

Abstract
Human immunodeficiency virus (HIV) infection leads to numerous perturbations of B cells through mechanisms that remain elusive. We performed DNA microarray, phenotypic, and functional analyses in an effort to elucidate mechanisms of B cell perturbation associated with ongoing HIV replication. 42 genes were up-regulated in B cells of HIV-viremic patients when compared with HIV-aviremic and HIV-negative patients, the majority of which were interferon (IFN)-stimulated or associated with terminal differentiation. Flow cytometry confirmed these increases and indicated that CD21low B cells, enhanced in HIV-viremic patients, were largely responsible for the changes. Increased expression of the tumor necrosis factor (TNF) superfamily (TNFSF) receptor CD95 correlated with increased susceptibility to CD95-mediated apoptosis of CD21low B cells, which, in turn, correlated with HIV plasma viremia. Increased expression of BCMA, a weak TNFSF receptor for B lymphocyte stimulator (BLyS), on CD21low B cells was associated with a concomitant reduction in the expression of the more potent BLyS receptor, BAFF-R, that resulted in reduced BLyS binding and BLyS-mediated survival. These findings demonstrate that altered expression of genes associated with IFN stimulation and terminal differentiation in B cells of HIV-viremic patients lead to an increased propensity to cell death, which may have substantial deleterious effects on B cell responsiveness to antigenic stimulation.
Keywords: terminal differentiation, interferon, immune activation, gene expression, microarray
Introduction
HIV infection is associated with numerous immunologic deficiencies, several of which are paradoxically associated with immune hyperactivation induced by ongoing viral replication. The aberrant immune activation induced by HIV represents a systemic effect that is best illustrated by the profound effects of HIV on B cells and CD8+ T cells (1–3), two populations of lymphocytes that are not direct targets for productive HIV replication. Although B cells have been shown to carry replication-competent HIV virions on their surface via CD21–C3d immune complex interactions (4), the relatively low frequency of such cells is far below the percentage of B cells whose function is altered in the presence of ongoing HIV replication, suggesting a preponderance of indirect over direct effects of the virus on B cells of infected individuals. The most prominent B cell defects associated with HIV infection include hypergammaglobulinemia (5–7), increased expression of markers of activation and terminal differentiation (8–10), increased secretion of immunoglobulins in vitro (11), decreased response to B cell activators in vitro and in vivo (12–16), and increased susceptibility to apoptosis (17, 18). With the availability of effective antiretroviral therapy, it has been possible to demonstrate that most of these B cell defects are directly associated with high levels of HIV plasma viremia. Several longitudinal and cross-sectional studies have shown that reduction of HIV plasma viremia leads to normalization of B cell activities, as evidenced by reductions in polyclonal and HIV-specific Ig levels (6, 9, 19–22), restoration of APC function and responsiveness to CD4+ T cell help (10, 16), and normalization of expression of B cell markers of activation and terminal differentiation (9, 10, 23).
Although there is widespread consensus that HIV infection leads to B cell dysfunction, the underlying mechanisms and nature of the dysregulation remain poorly understood. Our previous findings suggested that the hypergammaglobulinemia observed in HIV-infected viremic patients resulted from stimulation of B cell terminal differentiation by HIV-mediated immune activation (9). However, others have proposed that depletion of memory B cells and consequent enrichment of activated naive B cells are the basis for B cell immune dysfunction in HIV-infected patients (23). Our proposal for B cell terminal differentiation was based primarily on the observation that patients who did not suppress their HIV viremia had an increased percentage of B cells that expressed reduced levels of CD21, a marker that has been shown to decrease with differentiation toward the plasma cell phenotype (24). The subpopulation of B cells expressing reduced levels of CD21 was also found to possess plasmacytoid features not found in the CD21high fraction and was also shown to secrete high levels of immunoglobulins and respond poorly to B cell proliferative signals (9). The loss of cell surface CD21 was also associated with decreased levels of CD21 mRNA, further suggesting that the CD21low fraction was associated with B cell terminal differentiation (25), rather than ligand-induced receptor depletion. However, other features of B cell terminal differentiation, such as loss of surface Ig expression and other surface markers such as HLA-DR, CD20, and CD19 were not consistently observed on B cells of HIV-viremic patients, suggesting either that HIV-induced immune activation induces partial terminal differentiation or that a completely unique pathway of aberrant B cell activation occurs in HIV infection.
Considering the current uncertainties associated with HIV-mediated B cell dysfunction, we embarked on a study that began with the analysis of gene expression profiles in B cells of HIV-viremic patients, led to a comprehensive analysis of B cell markers associated with reduced CD21 expression, and culminated in analyses associated with altered expression of members of the TNF superfamily (TNFSF) of receptors. The data presented in this paper strongly suggest that HIV viremia leads to alterations in the expression of TNFSF receptors on B cells, rendering these cells more susceptible to cell death. These findings present a complex picture of survival and death in B cells and how HIV infection can shift this delicate balance and lead to profound dysfunction of the B cell arm of the immune response.
Materials and Methods
Study Subjects.
Three groups of individuals were investigated: patients chronically infected with HIV whose plasma viremia was >10,000 copies of HIV RNA/ml, patients chronically infected with HIV whose plasma viremia was <100 copies of HIV RNA/ml, and HIV-negative donors. HIV-viremic patients were either antiretroviral therapy-naive, not fully compliant with their antiretroviral regimen, or failing therapy, whereas the HIV-aviremic patients were receiving effective antiretroviral regimens. Leukapheresis and blood draws were conducted in accordance with protocols approved by the Institutional Review Boards of the University of Toronto and the National Institute of Allergy and Infectious Diseases (NIAID) at the National Institutes of Health.
