

Abstract
Recent advances in nanotechnology have enabled the development of nanoscale sensors that outperform conventional biosensors. This review summarizes the nanoscale biosensors that use aptamers as molecular recognition elements. The advantages of aptamers over antibodies as sensors are highlighted. These advantages are especially apparent with electrical sensors such as electrochemical sensors or those using field-effect transistors.
Figure.
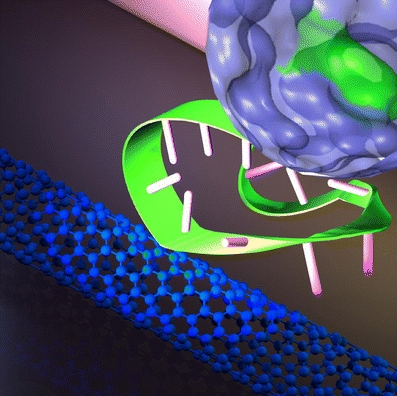
Feeling proteins with aptamer-functionalized carbon nanotubes
Keywords: Aptamers, Nanobiosensors, Electrochemical aptamer sensors, Field-effect transistor aptamer sensors
Introduction
With the completion of the Human Genome Project, we have a better understanding of disease-related biomaterials such as DNA, RNA, proteins and small biological molecules. Such knowledge can impact strongly on human health, because the identification of a disease-specific marker at an early stage of a disease can greatly improve the clinical success rate, and improve disease-related mortality. For early detection of disease, it is necessary to locate minute amounts of disease-related proteins, peptides or other small molecules in a patient’s bloodstream or body fluid. Nanotechnology combined with biotechnology is expected to provide paradigm-breaking solutions in this area, as these technologies are extremely sensitive [1]. Already, nanoscale materials such as quantum dots [2] and metallic nanoparticles [3, 4], as well as nanodevices such as the nanocantilever [5], and nano field-effect transistors (FETs) [6–9] show potential for detecting tiny molecular signals.
In this review, we concentrate on nanoscale electrical biosensors with aptamers as molecular recognition elements. Biosensors are devices that can be used to detect the presence of a target analyte. As shown in Fig. 1, biosensors are built up from a sample delivery component, a molecular recognition subsystem, a transducer changing the binding or reaction of target molecules into a measurable physical signal (mass, charge, heat or light) and a signal-processing unit. We are particularly interested here in electrical transducers, because they provide the most promising solutions for point-of-care disease detection. Biosensors using electrical transducers are rapid and convenient, and they do not require heavy optical instruments or expensive measuring devices.
Fig. 1.

A biosensor. MIP molecularly imprinted polymer
Aptamers as molecular recognition elements
For the highly sensitive detection of a target analyte, the selectivity of the recognition element is vital in sensor performance. Until recently, immunosensors using antibody–antigen recognition were employed for biodetection. However, highly sensitive and stable biosensors may use different recognition elements. In the 1980s, functional nucleic acids that can specifically bind and regulate viral or celluar proteins with high affinity were discovered [10]. The major breakthrough for functional nucleic acids occurred in 1990. First, the presence of small functional RNA molecules (TAR aptamers) that can inhibit viral replication [11] was revealed; they can effectively bind and inhibit the activity of viral protein, thereby raising the possibility that they may be used as therapeutic agents. In the second study in 1990, Tuerk and Gold [12] established the in vitro screening process termed "systematic evolution of ligand by exponential enrichment" (SELEX), to identify the RNA ligands with affinity for T4 DNA polymerase and various organic dyes [12]. Such nucleic acids were termed "aptamers" [from the Latin aptus (fit or appropriate)] by Ellington and Szostak [13].
