

Abstract
Expression of chemokine receptors by tumors, specifically CCR4 on cutaneous T cell lymphomas, is often associated with a poor disease outcome. To test the hypothesis that chemokine receptor-expressing tumors can be successfully controlled by delivering toxins through their chemokine receptors, we have generated fusion proteins designated chemotoxins: chemokines fused with toxic moieties that are nontoxic unless delivered into the cell cytosol. We demonstrate that chemokines fused with human RNase eosinophil-derived neurotoxin or with a truncated fragment of Pseudomonas exotoxin 38 are able to specifically kill tumors in vitro upon internalization through their respective chemokine receptors. Moreover, treatment with the thymus and activation-regulated chemokine (CCL17)-expressing chemotoxin efficiently eradicated CCR4-expressing cutaneous T cell lymphoma/leukemia established in NOD-SCID mice. Taken together, this work represents a novel concept that may allow control of growth and dissemination of tumors that use chemokine receptors to metastasize and circumvent immunosurveillance.
Chemokines are a group of small 8- to 15-kDa secreted and structurally related peptides that primarily regulate cell trafficking and diapedesis, although they exhibit a number of additional functions (1, 2, 3). To date, the group consists of ~50 chemokines classified into four superfamilies (two major types CC and CXC and two minor C and CX3C chemokines) on the basis of cysteine residues. They bind and signal through heterotrimeric Gi protein-coupled seven-transmembrane chemokine receptors that are differentially expressed on various subsets of immune cells (4–10). Chemokines and chemokine receptors appear to play a significant role in regulation of growth and metastatic spread of tumors, and their expression is often associated with a poor disease outcome. Since a first report that correlated breast cancer metastasis with expression of CXCR4, CCR7, and CCR10 (11), a number of other chemokine receptors were shown to be differentially expressed on tumors. For example, CXCR3 is found expressed on primary melanoma (12), breast cancer (13), and various lymphomas, such as T cell and NK cell lymphomas, chronic lymphocytic leukemia/small lymphocytic lymphoma, and splenic marginal zone B cell lymphoma (14–16). The metastatic migration of tumors to lymphoid organs was associated with their overexpression of CCR7 and CXCR4, whereas tumors homing into the skin were associated with expression of CCR4 (11, 17–19). Besides migration, chemokine/chemokine receptors also affect the viability and survival of tumor cells through the activation of their prosurvival and proliferation signaling cascades (20, 21). As a result, a prosurvival signal transmitted by the activated CCR1, CCR4, and CXCR4 leads to a greater risk of metastasis and poorer survival in patients with primary melanoma (11, 12) and colorectal cancer (22). Similarly, unfavorable outcome of the disease in patients with adult T cell leukemia/lymphoma (ATLL),3 mucosis fungoides, and Sézary syndrome was also associated with overexpression of CCR4 by malignant CD4+ T cells (1, 2, 19). In contrast, chemokines not only recruit tumors, but they can also induce infiltration of various immunosuppressive cells, such as T regulatory cells (Tregs), immunosuppressive inhibitory macrophages, and NK T cells, leading to escape from immunosurveillance and an unfavorable disease outcome. For example, CCR4+ Tregs were shown to be recruited to cutaneous lymphoma and ovarian cancer sites that expressed high levels of thymus and activation-regulated chemokine (TARC)/CCL17 or macrophage-derived chemokine/CCL22 (19, 23).
Immunotherapeutic interventions that block chemokine receptor signaling expressed by tumors remain an attractive, but insufficiently explored strategy. The CCR4-expressing tumors were successfully controlled in mice treated with Abs to CCR4, inducing NK-mediated Ab-dependent cellular cytotoxicity (1, 24). However, the efficacy of the approach can be affected by the host FcR genetic polymorphism, and its clinical potency in humans remains to be determined. It is tempting to speculate that the strategies that directly kill chemokine receptor-expressing tumors might elicit a higher degree of the disease control. To test this, we generated a formulation designated chemotoxin: chemokines fused with toxic moieties, such as RNases or toxins that are noncytotoxic unless delivered into the cell cytosol. The work has been inspired by our recent findings that chemokines can deliver exogenous Ags into cytosol to be processed and cross-presented to the MHC class I molecules (25). The process is very efficient because only nM amounts of chemokine-fused Ags are sufficient to induce cross-presentation capable of stimulating Ag-specific CTL. In concordance, as shown in this work, chemotoxins are able to specifically deliver toxins into cytosol of the targeted cells using their chemokine receptors. Our experiments using a mouse model for human cutaneous T cell leukemia/lymphoma demonstrate that the strategy is simple and potent, because treatment with TARC chemotoxin almost completely eradicated CCR4-expressing established T cell tumors, while leaving receptor-negative cells untouched. We think that the strategy may have significant clinical value for control of tumors that use chemokine receptors for metastatic spread or to circumvent immunosurveillance.
Materials and Methods
Cell lines and mice
Human acute T-lymphoblastic leukemia cell lines CCRF-CEM (CEM, CCL-119) and MOLT-4 (CRL-1582), and human embryonic kidney (HEK)-293 cells were purchased from American Type Culture Collection. HEK-293 cells stably transfected with human CCR8 (HEK/CCR8) were a gift from Z. Howard (Science Applications International, Frederick, MD). HEK-293 cells were cultured in DMEM (Invitrogen Life Technologies) containing 10% FBS. The same medium, but with 400 μg/ml G418 (Sigma-Aldrich), was used to maintain HEK/CCR8 cells. CEM and MOLT-4 cells were cultured in a standard RPMI 1640 medium (Invitrogen Life Technologies) supplemented with 10% FBS. Female 6- to 8-wk-old NOD/LtSz-scid/scid (NOD/SCID) mice were purchased from The Jackson Laboratory and kept under pathogen-free conditions at the National Institute on Aging animal facility. Animal care was provided in accordance with the procedures outlined in the “Guide for the Care and Use of Laboratory Animals” (National Institutes of Health Publication No. 86-23, 1985).
Plasmid constructs
Schema of constructs is shown in Fig. 1. Mature sequence for TARC/CCL17 was cloned using RT-PCR from human thymus RNA using the following pairs of primers: PRhTARCM-1 (ATCACCATG GCA CGA GGG ACC AAC GTG GGC CGG GAG T) and PRhTARC-GC-R3 (ATA CTC GAG GCT ACC CCC ACC GCC AGA GCC ACC ACC ACC AGA CCT CTC AAGGCTTTGCAGGTA); cutaneous T cell-attracting chemokine (CTACK)/CCL27 was cloned using primers PRhCCL27M-2 (TAT CATATGTTCCTACTGCCACCCAGCACTGCCT) and PRhCCL27 GS-R1 (TA CTC GAG GGA AGC TTC ACC GCC AGA GCCACCACC GCCCATTTTCCTTAG). Eosinophil-derived neurotoxin (EDN) was cloned using the following PCR primers: PREDN-1 (ATA CTC GAG GGT GGT GGT GGT TCT AAA CCG CCG CAG TTC ACT TGG GCT) and PREDN-R2 (CGC GGA TCC GAT GAT ACG GTC CAG ATG AAC CGG AAC) from the EDN-expressing plasmid described elsewhere (26). Bacterial expression vectors with CCR8 antagonist-encoding mature sequence of MC148 gene of Molluscum contagiosum virus (MC148) or mutant MC148 (MC148D, which could not bind CCR8 due to single amino acid residue replacement) have been described previously (27). A truncated (38-kDa) form of Pseudomonas exotoxin 38 (PE38) that does not have cell binding and internalization domain (28) was cloned from the pOND 21-2 plasmid (gift from I. Pastan, National Cancer Institute, Bethesda, MD) using following PCR primers: PRPE38-3 (ATAACCATG GAA GCT TCT GGA GGT CCC GAG GGC GGC AGC CTG GCC GCG CTGA) and PRPE38-R2 (TAT AGA TCT CTT CAG GTC CTC GCG CGG CGG TTT GCC GGG CTG GCT). To construct a chemotoxin-expressing vector, PCR fragments encoding for mature sequence of human TARC were cut with NcoI and XhoI enzymes and ligated in frame with XhoI- and BamHI-cut fragments encoding for EDN (TARC-EDN) and inserted into the bacterial expression plasmid pET11 opened with NcoI and BamHI (Novagen) that was modified to contain also c-myc and six His tags (29). To construct EDN fusions with wild-type MC148 or mutant MC148D, XhoI- and BamHI-cut EDN fragments were cloned into the XhoI- and BamHI-opened pMC148-VL315 and pMC148D-VL315 plasmids (27). To construct chemotoxins with PE38, PE38 fragment with HindIII (cut and blunted) and BglII ends was ligated into expression vectors for TARC, MC148, or CTACK/CCL27 opened with XhoI (blunted) and BamHI (TARC-PE38, MC148-PE38, and CTACK-PE38, respectively). The following control constructs were used: plasmids expressing MC148 or TARC fused with irrelevant tumor Ag such as oncofetal Ag (OFA)-immature laminin receptor protein (TARC-OFA) or plasmacytoma MOPC315-derived VL315 (MC148-VL315). A spacer fragment was inserted between chemoattractant and fusion moieties to enable proper folding of the protein. All constructs were verified by the DNA sequencing (Keck DNA Sequencing; Yale University).
