

Abstract
The medium subunit of neurofilament (NF-M) is extensively modified by phosphate and O-linked β-N-acetylglucosamine (O-GlcNAc). Phosphorylation of NF-M plays a critical role in regulating its translocation, filament formation, and function. However, the regulation of NF-M phosphorylation and the role of NF-M O-GlcNAcylation (a modification by which GlcNAc is attached to the serine/threonine residues of a protein via an O-linked glycosidic bond) are largely unknown. Here, we demonstrate that O-GlcNAcylation and phosphorylation of NF-M regulate each other reciprocally in cultured neuroblastoma cells and in metabolically active rat brain slices. In animal models of fasting rats, which mimicked the decreased glucose uptake/metabolism observed in brains of individuals with Alzheimer disease (AD), we found a decrease in O-GlcNAcylation and increase in phosphorylation of NF-M. We also observed decreased O-GlcNAcylation and an increased phosphorylation of NF-M in AD brain. These results suggest that O-GlcNAcylation and phosphorylation of NF-M are regulated reciprocally and that the hyperphosphorylation and accumulation of NF-M in AD brain might be caused by impaired brain glucose uptake/metabolism via down-regulation of NF-M O-GlcNAcylation.—Deng, Y., Li, B., Liu, F., Iqbal, K., Grundke-Iqbal, I., Brandt, R., Gong, C.-X. Regulation between O-GlcNAcylation and phosphoryla-tion of neurofilament-M and their dysregulation in Alzheimer disease.
Keywords: glycosylation, cytoskeleton, glucose metabolism
Neurofilaments (NFS) are the intermediate filaments of neurons and one of the major components of the neuronal cytoskeleton, which are essential for forming and maintaining cell shape and facilitating intracellular transport. NFs are formed by polymerization of three subunits, termed light (NF-L), medium (NF-M), and heavy (NF-H) chains, with the apparent molecular weights of 68, 160, and 200 kDa, respectively, in SDS-polyacrylamide gel electrophoresis (PAGE) (1). NF-M is critical to the cross-bridge formation, stabilization, and longitudinal extension of the filament network and plays a more profound role than NF-H in regulating the structure and function of NFs (2–4). NF-M is extensively modified by phosphates and O-linked β-N-acetyl-glucosamine (O-GlcNAc) at serine and threonine residues, especially those in the head domain and in the KSP repeats of the tail domain (1, 5–10). Many studies have demonstrated that phosphorylation of NF-M plays a critical role in regulating its translocation, filament formation, and function (1, 11). However, the role of NF-M O-GlcNAcylation, a modification by which O-GlcNAc is attached to the serine/threonine residues of a protein via an O-linked glycosidic bond, is not known.
Both phosphorylation and O-GlcNAcylation of proteins are dynamically regulated in the cell and, for many proteins, they regulate each other in a reciprocal manner (12). In the brains of patients with Alzheimer disease (AD), phosphorylation of NFs is dysregulated in such a way that they are hyperphosphorylated and accumulated (13–16). Hyperphosphorylation and accumulation of NFs may contribute to the retrograde degeneration of neurons in AD, but the mechanism leading to NF hyperphosphorylation is not understood. We recently found that phosphorylation of tau protein is inversely regulated by O-GlcNAcylation and that abnormal hyperphosphorylation of tau, which leads to neurofibrillary degeneration in AD (17), can be caused by decreased brain glucose metabolism via down-regulation of tau O-GlcNAcylation (18, 19). Because glucose uptake and metabolism are impaired (20) and protein O-GlcNAcylation level is decreased (18) in AD brain, it is intriguing to hypothesize that these abnormalities might also lead to hyperphosphorylation and, consequently, accumulation of NFs.
In this study, we investigated the regulation between O-GlcNAcylation and phosphorylation of NF-M and the molecular mechanism leading to the hyperphosphorylation and accumulation of NF-M in AD brain. We found that O-GlcNAcylation and phosphorylation of NF-M indeed regulated each other reciprocally both in cultured neuroblastoma cells and in the mammalian brain and that decreased brain glucose metabolism caused decreased O-GlcNAcylation and increased phosphorylation of NF-M. More importantly, we also observed decreased O-GlcNAcylation as well as hyper-phosphorylation of NF-M in AD brain. These findings, for the first time, suggest a unique mechanism of regulation of NF phosphorylation and provide a novel explanation on the hyperphosphorylation and accumulation of NFs in AD brain.
MATERIALS AND METHODS
Human brain tissue
Postmortem human brain tissue (postmortem delay <3 h) was obtained from the Sun Health Research Institute Brain Donation Program (Sun City, AZ, USA). The diagnosis of all cases was confirmed histopathologically. Medial temporal cortices of 7 AD (79.4±5.27 yr old, 4 male and 3 female) and 7 control (79.4±6.69 yr old, 4 male and 3 female) cases were homogenized in a buffer consisting of 60 mM Tris-HCl (pH 6.8), 3% SDS, 5% β-mecaptoethanol, 10% glycerol, 0.05% bromphenol blue, 0.5 mM phenylmethyl sulfonyl fluoride, 50 mM NaF, and 10 μg/ml each of leupeptin, pepstatin A, and aprotinin. Protein concentrations of the homogenates were determined by modified Lowry method (21).