The SELEX process is a technique for screening a very large library of oligonucleotides with random sequences by iterative cycles of selection and amplification. Figure 2 shows a schematic diagram of the SELEX process [14]. The starting pool must be large enough to generate a high probability of producing a desired aptamer (normally around 1015 different sequences). The initial library of random-sequence oligonucleotides, which is obtained through combinatorial chemical synthesis, is incubated with a target of interest. Oligonucleotides showing affinity for the target are partitioned by affinity chromatography or filtration, and then amplified by PCR (for DNA libraries) or reverse transcription PCR (for RNA libraries) to create a new pool enriched in those oligonucleotides having a higher affinity for the target. As this cycle of selection and amplification is repeated, the abundance of the high-affinity oligonucleotides increases exponentially. Negative selection and counterselection are often employed in order to remove aptamers which bind to supports and molecules similar to the target, respectively. Typically after eight to 15 cycles, cloning and sequencing of the enriched library are carried out, revealing the sequence of oligonucleotides highly specific to the target. Once the sequence information has been obtained, the desired aptamer can be readily produced by chemical synthesis. For a given target molecule, DNA aptamers and RNA aptamers can be generated. However, they are quite different in sequence and three-dimensional structure since DNA lacks the 2′ hydroxyl group of the RNA. For example, both the DNA version of the RNA aptamer for ATP and the RNA version of the DNA aptamer for ATP do not recognize ATP [15].
Fig. 2.

The systematic evolution of ligand by exponential enrichment (SELEX) process [14]
In Table 1, we summarize the advantages of aptamers over antibodies and the yellow-colored rows detail special advantages of aptamers as recognition elements [16]. First, aptamers possess high target affinity. It has been generally recognized that aptamer affinity is comparable to or even higher than that of antibodies, even though this is not always true. Unlike antibodies, aptamers can distinguish between targets even if the targets are in the same family. For example, anti-caffeine aptamers have lower affinity (by a factor of 104) for theophylline, despite the two compounds differing by only a single methyl group [17]. Because aptamers are selected using an in vitro selection process (SELEX), they are cost-effective, have uniform activity, and antitoxin aptamers can be readily synthesized. Antitoxin antibodies are more difficult to prepare, since they are cultured in animal cells. The immobilization of the recognition element can be crucial in biosensors, and this is easier with aptamers than with antibodies, because chemical modification of nucleic acids is simple and straightforward compared with that of antibodies. One of the biggest advantages of aptamers lies in their high stability. Being nucleic acids, aptamers are highly stable, and therefore have unlimited shelf lives. Also, because aptamers can undergo reversible changes of conformation with variations in temperature or salt concentration, aptamer-based sensors are potentially recyclable. The major limitation of aptamers (especially RNA aptamers) as molecular recognition elements is degradation by nucleases. However, it was shown that the problem can be overcome through chemical modifications of the sugar at the 2′-position [18] or use of mirror-image analogs [19]. Recently chemically modified nucleic acid aptamers for in vitro selections have been reviewed [20].
Table 1.
Advantages of aptamers over antibodies [16]

As aptamers show high target affinity and high target specificity, they can be used as therapeutic agents, in affinity columns and as recognition elements in sensors detecting a wide range of targets. There are virtually no limits of targets for aptamers; small molecules [14], proteins [15] and antibiotics [21]. The first use of an aptamer as a molecular recognition element in sensors was reported in 1996, when fluorescent-tagged aptamers were employed for optical detection of human neutrophil elastase [22]. Since then, various transducers employing aptamers for recognition have been reported. These include the quartz crystal microbalance [23], the surface plasmon resonance technique [24], evanescent-wave-induced fluorescence [25], microcantilevers [26], electrochemical sensors [27–35] and FETs fabricated with single-walled carbon nanotubes (SWNTs) [36, 37]. In this review, we summarize recent advances in electrochemical and FET types of aptamer sensors, focusing on the high sensitivity and recyclability of the aptamer-containing devices.