FIGURE 1.
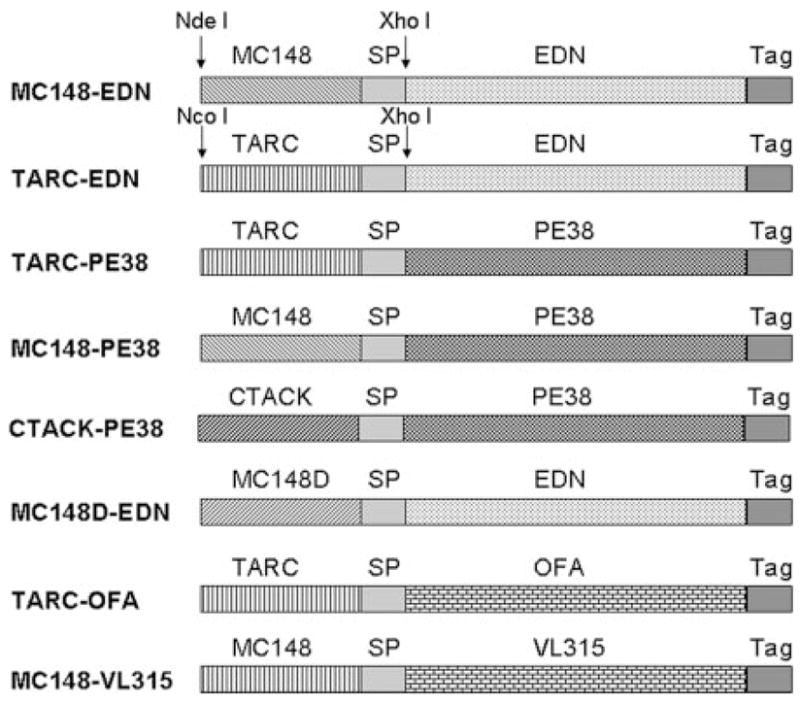
Schema of bacterial expression constructs used. Genes for the mature forms of viral chemokines MC148, TARC/CCL17, or CTACK/CCL27 were fused in frame with either EDN (MC148-EDN and TARC-EDN) or PE38 (TARC-PE38, MC148-PE38, and CTACK-PE38). Control constructs expressed chemokine fusions with the VL chain of mouse plasmacytoma MOPC315 (MC148-VL315) or with tumor Ag OFA (TARC-OFA), or mutant MC148 fused with EDN (MC148D-EDN). All constructs contained c-myc and His peptide tags to enable purification and detection (Tag), and a spacer peptide (SP) separating chemokines from Ags to enable proper protein folding.
Protein production and purification
Chemotoxin plasmids were transformed into BL21 (DE3) Escherichia coli cells (Stratagene). The cells were grown at 30°C in 2×YT medium supplemented with 100 μg/ml each ampicillin and carbenicillin and 1% dextrose. Chemotoxin production was induced for 6 h at 37°C with 1 mM isopropyl-β-D-thiogalactopyranoside in the same growth medium, except with 0.3% dextrose. Recombinant proteins were purified and refolded from bacterial inclusion bodies, as described previously (30). The integrity and purity of recombinant proteins were on average >90%, as tested by SDS-PAGE under reducing conditions and verified by Western blot hybridization with 9E10 anti-c-myc mAb (Sigma-Aldrich) or with rabbit polyclonal anti-EDN Ab (26).
RNase activity assay
RNase activity was determined at 37°C by monitoring the formation of perchloric acid-soluble nucleotides, as described (26). Each assay was repeated twice, and the data were pooled.
Chemotaxis assay
The migration of CEM and MOLT-4 cells was assessed using a 48-well microchemotaxis chamber (NeuroProbe) with a 5-μm polycarbonate filter (Osmonics), as described (31, 32). Briefly, the lower chambers were filled with 25 μl of complete RPMI 1640 medium containing titrated amounts of chemoattractants, or human TARC/CCL17 (R&D Systems), or binding medium alone (negative control). The top chambers were filled with 50 μl of cells (3 × 106); the two chambers were separated by a polycarbonate filter (5 μm pore size; Nucleopore) coated with 10 μg/ml fibronectin (Sigma-Aldrich). Cells were incubated at 37°C with 5% CO2 for 1.5 h. Nonmigrated cells were gently removed, and migrated cells were fixed and stained with H&E and counted under ×100 magnification in three randomly chosen fields. Results are expressed as number of cells per microscopic field and represent mean ± SD of three wells.
Chemokine receptor-binding internalization assays
Expression of CCR4 and CCR8 was tested using FITC-conjugated anti-human CCR4 and CCR8 Abs (R&D Systems), respectively, according to manufacturer instructions. The ligand-binding internalization assays were performed with 1 × 105 cells blocked with 10% mouse serum in PBS containing 2% BSA (PBS-B). Proteins (50 μg/ml) were incubated in cell culture medium for 1 h at 37°C or at 4°C, and, after extensive washings with PBS-B, incubated with either anti-c-myc mAb (1/100 dilution, 9E10) or anti-EDN polyclonal rabbit Ab (1/100 dilution). Then cells were incubated with respective secondary Abs conjugated with FITC, such as anti-mouse IgG FITC (Jackson ImmunoResearch Laboratories) and anti-rabbit IgG FITC (Sigma-Aldrich) Abs. The binding/internalization was assessed by flow cytometry on a FACScan (BD Biosciences) using CellQuest software. Alternatively, chemotoxin binding and internalization were assessed by fluorescent microscopy. Briefly, cells (1 × 105) were treated for 1 h at 37°C or at 4°C with 25 μg/ml fusion proteins in RPMI 1640 containing 10% FBS. Then cells were fixed with 3.7% formaldehyde, permeabilized with 0.2% Triton X-100 (5 min), and incubated with mouse anti-c-myc Ab for 1 h at 37°C, and for 30 min at 37°C with goat anti-mouse IgG Ab conjugated to Alexa Fluor 488 (Molecular Probes). Images were acquired with a ×40 objective on an Axiovert 200 microscope (Carl Zeiss Vision) and using Axiovison software (Carl Zeiss Vision).
Cell viability assays
Cell viability was assessed using cell proliferation reagent WST-1 (Roche Applied Science). A minimum of three independent assays was performed in triplicate for each cell type and protein. Cells (5 × 104 per well), plated in 96-well flat-bottom plates 1 day before the assay, were treated with titrated amounts of proteins in complete RPMI 1640 medium supplemented with 1% FBS (for HEK-293 cells or HEK/CCR8 cells) or 10% FBS (for CEM and MOLT-4 cells) and cultured for up to 7 days. Then, cell medium was replaced with 10% WST reagent, and cell viability was assessed after 2–4 h of incubation at 37°C. Results are expressed in percentage of OD450 values of PBS-treated cells. For in vitro cell outgrowth assay, 1 × 104 CEM cells were plated in 96-well plate and treated with either PBS or 10 μg/ml proteins, and number of viable cells was evaluated daily for 3 days using WST reagent. Results are expressed as percentage of OD450 values of PBS-treated cells. Cell apoptosis was tested by staining them with Annexin-V-Fluor Staining kit (Roche Applied Science), according to manufacturer instructions, and analyzed by flow cytometry.
CEM tumor growth in vivo and in vitro manipulations
Six- to 8-wk-old female NOD/SCID mice were challenged s.c. with 2 × 107 CEM cells. Twelve days later, the mice were checked for the presence of palpable tumors, and mice that did not develop tumors were excluded from the study. The remaining mice were randomized and intratumorally injected with either PBS, TARC-PE38, or TARC-OFA (25 μg/mice, each) daily for 5 days (n = 5 or 6 per treatment group). Tumor size was measured in perpendicular dimensions every other day, and tumor surface area was calculated. Mice were euthanized at day 27 or when tumor area reached 400 mm2. For histological analysis, tumor was fixed in 10% formalin and embedded in paraffin. Paraffin slides were stained with H&E and analyzed with light microscopy.