Antibodies
Monoclonal antibody NL6 was raised against a cytoskeletal fraction prepared from human neurons and recognizes an O-GlcNAcylated epitope at or close to the KSP region of the projection domain of human and rat NF-M (6). We further characterized its specificity and confirmed that this antibody recognizes NF-M in an O-GlcNAcylation-dependent manner and does not have any detectable cross-reaction to any proteins of human and rat brains (see Supplemental Fig. 1). Monoclonal antibody SMI31 against the phosphorylated epitopes of the KSP repeats of NF-M/H was purchased from Sternberger Monoclonals Inc. (Baltimore, MD, USA). Polyclonal antibody NF160 (ab9034) against NF-M in a phosphorylation- and O-GlcNAcylation-independent manner was purchased from Abcam Inc. (Cambridge, MA, USA). Polyclonal antibody R61d against all three subunits of NF was raised in our laboratories. Monoclonal DM1A against α-tubulin was obtained from Sigma-Aldrich Co. (St. Louis, MO, USA).
Western blots, immunocytochemistry, and immunohistochemistry
Samples were first resolved in 7.5% SDS-PAGE and electro-transferred onto Immobilon-P membrane (Millipore, Bedford, MA, USA). The blots were then probed with NL6 (1:2000), SMI31 (1:5000), R61d (1:2000), NF160 (1:5000), or DM1A (1:2000) and developed with horseradish peroxidase-conjugated secondary antibody and enhanced chemiluminescence kit (Pierce, Rockford, IL, USA). Immunostaining of SH-SY5Y cells was carried out by conventional immunocytochemical protocol using ABC staining kit (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA). Double immunofluorescence staining of rat brain tissue sections was carried out by using Alexa488-conjugated anti-mouse IgG and Alexa543-conjugated anti-rabbit IgG (Molecular Probes, Eugene, OR, USA). In some experiments, the tissue sections were counter-stained with TO-PRO3, a nucleic acid-specific marker, to visualize the nuclei at 633 nm excitation wavelength.
Cell culture and treatments
SH-SY5Y human neuroblastma cells (ATCC, Manassas, VA, USA) were propagated in a 1:1 mixture of Eagle’s Minimum Essential Medium and Ham’s F12 Medium supplemented with 10% heat-inactivated fetal bovine serum (FBS, Gibco BRL, New York, NY, USA), 100 U/ml penicillin, and 100 mg/ml streptomycin. Cells were plated at a density of ~5 × 106 cells/cm2 in six-well culture plates coated with poly-D-lysine (for Western blot analysis) or at a density of ~1.0 × 105 cells/cm2 on coated chamber slides (for immunocytochemis-try). After induction of differentiation for 12 h by withdrawal of fetal bovine serum, the cells were treated either with different concentrations of OA or with 0.1 μM OA plus 20 μM PUGNAc (Toronto Research Chemicals, Inc., Toronto, Canada) or 10 mM STZ (Sigma-Aldrich Corp.) for 3 h. At the end of treatments, the cells were either lysed for Western blot analysis or fixed for immunocytochemical staining.
Preparation and treatments of rat brain slices
Metabolically competent rat brain slices (400 μm thick) were prepared from 2- to 3-month-old male CD rats (Charles River Lab, Wilmington, MA, USA), as described (22). The slices were incubated with artificial cerebrospinal fluid containing 0, 1.0, or 5.0 mM OA at 33°C for 3 h, followed by homogenization for Western blot analysis and fixation for immunohistochemistry, respectively, as described previously (22). All experiments involving animals were approved by our Institutional Animal Welfare Committee, and the “Principles of Laboratory Animal Care” (NIH publication # 86–23, revised 1985) and the U.S. Law on the Protection of Animals were followed.
Fasting of rats
CD rats (female, 6 months old, from Charles River Laboratories, Wilmington, MA, USA) were caged singly at room temperature (23°C) and 12-h light–dark cycle. Ten rats were randomly divided into two groups. For the fasting group, food was removed from the cages, but the rats were allowed free access to water. After 72 h, all rats were killed by using carbon dioxide gas, and the brains were removed and divided into two hemispheres. The cerebral cortex and the hippocampus of the left hemispheres were homogenized separately in 50 mM Tris-HCl (pH 7.6) containing 0.1 mM phenylmethyl sulfonyl fluoride, 1.0 mM sodium orthovanadate, 1.0 mM NaF, 100 mM GlcNAc, 0.1 μM PUGNAc, and 2.0 mg/ml each of aprotinin, leupeptin, and pepstatin. The homogenates were stored at –70°C for Western blot analysis. The right hemispheres were fixed with 10% buffered formalin and embedded in paraffin. Tissue sections (10 μm) were sliced for triple immunofluorescence staining.
RESULTS
O-GlcNAcylation and phosphorylation of NF-M occur reciprocally in cultured cells
To investigate whether O-GlcNAcylation and phosphorylation of NF-M affect each other, we treated SH-SY5Y cells with okadaic acid (OA), the most commonly used protein phosphatase inhibitor (22, 23), to elevate phosphorylation of NF-M. As expected, OA induced an increase in NF-M phosphorylation in a dose-dependent manner, as determined by monoclonal antibody SMI31 (Fig. 1a, b), which recognizes only the phosphorylated tail domain of NF-M and NF-H (7, 24, 25). We also observed a dose-dependent decrease in O-GlcNAcylation of NF-M, as determined by monoclonal antibody NL6, which recognizes NF-M only if the tail domain is modified by O-GlcNAc (ref. 6 and Supplemental Fig. 1). Immunocytochemical staining of these cells indicated that OA treatment markedly increased SMI31 staining and caused accumulation of the phosphorylated NF in the cells (Fig. 1c). In the untreated SH-SY5Y cells, O-GlcNAcylated NF-M stained by NL6 condensed mainly in the cytoplasmic compartment. After treatment with OA, the NL6 immunostaining was dramatically decreased (Fig. 1c). These results suggest that phosphorylation of NF-M induces a decrease of its O-GlcNAcylation in SH-SY5Y cells.