Electrical aptamer sensors
Electrochemical aptamer sensors
Typical electrochemical sensors operate by reacting with an analyte of interest and producing an electrical signal proportional to the analyte concentration. Many papers have reported the fabrication of electrochemical aptamer sensors where specific binding of an analyte to an aptamer on an electrode can produce an electrical signal [27–35]. Some authors use sandwich assays, employing a secondary aptamer for signal enhancement [28, 29]. However, a technique that is label-free and fast in response is favored especially for electrical types of biosensors. A difficulty with sandwich assays has recently been removed by Xiao et al. [31], who developed a “signal-on” architecture. Earlier electrochemical aptamer sensors used a “signal-off” architecture [30], in which the binding of the target analyte resulted in a decrease in the signal. In such cases, no more than 100% of the signal can be suppressed by target binding, and hence the dynamic range of the sensor is limited. Also, it is difficult to distinguish a real binding event from a false signal originating from contaminants or sensor degradation. To fabricate the signal-on electrochemical aptamer sensor, Xiao et al. introduced a short DNA sequence tagged with methylene blue (MB) that can hybridize with an aptamer. A single-stranded DNA aptamer, which binds to thrombin, was chosen as a model system. Thrombin is a proteolytic enzyme which facilitates blood clotting by converting fibrinogen into fibrin and is also regarded as a tumor marker in the diagnosis of pulmonary metastasis [38]. Figure 3 shows a schematic diagram of the signal-on aptamer sensor. As shown, the sensor is constructed by immobilizing a thiolated thrombin aptamer to a gold electrode, and an MB-tagged partially complementary DNA is added to form a double-stranded complex. In this configuration, owing to the large persistence length of double-stranded DNA, the MB moiety is far away from the sensor surface, and only a small Faradaic current is observed. The persistence length, defined as the distance over which the direction of a polymer segment persists, provides a measure of elasticity in polymer chains. In the case of double-stranded DNA, the persistence length is about 50 nm, or 150 bp [39]. Since the thrombin aptamer is much shorter than 150 bp [40] (the recognition part is only 15 bp in length), the complex would be in a “standing” position. When thrombin is introduced to such a complex, the thrombin aptamer binds to thrombin, and the MB-tagged single-stranded DNA approaches the electrode surface, improving the electron transfer with an increase of current. The persistence length of a single-stranded DNA is 1.5 nm in 2 M NaCl solution, and 3 nm in 25 mM NaCl solution [41].
Fig. 3.

Thrombin binding with an engineered aptamer. MB methylene blue. (Reprinted with permission from [31], copyright 2005 American Chemical Society)
In the work of Xiao et al., a threefold signal gain was observed with 260 nM thrombin, while only a 40% signal decrease was observed using a signal-off sensor. The detection limit of the sensor thus rose to 3 nM as shown in Fig. 4. Such a high sensitivity is comparable even with that given by sensors using amplification by labeling [28, 29] or optical sensors using evanescent-wave-induced fluorescence anisotropy [25]. Table 2 summarizes the electrochemical aptamer sensors available for thrombin.
Fig. 4.

Electrochemical signal from the signal-on aptamer sensor. (Reprinted with permission from [31], copyright 2005 American Chemical Society)
Table 2.
The detection limits of an electrochemical aptamer sensor for thrombin
| Detection limit | Characteristics | Reference | |
|---|---|---|---|
| 1 | 3 nM | Uses an MB-tagged "holding" DNA sequence | [31] |
| 2 | 80 nM/3.5 nM | Peroxidase-labeled thrombin gives an 80 nM detection limit. Biotin-labeled aptamer with horse radish peroxidase labeled streptavidin gives a detection limit of 3.5 nM | [27] |
| 3 | 1 μM | Sandwich assay using two aptamers with different binding sites. Secondary aptamer labeled with glucose dehydrogenase | [28] |
| 4 | 10 nM | Same as for 3, but pyrroquinoline quinine glucose dehydrogenase is used for labeling | [29] |
| 5 | 6.4 nM | MB-tagged thrombin aptamer (signal-off sensor) | [30] |
| 6 | 0.1–0.15 nM | Thrombin aptamer with ferrocene moiety (square-wave voltametry or chronopotetiometry used for the measurement) | [32] |
| 7 | 11 nM | MB-intercalated thrombin aptamer | [25] |
MB methylene blue
Owing to the unique three-dimensional structure of an aptamer, it is not even necessary to use "holding" DNA sequences. Baker et al. [34] reported the rapid, label-free detection of cocaine in both adulterated samples and biological fluid. The cocaine aptamer engineered by Stojanovic et al. [42] was used in this work. This aptamer is thought to form a partially unfolded structure, with only one of its three double-stranded stems intact, when target molecules are absent. In the presence of target, the aptamer folds into the cocaine-binding three-way junction structure of Fig. 5a. With this unique approach, Baker et al. reported the detection of 500 μM cocaine in biological fluid, even when contaminants were present. The ability to detect cocaine in undiluted biological fluids (blood, for example) is crucial for simple forensic detection technology. The binding of small molecules to an aptamer is highly reversible, so it is possible to regenerate the sensor. Baker et al. [34] could regenerate the sensor to a degree of 99% by simply washing a used sensor with buffer.
