

Abstract
Heat shock proteins (hsps) are protective against the harmful effects of mutant expanded polyglutamine repeat proteins that occur in diseases such as Huntington’s, prompting the search for pharmacologic compounds that increase hsp expression in cells as potential treatments for this and related diseases. In this paper, we show that celastrol, a compound recently shown to up-regulate hsp gene expression, significantly decreases killing of cells expressing mutant polyglutamine protein. This effect requires the presence of the transcription factor responsible for mediating inducible hsp gene expression, HSF1, and is correlated with decreased amounts and increased sodium dodecyl sulfate (SDS) solubility of polyglutamine aggregates. These results suggest the potential of celastrol as a therapeutic agent in the treatment of human polyglutamine expansion diseases.
Electronic supplementary material
The online version of this article (doi:10.1007/s00109-007-0251-9) contains supplementary material, which is available to authorized users.
Keywords: Celastrol, HSF1, Polyglutamine, Aggregates, Heat shock proteins, hsp70
Introduction
A large number of neurodegenerative diseases, including Huntington’s and Kennedy’s diseases, are characterized by expanded polyglutamine repeats in certain proteins leading to their aggregation and toxic effects on the cells expressing them [1, 2]. A number of results suggest that polyglutamine-containing aggregates are important both for the initiation and progression of these diseases [3, 4]. Polyglutamine protein aggregates in brains of patients and transgenic animals have been found to contain various molecular chaperones, ubiquitin, and components of the 20S proteasome [5, 6]. This suggests that neuronal cells recognize the protein aggregates as abnormally folded and try to disaggregate or degrade the mutant proteins by recruiting chaperones and proteasomal components [7]. Consistent with this view, increased expression of hsp70 and other heat shock proteins (hsps) has been found to be effective in reducing the toxicity of mutant polyglutamine proteins, suggesting the potential of pharmacological up-regulation of hsp gene regulation as a means for treating polyglutamine expansion diseases such as Huntington’s [8–11].
Heat Shock Factor 1, HSF1, is the transcription factor responsible for up-regulating the expression of hsp70 and other hsp genes in response to cellular stress [12, 13]. HSF activation involves stress-induced conversion of this factor to its trimeric DNA-binding form [14–17]. The activated trimeric HSF1 then binds to heat shock elements in the promoters of hsp genes to up-regulate their transcription, ultimately leading to elevated levels of cytoprotective hsps in these cells.
The function of HSF1 as a key positive regulator of hsp expression, coupled to the observed ability of hsp proteins to protect cells from polyglutamine toxicity prompted the hypothesis that interventions that lead to activation of HSF1 could provide protection for cells against this toxicity and possibly represent agents that could be useful in treating polyglutamine expansion diseases such as Huntington’s [10, 18]. Consistent with this hypothesis, expression in cells of a constitutively active mutant HSF1 protein results in decreased polyglutamine toxicity and aggregate formation in both cell culture and animal models [19, 20]. These and other results have stimulated the search for pharmacological compounds capable of up-regulating hsp gene expression, as such compounds would theoretically have potential as treatments for these and other diseases of protein misfolding.
Recently, it was demonstrated that a compound called celastrol, which is found in the Celastraceae family of plants and is already being used to treat diseases such as rheumatoid arthritis, bacterial infection, and fever [21, 22], is able to activate HSF1 and up-regulate hsp gene expression [18]. Celastrol has previously been found to inhibit the aggregation of purified Q58-huntingtin 1-171 amino terminal fragment in an in vitro assay, although this study did not examine effects of celastrol on polyglutamine aggregation in cells [23]. Celastrol has also been found to inhibit the cytotoxicity of expanded polyglutamine form of huntingtin exon 1 [24] and androgen receptor [25], but these studies did not examine the mechanism by which celastrol protects cells from expanded polyglutamine protein cytotoxicity.
The results of the experiments presented in this paper demonstrate that celastrol is effective in preventing both the aggregation and toxicity of polyglutamine expression in cells and that it mediates these effects via the HSF1-mediated gene expression pathway. These results support the potential of this drug as a possible therapeutic agent for treating polyglutamine expansion diseases. The results also suggest that other drugs that stimulate HSF1 activity leading to hsp gene expression may also have beneficial activity against these disease states as well as other human diseases that are caused by protein misfolding.