Figure 1.

OA induces increased phosphorylation and decreased O-GlcNAcylation of NF-M in SH-SY5Y neuroblastoma cells. a) Cells after treatment with the indicated concentrations of OA for 3 h were analyzed by Western blots developed with antibody SMI31 to phosphorylated NF-M/H or NL6 to O-GlcNAcylated NF-M. Blots developed with antibody R61d to total NFs and DM1A to α-tubulin were included as loading controls. Arrowheads indicate the 160-kDa NF-M band. b) The phosphorylation (SMI31 immunoreactivity) and O-GlcNAcylation (NL6 immunoreactivity) levels of blots shown in a were quantified and normalized with the total NF-M level (R61d immunoreactivity). Data are presented as mean ± SE. c) Immunocytochemical staining of SH-SY5Y cells treated with 0 or 0.1 μM OA for 3 h. Scale bar = 30 μm.
Next, we studied whether alteration of O-GlcNAcylation modulates phosphorylation of NF-M. When O-(2-acetamido-2-deoxy-D-glucopyranosylidene) amino-N-phenyl carbonate (PUGNAc) or streptozotocin (STZ), both of which are cell-permeable, selective inhibitors of β-N-acetylglucosaminidase (also called O-GlcNAcase) and thus can elevate cellular protein O-GlcNAcylation (26, 27), was included in the culture medium along with OA, we observed a marked decrease in OA-induced phosphorylation of NF-M as compared to the cells treated with OA alone (Fig. 2). The OA-induced decrease in O-GlcNAcylation of NF-M was also largely prevented by PUGNAc and STZ. Taken together, these results demonstrate that O-GlcNAcylation and phosphor-ylation of NF-M are regulated reciprocally in SH-SY5Y cells.
Figure 2.
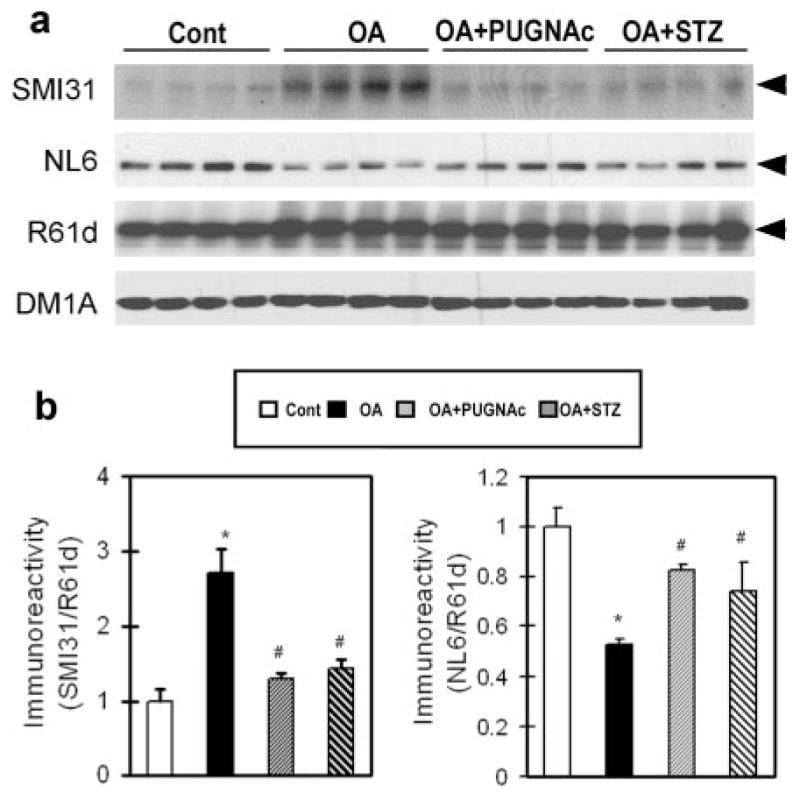
Effect of O-GlcNAcylation on OA-induced phos-phorylation of NF-M in SH-SY5Y cells. a) Cells were treated with either 0.1 μM OA alone or together with 20 μM PUGNAc or 10 μM STZ for 3 h, followed by Western blot analysis of the levels of phosphorylation and O-GlcNAcylation of NF-M with antibodies SMI31 and NL6, respectively. Blots developed with antibody R61d to total NFs and DM1A to α-tubulin were included as loading controls. Arrowheads indicate the 160 kDa NF-M band. Each lane was from an individual treatment. b) The phosphorylation and O-GlcNAcylation levels of NF-M of blots shown in a were densitometrically quantitated after being normalized with the total NF-M level of R61d blot. Data are presented as mean ± SE. *P < 0.05 vs. control group. #P < 0.05 vs. OA-treated group.