Fig. 5.

a Binding of coccaine with an MB-tagged aptamer. b Binding of platelet-derived growth factor (PDGF) with an MB-tagged aptamer. (a Reprinted with permission from [34], copyright 2006 American Chemical Society; b reprinted with permission from [35], copyright 2007 American Chemical Society)
Regeneration of an aptamer sensor is also possible after protein binding. Lai et al. [35] reported the detection of picomolar levels of platelet-derived growth factor (PDGF) directly in blood serum. PDGF is a protein that regulates cell growth and division, and is very important for blood vessel formation (angiogenesis). While generally undetectable in normal cells, PDGF is overexpressed in a variety of disease such as atherosclerosis, and human tumors including sarcomas and glioblastomas [14, 43]. As shown in Fig. 5b, specific binding of PDGF to the aptamer decreases the distance between MB and the electrode and consequently increases the electrical signal. The sensor could be also regenerated by 4-min incubation in 10% (w/v) sodium dodecyl sulfate.
In this section, we have shown that engineered aptamers, employed as recognition elements in electrochemical transducers, can greatly improve sensor performance. It is an advantage of aptamers over antibodies that aptamers can be engineered to either enhance or activate signals. Recently, aptazymes (aptamers with catalytic activities) have been reported, which combine an enzymatic role with a recognition function [44]. Carefully designed aptazymes can be developed as sensors that do not require labeling. An electronic (electrochemical) sensor approach with carefully designed aptamers offer potential for highly sensitive, selective and reusable sensor platforms.
Aptamer sensors with carbon nanotube field-effect transistor transducers
Since the first report on the biosensing ability of Si nanowire in 2001 [45] and the development of the chemical sensor operation of carbon nanotubes in 2000 [46], extensive research efforts have been initiated aimed at developing nanotube-based or nanowire-based sensors. Owing to the extremely large surface-to-volume ratio of one-dimensional nanostructures, it is possible to develop sensors with exquisite sensitivity. This is especially true in the case of carbon nanotubes, where all constituent atoms are at the surface. Any small disturbance of or adsorption to the surface could result in a large change in electrical conductance. Already, biosensors fabricated with semiconducting nanowires or nanotubes have been used as sensors that can detect a single virus [6], small molecules [47] or minute amounts of proteins in solution [7–9].
FET sensors measure a change in the surface potential occurring during the binding between an analyte and a recognition element. Figure 6 shows a schematic diagram of the working principle of carbon-nanotube-based FET sensors. FET sensors “feel” the charges when target molecules bind with probe molecules immobilized on the sensor surfaces. Such an effect is often referred to as an “electrostatic gating effect” since the bound target molecules behave as an additional gate electrode. In the case of carbon nanotubes, SWNTs behave as p-type semiconductors in ambient atmosphere. In p-type FETs, positive gate voltage decreases the conductance (depletion) and negative gate voltages increase the conductance (accumulation) from the device. Therefore, if positively charged molecules bind with probe molecules on SWNT-FETs, this event appears as a decrease of conductance, while binding of negatively charged molecules appears as an increase of conductance in SWNT-FETs as denoted in Fig. 6.