Materials and methods
Plasmids, cell culture, and celastrol
The expression constructs encoding Q19-YFP, Q57-YFP, and Q81-YFP were kindly provided by Dr. James Burke (Duke University). The polyglutamine-YFP (Qn-YFP) vectors were constructed using CAG repeats that were synthesized by polymerase chain reaction (PCR) from human dentatorubral pallidoluysian atrophy (DRPLA) cDNAs containing different CAG repeats. HeLa cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Cellgro) supplemented with 10% fetal bovine serum (FBS) and 50 μg/ml gentamicin. PC12 cells were cultured in DMEM medium (Cellgro) supplemented with 5% FBS and 10% heat-inactivated horse serum (Gibco), and 100 units/ml Penicillin-Streptomycin (Gibco). HSF1+/+ and HSF1−/− mouse embryo fibroblast (MEF) cells (kindly provided by Dr. Ivor Benjamin, University of Utah) were cultured in DMEM medium (Cellgro) supplemented with 10% FBS, 1× Penicillin-streptomycin (Gibco), and 1× Non-essential amino acids (Gibco). Transfection was performed using Effectene transfection reagent (Qiagen), following the manufacturer’s protocol. Celastrol (Calbiochem) was dissolved in dimethyl sulphoxide at a stock concentration of 5.54 mM.
Trypan blue cell viability assay
Cells were collected by centrifugation at 1,000 rpm for 10 min at 4°C, and the pellet was washed twice with 1× phosphate-buffered saline (PBS). The cell pellet was then resuspended in 1× PBS to a concentration of approximately 106 cells/ml. A 1:1 dilution of the suspension was prepared using a solution containing 0.4% trypan blue stain (Gibco), and the suspension was then loaded into the counting chamber of a hemocytometer. The number of stained cells as well as the total number of cells was counted, and the percentage of stained cells was taken to represent the percentage of cell death. Experiments were repeated three times.
Fluorescence microscopy
Cells were seeded on coverslips, and 48 h after transfection, Hoechst 33342 and verapamil were added to the medium to final concentrations of 5 and 50 μg/ml, respectively. After incubation at 37°C for 30 min, the medium was removed, and the coverslips washed with 1× PBS for 5 min. A solution containing 3.7% paraformaldehyde in 1× PBS was added, and after 20 min incubation at room temperature, coverslips were washed with 1× PBS for 5 min. Coverslips were washed briefly three times in distilled water and mounted onto a slide spotted with 15 μl Vectashield (Vector Laboratories). Excess fluid was wicked from the coverslip and the edges of the coverslip sealed with fingernail polish. The fluorescence was then visualized using a Nikon fluorescent microscope and pictures taken with a Nikon Spotcam digital-imaging camera. To quantify the formation of polyglutamine aggregates, visual fields which contained similar numbers of cells (based on the density of nuclei stained by Hoechst) were chosen under 20× objective, and then the number of cells that contained aggregates in each field of vision was counted. Three different visual fields were quantified in each case. There were approximately 300 cells in each visual field for the experiments involving HeLa cells and PC12 cells, and approximately 100 cells in the experiments involving MEF cells. We scored a cell as positive if it had any visible aggregates, and the aggregates in the majority of cells appeared to be intranuclear or perinuclear (example shown in Fig. S1).
Extract preparation and Western blot assay
Cell lysis was performed on ice for 30 min in 50 mM Tris–HCl [pH 8.0], 100 mM NaCl, 5 mM MgCl2, 0.5% NP40 lysis buffer [26] containing 1× protease inhibitor cocktail (Roche), and 1 mM phenylmethanesulfonyl fluoride added. To examine hsp70 level, the cell lysate was cleared by centrifugation at 1,000 rpm at 4°C for 10 min, and the protein concentration of the supernatant was then determined by BioRad assay. Five micrograms of protein extract was subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blot following standard procedures. The antibodies and dilutions used to probe the Western blots were as follows. Anti-hsp70 (stress-induced form; Stressgen) was used at 1:10,000, anti-β-actin antibody (Sigma) was used at 1:10,000, and the anti-green fluorescent protein (GFP) polyclonal antibody (Bethyl labs) was used at a dilution of 1:4,000. Hsp70 Western blots were quantified using ImageQuant software.
Filtration assay
Cell lysis was performed on ice for 30 min in the lysis buffer described above. The insoluble fraction was obtained by centrifugation of the cell lysate at 14,000 rpm at 4°C for 10 min. The pellet was resuspended in DNase buffer (20 mM Tris–HCl [pH 8.0], 15 mM MgCl2, and 0.5 mg/ml DNase I), followed by incubation at 37°C for 1 h. After DNase treatment, the protein concentration was determined by analyzing an aliquot using the BioRad assay. The incubation was terminated by adjusting the mixtures to 20 mM ethylenediamine tetraacetic acid (EDTA), 2% sodium dodecyl sulfate (SDS), and 50 mM dithiothreitol and boiling for 5 min. Thirty micrograms of protein was diluted into 200 μl 2% SDS, and this was then filtered through a 0.2-μm pore size cellulose acetate membrane (Schleicher & Schuell) that was equilibrated with 2% SDS using a Dot-Blot system (Schleicher & Schuell). Filters were washed twice with 200 μl 0.1% SDS and then subjected to anti-GFP Western blot [27].
