

SUMMARY
A key characteristic of stem cells and cancer cells is their ability to self-renew. To test if Wnt signaling can regulate the self-renewal of both stem cells and cancer cells in the hematopoietic system, we developed mice that lack β-catenin in their hematopoietic cells. Here we show that β-catenin-deficient mice can form HSCs, but that these cells are deficient in long-term growth and maintenance. Moreover, β-catenin deletion causes a profound reduction in the ability of mice to develop BCR-ABL-induced chronic myelogenous leukemia (CML), while allowing progression of acute lymphocytic leukemia (ALL). These studies demonstrate that Wnt signaling is required for the self-renewal of normal and neoplastic stem cells in the hematopoietic system.
INTRODUCTION
Wnt proteins are secreted signaling molecules that influence both development and cancer. In the context of development, Wnts regulate segment polarity in Drosophila (Siegfried and Perrimon, 1994), axis specification in Xenopus (Moon et al., 1997), and differentiation of limbs, brain, kidney, and the reproductive tract in mice (Liu et al., 1999; Miller and Sassoon, 1998; Monkley et al., 1996; Parr and McMahon, 1994; Yoshikawa et al., 1997). In addition to its importance in normal development, dysregulation of the Wnt pathway can have potent oncogenic effects. A causal role for this pathway in oncogenesis was originally demonstrated using mice in which overexpression of Wnt-1 led to mammary tumors (Tsukamoto et al., 1988). In humans, inactivating mutations in the Wnt pathway inhibitor APC are associated with the colon cancer susceptibility syndrome familial adenomatous polyposis (Kinzler et al., 1991). Mutations in APC as well as β-catenin, a key mediator of Wnt signaling, are also found in a majority of sporadic colon cancers (Giles et al., 2003), as well as in hepatocellular carcinoma (Satoh et al., 2000), thyroid cancer, and ovarian cancer (Gamallo et al., 1999; Garcia-Rostan et al., 1999).
The fact that Wnt signaling is dysregulated in multiple solid cancers together with its observed influence on hematopoietic stem and progenitor cells (reviewed in Reya and Clevers, 2005) suggests that the same pathway may become aberrantly activated to promote leukemia. Several studies support this possibility. For example, pre-B cell leukemia lines carrying the E2A-PbX translocation overexpress Wnt proteins (McWhirter et al., 1999), and survival of these cells in vitro can be inhibited by blocking Wnt signaling (Mazieres et al., 2005). Similarly, AML-associated fusion proteins enhance replating efficiency of hematopoietic stem cells (HSCs), and this is abrogated upon inhibition of Wnt signaling (Muller-Tidow et al., 2004; Zheng et al., 2004). Finally, cells from CML patients display activated Wnt signaling and a dependence on this pathway for growth in vitro (Jamieson et al., 2004). While these data suggest that activation of the Wnt pathway may contribute to leukemic cell growth in vitro, whether Wnt signaling is required for progression of leukemias in vivo is unknown.
To address this question, we generated conditional β-catenin null mice. Using these mice we show that loss of β-catenin affects both normal and malignant hematopoietic cells in vivo. While HSCs are established in the absence of β-catenin, they are impaired in long-term growth and maintenance following transplantation. Importantly, using a BCR-ABL model of leukemogenesis, we show that loss of β-catenin impairs the ability of mice to develop CML, due in part to decreased self-renewal of CML stem cells. In contrast, these mice are capable of developing ALL. Since CML is thought to be initiated in HSCs, and ALL in committed precursors, these data raise the intriguing possibility that β-catenin is preferentially required for BCR-ABL-induced leukemias that originate in stem cells. These data also suggest that the dependence of leukemias on Wnt signaling is dictated not only by the oncogene itself but also by the specific cell that it targets.
SIGNIFICANCE
The Wnt pathway has been implicated in the progression of many cancers, but its role in leukemia is unclear. Using a conditional β-catenin−/− mouse, we show that in vivo myeloid leukemia progression is dependent on β-catenin. Importantly, while loss of β-catenin dramatically reduces BCR-ABL-induced CML development, it allows ALL to proceed unimpaired. Given the evidence that CML may be initiated in HSCs, and ALL in committed precursors, these data suggest that β-catenin may be preferentially required for leukemias that originate in stem cells.
RESULTS
Generation of Conditional β-Catenin Null Mice
Conventional β-catenin−/− mice die in utero (Haegel et al., 1995; Huelsken et al., 2000). Therefore, to examine the role of β-catenin in adult hematopoietic stem cell function and leukemia progression, we generated conditional knockouts. Mice in which β-catenin is flanked by loxP sites (flox-β-catenin mice) (Brault et al., 2001) were crossed with mice in which the Cre recombinase is driven by vav regulatory elements (vav-Cre mice). Vav is an adaptor protein expressed mostly in the hematopoietic system, including in HSC-enriched fractions (Almarza et al., 2004; de Boer et al., 2003). Progeny from this cross would be expected to delete β-catenin predominantly in the hematopoietic system (Figure 1A).
Figure 1. Conditional Deletion of β-Catenin in the Hematopoietic System.

(A) The schematic indicates the strategy for deletion of β-catenin in hematopoietic cells using mice carrying a floxed β-catenin allele crossed to transgenic mice in which expression of the cre recombinase is directed by the vav promoter.
(B) Analysis of deletion efficiency by genomic PCR analysis. Genomic DNA from control and β-catenin−/− whole bone marrow cells was amplified using primer RM68/69, which does not generate a product for the floxed allele but generates a 631 bp product for the floxdel allele (left lanes; 45 relative fluorescence units for −/− and 0.64 RFU for control), and primer RM41/42/43, which generates a product of 324 bp for the floxed allele (1.5 RFU for control and 0.0006 RFU for −/−) and a 500 bp product for the floxdel allele (69 RFU for −/− and 0.32 for control) (Brault et al., 2001). Quantitation of the difference in product formation was carried out by real-time PCR analysis. Results are representative of two experiments.
(C) Whole bone marrow lysates from control (+/+) and β-catenin knockout (−/−) mice were analyzed by western blot for presence of β-catenin. Results shown are representative of four experiments.
(D) KLS cells were isolated from control (+/+), heterozygous (+/−), and conditional β-catenin knockout (−/−) mice, and RT-PCR analysis was performed to determine expression of β-catenin. Results are representative of three experiments.
To ensure that this strategy was effective, we performed genomic PCR (Brault et al., 2001). This showed that in β-catenin−/− mice (loxp/loxp, cre), the floxed allele was efficiently deleted and the undeleted allele could no longer be detected (70- to 2500-fold reduction by real-time PCR) (Figure 1B). In addition, no β-catenin protein could be detected in the bone marrow of conditional null mice (Figure 1C). Finally, β-catenin mRNA expression was undetectable in c-kit+Lin−Sca-1+ (KLS) cells, a population highly enriched for HSCs (Figure 1D). These data suggested that β-catenin was efficiently deleted in HSCs and that this model could be used to test the requirement of β-catenin in HSC renewal and leukemogenesis.
To determine whether loss of β-catenin leads to defects in HSC function in vivo, we stained for markers of immature and mature hematopoietic cells. The frequency of long-term stem cells (c-kit+Lin−/loSca-1+Flk2− cells, or KLSF cells) was equivalent in control and β-catenin−/− mice (Figures 2A and 2B). In addition, these cells were equivalently distributed among the different stages of the cell cycle (Figure S1 in the Supplemental Data available with this article online). β-catenin−/− mice also had a normal distribution of B, T, and myeloid cells (data not shown) as well as CMP, GMP, and CLPs (Figure S2). To test the long-term self-renewal capacity of HSCs from β-catenin−/− mice, we transplanted 500 KLSF cells from control and β-catenin−/− mice in the presence of competing control bone marrow cells and monitored long-term reconstitution ability. Mice reconstituted with β-catenin−/− KLSF cells displayed a 5-fold reduction in donor chimerism compared to mice transplanted with control KLSF cells (Figures 2C and 2D and Figure S3). Importantly, the lineage distribution within the control- and knockout-derived populations was similar (Figure 2E), indicating that loss of β-catenin does not affect lineage differentiation, but rather impairs long term-growth and maintenance of HSCs. In addition, we carried out competitive limiting-dilution experiments using a range of 10–50 cells per recipient and found a 4-fold reduction in the frequency of functional HSCs as determined by CRU analysis (Figure 2F and Figure S4A). Upon secondary transplants there was a 6.3-fold reduction in the frequency of functional HSCs in β-catenin-deficient bone marrow (Figure 2G and Figure S4B), supporting the conclusion that the defect was in long-term HSC self-renewal. Finally, at short time points after transplantation(Adams et al., 2006), the levels of chimerism were equivalent (Figure 2H), suggesting that the long-term HSC renewal defects observed were unlikely to be a consequence of impaired homing.