Phosphorylation of NF-M induces a decrease of its O-GlcNAcylation in metabolically active rat brain slices
We further investigated the regulation between phosphorylation and O-GlcNAcylation of NF-M in metabolically active rat brain slices that have been demonstrated to be an excellent in situ model to study protein phosphorylation of the mammalian brain (22, 28). When the brain slices were treated for 3 h with 1.0 or 5.0 μM OA, which inhibited ~70% protein phosphatase 2A activity (28), we observed increased phosphorylation of NF-M by both Western blots and immunohistochemical staining with antibody SMI31 (Fig. 3). When the samples were examined for O-GlcNAcylation of NF-M with antibody NL6, we found a marked decrease of the NL6 signal by both Western blots and immunohistochemical staining (Fig. 3). As seen in the cerebral cortex (Fig. 3b, c), the increased phosphorylation and decreased O-GlcNAcylation of NF-M were also seen immunohistochemically in the axons of the white matter (Supplemental Fig. 2) and the hippocampus (Supplemental Fig. 3) after OA treatment. These results suggest that phosphorylation of NF-M down-regulates its O-GlcNAcylation in the mammalian brain.
Figure 3.

OA induces increased phosphorylation and decreased O-GlcNAcylation of NF-M in rat brain slices. Metabolically active rat brain slices were incubated with artificial cerebrospinal fluid containing 0, 1.0, or 5.0 μM OA for 3 h, followed by analysis of phosphorylation (by SMI31), O-GlcNAcylation (by NL6), and the total level (by NF160 and R61d) of NF-M by Western blots (a) and by double immunofluorescence staining with SMI31/NF160 (b) and NL6/NF160 (c). In a, blot with DM1A against α-tubulin was included as a loading control, and the arrowheads indicate the NF-M band. b, c) Show the cerebral cortex of the rat brain slices. Scale bar = 20 μm.
Decreased glucose metabolism causes decreased O-GlcNAcylation and increased phosphorylation of NF-M
Impaired brain glucose uptake/metabolism is an early abnormality of AD (20), which could lead to decreased intraneuronal level of UDP-GlcNAc, a donor for protein O-GlcNAcylation. To investigate whether this impairment results in decreased O-GlcNAcylation and increased phosphorylation of NF-M in the brain, rats were fasted for 72 h to induce the decreased brain glucose uptake/metabolism. We found that fasting caused a 52% and 81% decrease in NF-M O-GlcNAcylation in cerebral cortex and hippocampus, respectively, as determined by Western blots probed with antibody NL6 (Fig. 4a, b). When the phosphorylation level of NF-M in the tissue was determined by using antibody SMI31, we found a marked increase in NF-M phosphorylation in the fasted rat brains, and the increase in the hippocampus appeared to be greater than in the cerebral cortex (Fig. 4a, b). These results indicate that the impaired brain glucose uptake/metabolism can cause hyperphosphorylation of NF-M via down-regulation of its O-GlcNAcylation in vivo and that the hippocampus is more vulnerable than the cerebral cortex to low brain glucose-induced damage.
Figure 4.

Fasting causes decreased O-GlcNAcylation and increased phosphorylation of NF-M. a) Homogenates of the cerebral cortex and the hippocampus from rats with or without fasting for 72 h were analyzed by Western blots developed with NL6 to O-GlcNAcylated NF-M, SMI31 to phosphorylated NF-M/H, or R61d to the total NF proteins. Each lane was from an individual rat brain (b). The blots, as shown in a, were quantitated densitometrically and normalized by the NF-M level of R61d blot. The data are mean ± SE. *P < 0.05 vs. control group. c) Confocal microscopy of triple immunofluorescence staining of the hippocampus CA1 sector of control and fasted rats. Green, NL6 to O-GlcNAcylated NF-M or SMI31 to phosphorylated NF-M/H. Red, NF160 to total NF-M. Blue, TO-PRO3 to nuclei. Scale bar = 50 μm.
We further studied the distribution of O-GlcNAcylated and phosphorylated NF-M in the brains of fasting rats. Because both NL6 and SMI31 are mouse monoclonal antibodies that made double/triple staining difficult, we used NF160, a rabbit polyclonal antibody recognizing NF-M in a phosphorylation/O-GlcNAcylation-independent manner, as a reference. We found that although the NF160 staining appears to be similar between control and fasted rats, the NL6 staining was decreased and the SMI31 staining increased throughout the brains of the fasted rats. These fasting-induced immunohistochemical changes were more obvious in the hippocampus (Fig. 4c and Supplemental Fig. 4) than in the cerebral cortex (Supplemental Fig. 5) and in the white matter (Supplemental Fig. 6). These results were consistent with the observations when using Western blots (Fig. 4a, b).
O-GlcNAcylation and phosphorylation of NF-M are dysregulated in AD brain
To investigate whether impaired brain uptake/metabolism might have contributed to hyperphosphorylation and accumulation of NF-M in AD, we determined the O-GlcNAcylation and phosphorylation level of the homogenates from AD and the age-matched control brains. We found that the O-GlcNAcylation level of NF-M (stained by NL6) was decreased by ~65% and the phosphoryaltion level of NF-M (stained by SMI31) was increased by ~50% in AD brains as compared to that in controls (Fig. 5a, b). Immunohistochemically, the NL6 staining was weaker and the SMI31 staining was stronger in both the cerebral cortex (Fig. 5c) and the white matter (Supplemental Fig. 7) of AD brain sections than the control brain sections. Accumulation of the phosphorylated NF-M was also seen in the neuronal cell bodies of AD brains, but not in controls (Fig. 5c, insert). These results suggest that down-regulation of O-Glc-NAcylation may contribute to hyperphosphorylation of NF-M in AD brain.