SDS solubility assay
To perform the SDS solubility assay, cell lysates were prepared as described above for the filtration assay, including the 14,000 rpm centrifugation of the cell lysate and DNase treatment and protein concentration determination by BioRad assay. Forty micrograms of protein was then adjusted to 2× SDS-PAGE sample buffer (62.5 mM Tris·HCl [pH 6.8], 10% glycerol, 2% SDS, 5% β-mercaptoethanol) and boiled for 5 min after which the sample was analyzed by SDS-PAGE and Western blot with anti-GFP polyclonal antibody.
Statistical analysis
Statistical significance was determined using the Student’s t test. A P value of <0.05 was considered to be statistically significant.
Results
Celastrol protects against polyglutamine toxicity
The experimental system we chose to test for effects of celastrol on polyglutamine aggregation and toxicity is transfection of a Q57-YFP fusion protein into cell lines (gift of Dr. James Burke), which is an established model for the aggregation of polyglutamine containing proteins in vivo [28]. Consistent with this, the Q57-YFP protein forms clearly visible aggregates upon expression in HeLa cells, while cells transfected with Q19-YFP exhibit a diffuse pattern (Fig. S1). A previous study showed that 8 h of celastrol treatment leads to increased levels of the hsp70 protein [18]. As the studies described in this paper include cell death and aggregation assays performed at 48 h after transfection with the Q57-YFP construct, we wanted to ensure that cells treated with celastrol treatment for longer times also exhibit elevated hsp70 expression. As shown in Fig. 1a (upper panel), celastrol treatment of HeLa cells for 24 or 48 h both result in a dose-dependent increase in expression of the hsp70 protein. Quantification of these hsp70 Western blot results indicates that the induction of hsp70 by each celastrol concentration is very similar at 24 and 48 h, showing that treatment with this drug results in a sustained increase in hsp70 levels (lower panel). Next, to test whether treatment with this drug confers protection from polyglutamine toxicity, HeLa cells were transfected with the Q57-YFP construct and then incubated in media containing different concentrations of celastrol. The results of this experiment, shown in Fig. 1b, reveal that celastrol treatment is associated with a significant decrease in death of these cells expressing this mutant polyglutamine protein.
Fig. 1.
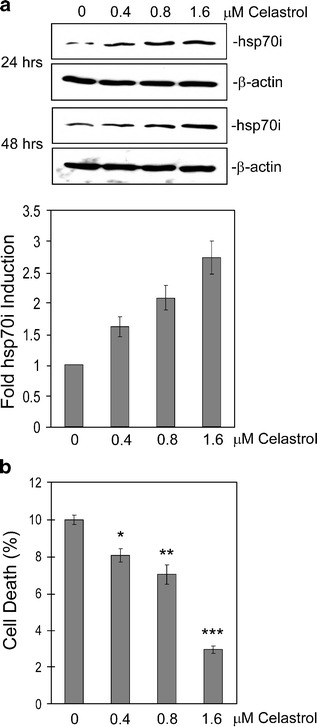
Celastrol treatment reduces Q57-YFP cytotoxicity. a Hsp70 protein level is increased by celastrol treatment. HeLa cells were treated with the indicated concentrations of celastrol for 24 or 48 h, after which cell extracts were made and subjected to Western blot using antibodies against hsp70 or β-actin (upper panel). These results were quantified using the ImageQuant program, and the values for the two treatment times, grouped by celastrol concentration, were graphed (lower panel). b HeLa cells were transfected with Q57-YFP along with celastrol treatment at the concentrations indicated. After 48 h, the amount of cell death was determined by trypan blue assay. Data are shown as means ± SE (*P < 0.007, **P < 0.004, ***P < 0.0001, for each celastrol concentration treatment vs no celastrol)
Celastrol protective effect are correlated with decreased number of cells containing polyglutamine aggregates
To determine whether the ability of celastrol to protect cells from polyQ toxicity could be mediated via effects on polyQ aggregates, we then examined whether celastrol treatment alters the number of polyQ aggregates in cells and/or their solubility. The results of these experiments show that treatment with celastrol is associated both with a decrease in the number of cells containing Q57-YFP aggregates, as quantified by fluorescence microscopy (Fig. 2a), and also with an increase in the amount of Q57-YFP that can be solubilized from aggregates by SDS treatment (Fig. 2b). SDS solubility of aggregates is related with their toxicity, and data from previous studies suggests that molecular chaperones may ameliorate the neurodegenerative effect of mutant polyglutamine protein, at least in part, by increasing the solubility of these proteins [9, 29]. Together, the results shown in Figs. 1 and 2 indicate that celastrol treatment decreases the death of cells expressing mutant polyglutamine protein and also decreases the number of cells containing Q57-YFP-aggregates and the insolubility of Q57-YFP-aggregates.
Fig. 2.