Figure 2. β-Catenin Is Required for Long-Term Stem Cell Maintenance and Transplantation In Vivo.
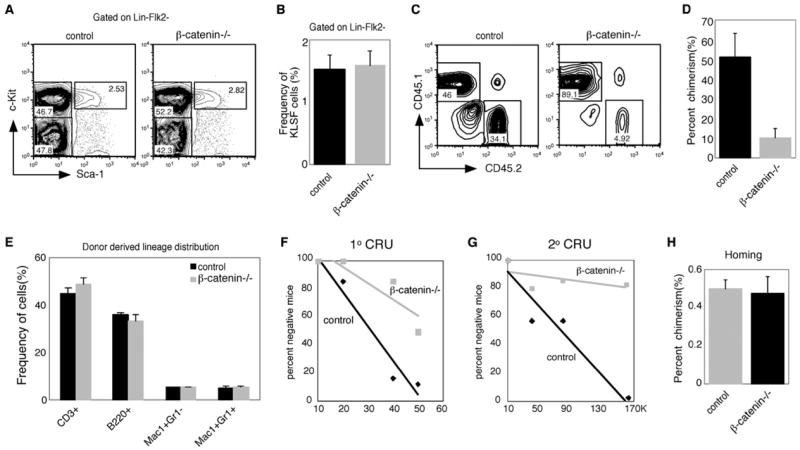
(A) Bone marrow cells from control or β-catenin knockout mice were analyzed for frequency of KLSF cells. Dot plots are shown for one representative control (control, left) and β-catenin knockout mouse (β-catenin−/−, right).
(B) Average frequency of KLSF cells in control and β-catenin knockout mice. n = 11 for control, n = 13 for knockout.
(C) Five-hundred KLSF cells from control or β-catenin knockout mice were transplanted together with competing bone marrow cells into lethally irradiated congenic recipients, and donor-derived chimerism was monitored for 20 weeks. Plots show representative donor-derived chimerism from individual mice at 20 weeks.
(D) Graph of average donor-derived chimerism after long-term reconstitution (5–7 mice in each cohort, p = 0.0079, n = 3). In five independent experiments, the difference in chimerism between control and knockout transplants was 5-fold (500 cells transplanted), 2.8-fold (2000 cells transplanted), and 1.4-fold (6000 cells transplanted).
(E) Contribution to differentiated lineages from control and β-catenin knockout cells following long-term bone marrow transplantation in peripheral blood. Results are representative of three independent bone marrow transplantation experiments, with 5–7 mice in each cohort.
(F) Limiting-dilution competitive repopulation analysis of control and β-catenin knockout mice. Eight mice were transplanted at each dose (10, 20, 40, and 50 cells/recipient for a total of 64 initial recipients for both genotypes). Peripheral blood cells of all surviving recipients were analyzed 10 weeks after transplantation, and data were analyzed by L-calc software. CRU difference is 4-fold between control and knockout.
(G) Secondary limiting-dilution competitive repopulation analysis of control and β-catenin knockout mice was carried out using the 50 cells/recipient group of primary transplanted mice at 21 weeks. Whole bone marrow cells were isolated from primary donors and transplanted into 5–7 recipients at each dose (10,000, 40,000, 80,000, and 160,000 cells/recipient for a total of 48 recipients). Peripheral blood cells of the secondary recipients were analyzed 10 weeks after transplantation, and data were analyzed by L-calc software. CRU difference is 6.3-fold between control and knockout.
(H) Relative homing ability of control and β-catenin−/− bone marrow cells. A total of 2 × 106 bone marrow cells from control or β-catenin−/− mice were transplanted into irradiated recipients (n = 4 in each group), and the presence of donor-derived cells was analyzed by FACS 6 hr posttransplant. In (B), (D), (E), and (H), error bars represent SEM.
Loss of β-Catenin Leads to Altered Characteristics of Leukemia Progression In Vivo
The influence of β-catenin on HSC function and the many parallels between the regulation of stem cells and cancer cells (Beachy et al., 2004; Reya et al., 2001; Wu et al., 2007) suggested that β-catenin may also affect leukemia formation. To test this we utilized a mouse model of leukemogenesis involving the BCR-ABL fusion protein. In humans, translocations involving the BCR and ABL genes lead to both CML and ALL (Ren, 2005). This disease can be recapitulated in mice by retrovirally introducing the p210 form of BCR-ABL into hematopoietic progenitors and transplanting these cells into lethally irradiated mice (Daley et al., 1990; Pear et al., 1998; Witte, 2001). We used this model to test the requirement of β-catenin in leukemia progression.
c-kit-enriched bone marrow cells from control or β-catenin−/− mice were infected with control retroviruses or viruses carrying BCR-ABL and transplanted (Figure 3A). Forty-eight hours after infection, control and β-catenin−/− cells expressed BCR-ABL transcripts (Figure 3B). In addition, the frequency of infected cells was similar in control and knockout cultures as determined by an IRES-GFP marker carried by the virus (data not shown). Mice transplanted with control BCR-ABL-infected cells became sick 20–24 days after transplantation (Figure 3C). In contrast, mice transplanted with β-catenin−/− BCR-ABL-infected cells showed a significant delay in onset of disease (75 days versus 46 days; Figures 3C and 5C).
Figure 3. Loss of β-Catenin Leads to Altered Characteristics of Leukemia Progression In Vivo.

(A) Experimental strategy to model BCR-ABL-induced leukemia. c-kit-enriched cells were isolated from control or β-catenin knockout mice and infected with BCR-ABL retroviruses for 48 hr prior to transplantation of 2–5 × 105 cells into lethally irradiated recipient mice. LTR, long terminal repeat.
(B) Control and β-catenin knockout c-kit-enriched bone marrow cells were infected with either vector control or BCR-ABL retroviruses, and expression levels of BCR-ABL were analyzed by real-time PCR. Results were normalized to the housekeeping gene β-actin. Data are an average of five independent samples. Error bars represent SEM.
(C) Survival curve of mice receiving BCR-ABL-infected control (+/+) or β-catenin knockout (−/−) c-kit-enriched bone marrow cells. Data shown are from one of four independent experiments, with 7–9 mice in each experimental group per experiment.
(D–F) Gross examination of mice transplanted with vector-infected control, BCR-ABL-infected control, and β-catenin knockout cells. A majority of mice transplanted with BCR-ABL-infected control cells became weak but did not develop paralysis, unlike several of those transplanted with knockout cells (left panel). Appearance of lung in mice transplanted with vector-infected control, BCR-ABL-infected control, or BCR-ABL-infected knockout cells (middle panel). Relative splenomegaly of mice transplanted with vector-infected control, BCR-ABL-infected control, or β-catenin knockout cells (right).
Figure 5. Loss of β-Catenin Impairs In Vivo Progression of CML but Allows Normal Development of ALL.

(A) Histologic appearance of leukemic cells in spleens of the majority of mice transplanted with BCR-ABL-infected control and β-catenin knockout cells. Arrowheads point to an increased frequency of immature and mature granulocytic cells in the spleens of mice transplanted with BCR-ABL-infected control cells, and increased lymphocytic cells in the spleens of mice transplanted with BCR-ABL-infected β-catenin knockout cells. Scale bar represents 100 μm.