Fig. 6.

The working principle of field-effect transistor (FET) sensors based on carbon nanotubes
Using FETs for immunosensing was first proposed by Shenk [48] in 1978. It was suggested that FETs could be used to detect surface polarization owing to the formation of an antibody and an antigen. This concept failed, however, because small ions in the solution caused a screening effect inhibiting detection of the changes [49]. Figure 7 shows the charged semiconductor surface in solution. As shown in Fig. 7, when a charged surface (this might be a metal electrode, a semiconductor or a charged dielectric) is brought into contact with an ionic solution, the surface charge potential is dissipated by redistribution of counter ions in the solution. The domains where the variation of electric potential occurs are called electrical double layers, and the depth of an electrical double layer can be determined by calculating the Debye screening length. The Debye screening length is the scale over which mobile charge carriers screen the electric field. In an electrolyte, the Debye screening length can be defined as follows:
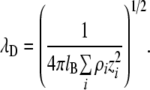 |
1 |
Fig. 7.

Electrical double layer at the sensor surface
Here the Bjerrum length lB is defined as
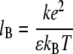 |
2 |
and is 0.7 nm at standard pressure and temperature. kB is the Boltzmann constant, k is defined as 1/4πɛ0, ρi is the number density of ions and the zi values are valencies of various ions.
The Debye screening length, therefore, is proportional to the inverse square root of the ion number density, i.e., to ionic strength. For pure water, the Debye screening length is about 1 μm, while it is only 0.3 nm in 1 M NaCl solutions. In 1 M NaCl solution, therefore, two ions or charged species separated by 1 nm do not interact with each other. If we consider the sensor surface, any interactions occurring outside the Debye screening length cannot be detected using FET sensors. The biggest disadvantage of FET sensors is that they may not work with blood samples or body fluids, because the ionic strength of such physiological solutions is about 150 mM, which yields a Debye screening length of about 1 nm.
Until recently, FET-type sensors have employed an antibody as the recognition receptor. However, the typical size of an antibody is about 10–15 nm [50, 51]. In that case, the binding of an antigen with an antibody occurs far outside the Debye screening length in physiological solutions. Molecular recognition elements with high specificity, selectivity and small sizes are essential for applications of FET-type sensors. In this respect, aptamers are highly suitable. Figure 8 shows the typical size ranges of antibodies and aptamers [52]. Because aptamers are mostly short nucleic acids ranging from 10 to 60 bp in length, the binding of target molecules with aptamers can occur inside the electrical double layer, even in 20–50 mM salt solutions.
Fig. 8.

Size comparison of an aptamer with an antibody. IgG immunoglobulin G. (Reprinted from [52], copyright 2006 Elsevier)
Such a biggest merit of aptamers in FET sensors was first reported by So et al. [36] using a carbon nanotube biosensor with thrombin aptamers as molecular recognition elements. They also demonstrated the sensitivity and selectivity of aptamers in SWNT-FETs. Anti-thrombin aptamers (5′-GGT TGG TGT GGT TGG-NH2-3′) were immobilized onto the surface of the SWNT-FET, fabricated using a patterned growth technique, with carbonyl diimidazole–Tween 20 as the linking molecules (Fig. 9a). The authors observed an abrupt decrease in conductance upon reaction with thrombin, while almost no changes were observed with elastase (Fig. 9b). Elastase is a member of the thrombin family and has almost the same molecular weight and isoelectric point as thrombin. Moreover, the sensor could be regenerated by simple washing with 6 M guanidine hydrochloride solution. The authors regenerated the sensor more than five times without significant loss of activity. As shown in Fig. 9c, the authors measured the sensitivities of SWNT-FET sensors with aptamers as recognition elements. Before each measurement, the sensor was "reset" by washing it with guanidine hydrochloride solution. The lowest detection limit was around 10 nM thrombin, and the sensor signal was saturated at about 100 nM thrombin.