Figure 5.

Alterations of O-GlcNAcylation and phosphorylation of NF-M in AD brain. a) The O-GlcNAcylation and phosphorylation levels of NF-M in the medial temporal cortices of AD and age-matched control brains were analyzed by Western blots developed with antibodies NL6 and SMI31, respectively. Blots developed with R61d for total NF-M and DM1A for α-tubulin were also included as loading controls. Each lane was from an individual case. b) The blots shown in a were quantitated after being normalized by total NF-M of the R61d blot. Data are shown as mean ± SE. *P < 0.05 vs. control group. c) The paraffin-embedded cortex sections of AD and age-matched controls were stained with NL6, SMI31, and NF160. Scale bar = 25 μm.
DISCUSSION
Both NF-M and NF-H are highly phosphorylated proteins. Phosphorylation of the tail domain of NF subunits plays a critical role in regulating their translocation, filament formation, and function (1, 11). Most of the phosphorylation sites of NF-M/H are located at the KSP repeats of the tail domain (10, 16), which appear to contribute to NF spacing due to charge repulsion (29). It has been suggested that phosphorylation of NF-M/H at these sites is regulated by cyclin-dependent kinase 5 (30, 31), mitogen-activated protein kinases (32, 33), and protein phosphatase 2A (34, 35). The KSP repeats of NF are also highly modified by O-GlcNAcylation (5, 6, 9). In the present study, by using monoclonal antibody SMI31 against the phosphorylated epitope and NL6 against the O-GlcNAcylated epitope of the KSP repeats of NF-M, we have demonstrated that O-GlcNAcylation and phosphorylation of NF-M regulate each other in a reciprocal manner. This type of inverse regulation between O-GlcNAcylation and phosphorylation has also been seen in other proteins such as c-Myc and tau protein (18, 36–38). Thus, protein O-GlcNAcylation and phosphorylation might serve as a metabolic switch that regulates protein functions in response to intracellular glucose metabolism. Because NF-H has many more KSP repeats than NF-M and they are also highly modified by both O-GlcNAcylation and phosphorylation, a similar reciprocal regulation between O-GlcNAcylation and phosphorylation is likely to occur in NF-H also. Hence, our findings demonstrate a new way of regulation of NF phosphorylation.
Increased phosphorylation of NFs has been shown to increase their resistance to proteolytic degradation (39) and to result in their accumulation in neurons (14, 35, 40). Hyperphosphorylation and accumulation of NFs have been observed in AD brain (13–16). It is, therefore, possible that the abnormal accumulation of NFs in AD brain is due to their hyperphosphorylation, as is believed for abnormal tau accumulation being due to its hyperphosphorylation (17, 41). However, the mechanism leading to NF hyperphosphorylation in AD brain is hardly understood. Down-regulation of protein phosphatase 2A (42–44), which participates in dephosphorylating NFs at the tail domain (34, 35), might contribute partially to the hyperphosphorylation of NFs in AD brain. We have found now that decreased brain glucose uptake/metabolism in fasting rats induced down-regulation of NF-M O-GlcNAcylation and concurrent increased phosphorylation of NF-M. The decreased O-GlcNAcylation and increased phosphorylation of NF-M were also seen in AD brain neocortex, where the glucose uptake/metabolism is severely impaired (17, 45–47). On the basis of these observations, we postulate that the hyperphosphorylation and accumulation of NFs in AD brain could result from impaired glucose uptake/metabolism via down-regulation of NF O-GlcNAcylation. Consistent with our hypothesis, increased phosphorylation of NFs has been observed in the brains and spinal cords of diabetic rats (48) and in the brains of transgenic mice with brain insulin deficiency (49), both of which are characterized by deficient glucose uptake/metabolism.
In conclusion, we have demonstrated a novel regulation of NF phosphorylation by O-GlcNAcylation in cultured cells, in metabolically competent brain slices, and in the brain. We also found that impaired brain glucose uptake/metabolism can lead to decreased O-GlcNAcylation and increased phosphorylation of NFs, which is similar to the NF alterations in AD brain. These findings led us to postulate the involvement of impaired glucose uptake/metabolism via decreased O-GlcNAcylation as a cause of hyperphosphorylation and pathological accumulation of NFs in AD brain.
Supplementary Material
Acknowledgments
This work was supported in part by funds from the New York State Office of Mental Retardation and Developmental Disabilities; NIH grants AG027429 and AG019158; a U.S. Alzheimer’s Association grant (IIRG-05–13095); and a fellowship from the Li Foundation, Inc., New York, NY, USA. We thank Dr. George Merz for his help in confocal microscopy, Dr. Eulalia Badmaev for her help in brain tissue sectioning, Ms. Janet Murphy for secretarial assistance, and Ms. Maureen Marlow for editorial suggestions. We are also grateful to the Sun Health Research Institute Brain Donation Program of Sun City, Arizona, USA, for the provision of postmortem human brain tissue. The Brain Donation Program is partially supported by a National Institute on Aging grant (P30 AG19610, AZ Alzheimer’s Disease Core Center). R.B. is a member of the Scientific Advisory Board and owns stock in KineMed, Inc., Emeryville, CA, USA.