(B) Bone marrow cells from mice receiving BCR-ABL-infected control or β-catenin knockout cells were analyzed by flow cytometry for increased presence of granulocytic (Mac-1+Gr-1+ cells shown in upper panels) or lymphocytic lineages (B220+ cells shown in lower panels).
(C) Cumulative data from four experiments indicating type of leukemia induced by BCR-ABL in mice transplanted with control or β-catenin knockout (−/−) cells. Each circle represents an individual mouse. CML, chronic myelogenous leukemia; ALL-B, B cell acute lymphocytic leukemia; ALL-T, T cell acute lymphoblastic leukemia.
(D) Distribution of CML and ALL induced by BCR-ABL when using control or β-catenin knockout c-kit-enriched cells. *p = 0.005 (t test from four experimental groups).
We first analyzed the leukemias induced by BCR-ABL-infected control cells and found that a majority of them resembled CML. These leukemias were associated with weight loss, lung hemorrhaging, and significant splenomegaly (Figure 3E) compared to vector-infected controls (Figure 3D). Closer histological examination revealed that the lung and the liver were extensively infiltrated with leukemic cells, consistent with CML-like disease (Figure 4, top and middle rows; black arrowheads indicate leukemic cells).
Figure 4. Loss of β-Catenin Leads to an Altered Pattern of Leukemic Cell Infiltration in the Lung and Liver.

Hematoxylin/eosin staining of tissue sections from lung (left panels) and liver (right panels) of mice transplanted with vector-infected control, BCR-ABL-infected control, or BCR-ABL-infected knockout cells. Arrowheads point to infiltration of leukemic cells into tissues. Mice transplanted with BCR-ABL-infected β-catenin knockout cells show either reduced infiltration (bottom row left [lung], black arrowhead) or a different pattern of infiltration (bottom row right [liver], black arrowhead). The scale bar represents 100 μm.
Mice transplanted with BCR-ABL-infected β-catenin−/− cells displayed a markedly different course of disease. Several displayed paralysis of the hind legs, indicative of tumors in the spinal cord (Figure 3F, left). These mice did not display the lung hemorrhaging nor the extent of splenomegaly observed with BCR-ABL-infected control transplants (Figure 3F, middle and right panels), and they lacked the extensive infiltration of the lungs and liver (Figure 4, bottom row).
Loss of β-Catenin Impairs In Vivo Progression of CML but Allows Normal Development of ALL
To determine the identity of the leukemic cells in mice transplanted with BCR-ABL-infected control and knockout cells, we carried out histological analysis of the spleen. This revealed extensive infiltration of both immature and mature granulocytes in control transplanted mice (Figure 5A, middle). In contrast, the majority of mice transplanted with BCR-ABL-infected β-catenin−/− cells displayed lymphocytic cell infiltrates (Figure 5A, right). FACS analysis confirmed that the majority of leukemic cells in control mice were granulocytes (Figure 5B, upper panel); in contrast, β-catenin−/− transplanted mice showed a significant expansion of B cells (Figure 5B, lower panels) that were IgM− (data not shown), indicating that the leukemia resembled pre-B-ALL. Cumulative analysis of four independent groups of transplants revealed that the majority of mice (82.6%, 19/23) transplanted with BCR-ABL-infected control cells displayed a CML-like disease and only a few mice (17.4%, 4/23) succumbed to ALL (Figure 5C, bottom row). In contrast only 19% (4/21) of mice transplanted with BCR-ABL-infected β-catenin−/− cells succumbed to CML (Figure 5C, top row; Figure 5D). Importantly, at early time points the level of chimerism of BCR-ABL-infected control and β-catenin null cells was equivalent, suggesting that the reduced frequency of CML was unlikely to be due to a homing or engraftment defect (Figure S5). The frequency of BCR-ABL-positive clones with unique integration sites was also similar in the BCR-ABL-infected control or β-catenin null cells, suggesting that an early defect in the presence of leukemia-initiating clones was unlikely (Figure S6A). Furthermore the leukemias that did fully form in both control and β-catenin null samples appeared to be oligoclonal, suggesting that an insertional mutagenesis event, which can sometimes act to cooperatively lead to oncogenesis, was unlikely to be the basis of the observed CMLs (Figure S6B). Interestingly, the absence of β-catenin did not hinder the progression to ALL, which in the absence of CML became the predominant disease in 81% (17/21) (Figures 5C and 5D) of the mice transplanted with BCR-ABL-infected knockout cells. Further, the ALLs could be serially transplanted, confirming that they had intact renewal capacity in vivo (Figures S7A–S7C). β-catenin expression was undetectable in leukemias from β-catenin null cells (Figures S8A–S8C), making it unlikely that they were progressing normally because of not having deleted β-catenin. These data cumulatively indicate a differential requirement for β-catenin in BCR-ABL-induced CML and ALL.
Loss of β-Catenin Impairs Self-Renewal of CML Stem Cells
We next investigated the cellular basis for the impaired ability of β-catenin−/− cells to allow CML progression in vivo. To test whether knockout cells displayed decreased survival following BCR-ABL infection, control and β-catenin−/− c-kit-enriched cells were infected with control vector or BCR-ABL, and survival analyzed (Figure 6A). BCR-ABL led to increased survival equivalently in both the control and β-catenin−/− cells (Figure 6B). To test whether the loss of β-catenin could lead to reduced CML formation by reducing long-term propagation of cancer cells, we first carried out a serial re-plating assay, which provides an indication of cancer cell self-renewal in vitro. Both control and knockout cells formed equal numbers of colonies following vector and BCR-ABL infection in the first plating (Figures 6C and 6D). By the second plating, the BCR-ABL-infected control cells had an increased ability to give rise to new colonies as compared to vector-infected cells (Figure 6C). In contrast, the ability of BCR-ABL to induce colony formation was reduced to control levels in the absence of β-catenin (Figure 6D). Further, the control BCR-ABL cells continued to generate more colonies than control vector-infected cells in the third (2.5-fold) and fourth platings (3.4-fold) (Figure 6C). Interestingly, by the third plating, there appeared to be colonies in the knockout cultures infected with BCR-ABL as well (Figure 6D); however, these colonies were morphologically different from the myeloid colonies in the control cultures and were composed primarily of B cells (Figure 6E). This result mirrored the in vivo finding that the β-catenin−/− cells can be transformed along the B lineage and suggested that β-catenin is critically needed for the increased self-renewal capacity bestowed by BCR-ABL on hematopoietic stem cells to allow transformation to occur along the myeloid lineage.
Figure 6. Loss of β-Catenin Impairs BCR-ABL-Induced Renewal of CML Stem Cells.

(A) A schematic of the strategy to determine relative survival and replating efficiency after BCR-ABL infection in control and β-catenin knockout cells.
(B) c-kit-enriched cells from control or β-catenin knockout mice were infected with vector or BCR-ABL and stained with annexin-V and 7-AAD. Annexin-V+7-AAD− cells were quantified to determine frequency of apoptotic cells. Results are representative of four independent samples.
(C and D) Long-term serial replating. c-kit-enriched control or β-catenin−/− cells were infected with vector or BCR-ABL, and 5000 GFP-positive cells were sorted into wells containing methylcellulose media to assess primary colony formation. Colony numbers were counted on days 8–10. Cells were then harvested and counted, and 5000 cells were replated for a second, third, and fourth time, and colonies were counted on days 8–10 after each replating. White bars represent B lineage colonies. Results shown are an average of three independent experiments. Error bars show SEM.
(E) BCR-ABL-infected control and β-catenin−/− colonies were collected after the third and fourth plating and stained with the B cell marker B220. The plot shown is a representative FACS from the fourth plating.
(F) In vivo self-renewal assay for CML stem cells. Irradiated recipient mice were transplanted with 10,000 KLSGFP+ cells (CML stem cells) sorted from control or β-catenin knockout CMLs. Survival curves show the frequency of recipients succumbing to disease after receiving KLS cells from wild-type CML (solid line) or β-catenin null CML (dashed line) stem cells (total number of mice transplanted = 12). In (B), (C), and (D), error bars represent SEM.