Fig. 9.

a Single-walled carbon nanotube (SWNT) FET sensor with aptamers as molecular recognition elements. b Selectivity of an anti-thrombin aptamer-functionalized SWNT-FET. c Sensitivity of an anti-thrombin aptamer-functionalized SWNT-FET. The sensor was reset by washing it with 6 M guanidine hydrochloride solution before each measurement. CDI carbonyl diimidazole. (Reprinted with permission from [36], copyright 2005 American Chemical Society)
Maehashi et al. [37] have recently addressed the size issue of recognition elements in FET-based sensors. In their work, immunoglobulin E (IgE) aptamer (5′-NH2-GCG CGG GGC ACG TTT ATC CGT CCC TCC TAG TGG CGT GCC CCG CGC-3′) and monoclonal anti-IgE antibody (IgE-mAb) were immobilized on carbon nanotubes, with the aid of the linking molecule 1-pyrenebutanoic acid succinimidyl ester. Figure 10 a and b shows the responses of the aptamer-functionalized and antibody-functionalized sensors, respectively, when they were exposed to known concentrations of target IgE in 10 mM phosphate-buffered saline (PBS). As shown in Fig. 10, the aptamer-functionalized sensor exhibited small changes with only 250 pM target protein molecules, while a negligible change in conductance was observed in the antibody-funtionalized sensor even with 140 nM IgE. In 10 mM PBS, the Debye screening length would be roughly 3 nm, and the binding of IgE with the aptamer would be safely inside the electrical double layer. With IgE-mAb, however, there is little hope that binding can be detected in the buffer solution used for the experiment, because the antibody molecule is about 10 nm long [53].
Fig. 10.

The sensor signal measured from a an aptamer-functionalized SWNT-FET and b an antibody-functionalized SWNT-FET. IgE immunoglobulin E, PBS phosphate-buffred saline. (Reprinted with permission from [37], copyright 2007 American Chemical Society)
As discussed, aptamers may allow a wider range of analytes in FET sensors owing to their small size. SWNT-FET sensors with aptamers as recognition elements showed high sensitivity and selectivity, and could be readily regenerated. Highly sensitive carbon nanotube sensors combined with small, economic, highly selective and stable aptamers could provide cost-effective point-of-care testing devices.
Concluding remarks
This review has focused on the advantages of electrochemical and FET sensor types with aptamers as recognition elements. Through use of aptamers for recognition, no labeling is required for electrochemical sensors, and signal-on architecture that is only possible with aptamers has made possible the sensitivity improvements. Also, as pointed out already, sensors can be recycled because of the reversibility of aptamer configurations. Alternatively, bound proteins may be simply washed off, without damaging the aptamers. In immunosensing, by contrast, it is practically impossible to remove bound antigens from antibodies without damaging the antibodies, because both antibodies and antigens are proteins in nature. Aptamers have proved their superiority over antibodies in nano-FET sensors. When no signal can be measured with an antibody–antigen pair because of the large size of the antibody, the binding of small aptamers to various targets can occur inside the electrical double layer where the nanotube can "feel" the change.
Owing to the rapid aging of society, there is great demand for paradigm-breaking biosensors to detect the onset of disease with speed, convenience and accuracy. In a few more years, electrical nanosensors, with engineered aptamers, will become prominent in the market. They will be small in size, high in sensitivity and competitive in price.