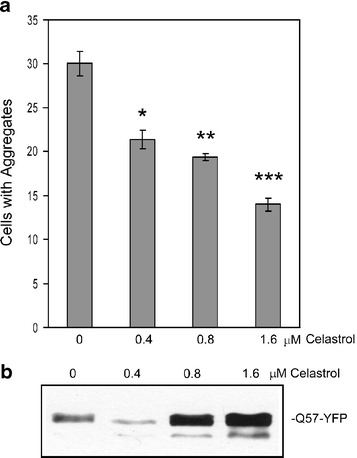
Celastrol treatment reduces number of cells containing Q57-YFP aggregates and increases Q57-YFP solubility. HeLa cells were transfected with Q57-YFP along with celastrol treatment at the concentrations indicated. a After 48 h of transfection, the formation of Q57-YFP aggregates was quantified using fluorescence microscopy. Visual fields which contained similar numbers of cells (based on the density of nuclei stained by Hoechst) were chosen under 20× objective, and then the number of aggregates in each field of vision was counted. Three different visual fields were quantified in each case, and data are shown as means ± SE (*P < 0.004, **P < 0.001, ***P < 0.0003, for each celastrol concentration treatment vs no celastrol). b To determine the amount of Q57-YFP monomer that could be solubilized from aggregates in lysates of the transfected cells by SDS treatment, the protein concentration of the insoluble fraction of the cell lysates was determined, and then 40 μg of protein was subjected to SDS solubilization treatment, followed by Western blot using anti-GFP antibody
Protective effects of celastrol treatment in PC12 cells
As the deleterious effects of expression of mutant polyglutamine proteins in vivo are observed primarily in cells of neuronal origin, we next sought to test the effect of celastrol treatment on polyglutamine toxicity and aggregation in PC12 cells. As shown in Fig. 3a, PC12 cells are very sensitive to killing by expression of the Q57-YFP protein, but as observed for HeLa cells in Fig. 1b above, celastrol treatment results in a significant decrease in death of these cells. As was observed in the experiments using HeLa cells, treatment of PC12 cells with celastrol resulted in a significant decrease in the number of polyglutamine aggregates in the cells (Fig. 3b) and an increase in the amount of Q57-YFP that can be solubilized from aggregates by SDS treatment (Fig. 3c). Interestingly, the concentrations of celastrol required to achieve these effects on polyglutamine toxicity and number and solubility properties of aggregates in PC12 cells were significantly lower than that needed for HeLa cells.
Fig. 3.
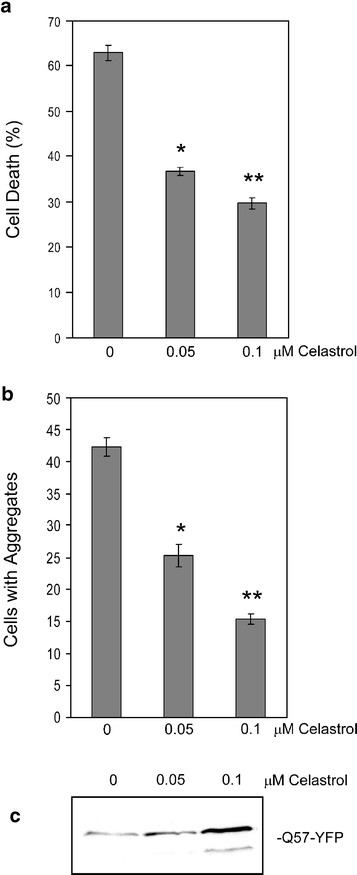
Celastrol effects on Q57-YFP toxicity and aggregates in PC12 cells. PC12 cells were transfected with Q57-YFP along with celastrol treatment at the concentrations indicated. After 48 h, the amount of cell death was determined by trypan blue assay (a), the number of cells containing Q57-YFP aggregates was quantified using fluorescence microscopy (b), and the amount of Q57-YFP monomer solubilized from aggregates by SDS treatment visualized by Western blot using anti-GFP antibody (c). In a and b, data are shown as means ± SE [*P < 0.0001 and **P < 0.0001 (a), *P < 0.001 and **P < 0.0001 (b), in each case for each celastrol concentration treatment vs no celastrol)
HSF1−/− cells exhibit increased polyglutamine aggregation and toxicity
Inducible hsp expression is thought to be important for the ability of cells to decrease polyglutamine toxicity and aggregation [8–11]. Consistent with this hypothesis, HSF1−/− MEFs [30] that are transfected with Q57-YFP exhibit a higher incidence of cell death than HSF1+/+ MEF cells (Fig. 4a). The HSF1−/− MEFs also show increased Q57-YFP aggregation, both as measured by numbers of aggregates using fluorescence microscopy of cells (Fig. 4b) and by amount of aggregated Q57-YFP protein present in cell extracts detected by filtration assay (Fig. 4c).
Fig. 4.