To more definitively test whether the CML impairment in the absence of β-catenin was due to a defect in cancer stem cell self-renewal in vivo, we isolated GFP+KLS cells from control or β-catenin−/− primary CML and transplanted them into secondary recipients. These cells have been shown previously to successfully propagate CML formation (Hu et al., 2006), thus broadly fitting the criteria for CML stem cells. Upon transplantation, CML stem cells from control mice successfully propagated leukemia such that 65% of the recipients succumbed to the disease (Figure 6F). In contrast, none of the recipients transplanted with CML stem cells from the β-catenin−/− mice developed the disease during the same time period. These data are consistent with the in vitro replating data and indicate that the loss of β-catenin impairs CML progression by reducing the self-renewal ability of CML stem cells.
We also carried out control experiments to rule out the possibility that the impaired CML progression was due to an altered frequency of cells that could serve as targets for BCR-ABL. FACS analysis revealed equivalent fractions of lineage-negative, long-term and short-term cells in both the control and β-catenin knockout c-kit+ population (Figures S9A–S9C). We also found that control and β-catenin knockout cells retained similar frequencies of lymphoid and myeloid lineage-committed cells following infection with BCR-ABL (Figure S9D). Further, when purified KLSF− cells were used to equalize the target population for BCR-ABL infection (Figure S9E), control cells gave rise to leukemia in 100% of the mice (CML in 7/8 and B-ALL in 1/8), while knockout cells generated significantly fewer CMLs (12.5%, only one case of CML). Finally, reintroduction of activated β-catenin complemented the defect in CML formation (Figures S10A–S10C). These data cumulatively indicate that the reduced ability of BCR-ABL to cause CML in the absence of β-catenin is unlikely to be due to differences in target populations infected between wild-type and mutant mice.
Differential Wnt Reporter Activity in BCR-ABL-Induced CML and ALL
We next investigated whether the differential dependence of CML versus ALL on β-catenin was due to differential utilization of the Wnt pathway in the two leukemias. We thus generated a lentiviral Wnt reporter, BATRED (see Experimental Procedures), that would fluoresce red upon activation of a Wnt signal and could be detected in the GFP-expressing BCR-ABL-infected leukemia cells. In control experiments, the BATRED reporter responded to β-catenin activation as expected (Figure S11). Reporter activity could be detected in 42% (188/448) of the CML cells isolated from control mice (Figure 7A). In some CML samples the reporter activity was particularly high in some cells and mid to low in other cells, but consistently above background control levels. In contrast, BATRED activity was undetectable or significantly lower in the cells derived from ALL samples (Figure 7A). As a control, we confirmed that the BATRED reporter had integrated into both ALL and CML cells (Figure 7B). Additionally, the BATRED reporter could be activated in ALL cells when Wnt signaling was forcibly turned on using lithium chloride (Figure 7C), suggesting it was unlikely that the reporter was silenced preferentially in ALL cells. These data indicate that Wnt reporter activity is higher in BCR-ABL-induced CML compared to ALL. This differential utilization is consistent with the differential requirement for β-catenin in CML and ALL.
Figure 7. Differential Wnt Reporter Activity in CML versus ALL.
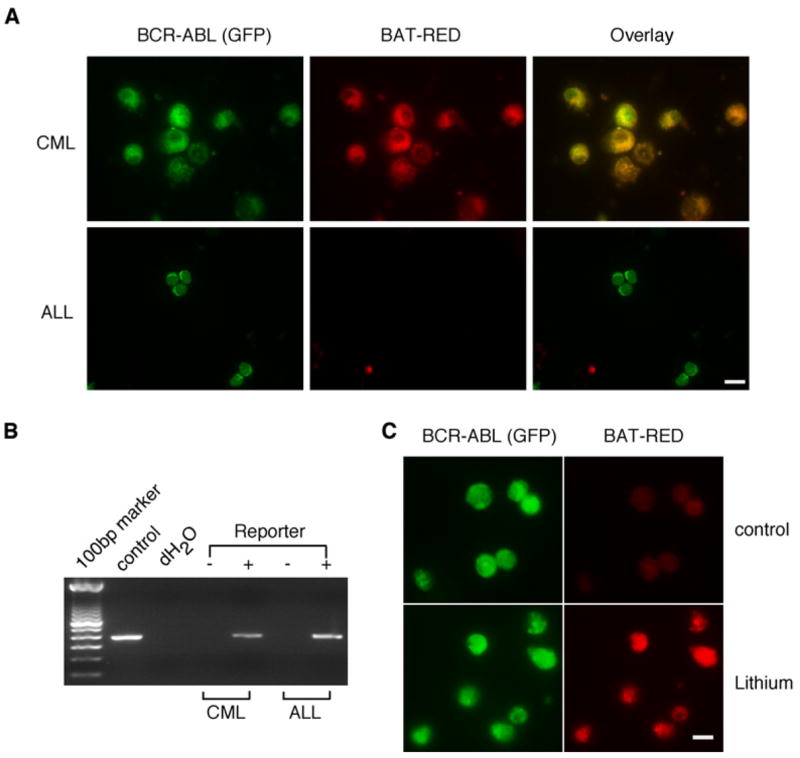
(A) Leukemic cells from BCR-ABL-induced CML or ALL were sorted for GFP and then infected with a lentiviral Wnt reporter expressing dsREd (BATRED) upon Wnt activation. Cytospins were prepared 48 hr later and observed by immunofluorescence microscopy.
(B) Genomic DNA was extracted from Wnt-reporter-infected CML or ALL cells, and viral integration was checked by PCR analysis using specific primers designed to amplify dsREd. The Wnt reporter plasmid served as a positive control. Data shown in both (A) and (B) are representative of three independent experiments.
(C) GFP+ leukemic cells from β-catenin−/− ALL were sorted, infected with the BATRED reporter virus, and treated with control media or 1 mM lithium chloride for 48 hr. Cells were then cytospun, and reporter activity was analyzed by immunofluorescence. Data are representative of two independent experiments. In (A) and (C), the scale bar represents 10 μm and 3 μm, respectively.
BCR-ABL-Induced Phosphorylation Is Reduced in the Absence of β-Catenin
Finally, we investigated the specific molecular changes that occur with the loss of β-catenin that may contribute to the impaired ability of BCR-ABL to transform cells along the myeloid lineage. We first examined whether the β-catenin−/− cells have reduced levels of genes that confer myeloid identity but did not find any changes in the expression of CEBPalpha, Id1, or Pax5 (Figure S12). Lack of β-catenin also did not significantly change the expression of cyclinD1, cyclinD2, and c-Myb (data not shown). It has previously been shown that CML progression, but not ALL progression, is impaired in the absence of Stat5α (Ye et al., 2006; Sexl et al., 2000), which is normally phosphorylated and thereby activated by BCR-ABL. We thus tested if phosphorylated Stat5α was reduced in the absence of β-catenin, and found that p-Stat5α was reduced by approximately 4-fold in the β-catenin null cells following BCR-ABL transduction (Figures 8A and 8B). While there was some reduction in total Stat5 protein as well, it was not as marked as the reduction in phosphorylated Stat5 and did not appear to be responsible for the reduced levels of p-Stat5α observed (Figures 8C and 8D). That the loss of Stat5α leads to a differential impact on CML and ALL together with our data that loss of β-catenin reduces BCR-ABL induced p-Stat5α provides support, at a molecular level, for the idea that β-catenin is differentially required in CML and ALL. In addition to the phosphorylation defect of Stat5α, we also observed a global decrease in protein tyrosine phosphorylation in established CML, but not ALL, derived from β-catenin−/− cells (2.6- to 4-fold reduction; Figure 8E). While the transcriptional levels of BCR-ABL were relatively equivalent in the leukemias from control and β-catenin−/− cells (Figure 8F), the levels of BCR-ABL protein was significantly reduced in the absence of β-catenin in CML cells but not ALL cells (Figure 8G). In light of the fact that BCR-ABL and β-catenin can form a complex in vivo (Coluccia et al., 2007; and data not shown), our data raise the intriguing possibility that the loss of β-catenin leads to reduced levels of BCR-ABL and reduced phosphorylation of relevant substrates; this reduced activation of downstream targets may lead to a consequent decrease in CML self-renewal ability and progression.