To study the sensitivity to TARC-PE38-induced cell death, tumor cells from PBS- and TARC-PE38-treated mice were isolated and cultured ex vivo for 7 days. Then, these tumor-derived cells were seeded in 96-well plates (5 × 104 cells/well) and treated for 2 days with 10 μg/ml TARC-PE38. Parental CEM cells (purchased from American Type Culture Collection) were used as control. Cell viability was assessed with WST reagent, as described. Data were expressed as percentage of OD450 values of corresponding control untreated cells. To study the effect of repeated treatments with TARC-PE38 on CCR4 expression and resistance to killing, CEM cells were treated once or five times (with 2-day intervals) with TARC-PE38 (10 μg/ml) and then were cultured for 4 wk to expand remaining cells. Control cells were treated with PBS or TARC-OFA in the same way. Cells were stained with anti-CCR4 Ab and analyzed by FACS. Some cells were plated in 96-well plates and were treated with TARC-PE38 (10 μg/ml), and the cell viability was assessed 2 days thereafter.
To study the effect of chemotoxins after systemic injections, NOD/SCID mice (n = 6) were injected with one million CEM cells via the tail vein. To determine the presence of CEM cells, mice were bled after 16 days of challenge with tumor cells, and the proportion of cells expressing human CD45-FITC Ab (R&D Systems) in peripheral blood was determined by FACS. At day 20 posttumor challenge, mice were randomized and i.v. injected with 10 μg of TARC-PE38 or CTACK-PE38 once daily for 4 consecutive days. At day 25, mice were culled and spleens were removed to assess presence of CEM cells. Spleen-derived mononuclear cells (5 × 106) were cultured ex vivo in RPMI 1640 supplemented with 10% FBS for 10 days. Cells were either stained with anti-human CD45 Ab and analyzed by FACS or were stained with trypan blue to evaluate the number of viable cells.
Statistical analysis
All data are expressed as means ± SD. Data were analyzed using a computer-based software system (StatView 5.0.1.; SAS Institute). Differences were tested by Student’s t test or ANOVA, followed by post hoc Scheffe’s F test.
Results
Proof of concept: Chemokines fused with toxic moieties bind their respective chemokine receptors and induce cell killing
We have recently reported that exogenous Ags, if linked (fused) with chemokines, can be efficiently delivered to the cell cytosol and presented to MHC class I molecules using the classical proteasome-mediated pathway (25). We hypothesized that chemokines might also deliver cytotoxic moieties to enable a preferential killing of the chemokine receptor-expressing cells. To test this, we have used toxins that do not bind and kill cells by themselves unless delivered to the cell cytosol. Chemokines were genetically fused with human pancreatic eosinophilic RNase, EDN (MC148-EDN and TARC-EDN), or a truncated form of PE38 (TARC-PE38, MC148-PE38, and CTACK-PE38; Fig. 1). The idea was that MC148 and TARC chemotoxins would preferentially kill CCR8- and CCR4-expressing cells, respectively. Control constructs expressed MC148 and TARC fusions with irrelevant non-toxic tumor Ags (MC148-Vl315 and TARC-OFA, respectively; Fig. 1) (27, 33). In addition, toxins were also fused with a mutant MC148 that lost the ability to bind CCR8 (MC148D-EDN; Fig. 1) (27). Chemotoxins retained the expected functional properties of the fused moieties. Although no RNase activity was exhibited by controls (MC148-VL315; Fig. 2A), MC148-EDN and TARC-EDN (Fig. 2A) exhibited significant RNase activity in vitro, although several fold less than free EDN. Moreover, it appears that the toxin moiety does not affect the ability of the chemokines to chemotax, because both TARC-EDN and TARC-PE38 specifically attracted CCR4-expressing CEM cells with comparable potency as TARC/CCL17 alone at equal molar ratios (Fig. 2B). Of note: the fusions have ~6 times larger size than free chemokine, and a lesser chemotaxis may be due to the fact that our samples may also contain some amounts of inactive and improperly folded fusions (our unpublished observation). Furthermore, MC148 and TARC chemotoxins could specifically bind CCR8- or CCR4-expressing HEK/CCR8 and CEM cells, respectively (Fig. 2, C–F). Control cells, HEK-293 (Fig. 2C) and MOLT-4 (data not shown), that did not express CCR8 and CCR4 failed to bind chemotoxins. Importantly, chemotoxins were efficiently internalized upon binding with their receptors. Although chemotoxins were mostly detected on the cell surface when incubated at the conditions that sequester receptor internalization (4°C; Fig. 2, D and F), they were internalized within a few minutes of incubation at the permissive temperature (37°C, TARC-EDN; Fig. 2D) and found in the cell cytosol (37°C, TARC-PE38; Fig. 2F). The internalization was through the respective chemokine receptor, because CCR4 was also down-regulated upon incubation with TARC-EDN at 37°C, but not at 4°C (Fig. 2E). Controls, unlinked free toxins such as EDN (Fig. 2E) or toxins fused with mutant chemokine (data not shown), did not bind and affect expression of CCR4.
FIGURE 2.
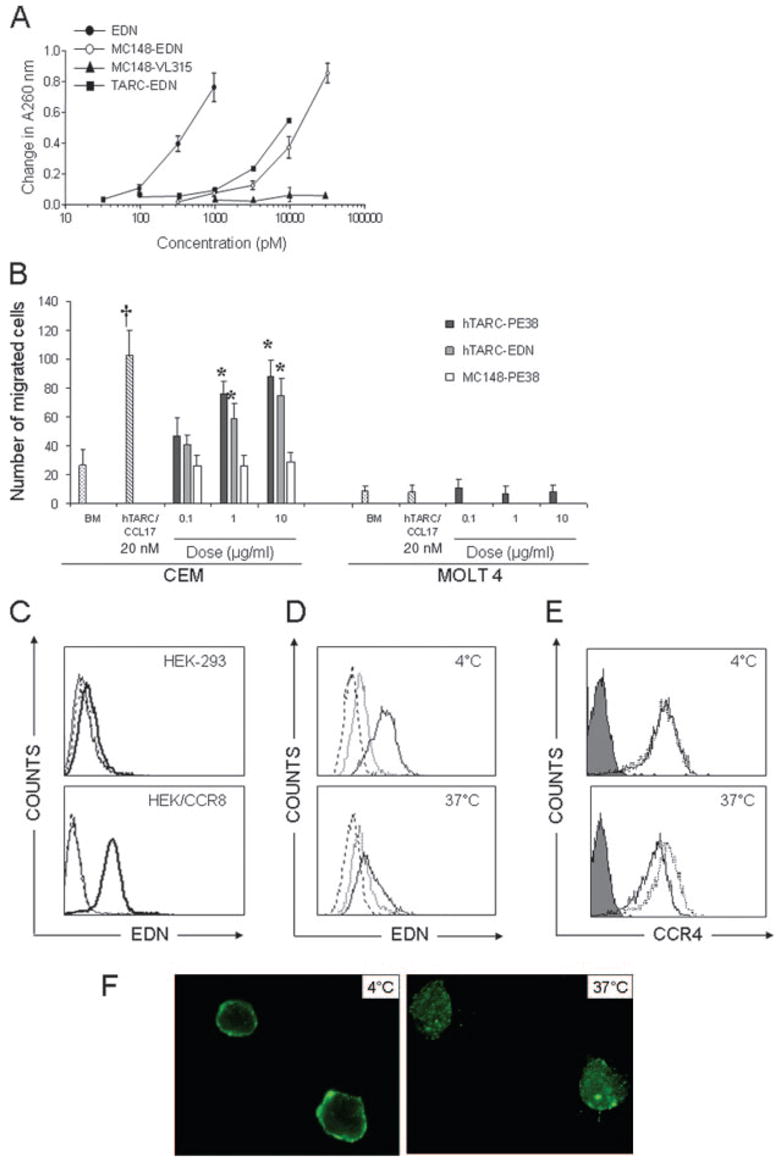
Chemokine fusions retain properties of moieties that were fused. A, MC148-EDN and TARC-EDN, but not MC148-VL315, retain RNase activity. Data are mean ± SD of representative two independent experiments. B, TARC-EDN and TARC-PE38 chemoattract CCR4-positive CEM, but not CCR4-negative MOLT-4 cells. Chemotaxis was performed, as described in Materials and Methods. Results are expressed as a number of cells per microscopic field and represent mean ± SD of migrated cells of three independent wells. *, p < 0.01 is for comparisons with MC148-PE38; †, p < 0.01 is for comparisons between hTARC/CCL17 (20 nM) and TARC-PE38 or TARC-EDN at 1 μg/ml (20 nM) dose. C, MC148-EDN binds to CCR8-positive HEK-293/CCR8, but not to CCR8-negative parental HEK-293 cells. Cells were incubated for 30 min at 4°C in the presence of either EDN (dashed line), or TARC-EDN (thick solid line), or saline (PBS, thin solid line). D, CEM cells internalize TARC-EDN (solid line). Controls for the binding were EDN (dotted line) and PBS (dashed line). Cells were incubated with 50 μg/ml proteins for 30 min at either 4°C to detect binding or 37°C to detect internalization. TARC-EDN and EDN were detected using rabbit polyclonal anti-EDN Ab and FITC-conjugated secondary Ab. E, The TARC-EDN internalization is accompanied by CCR4 down-regulation (solid line, 37°C). Incubations with EDN (dotted line) did not affect CCR4 expression. Expression of CCR4 was detected with FITC-conjugated anti-human CCR4 Ab; FITC-conjugated isotype-matched IgG (filled line) was used as control. F, TARC-PE38 is sequestered on the surface of CEM cells at 4°C, but is internalized at 37°C. Cells were incubated with 25 μg/ml TARC-PE38 for 1 h at either 4 or 37°C, followed by the staining with anti-c-myc Ab (9E10) and FITC-conjugated secondary Ab. Cells were visualized using fluorescence microscopy. All figures show representative data from at least three independent experiments with similar results.