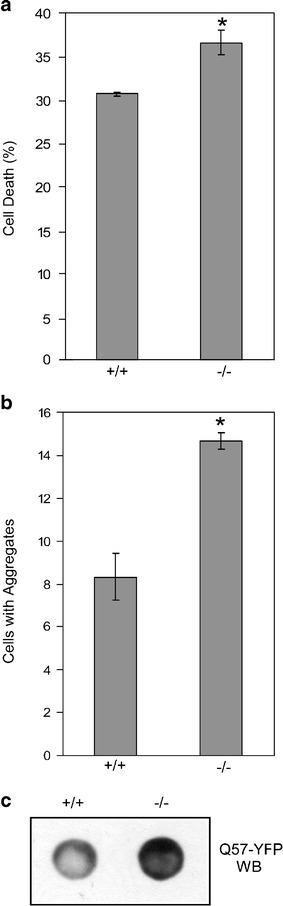
HSF1−/− cells exhibit higher Q57-YFP aggregation and cell death. HSF1−/− and wild-type MEF cells were transfected with Q57-YFP, and after 48 h, cell death was examined by trypan blue assay (a), and the number of cells containing Q57-YFP aggregates was quantified using fluorescence microscopy (b), or filtration assay, in which 30 μg of the insoluble fraction was filtered through 0.2 μm cellulose acetate membrane, and the aggregates retained on the membrane were immunoblotted using anti-GFP antibody (c). In a and b, data are shown as means ± SE. *P < 0.008 (a); *P < 0.003 (b)
Protective effects of celastrol require HSF1
Based on previous results showing the ability of celastrol to activate HSF1 leading to increased hsp gene expression [18], we hypothesized that this drug’s ability to decrease polyglutamine toxicity and aggregation shown by the results of Figs. 1, 2, and 3 above could be mediated via the HSF1-regulated gene expression pathway. To test this hypothesis, we compared the effect of celastrol treatment on polyglutamine toxicity and aggregation in the HSF1+/+ vs HSF1−/− MEF cells. Celastrol does not exhibit any toxicity in HeLa cells at 1.6 μM, but we found its optimal concentration in MEF cells to be lower than that of HeLa cells. Thus, for these experiments, we used 0.4 μM celastrol treatment, which has no toxic effect in either HSF1−/− or wild-type MEFs (data not shown). First, Western blot analysis shows that the HSF1−/− cells are unable to up-regulate hsp70 protein expression in response to celastrol treatment, demonstrating that celastrol-induced up-regulation of hsp70 expression is mediated by the HSF1-regulated gene expression pathway (Fig. 5a).
Fig. 5.
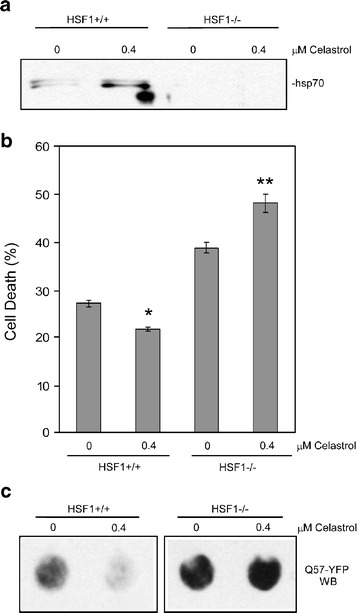
Celastrol decreases Q57-YFP toxicity and aggregation in wild-type but not HSF1−/− MEF cells. a Celastrol treatment does not induce hsp70 expression in HSF1−/− cells. Wild-type and HSF1−/− MEF cells were treated with no celastrol or 0.4 μM celastrol, and after 12 h, cell extracts were made, and 5 μg protein was loaded into each lane for immunoblotting with anti-hsp70 antibodies. b and c Wild-type and HSF1−/− MEF cells were transfected with Q57-YFP, with celastrol at the indicated concentrations added at the same time. After 48 h, cell death was examined by trypan blue assay (b), with data shown as means ± SE (*P < 0.002 for HSF2+/+ 0.4 μM celastrol vs no celastrol; *P < 0.002 for HSF2−/− 0.4 μM celastrol vs no celastrol), and amount of aggregated Q57-YFP was determined by filtration assay followed by anti-GFP Western blot (c)
The results shown above (Figs. 1, 2, 3, and 5a) indicate that celastrol treatment is effective in preventing polyglutamine aggregation and toxicity and that this drug up-regulates hsp70 expression by an HSF1-dependent mechanism. These results suggest that celastrol treatment protects cells by stimulating HSF1-dependent expression of hsps. However, celastrol could also modulate other cellular activities that could contribute to these protective effects. Thus, to test the contribution of HSF1-regulated pathways in these protective effects, we transfected HSF1+/+ and HSF1−/− MEF cells with Q57-YFP in combination with celastrol treatment and then measured the amount of cell death and levels of aggregated Q57-YFP. The results of this experiment show that, as it did for the HeLa and PC12 cells in the experiments of Figs. 1 and 3 above, celastrol treatment resulted in decreased death of HSF1+/+ MEF cells transfected with Q57-YFP, but treatment with this drug did not protect the HSF1−/− cells (Fig. 5b). Consistent with these results, the HSF1+/+ cells, but not the HSF1−/− cells, showed decreased levels of aggregated Q57-YFP upon celastrol treatment, as measured by the filtration assay (Fig. 5c). These results suggest that the HSF1 protein plays an important role in the protective effects of celastrol against polyglutamine toxicity and aggregation.