Figure 8. BCR-ABL-Induced Phosphorylation of Stat5α Is Reduced in the Absence of β-Catenin.

(A and C) c-kit enriched control or β-catenin−/− cells were infected with BCR-ABL for 48 hr. Infected cells (GFP positive) were sorted and cultured for another 3–4 days, and then cytospins were prepared and stained with indicated antibodies. Data are representative of three independent experiments. In (A) and (C), the scale bar represents 10 μm. (B and D) Quantification of fluorescence intensities/cell for phosphorylated (B) and total Stat5α (D) using Metamorph software. In control cells, phosphorylation of Stat5α was significantly induced following BCR-ABL transduction (p = 0.003). In contrast, BCR-ABL transduction in β-catenin−/− cells did not induce Stat5α phosphorylation over vector and showed reduced phosphorylation compared to BCR-ABL-infected control cells (p = 0.008). (E) Relative levels of protein tyrosine phosphorylation in CML and ALL cells from control or β-catenin−/− leukemias. GFP+ leukemia cells from CML or ALL of each genotype were sorted, and protein lysates were analyzed by western blotting with an anti-phosphotyrosine antibody. (F) Real-time PCR analysis of BCR-ABL mRNA levels in control and β-catenin−/− leukemia cells. (G) Relative levels of BCR-ABL protein in CML and ALL from control or β-catenin−/− leukemias. GFP+ leukemia cells from CML or ALL of each genotype were sorted, and protein lysate was analyzed by western blotting with an anti-abl antibody. Data are representative of two to three independent experiments. In (B), (D), and (F), error bars represent SEM.
DISCUSSION
Our studies indicate that deletion of β-catenin in the hematopoietic system impairs normal HSC function and leukemia progression in vivo. During normal hematopoiesis, while the loss of β-catenin did not affect the initial establishment of HSCs and hematopoietic differentiation, it led to a clear reduction of long-term maintenance as assayed by transplantation. These data overall strengthen the findings that retroviral modulation of Wnt signaling in HSCs can influence proliferation and reconstitution in mice (Reya et al., 2003; Willert et al., 2003; Baba et al., 2005, 2006) as well as the observation that delivery of Wnt protein can enhance human HSC reconstitution in a xenograft model (Murdoch et al., 2003). However, an IFN-inducible β-catenin−/− mouse was reported to have no defects in the hematolymphoid or any other system (Cobas et al., 2004). It is possible that the differences in phenotypes observed may be due to the approach by which β-catenin was deleted. Using IFN induction to induce deletion in adulthood may set up a different context for testing requirements than using a transgenic in which cre recombinase is driven by a promoter starting in embryonic life, as was done in our case as well as in experiments demonstrating a requirement for β-catenin in development of multiple major organs (Brault et al., 2001; Cattelino et al., 2003; Dessimoz et al., 2005; Xu et al., 2003).
One of the most important findings that we report here is that loss of β-catenin affects myeloid leukemia progression in vivo. Several studies have suggested that Wnt signaling is active in many leukemic cell lines and patient samples (reviewed in Reya and Clevers, 2005). However, whether this pathway is required for in vivo leukemia progression was unknown. Our data clearly demonstrate that deletion of β-catenin leads to a reduced ability of BCR-ABL to induce CML in vivo. Further we found that the impaired CML incidence could be rescued by ectopic β-catenin expression (Figure S10), supporting the notion that the loss of β-catenin directly impacts CML progression. Interestingly, only a low dose of β-catenin was effective in this rescue, while a high dose appeared to interfere with leukemogenesis. These data indicate that the precise levels of β-catenin are critical to determining its influence on hematopoietic cells, and they may help explain the data observed with transgenic mice overexpressing high levels of β-catenin in which an expansion of phenotypic HSCs is followed by loss of differentiated hematopoietic cells (Kirstetter et al., 2006; Scheller et al., 2006).
Our data also provide insight into the cellular and molecular basis for impaired CML progression in the absence of β-catenin. At a cellular level, the serial replating and CML serial transplantation assays suggest that the decreased progression of CML is due to a reduced ability of BCR-ABL to sustain long-term renewal of CML stem cells in the absence of β-catenin. At a molecular level, our data show that the loss of β-catenin reduces the levels of BCR-ABL and impairs phosphorylation of proteins in BCR-ABL-induced CML. The fact that the loss of β-catenin reduces phosphorylation of Stat5α and other substrates raises the possibility that Wnt signaling may regulate CML progression by allowing the maintenance of normal levels of BCR-ABL protein, which in turn phosphorylates Stat5α and other substrates needed for optimal transformation along the myeloid lineage.
Remarkably, while CML formation was reduced in the absence of β-catenin, ALL formation could proceed relatively unimpaired. It has been proposed that CML and ALL may arise from distinct cells of origin. Specifically, some data indicate that the cell of origin for CML is a stem cell (Li et al., 1999; Takahashi et al., 1998), while the cell of origin for ALL is a committed B cell precursor (Li et al., 1999). In our experiments presented here, we find that CML occurs almost exclusively when KLSF cells are used as the cell of origin (Figure S9). Furthermore, we find that when progenitor B220+IgM− cells are used as the cell of origin, the leukemias that are generated are B-ALL (Figure S13). These data indicate that CML is the predominant disease when starting with an uncommitted HSC-enriched fraction and that B-ALL is predominant when committed lymphoid progenitor cells are targeted. The fact that we do not observe significant ALL from BCR-ABL-infected HSCs suggests that the cells in which the leukemia initiates may determine the outcome of the leukemia subtype. This is similar to patients in whom the BCR-ABL translocation can be found in multiple lineages (suggesting a stem cell hit) but only CML occurs. Thus it is possible that the requirement for β-catenin is different in the two leukemias precisely because of the different transcriptional contexts in the two different cells of origin. If this is the case, the low frequency of B-ALL from KLSF may indicate a low level of contamination of progenitors in the KLSF fractions or some level of differentiation during the BCR-ABL infection process in vitro. On the other hand, the fact that we do observe some frequency of B-ALL even when starting with KLSF cells may indicate that ALL can be initiated in either uncommitted or committed progenitors. If this is the case, it is possible that the differential need for β-catenin is not because the initiating cell is different for CML and ALL but because the leukemias are ultimately propagated along different lineages, which have different transcriptional contexts.
Several genetic changes have been shown to lead to altered ratios of CML and ALL. For example, the combined loss of the src kinases Lyn, Hck, and Fgr preferentially reduces the incidence of B-ALL relative to CML (Hu et al., 2004), while reduced activity of the tumor suppressor Arf preferentially accelerates ALL formation (Williams et al., 2006). Insight into the molecular changes that lead to a preferential reduction of CML incidence has come primarily from studies of mutations of the abl kinase domain. Specifically, mutations in the SH2 domain as well as in the Grb2-binding domain of BCR-ABL lead to reduced formation of CML but allow normal progression of ALL (Million and Van Etten, 2000; Roumiantsev et al., 2001). However, downstream pathways or cascades that may mediate the differential effects of these mutations remain less well elucidated. This raises the possibility that these mutations may affect the ability of BCR-ABL to influence the Wnt pathway. Alternatively, Wnt signaling may be an independent modulator of the differential signaling requirements between CML and ALL progression.