Taken together, these data indicate that chemotoxins were efficiently delivered into the cytosol using their respective chemokine receptors. Next, we tested whether this would render them cytotoxic. HEK/CCR8 cells were incubated with titrated amounts of MC148-EDN, or control EDN alone and MC148-VL315. As shown in Fig. 3A, HEK/CCR8 cells were only killed when incubated with MC148-EDN, but not with EDN alone or MC148-VL315. The cytotoxicity was specific to the CCR8-expressing cells, because MC148-EDN failed to kill the CCR8-negative parental HEK-293 cells (data not shown). However, despite the fact that significant cell death was induced by the treatment with a relatively low amount of EDN chemotoxin (starting from 3 μg/ml/100 nM; Fig. 3A), it was only detectable after a prolonged incubation up to 7 days.
FIGURE 3.
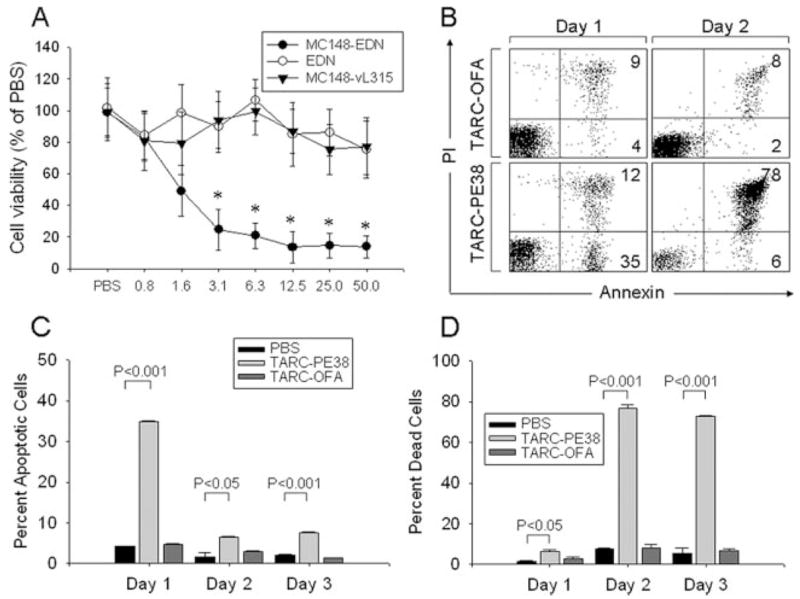
EDN and PE38 fused with chemokines induce cell death. A, MC148-EDN induces death of CCR8-expressing HEK-293/CCR8 cells. Control treatments were EDN alone and MC148-VL315. Cell viability (mean ± SD) was measured 7 days after incubation with titrated amounts of proteins (μg/ml, x-axis) by WST assay. Results are presented in percentage from values of PBS-treated cells. *, p < 0.05 is for comparisons with the PBS group. B–D, TARC-PE38 induces apoptosis and death of CEM cells. B, Representative dot plots of annexin- and propidium iodide-stained CEM cells treated with 10 μg/ml TARC-PE38 or TARC-OFA for 1 and 2 days. Numbers represent percentage of positive cells in the corresponding quadrants. Proportions of apoptotic cells (annexin positive, C) and dead cells (double positive for annexin/propidium iodide, D) after incubation with 10 μg/ml TARC-PE38 or TARC-OFA for up to 3 days are shown. Data are mean ± SD of triplicates.
Bacterial toxin-containing chemotoxins specifically and efficiently induce cell death in vitro
To search for chemotoxins with a faster rate of activity, we tested chemokines fused with the exotoxin fragment PE38 from Pseudomonas aeruginosa that was reported to kill mammalian cells within a short period of time by inhibiting protein synthesis via the ADP ribosylation of elongation initiation factor 2 (34, 35). Indeed, significant apoptosis was elicited in cells treated overnight with TARC-PE38 (Fig. 3, B and C), leading to mostly dead cells (>80%) within 2 days of incubation (Fig. 3, B and D). Similar to EDN chemotoxins, TARC-PE38 also acted through CCR4, because it only killed the CCR4-expressing CEM, but not CCR4-negative MOLT-4 cells (Fig. 4A). Controls, such as TARC-OFA and TGFα-PE38 (Fig. 4B), or CTACK-PE38 that acts through CCR10 (data not shown), failed to induce cell death, further supporting that the process was specific and required CCR4. TARC-PE38 was a very potent chemotoxin, because a single treatment with as low as 2–8 nM TARC-PE38 (0.1–0.4 μg/ml; Fig. 4A) was sufficient to kill CEM cells. In fact, a single treatment with TARC-PE38 significantly suppressed expansion of fast-growing CEM tumor cells in vitro, leaving only a few viable cells (Fig. 4C). Of note: the remaining cells are presumably derived from a small proportion (3%) of CCR4-negative CEM cells that was present before the treatment. The CCR4-negative cells are no longer susceptible to TARC-PE38 and eventually grow back, as reflected by a slight, but significant increase in OD450 values by day 3 treatment (47 ± 4) when compared with day 2 (20 ± 4, p < 0.01; Fig. 4C).
FIGURE 4.
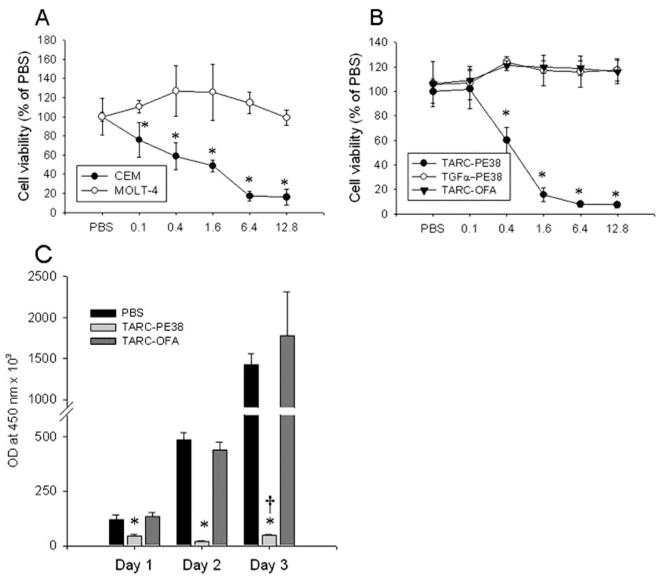
Specificity of TARC-PE38 killing. A, TARC-PE38 induces death of CCR4+ CEM cells, but not CCR4− MOLT-4 cells. Cells were incubated with titrated amounts of TARC-PE38 (μg/ml, x-axis) for 2 days, and cell viability was measured using WST assay. Results are presented as percentage of viable PBS-treated cells.*, p < 0.05 is for comparisons between CEM and MOLT-4 at the indicated concentrations of TARC-PE38. B, CEM cells are only killed with TARC-PE38, but not with TGFα-PE38 or TARC-OFA. Protein concentrations shown are μg/ml (x-axis).*, p < 0.05 is for comparisons between the TARC-PE38 and TARC-OFA at the indicated concentrations. C, Outgrowth kinetics of CEM cells cultured in the presence of either 10 μg/ml TARC-PE38 or TARC-OFA, or PBS.*, p < 0.05 is for comparisons between the TARC-PE38 and TARC-OFA. †, p < 0.01 is for comparison with day 2 of TARC-PE38-treated cells. The viability was assessed using WST assay. Data (mean ± SD) results are presented as OD values at 450 nm. All figures show representative data from at least three independent experiments with similar results.
TARC-PE38 effectively eradicates established CCR4+ tumors in mice
CCR4 expression by cutaneous T cell leukemia/lymphomas is an indicator for the patients’ poor clinical outcome (1, 19). Due to the lack of murine models for cutaneous leukemia, we have experimented with s.c. established human T cell lymphoblastoid CEM tumor cells in NOD-SCID mice. Mice with 12-day growing CEM tumors were injected intratumorally with 25 μg of TARC-PE38 or TARC-OFA, or mock treated with saline once daily for 5 consecutive days. Injections of TARC-OFA or PBS did not affect tumor growth and mice had to be sacrificed by day 27 when tumor size reached ~400 mm2 (Fig. 5, A and E). In contrast, almost complete tumor suppression was observed in mice treated with TARC-PE38 (Fig. 5, B and E), which was also associated with appearance of substantial necrotic lesions at the tumor challenge site (Fig. 5B). The data were supported by histological analysis taken at 27 days after tumor challenge; the TARC-PE38-treated tumors were mostly necrotized (data not shown) and tumor cells were detected only at the margin of necrotic areas (Fig. 5D). In contrast, control-treated samples primarily consisted of tumor cells (cells with large and dense nuclei; Fig. 5C) infiltrated in the dermis and spread into s.c. tissue.