References
- 1.National Cancer Institute (2004) Cancer nanotechnology plan. National Cancer Institute, Bethseda
- 2.Smith AM, Dave S, Nie S, True L, Gao X (2006) Expert Rev Mol Diagn 6:231–244 [DOI] [PubMed]
- 3.Nath N, Chilkoti A (2004) J Fluorescence 14:377–389 [DOI] [PubMed]
- 4.Han MS, Lytton-Jean AKR, Oh B-K, Heo J, Mirkin CA (2006) Angew Chem Int Ed 45:1807–1810 [DOI] [PubMed]
- 5.Shekawat G, Tark S-H, Dravid VP (2006) Science 311:1592–1595 [DOI] [PubMed]
- 6.Patolsky F, Zheng G, Hayden O, Lakadamyali M, Zhuang X, Lieber CM (2004) Proc Natl Acad Sci USA 101:14017–14022 [DOI] [PMC free article] [PubMed]
- 7.Zheng G, Patolsky F, Cui Y, Wang WU, Lieber CM (2005) Nat Biotechnol 23:1294–1301 [DOI] [PubMed]
- 8.Park D-W, Kim Y-H, Kim BS, So H-M, Won K, Lee J-O, Kong K, Chang H (2006) J Nanosci Nanotechnol 6:3499–3502 [DOI] [PubMed]
- 9.Besteman K, Lee J-O, Wiertz FGM, Heering HA, Dekker C (2003) Nano Lett 3:727–730 [DOI]
- 10.Cullen BR, Greene WC (1989) Cells 58:423–426 [DOI] [PubMed]
- 11.Marciniak RA, Garcia-Blanco MA, Sharp PA (1990) Proc Natl Acad Sci USA 87:3624–3628 [DOI] [PMC free article] [PubMed]
- 12.Tuerk C, Gold L (1990) Science 249:505–510 [DOI] [PubMed]
- 13.Ellington AD, Szostak JW (1990) Nature 346:818–822 [DOI] [PubMed]
- 14.Nimjee SM, Rusconi CP, Sullenger BA (2006) In Klussmann S (ed) The aptamer handbook. Wiley-VCH, Weinheim, pp 131–166
- 15.Fickert H, Fransson IG, Hahn U (2006) In Klussmann S (ed) The aptamer handbook. Wiley-VCH, Weinheim, pp 95–115
- 16.Nimjee SM, Rusconi CP, Sullenger BA (2005) Annu Rev Med 56:555–583 [DOI] [PubMed]
- 17.Zimmermann GR, Wick CL, Shields TP, Jenison RD, Pardi A (2000) RNA 6:659–667 [DOI] [PMC free article] [PubMed]
- 18.Pagratis NC, Bell C, Chang Y-F, Jennings S, Fitzwater T, Jellinek D, Dang C (1997) Nat Biotechnol 15:68–73 [DOI] [PubMed]
- 19.Nolte A, Kluβmann S, Bald R, Erdmann VA, Fürste JP (1996) Nat Biotechnol 14:1116–1119 [DOI] [PubMed]
- 20.Kusser W (2000) Rev Mol Biotechnol 74:27–38 [DOI] [PubMed]
- 21.Lorentz C, Schroeder R (2006) In Klussmann S (ed) The aptamer handbook. Wiley-VCH, Weinheim, pp 116–130
- 22.Davis KA, Abrams B, Lin Y, Jayasena SD (1996) Nucleic Acids Res 24:702–706 [DOI] [PMC free article] [PubMed]
- 23.Liss M, Petersen B, Wolf H, Prohaska E (2002) Anal Chem 74:4488–4495 [DOI] [PubMed]
- 24.Kawakami J, Imanaka H, Yokota Y, Sugimoto N (2000) J Inorg Biochem 82:197–206 [DOI] [PubMed]
- 25.Potyrailo RA, Conrad RC, Ellington AD, Hieftje GM (1998) Anal Chem 70:3419–3425 [DOI] [PubMed]
- 26.Savran CA, Knudsen SM, Ellington AD, Manalis SR (2004) Anal Chem 76:3194–3198 [DOI] [PubMed]
- 27.Mir M, Vreeke M, Katakis I (2006) Electrochem Commun 8:505–511 [DOI]