Overall, our work supports data showing that Wnt signaling is active in and important for human CML. Specifically it has been shown that granulocyte-macrophage progenitors from chronic myelogenous leukemia patients and blast crisis cells from patients resistant to therapy display activated Wnt signaling (Jamieson et al., 2004). Additionally, inhibition of β-catenin through ectopic expression of axin decreases the replating capacity of leukemic cells, suggesting that human chronic myelogenous leukemia precursors are dependent on Wnt signaling for growth and renewal in vitro. Our data using a mouse model of BCR-ABL-induced CML are consistent with these findings and demonstrate that progression of CML in vivo is also critically dependant on intact β-catenin. The work in the human system together with that in our mouse model collectively suggests that Wnt signaling could prove to be a relevant therapeutic target. Thus, in the long-term, it will be important to explore whether blocking Wnt signaling either alone or together with inhibition of BCR-ABL kinase activity may be an effective avenue to halt CML progression.
EXPERIMENTAL PROCEDURES
Mice
The loxP-β-catenin and Vav-cre transgenic mice used were in the C57Bl/6J background. Transplant recipients (C57Bl/Ka CD45.1) were 8–10 weeks of age. All mice were bred and maintained on acidified water in the animal care facility at Duke University Medical Center. All animal experiments were performed according to protocols approved by the Duke University Institutional Animal Care and Use Committee.
HSC Isolation and Analysis
Isolation of HSCs from bone marrow and their transplantation for in vivo analysis of function were performed as described (Domen et al., 2000; Duncan et al., 2005). For the competitive repopulation unit (CRU) assay, 10, 40, and 50 sorted KLSCD34− control or β-catenin−/− bone marrow cells were mixed with 200,000 competing bone marrow cells and injected into lethally irradiated CD45.1 recipients. Multilineage repopulation was assessed at 10 weeks after transplantation. Secondary limiting-dilution competitive repopulation analysis was carried out as described (Ema et al., 2005). Whole bone marrow cells were isolated from primary recipients originally transplanted with control or β-catenin−/− KLSCD34− cells for 21 weeks and transplanted into 5–7 recipients at each dose for each genotype (10,000, 40,000, 80,000, and 160,000 cells/recipient). Peripheral blood cells of the secondary recipients were analyzed 10 weeks after transplantation. CRU was calculated using L-Cal software (StemCell Technologies). To assess homing in vivo, 2 × 106 bone marrow cells from control or β-catenin−/− mice were transplanted into lethally irradiated 45.1 recipients (4 mice/group). The recipient mice were sacrificed 6 hr posttransplantation, and the presence of donor cells was analyzed by FACS. For leukemia stem cell self-renewal analysis in vivo, CML stem cells were isolated from CMLs of each genotype as described before (Hu et al., 2006). To retrieve sufficient numbers of CML stem cells (from pooled leukemias) to cause secondary disease, the primary CML was induced with high numbers of KLSF cells (50,000 at 50% transduction efficiency). Ten thousand freshly isolated CML stem cells were transplanted into lethally irradiated recipient mice together with 200,000 competitive whole bone marrow cells (n = 6 per group).
Construction and Use of Lentiviral Wnt Reporter
To construct a lentiviral Wnt reporter that would fluoresce red upon activation of the Wnt signal, we used the reporter elements of the BATGAL construct in which seven tandem Lef/Tcf sites were cloned upstream of the minimal promoter TATA box of the Siamois gene (Maretto et al., 2003). These “BAT” elements were cloned in front of a ds“RED”.T4 element (Bevis and Glick, 2002). Second, the XmaI and BsrG1 fragment of lenti-OT-GFP (Reya et al., 2003) was replaced by the seven tandem Lef/Tcf sites, minimal promoter TATA, and dsRED.T4, resulting in the “BATRED” reporter. For detecting Wnt signaling by dsREd lentiviral Wnt reporter, GFP-positive leukemia cells from CML or ALL mice were sorted and infected with lentiviral reporter for 48 hr, and cytospins were generated. Confocal images were obtained with a Zeiss 410 Axiovert Microscope.
Analysis of Diseased Mice
After transplantation, recipient mice were evaluated daily for signs of morbidity, weight loss, failure to thrive, and splenomegaly. Premorbid animals were sacrificed and relevant tissues harvested and analyzed by flow cytometry and histopathology. For flow cytometric analysis, leukemic cell populations were incubated with antibodies to murine CD3, B220, Gr-1, and Mac-1 (eBiosciences) and analyzed on FAC-Vantage SE (BD) with FlowJo software (Tree Star, Inc). Mice that died prior to analysis (three in the control group, and one in the −/− group) were excluded from Figure 4C.
Cell Culture and Methylcellulose Colony Formation
For liquid culture, control or BCR-ABL-infected c-kit-enriched cells were sorted directly into 96-well U bottom plate with the same medium used for prestimulation of cells (Supplemental Experimental Procedures); for methylcellulose assays, 1000–5000 GFP-positive cells were sorted directly into 12-well plates with complete methylcellulose medium (StemCell Technologies, catalog number M3434). Colony numbers were initially counted 8–10 days after plating. Subsequently, cells were harvested and counted, and 1000–5000 cells were replated into new 12-well plates. Apoptosis assays were performed by staining cells with annexin-V and 7-AAD (BD, Biosciences).
Immunofluorescence Staining
Vector or BCR-ABL-infected c-kit-enriched cells were sorted for GFP by FACS and cytospun, and fixed in 4% paraformaldehyde. Primary antibodies used were goat anti-phosphorylated Stat5α (sc-11761, Santa Cruz) or rabbit anti-Stat5α (sc-1081, Santa Cruz), and secondary antibodies were rabbit anti-goat-PE or donkey anti-rabbit-PE. DAPI (Molecular Probes) was added as a nuclear counter-stain.
Supplementary Material
The Supplemental Data include Supplemental Experimental Procedures and thirteen supplemental figures and can be found with this article online at http://www.cancercell.org/cgi/content/full/12/6/528/DC1/.
Acknowledgments
We are very grateful to Ann Marie Pendergast and Warren Pear for reagents, advice, and comments on the manuscript, Dimitris Kioussis for the vav-Cre transgenic line, Anne Lai and Motonari Kondo for help with the CLP, CMP, and GMP analysis, Beth Harvat for cell sorting, Daniel Neef for help with northern blotting, Donald McDonnell for use of the icycler, Nick D’Amato for help with PCR analysis, and Sandria Elrod-Iqbal for help with preparation of the manuscript. TR is a recipient of a Cancer Research Institute Investigator Award, an Ellison Medical Foundation New Scholar Award, and a Leukemia and Lymphoma Society Scholar Award. This work was also supported by NIH grants DK63031, DK072234, and AI067798.