FIGURE 5.
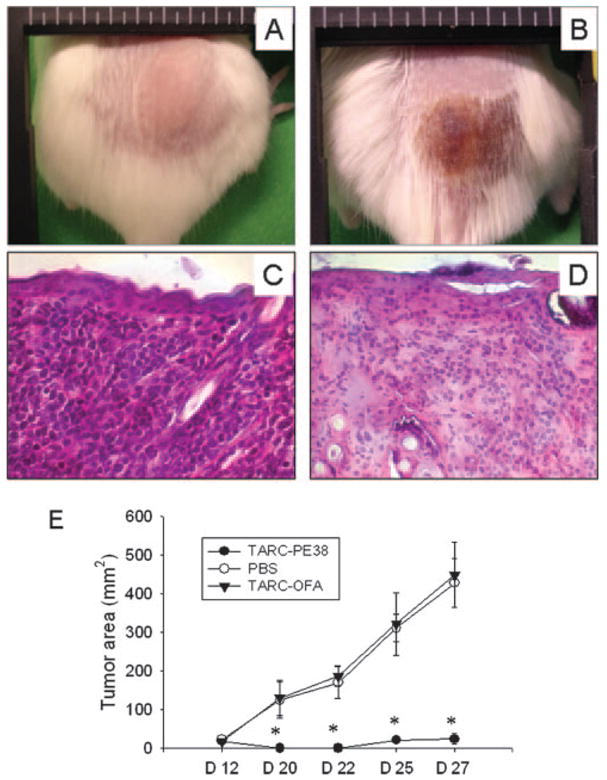
Eradication of CEM tumors established in NOD-SCID mice. A and B, Macroscopic appearance of tumors treated with TARC-OFA (A) or TARC-PE38 (B) at day 27 posttumor challenge. C and D, Microscopic appearance (H&E-stained slides, original magnification, ×200) of tumors treated with TARC-OFA (C) and TARC-PE38 (D, margin of necrotic area) at day 27 posttumor challenge. E, Tumor growth plot of mice treated with TARC-PE38, TARC-OFA, or PBS.*, p < 0.05 is for comparisons between the TARC-PE38 and TARC-OFA groups at the days indicated. Representative data from two independent experiments with comparable data with eight mice per group.
Tumor relapse is due to survival of receptor-negative cells in NOD-SCID mice
Despite the potency of the treatment and the fact that the TARC-PE38 treatment eliminated any palpable signs of tumor in mice, the tumor eventually relapsed and a new tumor growth could be detected at the margins of the necrotic area and surrounding skin. In addition, the tumor no longer responded to the TARC-PE38 treatment, even if treated multiple times by higher doses (100 μg/ml; data not shown). Therefore, we hypothesized that these tumors might represent an outgrowth of resistant cells that had lost expression of CCR4. In support, the relapsed tumor cells from TARC-PE38-treated mice expressed significantly reduced surface CCR4 compared with tumors grown in mock-treated mice (CCR4 mean fluorescence intensity, 92.5 ± 1.3% vs 147.2 ± 16.4%, respectively, p < 0.05; Fig. 6A). Thus, these data indicate that the TARC-PE38 resistant and relapsed tumors are mostly represented by the cells that either do not signal via CCR4 or lost its surface expression. For example, cells isolated from TARC-PE38-treated and relapsed tumors were significantly less susceptible to additional ex vivo treatments with TARC-PE38 ( p < 0.001, •; Fig. 6B). In contrast, TARC-PE38 killed both control ex vivo cultured cells from PBS-treated mice as efficiently as parental in vitro cultured CEM cells (○ and ▵, respectively; Fig. 6B). To further assess this, the parental in vitro cultured CEM cells were pretreated ether once or five times (with 2-day intervals) with 10 μg/ml TARC-PE38 or TARC-OFA, or PBS. Then, the cells were cultured for 4 wk without any treatments. The majority of control CEM cells pretreated with TARC-OFA or PBS were viable (see also Fig. 4C) and expressed CCR4 (97%; Fig. 6C). In contrast, a single pretreatment with TARC-PE38 yielded dramatic cell death, leaving only a small number of viable cells (<5%, data not shown; also see Fig. 4C) that contained significantly reduced proportion of CCR4-expressing cells (48%, p < 0.001; Fig. 6C). The proportion of CCR4-negative cells was further increased (78%; Fig. 6C) when cells were pretreated with TARC-PE38 for five times. Importantly, these cells not only became CCR4 negative, but also acquired resistance to subsequent treatments with TARC-PE38 (Fig. 6D). Control pretreatments with TARC-OFA or PBS did not affect either the proportion of CCR4-expressing cells (Fig. 6C), or sensitivity to TARC-PE38 (Fig. 6D).
FIGURE 6.
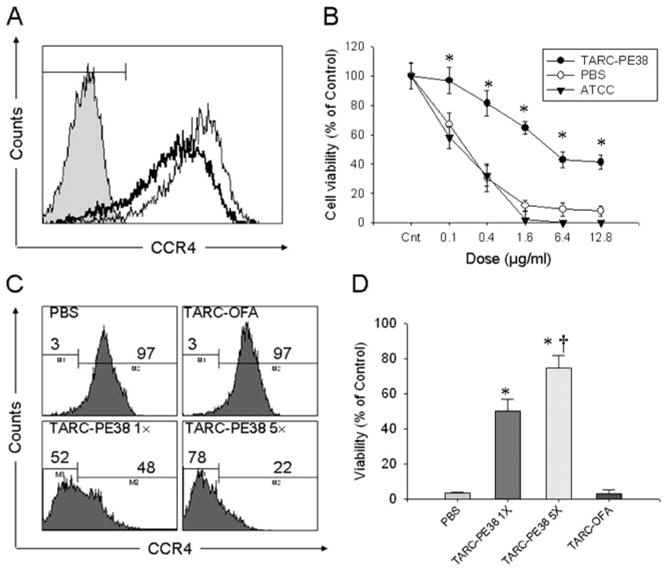
Mechanism of tumor escape in NOD-SCID mice: TARC-PE38 kills selectively CCR4-positive tumors, but does not prevent growth of escapees that do not express CCR4. A, CCR4 expression in tumor cells isolated from PBS (thin line)- or TARC-PE38 (thick line)-treated mice. Cells were stained with anti-human CD45-PE and anti-human CCR4-FITC Abs and were analyzed by FACS. Histogram shows CCR4 expression in CD45-gated cells. Filled line: isotype-matched FITC-conjugated IgG. B, Effect of TARC-PE38 on viability of tumor cells derived from PBS- or TARC-PE38-treated mice. Parental in vitro cultured CEM cells from American Type Culture Collection were used as control.*, p < 0.01 is for comparisons with the parental CEM cells treated with TARC-PE38 at the indicated doses. C, CCR4 expression on CEM cells treated with 10 μg/ml TARC-PE38 once (TARC-PE38 1×) or five times (TARC-PE38 5×) and cultured thereafter for 4 wk. Controls were treated with TARC-OFA or PBS. D, Effect of TARC-PE38 on viability of CEM cells treated and cultured, as described for C. Cells were overnight treated again with 10 μg/ml TARC-PE38 to test their resistance (viability).*, p < 0.01 is for comparisons with the TARC-OFA treatment. Representative data from at least two independent experiments with comparable data.
Systemic treatments with TARC-PE38 are safe and effective in mice
Next, we tested whether tumor dissemination can be controlled by the systemic administrations of TARC-PE38. Twenty days after i.v. injection of CEM cells, NOD-SCID mice were treated with 10 μg of TARC-PE38 (i.v., once per day) for 4 consecutive days. Control mice were treated with CTACK-PE38, a PE38 fusion with chemokine that specifically binds CCR10. As shown in Fig. 7A, only <1% of mononuclear cells isolated from spleens of the TARC-PE38-treated mice can be associated with CEM tumors, because they expressed human CD45. In contrast, significantly higher amounts of tumors were found in control CTACK-PE38-treated mouse spleens (9.5%; Fig. 7A). Cells isolated from both TARC-PE38- and CTACK-PE38-treated mice were able to grow ex vivo and became almost 100% positive for human CD45 (data not shown) after 10 days of culture. However, the number of cells expanded from spleens of the TARC-PE38-treated mice was 20-fold less than that of the CTACK-PE38-treated mice (Fig. 7B). Taken together, our data indicate that chemotoxins can efficiently regress the CCR4-positive T cell tumors established in mice, suggesting their potential usefulness in the control of cutaneous leukemias that express CCR4. Although the CCR4-negative escapees had significantly slower growth rate (data not shown), their impact remains unknown due to the limits of the model used.