- 28.Ikebukuro K, Kiyohara C, Sode K (2004) Anal Lett 37:2901–2909 [DOI]
- 29.Ikebukuro K, Kiyohara C, Sode K (2005) Biosens Bioelectron 20:2168–2172 [DOI] [PubMed]
- 30.Xiao Y, Lubin AA, Heeger AJ, Plaxco KW (2005) Angew Chem Int Ed 117:5592–5595 [DOI] [PubMed]
- 31.Xiao Y, Piorek BD, Heeger AJ, Plaxco KW (2005) J Am Chem Soc 127:17990–17991 [DOI] [PubMed]
- 32.Sánchez JLA, Baldrich E, Radi AEG, Dondapati S, Sanchez PL, Katakis I, O’sullivan CK (2006) Electroanalysis 18:1957–1962 [DOI]
- 33.Bang GS, Cho S, Kim B-G (2005) Biosens Bioelectron 21:863–870 [DOI] [PubMed]
- 34.Baker BR, Lai RY, Wood MS, Doctor EH, Heeger AJ, Plaxco KW (2006) J Am Chem Soc 128:3138–3139 [DOI] [PubMed]
- 35.Lai RY, Plaxco KW, Heeger AJ (2007) Anal Chem 79:229–233 [DOI] [PubMed]
- 36.So H-M, Won K, Kim YH, Kim B-K, Ryu BH, Na PS, Kim H, Lee J-O (2005) J Am Chem Soc 127:11906–11907 [DOI] [PubMed]
- 37.Maehashi K, Katsura T, Kerman K, Takamura Y, Matsumoto K, Tamiya E (2007) Anal Chem 79:782–787 [DOI] [PubMed]
- 38.Nierodzik ML, Karpatkin S (2006) Cancer Cell 10:355–362 [DOI] [PubMed]
- 39.Smith SB, Finzi L, Bustamante C (1992) Science 258:1122–1126 [DOI] [PubMed]
- 40.Tasset DM, Kubik MF, Steiner W (1997) J Mol Biol 272:688–698 [DOI] [PubMed]
- 41.Murphy MC, Rasnik I, Cheng W, Lohman TM, Ha T (2004) Biophys J 86:2530–2537 [DOI] [PMC free article] [PubMed]
- 42.Stojanovic MN, de Prada P, Landry DW (2001) J Am Chem Soc 123:4928–4931 [DOI] [PubMed]
- 43.Cao Z, Suljak SW, Tan W (2005) Curr Proteom 2:31–40 [DOI]
- 44.Seetharaman S, Zivarts M, Sudarsan N, Breaker RR (2001) Nat Biotechnol 19:336–341 [DOI] [PubMed]
- 45.Cui Y, Wei Q, Park H, Lieber CM (2001) Science 293:1289–1292 [DOI] [PubMed]
- 46.Kong J, Franklin NR, Zhou C, Chapline MG, Peng S, Cho K, Dai H (2000) Science 287:622–625 [DOI] [PubMed]
- 47.Wang WU, Chen C, Lin K, Fang Y, Lieber CM (2005) Proc Natl Acad Sci USA 102:3208–3212 [DOI] [PMC free article] [PubMed]
- 48.Shenck JF (1978) In Chung PW (ed) Theory, design and biomedical applications of solid state chemical sensors. CRC, Boca Raton pp 165–173
- 49.Schasfoort RBM, Kooyman RPH, Bergveld P, Greve J (1990) Biosens Bioelectron 5:103–124 [DOI] [PubMed]
- 50.Rudikoff S, Potter M (1976) Proc Natl Acad Sci USA 73:2109–2112 [DOI] [PMC free article] [PubMed]
- 51.Teillaud JL, Desaymard C, Giusti AM, Haseltine B, Pollock RR, Yelton DE, Zack DJ, Scharff MD (1983) Science 222:721–726 [DOI] [PubMed]
- 52.Lee JF, Stovall GM, Ellington AD (2006) Curr Opin Chem Biol 10:282–289 [DOI] [PubMed]
- 53.Kane PM, Holowka D, Baird B (1988) J Cell Biol 107:969–980 [DOI] [PMC free article] [PubMed]