by interleukin 4. J Exp Med. 1998;188:1039–46. doi: 10.1084/jem.188.6.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fukushi JMT, Shono T, Nishie A, Torisu H, Ono M, Kuwano M. Novel biological functions of interleukin-4: formation of tube-like structures by vascular endothelial cells in vitro and angiogenesis in vivo. Biochem Biophys Res Commun. 1998;250:444–8. doi: 10.1006/bbrc.1998.9334. [DOI] [PubMed] [Google Scholar]
- 41.Shimamura T, Husain SR, Puri RK. The IL-4 and IL-13 Pseudomonas exotoxins: new hope for brain tumor therapy. Neurosurg Focus. 2006;20:E11. doi: 10.3171/foc.2006.20.4.6. [DOI] [PubMed] [Google Scholar]
- 42.Okada H, Lieberman FS, Edington HD, et al. Autologous glioma cell vaccine admixed with interleukin-4 gene transfected fibroblasts in the treatment of recurrent glioblastoma: preliminary observations in a patient with a favorable response to therapy. J Neurooncol. 2003;64:13–20. doi: 10.1007/BF02700016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).