FIGURE 7.
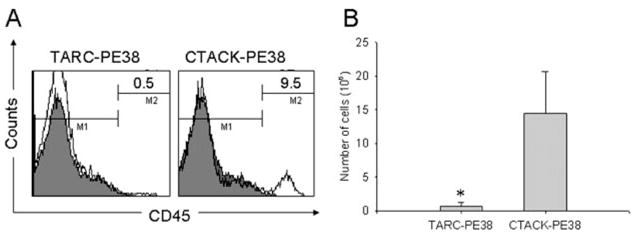
Dissemination of CEM cells in NOD-SCID mice can be controlled by the systemic injections of TARC-PE38. A, Proportion of human CD45-positive cells (tumor cells) in spleens of mice injected i.v. with CEM cells and treated with TARC-PE38 or CTACK-PE38. Numbers in histograms indicate the percentage of cells stained with anti-human CD45 Ab (open lines). The staining with isotype-matched control IgG is shown (filled lines). B, Total number of tumor cells expanded from spleens of TARC-PE38- and CTACK-PE38-treated mice after 10 days of ex vivo culture. Data are mean ± SD of three mice per group, and*, p < 0.001 is for comparisons with CTACK-PE38 treatment.
Discussion
Chemokines and chemokine receptors expressed by tumors or tumor-containing tissues regulate a number of important functions that eventually affect disease outcome. They can directly induce cell proliferation (MIP1α/CCL3, I-309/CCL1, stromal cell-derived factor 1/CXCL12, TARC/CCL17) (20, 21) and regulate angiogenesis and infiltration of immune cells, etc. (see reviews (3, 36, 37)). Tumor-expressed chemokines attract inhibitory macrophages, NK T cells, and CD25+CD4+FoxP3+ Tregs, leading to severe immunosuppression and poor disease outcome in a number of cancers (23, 38). For example, infiltration of suppressive CCR4-expressing CD4+CD25high Tregs is thought to be caused by CCL17 or CCL22 produced at the tumor site (1, 38). In contrast, tumors by themselves produce various immunosuppressive factors, or even, in the case of ATLL, some of them might originate from transformed immunosuppressive cells (1). These features are thought to negatively affect vaccine-induced antitumor cellular responses, leading to tumor escape despite the presence of tumor-specific cytolytic CD8+ T cells (39, 40). Therefore, alternative strategies that would directly kill malignant cells had been extensively explored. A surge of Ab-mediated therapeutics was initiated by the successful clinical use of rituximab, CD20-targeting Ab (for review, see Ref. 41). Tumor cells from ATLL patients were shown to be killed by incubation with mAb to CCR4, and mice injected with anti-CCR4 Ab were protected from CCR4-expressing tumors (1). This protection was based on induction of the Ab-dependent cellular cytotoxicity-mediated cell killing, and the CCR4-expressing human tumors were not uniformly killed in vitro (1), suggesting that clinical efficiency of anti-CCR4 Abs may be affected by the host FcR polymorphism (42, 43).
In this study, we report a different strategy that uses formulations designated chemotoxins, to preferentially and specifically eliminate chemokine receptor-expressing cells. As we have shown for chemokines fused with tumor Ags, the strategy was based on the ability of chemokines to deliver toxins into the cell cytosol. We recently reported that nonimmunogenic tumor Ags were rendered immunogenic and elicited therapeutic antitumor immunity, if they were targeted to chemokine receptors to use their internalization machinery (25, 33). For example, chemokine fusions that were targeted to CCR6 and CCR8 were internalized into endo/lysosomal compartments, where some of them were degraded and presented to MHC class II molecules, whereas the remaining ligands were able to efficiently escape into cytosol to be degraded by proteasomes for MHC class I cross-presentation (25, 33). Our present data indicate that chemokines could also deliver RNases (EDN) to the cell cytosol and induce the death of cells that expressed the respective chemokine receptor. However, EDN chemotoxin in the present form may not be useful for the treatment of fast-growing malignancies, as its killing dynamics were significantly slow, presumably reflecting the nature of the EDN-induced apoptosis. It remains open to question whether activity of RNase-expressing chemotoxins would be augmented by increasing their RNase activity, protecting them from the intracellular proteases, or improving their delivery into the cytosol. It is tempting to speculate that the reduced RNase activity of chemokine EDN may be improved by generation of EDN-chemokine fusions, so as to free the N terminus of EDN to preserve its activity (our unpublished observation). In contrast, we hypothesized that the efficacy might be improved by use of faster acting moieties such as the exotoxin fragment PE38 of P. aeruginosa. The exotoxin was reported to kill mammalian cells within a short period of time by inhibiting protein synthesis via the ADP ribosylation of elongation initiation factor 2 (34). Importantly, PE38 appears to be nontoxic unless delivered into the cell cytosol (28, 35), and PE38 fusions with Abs to various cell surface Ags, such as CD25, CD22, and IL-4R, were successfully tested for treatment of hematological malignancies (44, 45) (for review, see Ref. 46). Interestingly, although CD22 is expressed on normal B cells and the majority of B cell malignancies, the anti-CD22 immunotoxin treatments only benefited patients with hairy cell leukemia, but not patients with non-Hodgkin’s lymphoma (NHL) and chronic lymphocytic leukemia (45). Similarly, immunotoxins that target CD25 were shown to be moderately effective in patients with NHL and cutaneous T cell lymphoma (46). It is noteworthy that CD25, IL-2Rα, is a necessary and important survival receptor for cells that depend on IL 2, including immune T cells. Thus, these cells would be also depleted by the anti-CD25 treatment, and beneficial effects of CD25-targeting immunotoxins might be contradicted by the loss of tumor-specific T cells needed for eradication of residual or escaped tumors.
We have wanted to develop a less harmful and specific strategy to eradicate hematological tumors, such as NHL, chronic lymphocytic leukemia, cutaneous T cell lymphoma, and ATLL, delivering toxins via chemokine receptors. Although CCR4 is also expressed by ~20% of human peripheral blood CD4+ T cells (D. Baatar, unpublished data), depletion of CCR4-expressing cells by the systemic injections of TARC-PE38 did not cause apparent and adverse effects in C57BL/6 and BALB/c mice, because they were able to mount Ag-specific humoral and cellular immune responses (our unpublished data). In concordance, others recently reported that the Ab-mediated depletion of CCR4-expressing tumors (and normal cells) was well tolerated in mice (1). Recently, we have shown that the majority of human peripheral blood Tregs expressed CCR4, and their depletion using TARC-PE38 was sufficient to revert the suppressive state of CD8+ T cells (47). Therefore, the TARC-PE38 treatment would not only directly kill the CCR4-expressing leukemia cells, but also would eliminate Tregs to build beneficial conditions for activation of antitumor T cell responses, a necessary step for the elimination of the remaining escapees and residual disease. Of note: the efficacy to bind and kill murine CCR4-expressing cells was comparable regardless of the origin of TARC; both mouse and human TARC fusions with PE38 worked well and human TARC-PE38 effectively depleted murine CCR4+ CD4 T cells in immunocompetent BALB/c or C57BL/6 mice (data not shown and also see Ref. 47). Experimenting with human T cell tumors established in NOD-SCID mice, we demonstrated that TARC-PE38 can efficiently regress the CCR4-expressing cutaneous tumors in mice. We have also tested whether chemotoxins injected systemically can affect dissemination of tumors, although their therapeutic efficacy may be reduced by a relative short plasma t1/2 time. Intravenous injections of TARC-PE38, but not control CTACK-PE38, dramatically reduced the number of spleen-infiltrating CEM tumor cells, indicating that chemotoxins may be also useful for systemic treatment of nonsolid tumors. Because expression of CCR4 is often associated with a poor disease outcome, this may be an attractive strategy to control and combat the disease.