References
- 1.Liu Q, Xie F, Siedlak SL, Nunomura A, Honda K, Moreira PI, Zhua X, Smith MA, Perry G. Neurofilament proteins in neurodegenerative diseases. Cell Mol Life Sci. 2004;61:3057–3075. doi: 10.1007/s00018-004-4268-8. [DOI] [PubMed] [Google Scholar]
- 2.Elder GA, Friedrich VL, Jr, Bosco P, Kang C, Gourov A, Tu PH, Lee VM, Lazzarini RA. Absence of the mid-sized neurofilament subunit decreases axonal calibers, levels of light neurofilament (NF-L), and neurofilament content. J Cell Biol. 1998;1141:727–739. doi: 10.1083/jcb.141.3.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Elder GA, Friedrich VL, Jr, Pereira D, Tu PH, Zhang B, Lee VM, Lazzarini RA. Mice with disrupted midsized and heavy neurofilament genes lack axonal neurofilaments but have unaltered numbers of axonal microtubules. J Neurosci Res. 1999;57:23–32. doi: 10.1002/(SICI)1097-4547(19990701)57:1<23::AID-JNR3>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 4.Garcia ML, Lobsiger CS, Shah SB, Deerinck TJ, Crum J, Young D, Ward CM, Crawford TO, Gotow T, Uchiyama Y, et al. NF-M is an essential target for the myelin-directed “outside-in” signaling cascade that mediates radial axonal growth. J Cell Biol. 2003;163:1011–1020. doi: 10.1083/jcb.200308159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dong DL, Xu ZS, Chevrier MR, Cotter RJ, Cleveland DW, Hart GW. Glycosylation of mammalian neurofilaments. Localization of multiple O-linked N-acetylglu-cosamine moieties on neurofilament polypeptides L and M. J Biol Chem. 1993;268:16679–16687. [PubMed] [Google Scholar]
- 6.Ludemann N, Clement A, Hans VH, Leschik J, Behl C, Brandt R. O-glycosylation of the tail domain of neurofilament protein M in human neurons and in spinal cord tissue of a rat model of amyotrophic lateral sclerosis (ALS) J Biol Chem. 2005;280:31648–31658. doi: 10.1074/jbc.M504395200. [DOI] [PubMed] [Google Scholar]
- 7.Lee VM, Otvos L, Carden MJ, Hollosi M, Dietzschold B, Lazzarini RA. Identification of the major mul-tiphosphorylation site in mammalian Neurofilaments. Proc Natl Acad Sci U S A. 1988;85:1998–2002. doi: 10.1073/pnas.85.6.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu ZS, Liu WS, Willard MB. Identification of six phosphorylation sites in the COOH-terminal tail region of the rat neurofilament protein M. J Biol Chem. 1992;267:4467–4471. [PubMed] [Google Scholar]
- 9.Dong DL, Xu ZS, Hart GW, Cleveland DW. Cytoplasmic O-GlcNAc modification of the head domain and the KSP repeat motif of the neurofilament protein neurofilament-H. J Biol Chem. 1996;271:20845–20852. doi: 10.1074/jbc.271.34.20845. [DOI] [PubMed] [Google Scholar]
- 10.Trimpin S, Mixon AE, Stapels MD, Kim MY, Spencer PS, Deinzer ML. Identification of endogenous phosphorylation sites of bovine medium and low molecular weight neurofilament proteins by tandem mass spectrometry. Biochemistry. 2004;43:2091–2105. doi: 10.1021/bi030196q. [DOI] [PubMed] [Google Scholar]
- 11.Petzold A. Neurofilament phosphoforms: surrogate markers for axonal injury, degeneration, and loss. J Neurol Sci. 2005;233:183–198. doi: 10.1016/j.jns.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 12.Kamemura K, Hart GW. Dynamic interplay between O-glycosylation and O-phosphorylation of nucleocytoplasmic proteins: a new paradigm for metabolic control of signal transduction and transcription. Prog Nucleic Acids Res Mol Biol. 2003;73:107–136. doi: 10.1016/s0079-6603(03)01004-3. [DOI] [PubMed] [Google Scholar]
- 13.Sternberger NH, Sternberger LA, Ulrich J. Aberrant neurofilament phosphorylation in Alzheimer disease. Proc Proc Natl Acad Sci U S A. 1985;82:4274–4276. doi: 10.1073/pnas.82.12.4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang J, Tung YC, Wang Y, Li XT, Iqbal K, Grundke-Iqbal I. Hyperphosphorylation and accumulation of neurofilament proteins in Alzheimer disease brain and in okadaic acid-treated SY5Y cells. FEBS Lett. 2001;507:81–87. doi: 10.1016/s0014-5793(01)02944-1. [DOI] [PubMed] [Google Scholar]
- 15.Perry G, Rizzuto N, Autilio-Gambetti L, Gambetti P. Paired helical filaments from Alzheimer disease patients contain cytoskeletal components. Proc Natl Acad Sci U S A. 1985;82:3916–3920. doi: 10.1073/pnas.82.11.3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee VM, Otvos L, Jr, Schmidt ML, Trojanowski JQ. Alzheimer disease tangles share immunological similarities with multiphosphorylation repeats in the two large neurofilament proteins. Proc Natl Acad Sci U S A. 1988;85:7384–7388. doi: 10.1073/pnas.85.19.7384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gong CX, Liu F, Grundke-Iqbal I, Iqbal K. Impaired brain glucose metabolism leads to Alzheimer neurofibrillary degeneration through a decrease in tau O-GlcNAcylation. J Alzheimers Dis. 2006;9:1–12. doi: 10.3233/jad-2006-9101. [DOI] [PubMed] [Google Scholar]