Discussion
The results described above indicate that the drug celastrol is able to decrease polyglutamine toxicity, supporting the proposal that this drug could potentially be useful in the treatment of Huntington’s disease and possibly other human polyglutamine expansion disorders. In support of this possibility, it has been found that celastrol treatment of mice results in elevation of hsp70 levels in neurons in the brains of these mice [31]. The results also show that lower concentrations of celastrol are required for its protective effects against polyglutamine toxicity in PC12 cells, cells with neuronal characteristics, compared to other cell types. A particularly beneficial aspect of this drug is that it is already being used to treat people with other disorders [21, 22], and thus it could likely be adapted more quickly for use in treating polyglutamine expansion diseases than other drugs not currently being used in humans.
Results presented in this paper indicate that the protective effect of celastrol against polyglutamine toxicity is associated with decreased numbers of cells containing aggregates as well as increased SDS-solubility of the mutant polyglutamine protein. This might seem to be at odds with findings that formation of polyglutamine aggregates can be protective for cells expressing these mutant proteins [32–34]. However, our results are consistent with the findings of a number of studies which indicate that molecular chaperones can reduce the formation of polyglutamine aggregates and increase the solubility of expanded polyglutamine proteins [8–11, 19, 20]. While one study found that hsp40 overexpression, but not hsp70 overexpression, was associated with reduced aggregation [35], the results of a number of studies do suggest that chaperones are able to reduce aggregation of mutant polyglutamine proteins. Molecular chaperones could be acting at multiple levels to reduce polyglutamine toxicity, but one possibility that has been proposed is that these chaperones, and by extension celastrol through its HSF1-mediated up-regulation of chaperones, may be acting at an early step to prevent formation of toxic intermediates before they can become part of larger aggregates [8–11, 19, 20]. This would explain both how celastrol treatment protects cells from polyglutamine toxicity and the effects we observe on polyglutamine aggregates in the celastrol-treated cells. An alternative explanation that must be considered, in keeping with previous studies indicating that aggregate formation can be protective for cells [32–34], is that the results we observe relating celastrol with the numbers of cells containing polyglutamine aggregates could be due, at least in part, to a potential ability of celastrol to enhance the survival of cells that do not contain aggregates, which would then lead to a perceived decrease in the numbers of cells containing aggregates in our experiments. Our data do indicate, however, the protective effects of celastrol with respect to polyglutamine toxicity.
In addition to its ability to its effects on HSF1 activity, celastrol has been found to affect other pathways in the cell, including the NF–κB pathway [18, 21, 22, 36]. However, our results suggest that the beneficial effects of celastrol treatment in decreasing polyglutamine toxicity are mediated through the action of the HSF1-regulated gene expression pathway, based on the finding that celastrol is not protective in Q57-YFP transfected HSF1−/− MEF cells (Fig. 5b). This finding also suggests that other drugs that activate HSF1 could also have potential as candidate therapeutic agents for treating polyglutamine expansion diseases.
Electronic supplementary material
Acknowledgements
We would like to thank Dr. James Burke for providing polyglutamine-YFP plasmid constructs, Dr. Ivor Benjamin for providing the HSF1+/+ and HSF1−/− MEF cells, and Dr. Doug Andres for providing PC12 cells. We also thank the other members of our laboratory for insightful discussions during the course of this work. This work was supported by NIH grants GM61053 and GM64606 to K.D.S.
Biographies
Yu-Qian Zhang
received her Bachelor degree in Molecular and Cellular Biology from the University of Science and Technology of China in 2003. She is currently a Ph.D. candidate in the Department of Molecular and Cellular Biochemistry at the University of Kentucky College of Medicine. Her research interests include the heat shock response in neurodegenerative diseases and cancer, protein sumoylation, and tumor immunology.

Kevin D. Sarge
received his Ph.D. in Biochemistry from North Carolina State University in Raleigh, NC, followed by postdoctoral training at Northwestern University in Evanston, IL. He is currently Professor of Molecular and Cellular Biochemistry at the University of Kentucky, Lexington, KY. His research interests include regulation of stress gene expression, gene bookmarking, and protein sumoylation.