However, despite the fact that the TARC-PE38 treatment eliminated any signs of detectable/palpable tumor, the malignancy eventually relapsed, but with cells that no longer responded to additional and repeated treatments of TARC chemotoxin. Our data indicate that the resistance to TARC-PE38 correlates with the cells that do not express CCR4, although we cannot rule out the possibility that the treatment also selects variants that aberrantly express CCR4 that cannot internalize. Moreover, TARC-PE38 does not induce loss of CCR4 per se, but rather it is unable to affect the growth of the receptor-negative tumor variants, which represented a minor (≤3–5%) population that existed before the treatment. Therefore, if a malignancy is heterogeneous, the treatment may lead to outgrowth of receptor-negative tumors, particularly in hosts with aberrant T cell immune responses. It remains to be tested whether escapees can survive and progress in hosts with an adequate immune system that would be activated during chemotoxin-induced massive cell death. Unfortunately, limitations of the model system used in this study do not allow us to address this question. Taken together, we present a novel concept that tumors can specifically be eradicated by delivering toxins through their chemokine receptors. This very simple and potent strategy can presumably be used for treatment of a wide variety of tumors, particularly hematological malignancies, which use chemokine receptors to metastasize or circumvent immunosurveillance. However, a complete eradication of tumors would probably be achieved by use of the strategy in combination with other treatment modalities such as chemotherapy. Finally, chemotoxins may also serve as a useful tool for understanding of biology and functions of various immune cells through their preferential and transient depletion, as it was recently used to elucidate biology of the skin-homing T regulatory cells (47).
Acknowledgments
We are grateful to Drs. Dan Longo and Ashani Weeraratna for helpful comments and suggestions; Ana Lustig (National Institute on Aging/National Institutes of Health) for critical reading of the manuscript; Dr. Ira Pastan (National Cancer Institute/National Institutes of Health) for the gift of PE38 cDNA and anti-TGFα-PE38; and Dot Bertak (National Institute on Aging/National Institutes of Health) for help with immunohistochemistry.
Footnotes
This research was supported by the Intramural Research Program of the National Institute on Aging, National Institutes of Health.
Abbreviations used in this paper: ATLL, adult T cell leukemia/lymphoma; CTACK, cutaneous T cell-attracting chemokine; EDN, eosinophil-derived neurotoxin; HEK, human embryonic kidney; NHL, non-Hodgkin’s lymphoma; OFA, oncofetal Ag; PE38, Pseudomonas exotoxin 38; TARC, thymus and activation-regulated chemokine; Tregs, T regulatory cells.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Ishida T, Iida S, Akatsuka Y, Ishii T, Miyazaki M, Komatsu H, Inagaki H, Okada N, Fujita T, Shitara K, et al. The CC chemokine receptor 4 as a novel specific molecular target for immunotherapy in adult T-cell leukemia/lymphoma. Clin Cancer Res. 2004;10:7529–7539. doi: 10.1158/1078-0432.CCR-04-0983. [DOI] [PubMed] [Google Scholar]
- 2.Yoshie O, Fujisawa R, Nakayama T, Harasawa H, Tago H, Izawa D, Hieshima K, Tatsumi Y, Matsushima K, Hasegawa H, et al. Frequent expression of CCR4 in adult T-cell leukemia and human T-cell leukemia virus type 1-transformed T cells. Blood. 2002;99:1505–1511. doi: 10.1182/blood.v99.5.1505. [DOI] [PubMed] [Google Scholar]
- 3.Proudfoot AE. Chemokine receptors: multifaceted therapeutic targets. Nat Rev Immunol. 2002;2:106–115. doi: 10.1038/nri722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sallusto F, Palermo B, Lenig D, Miettinen M, Matikainen S, Julkunen I, Forster R, Burgstahler R, Lipp M, Lanzavecchia A. Distinct patterns and kinetics of chemokine production regulate dendritic cell function. Eur J Immunol. 1999;29:1617–1625. doi: 10.1002/(SICI)1521-4141(199905)29:05<1617::AID-IMMU1617>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 5.Dieu MC, Vanbervliet B, Vicari A, Bridon JM, Oldham E, Ait-Yahia S, Briere F, Zlotnik A, Lebecque S, Caux C. Selective recruitment of immature and mature dendritic cells by distinct chemokines expressed in different anatomic sites. J Exp Med. 1998;188:373–386. doi: 10.1084/jem.188.2.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Campbell MJ, Esserman L, Byars NE, Allison AC, Levy R. Idiotype vaccination against murine B cell lymphoma: humoral and cellular requirements for the full expression of antitumor immunity. J Immunol. 1990;145:1029–1036. [PubMed] [Google Scholar]
- 7.Sebastiani S, Allavena P, Albanesi C, Nasorri F, Bianchi G, Traidl C, Sozzani S, Girolomoni G, Cavani A. Chemokine receptor expression and function in CD4+ T lymphocytes with regulatory activity. J Immunol. 2001;166:996–1002. doi: 10.4049/jimmunol.166.2.996. [DOI] [PubMed] [Google Scholar]
- 8.Iellem A, Mariani M, Lang R, Recalde H, Panina-Bordignon P, Sinigaglia F, D’Ambrosio D. Unique chemotactic response profile and specific expression of chemokine receptors CCR4 and CCR8 by CD4+CD25+ regulatory T cells. J Exp Med. 2001;194:847–853. doi: 10.1084/jem.194.6.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stine JT, Wood C, Hill M, Epp A, Raport CJ, Schweickart VL, Endo Y, Sasaki T, Simmons G, Boshoff C, et al. KSHV-encoded CC chemokine vMIP-III is a CCR4 agonist, stimulates angiogenesis, and selectively chemoattracts TH2 cells. Blood. 2000;95:1151–1157. [PubMed] [Google Scholar]
- 10.Bendall L. Chemokines and their receptors in disease. Histol Histopathol. 2005;20:907–926. doi: 10.14670/HH-20.907. [DOI] [PubMed] [Google Scholar]
- 11.Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50–56. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- 12.Longo-Imedio MI, Longo N, Trevino I, Lazaro P, Sanchez-Mateos P. Clinical significance of CXCR3 and CXCR4 expression in primary melanoma. Int J Cancer. 2005;117:861–865. doi: 10.1002/ijc.21269. [DOI] [PubMed] [Google Scholar]
- 13.Walser TC, Rifat S, Ma X, Kundu N, Ward C, Goloubeva O, Johnson MG, Medina JC, Collins TL, Fulton AM. Antagonism of CXCR3 inhibits lung metastasis in a murine model of metastatic breast cancer. Cancer Res. 2006;66:7701–7707. doi: 10.1158/0008-5472.CAN-06-0709. [DOI] [PubMed] [Google Scholar]
- 14.Ishida T, Inagaki H, Utsunomiya A, Takatsuka Y, Komatsu H, Iida S, Takeuchi G, Eimoto T, Nakamura S, Ueda R. CXC chemokine receptor 3 and CC chemokine receptor 4 expression in T-cell and NK-cell lymphomas with special reference to clinicopathological significance for peripheral T-cell lymphoma, unspecified. Clin Cancer Res. 2004;10:5494–5500. doi: 10.1158/1078-0432.CCR-04-0371. [DOI] [PubMed] [Google Scholar]
- 15.Jones D, Benjamin RJ, Shahsafaei A, Dorfman DM. The chemokine receptor CXCR3 is expressed in a subset of B-cell lymphomas and is a marker of B-cell chronic lymphocytic leukemia. Blood. 2000;95:627–632. [PubMed] [Google Scholar]
- 16.Trentin L, Agostini C, Facco M, Piazza F, Perin A, Siviero M, Gurrieri C, Galvan S, Adami F, Zambello R, Semenzato G. The chemokine receptor CXCR3 is expressed on malignant B cells and mediates chemotaxis. J Clin Invest. 1999;104:115–121. doi: 10.1172/JCI7335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cabioglu N, Yazici MS, Arun B, Broglio KR, Hortobagyi GN, Price JE, Sahin A. CCR7 and CXCR4 as novel biomarkers predicting axillary lymph node metastasis in T1 breast cancer. Clin Cancer Res. 2005;11:5686–5693. doi: 10.1158/1078-0432.CCR-05-0014. [DOI] [PubMed] [Google Scholar]
- 18.Gunther K, Leier J, Henning G, Dimmler A, Weissbach R, Hohenberger W, Forster R. Prediction of lymph node metastasis in colorectal carcinoma by expression of chemokine receptor CCR7. Int J Cancer. 2005;116:726–733. doi: 10.1002/ijc.21123. [DOI] [PubMed] [Google Scholar]