- 18.Liu F, Iqbal K, Grundke-Iqbal I, Hart GW, Gong CX. O-GlcNAcylation regulates phosphorylation of tau: a mechanism involved in Alzheimer’s disease. Proc Natl Acad Sci U S A. 2004;101:10804–10809. doi: 10.1073/pnas.0400348101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li X, Lu F, Wang JZ, Gong CX. Concurrent alterations of O-GlcNAcylation and phosphorylation of tau in mouse brains during fasting. Eur J Neurosci. 2006;23:2078–2086. doi: 10.1111/j.1460-9568.2006.04735.x. [DOI] [PubMed] [Google Scholar]
- 20.Hoyer S. Brain glucose and energy metabolism abnormalities in sporadic Alzheimer disease. Causes and consequences: an update. Exp Gerontol. 2000;35:1363–1372. doi: 10.1016/s0531-5565(00)00156-x. [DOI] [PubMed] [Google Scholar]
- 21.Bensadoun A, Weinstein D. Assay of proteins in the presence of interfering materials. Anal Biochem. 1976;70:241–250. doi: 10.1016/s0003-2697(76)80064-4. [DOI] [PubMed] [Google Scholar]
- 22.Gong CX, Lidsky T, Wegiel J, Grundke-Iqbal I, Iqbal K. Metabolically active rat brain slices as a model to study the regulation of protein phosphorylation in mammalian brain. Brain Res Brain Res Protoc. 2001;6:134–140. doi: 10.1016/s1385-299x(00)00046-5. [DOI] [PubMed] [Google Scholar]
- 23.Fujiki H, Suganuma M. Tumor promotion by inhibitors of protein phosphatases 1 and 2A: the okadaic acid class of compounds. Adv Cancer Res. 1993;61:143–194. doi: 10.1016/s0065-230x(08)60958-6. [DOI] [PubMed] [Google Scholar]
- 24.Sternberger LA, Sternberger NH. Monoclonal antibodies distinguish phosphorylated and nonphosphorylated forms of neurofilaments in situ. Proc Natl Acad Sci U S A. 1983;80:6126–6130. doi: 10.1073/pnas.80.19.6126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lichtenberg-Kraag B, Mandelkow EM, Biernat J, Steiner B, Schroter C, Gustke N, Meyer HE, Mandelkow E. Phosphorylation-dependent epitopes of neurofilament antibodies on tau protein and relationship with Alzheimer tau. Proc Natl Acad Sci U S A. 1992;89:5384–5388. doi: 10.1073/pnas.89.12.5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haltiwanger RS, Grove K, Philipsberg GA. Modulation of O-linked N-acetylglucosamine levels on nuclear and cytoplasmic proteins in vivo using the peptide O-GlcNAc-beta-N-acetylglucosaminidase inhibitor O-(2-acetamido-2-deoxy-D-glucopyranosylidene)amino-N-phenylcarbamate. J Biol Chem. 1998;273:3611–3617. doi: 10.1074/jbc.273.6.3611. [DOI] [PubMed] [Google Scholar]
- 27.Roos MD, Xie W, Su K, Clark JA, Yang X, Chin E, Paterson AJ, Kudlow JE. Streptozotocin, an analog of N-acetylglucosamine, blocks the removal of O-GlcNAc from intracellular proteins. Proc Assoc Am Physicians. 1998;110:422–432. [PubMed] [Google Scholar]
- 28.Gong CX, Lidsky T, Wegiel J, Zuck L, Grundke-Iqbal I, Iqbal K. Phosphorylation of microtubule-associated protein tau is regulated by protein phosphatase 2A in mammalian brain. Implications for neurofibrillary degeneration in Alzheimer’s disease. J Biol Chem. 2000;275:5535–5344. doi: 10.1074/jbc.275.8.5535. [DOI] [PubMed] [Google Scholar]
- 29.de Waegh SM, Lee VM, Brady ST. Local modulation of neurofilament phosphorylation, axonal caliber, and slow axonal transport by myelinating Schwann cells. Cell. 1992;68:451–463. doi: 10.1016/0092-8674(92)90183-d. [DOI] [PubMed] [Google Scholar]
- 30.Li BS, Zhang L, Gu J, Amin ND, Pant HC. Integrin alpha(1) beta(1)-mediated activation of cyclin-dependent kinase 5 activity is involved in neurite outgrowth and human neurofilament protein H Lys-Ser-Pro tail domain phosphorylation. J Neurosci. 2000;20:6055–6062. doi: 10.1523/JNEUROSCI.20-16-06055.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kesavapany S, Li BS, Pant HC. Cyclin-dependent kinase 5 in neurofilament function and regulation. Neurosignals. 2003;12:252–264. doi: 10.1159/000074627. [DOI] [PubMed] [Google Scholar]
- 32.Veeranna Amin ND, Ahn NG, Jaffe H, Winters CA, Grant P, Pant HC. Mitogen-activated protein kinases (Erk1,2) phosphorylate Lys-Ser-Pro (KSP) repeats in neurofilament proteins NF-H and NF-M. J Neurosci. 1998;18:4008–4021. doi: 10.1523/JNEUROSCI.18-11-04008.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li BS, Daniels MP, Pant HC. Integrins stimulate phosphorylation of neurofilament NF-M subunit KSP repeats through activation of extracellular regulated-kinases (Erk1/Erk2) in cultured motoneurons and transfected NIH 3T3 cells. J Neurochem. 2001;76:703–710. doi: 10.1046/j.1471-4159.2001.00064.x. [DOI] [PubMed] [Google Scholar]