References
- 1.Bates G. Huntingtin aggregation and toxicity in Huntington’s disease. Lancet. 2003;361:1642–1644. doi: 10.1016/S0140-6736(03)13304-1. [DOI] [PubMed] [Google Scholar]
- 2.Ross CA. Polyglutamine pathogenesis: emergence of unifying mechanisms for Huntington’s disease and related disorders. Neuron. 2002;35:819–822. doi: 10.1016/S0896-6273(02)00872-3. [DOI] [PubMed] [Google Scholar]
- 3.Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–548. doi: 10.1016/S0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- 4.Yamamoto A, Lucas JJ, Hen R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington’s disease. Cell. 2000;101:57–66. doi: 10.1016/S0092-8674(00)80623-6. [DOI] [PubMed] [Google Scholar]
- 5.Perez MK, Paulson HL, Pendse SJ, Saionz SJ, Bonini NM, Pittman RN. Recruitment and the role of nuclear localization in polyglutamine-mediated aggregation. J Cell Biol. 1998;143:1457–1470. doi: 10.1083/jcb.143.6.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cummings CJ, Mancini MA, Antalffy B, DeFranco DB, Orr HT, Zoghbi HY. Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat Genet. 1998;19:148–154. doi: 10.1038/502. [DOI] [PubMed] [Google Scholar]
- 7.Sittler A, Lurz R, Lueder G, Priller J, Lehrach H, Hayer-Hartl MK, Hartl FU, Wanker EE. Geldanamycin activates a heat shock response and inhibits huntingtin aggregation in a cell culture model of Huntington’s disease. Hum Mol Genet. 2001;10:1307–1315. doi: 10.1093/hmg/10.12.1307. [DOI] [PubMed] [Google Scholar]
- 8.Meriin AB, Sherman MY. Role of molecular chaperones in neurodegenerative disorders. Int J Hyperthermia. 2005;21:403–419. doi: 10.1080/02656730500041871. [DOI] [PubMed] [Google Scholar]
- 9.Muchowski PJ, Wacker JL. Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci. 2005;6:11–22. doi: 10.1038/nrn1587. [DOI] [PubMed] [Google Scholar]
- 10.Westerheide SD, Morimoto RI. Heat shock response modulators as therapeutic tools for diseases of protein conformation. J Biol Chem. 2005;280:33097–33100. doi: 10.1074/jbc.R500010200. [DOI] [PubMed] [Google Scholar]
- 11.Sakahira H, Breuer P, Hayer-Hartl MK, Hartl FU. Molecular chaperones as modulators of polyglutamine protein aggregation and toxicity. Proc Natl Acad Sci. 2002;99:16412–16418. doi: 10.1073/pnas.182426899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Voellmy R. On mechanisms that control heat shock transcription factor activity in metazoan cells. Cell Stress Chaperones. 2004;9:122–133. doi: 10.1379/CSC-14R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pirkkala L, Nykanen P, Sistonen L. Roles of the heat shock transcription factors in regulation of the heat shock response and beyond. FASEB J. 2001;15:1118–1131. doi: 10.1096/fj00-0294rev. [DOI] [PubMed] [Google Scholar]
- 14.Rabindran SK, Haroun RI, Clos J, Wisniewski J, Wu C. Regulation of heat shock factor trimer formation: role of a conserved leucine zipper. Science. 1993;259:230–234. doi: 10.1126/science.8421783. [DOI] [PubMed] [Google Scholar]
- 15.Sarge KD, Murphy SP, Morimoto RI. Activation of heat shock gene transcription by heat shock factor 1 involves oligomerization, acquisition of DNA-binding activity, and nuclear localization and can occur in the absence of stress. Mol Cell Biol. 1993;13:1392–1407. doi: 10.1128/mcb.13.3.1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baler R, Dahl G, Voellmy R. Activation of human heat shock genes is accompanied by oligomerization, modification, and rapid translocation of heat shock transcription factor HSF1. Mol Cell Biol. 1993;13:2486–2496. doi: 10.1128/mcb.13.4.2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Westwood JT, Wu C. Activation of Drosophila heat shock factor: conformational change associated with a monomer-to-trimer transition. Mol Cell Biol. 1993;13:3481–3486. doi: 10.1128/mcb.13.6.3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Westerheide SD, Bosman JD, Mbadugha BN, Kawahara TL, Matsumoto G, Kim S, Gu W, Devlin JP, Silverman RB, Morimoto RI. Celastrols as inducers of the heat shock response and cytoprotection. J Biol Chem. 2004;279:56010–56053. doi: 10.1074/jbc.M409267200. [DOI] [PubMed] [Google Scholar]
- 19.Rimoldi M, Servadio A, Zimarino V. Analysis of heat shock transcription factor for suppression of polyglutamine toxicity. Brain Res Bull. 2001;56:353–362. doi: 10.1016/S0361-9230(01)00602-5. [DOI] [PubMed] [Google Scholar]