- 19.Ishida T, Utsunomiya A, Iida S, Inagaki H, Takatsuka Y, Kusumoto S, Takeuchi G, Shimizu S, Ito M, Komatsu H, et al. Clinical significance of CCR4 expression in adult T-cell leukemia/lymphoma: its close association with skin involvement and unfavorable outcome. Clin Cancer Res. 2003;9:3625–3634. [PubMed] [Google Scholar]
- 20.Louahed J, Struyf S, Demoulin JB, Parmentier M, Van Snick J, Van Damme J, Renauld JC. CCR8-dependent activation of the RAS/MAPK pathway mediates anti-apoptotic activity of I-309/CCL1 and vMIP-I. Eur J Immunol. 2003;33:494–501. doi: 10.1002/immu.200310025. [DOI] [PubMed] [Google Scholar]
- 21.Lentzsch S, Gries M, Janz M, Bargou R, Dorken B, Mapara MY. Macrophage inflammatory protein 1-α (MIP-1α) triggers migration and signaling cascades mediating survival and proliferation in multiple myeloma (MM) cells. Blood. 2003;101:3568–3573. doi: 10.1182/blood-2002-08-2383. [DOI] [PubMed] [Google Scholar]
- 22.Kim J, Takeuchi H, Lam ST, Turner RR, Wang HJ, Kuo C, Foshag L, Bilchik AJ, Hoon DS. Chemokine receptor CXCR4 expression in colorectal cancer patients increases the risk for recurrence and for poor survival. J Clin Oncol. 2005;23:2744–2753. doi: 10.1200/JCO.2005.07.078. [DOI] [PubMed] [Google Scholar]
- 23.Scrivener S, Goddard RV, Kaminski ER, Prentice AG. Abnormal T-cell function in B-cell chronic lymphocytic leukemia. Leuk Lymphoma. 2003;44:383–389. doi: 10.1080/1042819021000029993. [DOI] [PubMed] [Google Scholar]
- 24.Niwa R, Shoji-Hosaka E, Sakurada M, Shinkawa T, Uchida K, Nakamura K, Matsushima K, Ueda R, Hanai N, Shitara K. Defucosylated chimeric anti-CC chemokine receptor 4 IgG1 with enhanced antibody-dependent cellular cytotoxicity shows potent therapeutic activity to T-cell leukemia and lymphoma. Cancer Res. 2004;64:2127–2133. doi: 10.1158/0008-5472.can-03-2068. [DOI] [PubMed] [Google Scholar]
- 25.Schiavo R, Baatar D, Olkhanud P, Indig FE, Restifo N, Taub D, Biragyn A. Chemokine receptor targeting efficiently directs antigens to MHC class I pathways and elicits antigen-specific CD8+ T-cell responses. Blood. 2006;107:4597–4605. doi: 10.1182/blood-2005-08-3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Newton DL, Nicholls PJ, Rybak SM, Youle RJ. Expression and characterization of recombinant human eosinophil-derived neurotoxin and eosinophil-derived neurotoxin-anti-transferrin receptor sFv. J Biol Chem. 1994;269:26739–26745. [PubMed] [Google Scholar]
- 27.Ruffini PA, Biragyn A, Coscia M, Harvey LK, Cha SC, Bogen B, Kwak LW. Genetic fusions with viral chemokines target delivery of nonimmunogenic antigen to trigger antitumor immunity independent of chemotaxis. J Leukocyte Biol. 2004;76:77–85. doi: 10.1189/jlb.1003481. [DOI] [PubMed] [Google Scholar]
- 28.Onda M, Wang QC, Guo HF, Cheung NK, Pastan I. In vitro and in vivo cytotoxic activities of recombinant immunotoxin 8H9(Fv)-PE38 against breast cancer, osteosarcoma, and neuroblastoma. Cancer Res. 2004;64:1419–1424. doi: 10.1158/0008-5472.can-03-0570. [DOI] [PubMed] [Google Scholar]
- 29.Biragyn A, Tani K, Grimm MC, Weeks SD, Kwak LW. Genetic fusion of chemokines to a self tumor antigen induces protective, T-cell dependent antitumor immunity. Nat Biotechnol. 1999;17:253–258. doi: 10.1038/6995. [DOI] [PubMed] [Google Scholar]
- 30.Biragyn A, Surenhu M, Yang D, Ruffini PA, Haines BA, Klyushnenkova E, Oppenheim JJ, Kwak LW. Mediators of innate immunity that target immature, but not mature, dendritic cells induce antitumor immunity when genetically fused with nonimmunogenic tumor antigens. J Immunol. 2001;167:6644–6653. doi: 10.4049/jimmunol.167.11.6644. [DOI] [PubMed] [Google Scholar]
- 31.Falk W, Goodwin RHJ, Leonard EJ. A 48-well micro chemotaxis assembly for rapid and accurate measurement of leukocyte migration. J Immunol Methods. 1980;33:239–247. doi: 10.1016/0022-1759(80)90211-2. [DOI] [PubMed] [Google Scholar]
- 32.Yang D, Chen Q, Stoll S, Chen X, Howard OM, Oppenheim JJ. Differential regulation of responsiveness to fMLP and C5a upon dendritic cell maturation: correlation with receptor expression. J Immunol. 2000;165:2694–2702. doi: 10.4049/jimmunol.165.5.2694. [DOI] [PubMed] [Google Scholar]
- 33.Biragyn A, Ruffini PA, Coscia M, Harvey LK, Neelapu SS, Baskar S, Wang JM, Kwak LW. Chemokine receptor-mediated delivery directs self-tumor antigen efficiently into the class II processing pathway in vitro and induces protective immunity in vivo. Blood. 2004;104:1961–1969. doi: 10.1182/blood-2004-02-0637. [DOI] [PubMed] [Google Scholar]
- 34.Allured VS, Collier RJ, Carroll SF, McKay DB. Structure of exotoxin A of Pseudomonas aeruginosa at 3.0-Angstrom resolution. Proc Natl Acad Sci USA. 1986;83:1320–1324. doi: 10.1073/pnas.83.5.1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li M, Dyda F, Benhar I, Pastan I, Davies DR. The crystal structure of Pseudomonas aeruginosa exotoxin domain III with nicotinamide and AMP: conformational differences with the intact exotoxin. Proc Natl Acad Sci USA. 1995;92:9308–9312. doi: 10.1073/pnas.92.20.9308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coscia M, Biragyn A. Cancer immunotherapy with chemoattractant peptides. Semin Cancer Biol. 2004;14:209–218. doi: 10.1016/j.semcancer.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 37.Godessart N. Chemokine receptors: attractive targets for drug discovery. Ann NY Acad Sci. 2005;1051:647–657. doi: 10.1196/annals.1361.109. [DOI] [PubMed] [Google Scholar]
- 38.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 39.Overwijk WW, Theoret MR, Finkelstein SE, Surman DR, de Jong LA, Vyth-Dreese FA, Dellemijn TA, Antony PA, Spiess PJ, Palmer DC, et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198:569–580. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wei S, Kryczek I, Zou L, Daniel B, Cheng P, Mottram P, Curiel T, Lange A, Zou W. Plasmacytoid dendritic cells induce CD8+ regulatory T cells in human ovarian carcinoma. Cancer Res. 2005;65:5020–5026. doi: 10.1158/0008-5472.CAN-04-4043. [DOI] [PubMed] [Google Scholar]
- 41.Maloney DG. Immunotherapy for non-Hodgkin’s lymphoma: monoclonal antibodies and vaccines. J Clin Oncol. 2005;23:6421–6428. doi: 10.1200/JCO.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 42.Boot JH, Geerts ME, Aarden LA. Functional polymorphisms of Fc receptors in human monocyte-mediated cytotoxicity towards erythrocytes induced by murine isotype switch variants. J Immunol. 1989;142:1217–1223. [PubMed] [Google Scholar]
- 43.Kim DH, Jung HD, Kim JG, Lee JJ, Yang DH, Park YH, Do YR, Shin HJ, Kim MK, Hyun MS, Sohn SK. FCGR3A gene polymorphisms may correlate with response to frontline R-CHOP therapy for diffuse large B-cell lymphoma. Blood. 2006;108:2720–2725. doi: 10.1182/blood-2006-01-009480. [DOI] [PubMed] [Google Scholar]
- 44.Arons E, Sorbara L, Raffeld M, Stetler-Stevenson M, Steinberg SM, Liewehr DJ, Pastan I, Kreitman RJ. Characterization of T-cell repertoire in hairy cell leukemia patients before and after recombinant immunotoxin BL22 therapy. Cancer Immunol Immunother. 2005;55:1100–1110. doi: 10.1007/s00262-005-0099-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kreitman RJ, Squires DR, Stetler-Stevenson M, Noel P, FitzGerald DJ, Wilson WH, Pastan I. Phase I trial of recombinant immunotoxin RFB4(dsFv)-PE38 (BL22) in patients with B-cell malignancies. J Clin Oncol. 2005;23:6719–6729. doi: 10.1200/JCO.2005.11.437. [DOI] [PubMed] [Google Scholar]
- 46.Kreitman RJ. Recombinant immunotoxins for the treatment of hematological malignancies. Exp Opin Biol Ther. 2004;4:1115–1128. doi: 10.1517/14712598.4.7.1115. [DOI] [PubMed] [Google Scholar]
- 47.Baatar D, Olkhanud P, Sumitomo K, Taub D, Gress R, Biragyn A. Human peripheral blood T regulatory cells (Tregs), functionally primed CCR4+ Tregs and unprimed CCR4− Tregs, regulate effector T cells using FasL. J Immunol. 2007;178:4891–4900. doi: 10.4049/jimmunol.178.8.4891. [DOI] [PMC free article] [PubMed] [Google Scholar]