References
- Adams GB, Chabner KT, Alley IR, Olson DP, Szczepiorkowski ZM, Poznansky MC, Kos CH, Pollak MR, Brown EM, Scadden DT. Stem cell engraftment at the endosteal niche is specified by the calcium-sensing receptor. Nature. 2006;439:599–603. doi: 10.1038/nature04247. [DOI] [PubMed] [Google Scholar]
- Almarza E, Segovia JC, Guenechea G, Gomez SG, Ramirez A, Bueren JA. Regulatory elements of the vav gene drive transgene expression in hematopoietic stem cells from adult mice. Exp Hematol. 2004;32:360–364. doi: 10.1016/j.exphem.2004.01.005. [DOI] [PubMed] [Google Scholar]
- Baba Y, Garrett KP, Kincade PW. Constitutively active beta-catenin confers multilineage differentiation potential on lymphoid and myeloid progenitors. Immunity. 2005;23:599–609. doi: 10.1016/j.immuni.2005.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba Y, Yokota T, Spits H, Garrett KP, Hayashi S, Kincade PW. Constitutively active beta-catenin promotes expansion of multipotent hematopoietic progenitors in culture. J Immunol. 2006;177:2294–2303. doi: 10.4049/jimmunol.177.4.2294. [DOI] [PubMed] [Google Scholar]
- Beachy PA, Karhadkar SS, Berman DM. Tissue repair and stem cell renewal in carcinogenesis. Nature. 2004;432:324–331. doi: 10.1038/nature03100. [DOI] [PubMed] [Google Scholar]
- Bevis BJ, Glick BS. Rapidly maturing variants of the Discosoma red fluorescent protein (DsRed) Nat Biotechnol. 2002;20:83–87. doi: 10.1038/nbt0102-83. [DOI] [PubMed] [Google Scholar]
- Brault V, Moore R, Kutsch S, Ishibashi M, Rowitch DH, McMahon AP, Sommer L, Boussadia O, Kemler R. Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development. 2001;128:1253–1264. doi: 10.1242/dev.128.8.1253. [DOI] [PubMed] [Google Scholar]
- Cattelino A, Liebner S, Gallini R, Zanetti A, Balconi G, Corsi A, Bianco P, Wolburg H, Moore R, Oreda B, et al. The conditional inactivation of the beta-catenin gene in endothelial cells causes a defective vascular pattern and increased vascular fragility. J Cell Biol. 2003;162:1111–1122. doi: 10.1083/jcb.200212157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobas M, Wilson A, Ernst B, Mancini SJ, MacDonald HR, Kemler R, Radtke F. Beta-catenin is dispensable for hematopoiesis and lymphopoiesis. J Exp Med. 2004;199:221–229. doi: 10.1084/jem.20031615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coluccia AM, Vacca A, Dunach M, Mologni L, Redaelli S, Bustos VH, Benati D, Pinna LA, Gambacorti-Passerini C. Bcr-Abl stabilizes beta-catenin in chronic myeloid leukemia through its tyrosine phosphorylation. EMBO J. 2007;26:1456–1466. doi: 10.1038/sj.emboj.7601485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daley GQ, Van Etten RA, Baltimore D. Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science. 1990;247:824–830. doi: 10.1126/science.2406902. [DOI] [PubMed] [Google Scholar]
- de Boer J, Williams A, Skavdis G, Harker N, Coles M, Tolaini M, Norton T, Williams K, Roderick K, Potocnik AJ, Kioussis D. Transgenic mice with hematopoietic and lymphoid specific expression of Cre. Eur J Immunol. 2003;33:314–325. doi: 10.1002/immu.200310005. [DOI] [PubMed] [Google Scholar]
- Dessimoz J, Bonnard C, Huelsken J, Grapin-Botton A. Pancreas-specific deletion of beta-catenin reveals Wnt-dependent and Wnt-independent functions during development. Curr Biol. 2005;15:1677–1683. doi: 10.1016/j.cub.2005.08.037. [DOI] [PubMed] [Google Scholar]
- Domen J, Cheshier SH, Weissman IL. The role of apoptosis in the regulation of hematopoietic stem cells: overexpression of Bcl-2 increases both their number and repopulation potential. J Exp Med. 2000;191:253–264. doi: 10.1084/jem.191.2.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan AW, Rattis FM, DiMascio LN, Congdon KL, Pazianos G, Zhao C, Yoon K, Cook JM, Willert K, Gaiano N, Reya T. Integration of Notch and Wnt signaling in hematopoietic stem cell maintenance. Nat Immunol. 2005;6:314–322. doi: 10.1038/ni1164. [DOI] [PubMed] [Google Scholar]
- Ema H, Sudo K, Seita J, Matsubara A, Morita Y, Osawa M, Takatsu K, Takaki S, Nakauchi H. Quantification of self-renewal capacity in single hematopoietic stem cells from normal and Lnk-deficient mice. Dev Cell. 2005;8:907–914. doi: 10.1016/j.devcel.2005.03.019. [DOI] [PubMed] [Google Scholar]
- Gamallo C, Palacios J, Moreno G, Calvo de Mora J, Suarez A, Armas A. beta-catenin expression pattern in stage I and II ovarian carcinomas: relationship with beta-catenin gene mutations, clinicopathological features, and clinical outcome. Am J Pathol. 1999;155:527–536. doi: 10.1016/s0002-9440(10)65148-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Rostan G, Tallini G, Herrero A, D’Aquila TG, Carcangiu ML, Rimm DL. Frequent mutation and nuclear localization of beta-catenin in anaplastic thyroid carcinoma. Cancer Res. 1999;59:1811–1815. [PubMed] [Google Scholar]
- Giles RH, van Es JH, Clevers H. Caught up in a Wnt storm: Wnt signaling in cancer. Biochim Biophys Acta. 2003;1653:1–24. doi: 10.1016/s0304-419x(03)00005-2. [DOI] [PubMed] [Google Scholar]
- Haegel H, Larue L, Ohsugi M, Fedorov L, Herrenknecht K, Kemler R. Lack of beta-catenin affects mouse development at gastrulation. Development. 1995;121:3529–3537. doi: 10.1242/dev.121.11.3529. [DOI] [PubMed] [Google Scholar]
- Hu Y, Liu Y, Pelletier S, Buchdunger E, Warmuth M, Fabbro D, Hallek M, Van Etten RA, Li S. Requirement of Src kinases Lyn, Hck and Fgr for BCR-ABL1-induced B-lymphoblastic leukemia but not chronic myeloid leukemia. Nat Genet. 2004;36:453–461. doi: 10.1038/ng1343. [DOI] [PubMed] [Google Scholar]
- Hu Y, Swerdlow S, Duffy TM, Weinmann R, Lee FY, Li S. Targeting multiple kinase pathways in leukemic progenitors and stem cells is essential for improved treatment of Ph+ leukemia in mice. Proc Natl Acad Sci USA. 2006;103:16870–16875. doi: 10.1073/pnas.0606509103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huelsken J, Vogel R, Brinkmann V, Erdmann B, Birchmeier C, Birchmeier W. Requirement for beta-catenin in anterior-posterior axis formation in mice. J Cell Biol. 2000;148:567–578. doi: 10.1083/jcb.148.3.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamieson CH, Ailles LE, Dylla SJ, Muijtjens M, Jones C, Zehnder JL, Gotlib J, Li K, Manz MG, Keating A, et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med. 2004;351:657–667. doi: 10.1056/NEJMoa040258. [DOI] [PubMed] [Google Scholar]
- Kinzler KW, Nilbert MC, Su LK, Vogelstein B, Bryan TM, Levy DB, Smith KJ, Preisinger AC, Hedge P, McKechnie D, et al. Identification of FAP locus genes from chromosome 5q21. Science. 1991;253:661–665. doi: 10.1126/science.1651562. [DOI] [PubMed] [Google Scholar]
- Kirstetter P, Anderson K, Porse BT, Jacobsen SE, Nerlov C. Activation of the canonical Wnt pathway leads to loss of hematopoietic stem cell repopulation and multilineage differentiation block. Nat Immunol. 2006;7:1048–1056. doi: 10.1038/ni1381. [DOI] [PubMed] [Google Scholar]
- Li S, Ilaria RL, Jr, Million RP, Daley GQ, Van Etten RA. The P190, P210, and P230 forms of the BCR/ABL oncogene induce a similar chronic myeloid leukemia-like syndrome in mice but have different lymphoid leukemogenic activity. J Exp Med. 1999;189:1399–1412. doi: 10.1084/jem.189.9.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Wakamiya M, Shea MJ, Albrecht U, Behringer RR, Bradley A. Requirement for Wnt3 in vertebrate axis formation. Nat Genet. 1999;22:361–365. doi: 10.1038/11932. [DOI] [PubMed] [Google Scholar]