- 20.Fujimoto M, Takaki E, Hayashi T, Kitaura Y, Tanaka Y, Inouye S, Nakai A. Active HSF1 significantly suppresses polyglutamine aggregate formation in cellular and mouse models. J Biol Chem. 2005;280:34908–34916. doi: 10.1074/jbc.M506288200. [DOI] [PubMed] [Google Scholar]
- 21.Li RL, Shu DF. Investigations and Clinical Applications of Trysterygium wilfordü hook F. Beijing, China: China Science and Technology Press; 1989. [Google Scholar]
- 22.Gunatilaka AAL, Herz W, Kirby GW, Moore RE, Steglich W, Tamm C. Triterpenoid quinonemethides and related compounds (Celastroids) Vienna: Springer; 1996. [Google Scholar]
- 23.Wang J, Gines S, MacDonald ME, Gusella JF. Reversal of a full-length mutant huntingtin neuronal cell phenotype by chemical inhibitors of polyglutamine-mediated aggregation. BMC Neurosci. 2005;6:1–12. doi: 10.1186/1471-2202-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aiken CT, Tobin AJ, Schweitzer ES. A cell-based screen for drugs to treat Huntington’s disease. Neurobiol Dis. 2004;16:546–555. doi: 10.1016/j.nbd.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 25.Piccioni F, Roman BR, Fischbeck KH, Taylor JP. A screen for drugs that protect against the cytotoxicity of polyglutamine-expanded androgen receptor. Hum Mol Genet. 2004;13:437–446. doi: 10.1093/hmg/ddh045. [DOI] [PubMed] [Google Scholar]
- 26.Sittler A, Walter S, Wedemeyer N, Hasenbank R, Scherzinger E, Eickhoff H, Bates GP, Lehrach H, Wanker EE. SH3GL3 associates with the Huntingtin exon 1 protein and promotes the formation of polygln-containing protein aggregates. Mol Cell. 1998;2:427–436. doi: 10.1016/S1097-2765(00)80142-2. [DOI] [PubMed] [Google Scholar]
- 27.Wanker EE, Scherzinger E, Heiser V, Sittler A, Eickhoff H, Lehrach H. Membrane filter assay for detection of amyloid-like polyglutamine-containing protein aggregates. Methods Enzymol. 1999;309:375–386. doi: 10.1016/S0076-6879(99)09026-6. [DOI] [PubMed] [Google Scholar]
- 28.Onodera O, Burke JR, Miller SE, Hester S, Tsuji S, Roses AD, Strittmatter WJ. Oligomerization of expanded-polyglutamine domain fluorescent fusion proteins in cultured mammalian cells. Biochem Biophys Res Commun. 1997;238:599–605. doi: 10.1006/bbrc.1997.7337. [DOI] [PubMed] [Google Scholar]
- 29.Chan HY, Warrick JM, Gray-Board GL, Paulson HL, Bonini NM. Mechanisms of chaperone suppression of polyglutamine disease: selectivity, synergy and modulation of protein solubility in Drosophila. Hum Mol Genet. 2000;9:2811–2820. doi: 10.1093/hmg/9.19.2811. [DOI] [PubMed] [Google Scholar]
- 30.McMillan DR, Xiao X, Shao L, Graves K, Benjamin IJ. Targeted disruption of heat shock transcription factor 1 abolishes thermotolerance and protection against heat-inducible apoptosis. J Biol Chem. 1998;273:7523–7528. doi: 10.1074/jbc.273.13.7523. [DOI] [PubMed] [Google Scholar]
- 31.Cleren C, Calingasan NY, Chen J, Beal MF. Celastrol protects against MPTP- and 3-nitropropionic acid-induced neurotoxicity. J Neurochem. 2005;94:995–1004. doi: 10.1111/j.1471-4159.2005.03253.x. [DOI] [PubMed] [Google Scholar]
- 32.Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–810. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- 33.Ryan AB, Zeitlin SO, Scrable H. Genetic interaction between expanded murine Hdh alleles and p53 reveal deleterious effects of p53 on Huntington’s disease pathogenesis. Neurobiol Dis. 2006;24:419–427. doi: 10.1016/j.nbd.2006.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bodner RA, Housman DE, Kazantsev AG. New directions for neurodegenerative disease therapy: using chemical compounds to boost the formation of mutant protein inclusions. Cell Cycle. 2006;5:1477–1480. doi: 10.4161/cc.5.14.2929. [DOI] [PubMed] [Google Scholar]
- 35.Zhou H, Li SH, Li XJ. Chaperone suppression of cellular toxicity of huntingtin is independent of polyglutamine aggregation. J Biol Chem. 2001;276:48417–48424. doi: 10.1074/jbc.M104140200. [DOI] [PubMed] [Google Scholar]
- 36.Lee JH, Koo TH, Yoon H, Jung HS, Jin HZ, Lee K, Hong YS, Lee JJ. Inhibition of NF-kappaB activation through targeting IkappaB kinase by celastrol, a quinone methide triterpenoid. Biochem Pharmacol. 2006;72:1311–1321. doi: 10.1016/j.bcp.2006.08.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.