- 34.Veeranna Shetty KT, Link WT, Jaffe H, Wang J, Pant HC. Neuronal cyclin-dependent kinase-5 phosphorylation sites in neurofilament protein (NF-H) are dephosphorylated by protein phosphatase 2A. J Neurochem. 1995;64:2681–2690. doi: 10.1046/j.1471-4159.1995.64062681.x. [DOI] [PubMed] [Google Scholar]
- 35.Gong CX, Wang JZ, Iqbal K, Grundke-Iqbal I. Inhibition of protein phosphatase 2A induces phosphorylation and accumulation of neurofilaments in metabolically active rat brain slices. Neurosci Lett. 2003;340:107–110. doi: 10.1016/s0304-3940(03)00096-x. [DOI] [PubMed] [Google Scholar]
- 36.Kelly WG, Dahmus ME, Hart GW. RNA polymerase II is a glycoprotein. Modification of the COOH-terminal domain by O-GlcNAc. J Biol Chem. 1993;268:10416–10424. [PubMed] [Google Scholar]
- 37.Chou TY, Dang CV, Hart GW. Glycosylation of the c-Myc transactivation domain (O-linked N-acetylglu-cosamine/nuclear glycosylation/protein-protein interaction/ protooncogene) Proc Natl Acad Sci U S A. 1995;92:4417–4421. doi: 10.1073/pnas.92.10.4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Comer FI, Hart GW. O-Glycosylation of nuclear and cytosolic proteins: dynamic interplay between O-GlcNAc and O-phosphate. J Biol Chem. 2000;275:29179–29182. doi: 10.1074/jbc.R000010200. [DOI] [PubMed] [Google Scholar]
- 39.Goldstein ME, Sternberger NH, Sternberger LA. Phosphorylation protects neurofilaments against proteolysis. J Neuroimmunol. 1987;14:149–160. doi: 10.1016/0165-5728(87)90049-x. [DOI] [PubMed] [Google Scholar]
- 40.Sanchez I, Hassinger L, Sihag RK, Cleveland DW, Mohan P, Nixon RA. Local control of neurofilament accumulation during radial growth of myelinating axons in vivo. Selective role of site-specific phosphorylation. J Cell Biol. 2000;151:1013–1024. doi: 10.1083/jcb.151.5.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Iqbal K, Alonso Adel C, Chen S, Chohan MO, El-Akkad E, Gong C-X, Khatoon S, Li B, Liu F, Rahman A, et al. Tau pathology in Alzheimer disease and other tauopathies. Biochim Biophys Acta. 2005;1739:198–210. doi: 10.1016/j.bbadis.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 42.Gong CX, Singh TJ, Grundke-Iqbal I, Iqbal K. Phosphoprotein phosphatase activities in Alzheimer disease brain. J Neurochem. 1993;61:921–927. doi: 10.1111/j.1471-4159.1993.tb03603.x. [DOI] [PubMed] [Google Scholar]
- 43.Gong CX, Shaikh S, Wang JZ, Zaidi T, Grundke-Iqbal I, Iqbal K. Phosphatase activity toward abnormally phosphorylated tau: decrease in Alzheimer disease brain. J Neurochem. 1995;65:732–738. doi: 10.1046/j.1471-4159.1995.65020732.x. [DOI] [PubMed] [Google Scholar]
- 44.Liu F, Grundke-Iqbal I, Iqbal K, Gong CX. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur J Neurosci. 2005;22:1942–1950. doi: 10.1111/j.1460-9568.2005.04391.x. [DOI] [PubMed] [Google Scholar]
- 45.Hoyer S. Causes and consequences of disturbances of cerebral glucose metabolism in sporadic Alzheimer disease: therapeutic implications. Adv Exp Med Biol. 2004;541:135–152. doi: 10.1007/978-1-4419-8969-7_8. [DOI] [PubMed] [Google Scholar]
- 46.Reiman EM, Caselli RJ, Chen K, Alexander GE, Bandy D, Frost J. Declining brain activity in cognitively normal apolipoprotein E epsilon 4 heterozygotes: A foundation for using positron emission tomography to efficiently test treatments to prevent Alzheimer’s disease. Proc Natl Acad Sci U S A. 2001;98:3334–3339. doi: 10.1073/pnas.061509598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alexander GE, Chen K, Pietrini P, Rapoport SI, Reiman EM. Evaluation of cerebral metabolic decline in dementia: a potential outcome measure in Alzheimer’s disease treatment studies. Am J Psych. 2002;159:738–745. doi: 10.1176/appi.ajp.159.5.738. [DOI] [PubMed] [Google Scholar]
- 48.McLean WG, Pekiner C, Cullum NA, Casson IF. Posttranslational modifications of nerve cytoskeletal proteins in experimental diabetes. Mol Neurobiol. 1992;6:225–237. doi: 10.1007/BF02780555. [DOI] [PubMed] [Google Scholar]
- 49.Schechter R, Beju D, Miller KE. The effect of insulin deficiency on tau and neurofilament in the insulin knockout mouse. Biochem Biophys Res Commun. 2005;334:979–986. doi: 10.1016/j.bbrc.2005.07.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.