- Maretto S, Cordenonsi M, Dupont S, Braghetta P, Broccoli V, Hassan AB, Volpin D, Bressan GM, Piccolo S. Mapping Wnt/beta-catenin signaling during mouse development and in colorectal tumors. Proc Natl Acad Sci USA. 2003;100:3299–3304. doi: 10.1073/pnas.0434590100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazieres J, You L, He B, Xu Z, Lee AY, Mikami I, McCormick F, Jablons DM. Inhibition of Wnt16 in human acute lymphoblastoid leukemia cells containing the t(1;19) translocation induces apoptosis. Oncogene. 2005;24:5396–5400. doi: 10.1038/sj.onc.1208568. [DOI] [PubMed] [Google Scholar]
- McWhirter JR, Neuteboom ST, Wancewicz EV, Monia BP, Downing JR, Murre C. Oncogenic homeodomain transcription factor E2A-Pbx1 activates a novel WNT gene in pre-B acute lymphoblastoid leukemia. Proc Natl Acad Sci USA. 1999;96:11464–11469. doi: 10.1073/pnas.96.20.11464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller C, Sassoon DA. Wnt-7a maintains appropriate uterine patterning during the development of the mouse female reproductive tract. Development. 1998;125:3201–3211. doi: 10.1242/dev.125.16.3201. [DOI] [PubMed] [Google Scholar]
- Million RP, Van Etten RA. The Grb2 binding site is required for the induction of chronic myeloid leukemia-like disease in mice by the Bcr/Abl tyrosine kinase. Blood. 2000;96:664–670. [PubMed] [Google Scholar]
- Monkley SJ, Delaney SJ, Pennisi DJ, Christiansen JH, Wainwright BJ. Targeted disruption of the Wnt2 gene results in placentation defects. Development. 1996;122:3343–3353. doi: 10.1242/dev.122.11.3343. [DOI] [PubMed] [Google Scholar]
- Moon RT, Brown JD, Torres M. WNTs modulate cell fate and behavior during vertebrate development. Trends Genet. 1997;13:157–162. doi: 10.1016/s0168-9525(97)01093-7. [DOI] [PubMed] [Google Scholar]
- Muller-Tidow C, Steffen B, Cauvet T, Tickenbrock L, Ji P, Diederichs S, Sargin B, Kohler G, Stelljes M, Puccetti E, et al. Translocation products in acute myeloid leukemia activate the Wnt signaling pathway in hematopoietic cells. Mol Cell Biol. 2004;24:2890–2904. doi: 10.1128/MCB.24.7.2890-2904.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murdoch B, Chadwick K, Martin M, Shojaei F, Shah KV, Gallacher L, Moon RT, Bhatia M. Wnt-5A augments repopulating capacity and primitive hematopoietic development of human blood stem cells in vivo. Proc Natl Acad Sci USA. 2003;100:3422–3427. doi: 10.1073/pnas.0130233100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parr BA, McMahon AP. Wnt genes and vertebrate development. Curr Opin Genet Dev. 1994;4:523–528. doi: 10.1016/0959-437x(94)90067-d. [DOI] [PubMed] [Google Scholar]
- Pear WS, Miller JP, Xu L, Pui JC, Soffer B, Quackenbush RC, Pendergast AM, Bronson R, Aster JC, Scott ML, Baltimore D. Efficient and rapid induction of a chronic myelogenous leukemia-like myeloproliferative disease in mice receiving P210 bcr/abl-transduced bone marrow. Blood. 1998;92:3780–3792. [PubMed] [Google Scholar]
- Ren R. Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat Rev Cancer. 2005;5:172–183. doi: 10.1038/nrc1567. [DOI] [PubMed] [Google Scholar]
- Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005;434:843–850. doi: 10.1038/nature03319. [DOI] [PubMed] [Google Scholar]
- Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- Reya T, Duncan AW, Ailles L, Domen J, Scherer DC, Willert K, Hintz L, Nusse R, Weissman IL. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature. 2003;423:409–414. doi: 10.1038/nature01593. [DOI] [PubMed] [Google Scholar]
- Roumiantsev S, de Aos IE, Varticovski L, Ilaria RL, Van Etten RA. The src homology 2 domain of Bcr/Abl is required for efficient induction of chronic myeloid leukemia-like disease in mice but not for lymphoid leukemogenesis or activation of phosphatidylinositol 3-kinase. Blood. 2001;97:4–13. doi: 10.1182/blood.v97.1.4. [DOI] [PubMed] [Google Scholar]
- Satoh S, Daigo Y, Furukawa Y, Kato T, Miwa N, Nishiwaki T, Kawasoe T, Ishiguro H, Fujita M, Tokino T, et al. AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nat Genet. 2000;24:245–250. doi: 10.1038/73448. [DOI] [PubMed] [Google Scholar]
- Scheller M, Huelsken J, Rosenbauer F, Taketo MM, Birchmeier W, Tenen DG, Leutz A. Hematopoietic stem cell and multilineage defects generated by constitutive beta-catenin activation. Nat Immunol. 2006;7:1037–1047. doi: 10.1038/ni1387. [DOI] [PubMed] [Google Scholar]
- Sexl V, Piekorz R, Moriggl R, Rohrer J, Brown MP, Bunting KD, Rothammer K, Roussel MF, Ihle JN. Stat5a/b contribute to interleukin 7-induced B-cell precursor expansion, but abl- and bcr/abl-induced transformation are independent of stat5. Blood. 2000;96:2277–2283. [PubMed] [Google Scholar]
- Siegfried E, Perrimon N. Drosophila wingless: a paradigm for the function and mechanism of Wnt signaling. Bioessays. 1994;16:395–404. doi: 10.1002/bies.950160607. [DOI] [PubMed] [Google Scholar]
- Takahashi N, Miura I, Saitoh K, Miura AB. Lineage involvement of stem cells bearing the philadelphia chromosome in chronic myeloid leukemia in the chronic phase as shown by a combination of fluorescence-activated cell sorting and fluorescence in situ hybridization. Blood. 1998;92:4758–4763. [PubMed] [Google Scholar]
- Tsukamoto AS, Grosschedl R, Guzman RC, Parslow T, Varmus HE. Expression of the int-1 gene in transgenic mice is associated with mammary gland hyperplasia and adenocarcinomas in male and female mice. Cell. 1988;55:619–625. doi: 10.1016/0092-8674(88)90220-6. [DOI] [PubMed] [Google Scholar]
- Willert K, Brown JD, Danenberg E, Duncan AW, Weissman IL, Reya T, Yates JR, III, Nusse R. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature. 2003;423:448–452. doi: 10.1038/nature01611. [DOI] [PubMed] [Google Scholar]
- Williams RT, Roussel MF, Sherr CJ. Arf gene loss enhances oncogenicity and limits imatinib response in mouse models of Bcr-Abl-induced acute lymphoblastic leukemia. Proc Natl Acad Sci USA. 2006;103:6688–6693. doi: 10.1073/pnas.0602030103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witte O. The role of Bcr-Abl in chronic myeloid leukemia and stem cell biology. Semin Hematol. 2001;38:3–8. doi: 10.1016/s0037-1963(01)90111-8. [DOI] [PubMed] [Google Scholar]
- Wu MF, Young HK, Rattis FM, Ashkenazi R, Jackson TL, Gaiano N, Oliver T, Reya T. Imaging hematopoietic precursor division in real time. Cell Stem Cell. 2007;1:541–554. doi: 10.1016/j.stem.2007.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Banerjee D, Huelsken J, Birchmeier W, Sen JM. Deletion of beta-catenin impairs T cell development. Nat Immunol. 2003;4:1177–1182. doi: 10.1038/ni1008. [DOI] [PubMed] [Google Scholar]
- Ye D, Wolff N, Li L, Zhang S, Ilaria RL., Jr STAT5 signaling is required for the efficient induction and maintenance of CML in mice. Blood. 2006;107:4917–4925. doi: 10.1182/blood-2005-10-4110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa Y, Fujimori T, McMahon AP, Takada S. Evidence that absence of Wnt-3a signaling promotes neuralization instead of paraxial mesoderm development in the mouse. Dev Biol. 1997;183:234–242. doi: 10.1006/dbio.1997.8502. [DOI] [PubMed] [Google Scholar]
- Zheng X, Beissert T, Kukoc-Zivojnov N, Puccetti E, Altschmied J, Strolz C, Boehrer S, Gul H, Schneider O, Ottmann OG, et al. Gamma-catenin contributes to leukemogenesis induced by AML-associated translocation products by increasing the self-renewal of very primitive progenitor cells. Blood. 2004;103:3535–3543. doi: 10.1182/blood-2003-09-3335. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The Supplemental Data include Supplemental Experimental Procedures and thirteen supplemental figures and can be found with this article online at http://www.cancercell.org/cgi/content/full/12/6/528/DC1/.