Cell Preparation and Cell Surface Marker Analyses.
PBMCs were obtained from leukapheresis or blood draws by Ficoll density gradient centrifugations. B cells were isolated from PBMCs by negative selection using an immunomagnetic column-based technique (StemCell Technologies Inc.). The purity of each B cell preparation, typically >95%, was verified as described previously (4). PE-labeled mAbs were purchased from BD Biosciences or R&D Systems. FITC-labeled anti-CD21 was purchased from Beckman Coulter. Streptavidin-PE, biotin-labeled anti-BCMA, and anti-TACI, as well as unlabeled goat anti-BAFF-R Abs, were purchased from R&D Systems. PE-labeled swine anti–goat was purchased from Caltag. B lymphocyte stimulator (BLyS; Human Genome Sciences) was biotinylated with NHS-LC-biotin (Pierce Chemical Co.) according to the manufacturer's recommendations. Negative controls included isotyped-matched mAbs for direct stains and omission of the first Ab for indirect stains. Stained cells were analyzed on a FACSCalibur flow cytometer (BD Biosciences).
DNA Microarray Analyses of Gene Expression.
Total RNA was isolated from 5–10 × 106 purified B cells using TRIzol (Invitrogen) according to the manufacturer's specifications. Hybridization and DNA microarray analyses were performed as described previously (26). 33P-labeled first-strand cDNA was synthesized from 5 μg of total RNA. cDNA probes were hybridized to DNA arrays comprised of 6,476 genes enriched in clones that had high-quality RefSeq matches. After extensive washing under stringent conditions, the hybridization signal was detected after a 60-h exposure using a FUJI BAS 2500 phosphorimaging system and captured and quantitated using ImaGene 4.0 software (BioDiscovery). After linear normalization to total signal, low expression values were assigned a low signal of 35, which represents the 98th percentile of blank spots, to avoid potential overinterpretation of low-signal changes. Only the data points with a coefficient of variation <0.8 for duplicate spots were included in further analyses. The data were log(2) transformed, and each gene expression profile among all HIV-viremic, HIV-aviremic, and HIV-negative patients was median centered.
B Cell Apoptosis Assay.
Freshly isolated B cells, or enriched CD21low and CD21high subsets (9), were maintained at 4°C or were incubated at 37°C for 2 and 16 h in RPMI 1640 medium plus 10% FCS in the presence or absence of 1 μg/ml FLAG-tagged Fas ligand (FasL; Kamiya Biomedical) or 800 ng/ml BLyS. Cells were collected, washed, resuspended in annexin V binding buffer (BD Biosciences), stained with FITC-labeled annexin V, and PE-labeled anti-CD21 mAb (Biosource International) according to the manufacturer's specifications for Apoptosis Detection Kit 1 (BD Biosciences).
For effect of HIV on Fas-mediated apoptosis, autologous virus was recovered from CD4+ T cells of HIV-viremic patients by coculture with PBMCs from HIV-negative donors as described previously (4), and the virus was clarified and concentrated by ultrafiltration. B cells were incubated with or without FasL in the presence of concentrated virus or concentrated supernatant from PBMCs alone to control for non-HIV effects.
Statistical Analysis.
Group comparisons for surface marker expression on bulk B cells were performed using the nonparametric analysis of variance Kruskal-Wallis test with Dunn's multiple comparison tests for medians. The Spearman rank method was used to test for correlation and adjustment of p-values for multiple testing was done with the Bonferroni method. The differences in cell surface marker expression and BLyS-binding effects relative to CD21 were compared by the two-tailed Wilcoxon signed-rank test.
Online Supplemental Material.
Additional B cell surface marker staining and comprehensive analyses of all markers are presented in Fig. S1 and Table S1. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20032236/DC1.
Results
Up-Regulation of Genes Associated with IFN Stimulation and Terminal Differentiation in B Cells of HIV-viremic Patients.
To establish B cell gene expression profiles associated with HIV infection, we analyzed PBMC-derived B cells from three different groups of individuals: 10 HIV-infected patients who controlled their plasma viremia with antiretroviral therapy (HIV-aviremic patients), 10 HIV-infected patients with similar CD4+ T cell counts but who did not control their plasma viremia (HIV-viremic patients), and 10 HIV-negative healthy donors. The DNA microarray data were analyzed by establishing the genes that were differentially regulated in one group relative to the two other groups. By far, the highest number of differentially expressed genes was observed when B cells of HIV-viremic patients were compared with the B cells of the HIV-aviremic and the HIV-negative patients. Those genes found to be up-regulated in B cells of HIV-viremic patients compared with the two other groups are shown in Fig. 1. Approximately 75% of the genes listed in Fig. 1 can be categorized into one of two functional groups: IFN-stimulated genes (ISGs) and genes associated with B cell activities. Of the 14 B cell–related genes, 10 were associated with B cell terminal differentiation and, whereas two of the remaining genes, Fλ1 and the IgH enhancer sequence found in sterile transcripts preceding gene rearrangement (27), would appear consistent with a more immature phenotype, both have been associated with mature B cell activities. Increased Fλ1 transcription has been observed during the germinal center reaction (28, 29), whereas IgH enhancer-containing transcripts have been associated with class switching (30) and isolated from germinal center–derived B cells (GenBank/EMBL/DDBJ accession nos. AW402612 and AA280858). As for the other two B cell–related genes, CD11b and synapsin III, both have been associated with B cell activities (28, 31), although it is currently unknown why they would be overexpressed in B cells of HIV-viremic patients. Overall, these data illustrate both the classic effects of a viral infection on cells of the immune system (32) as shown by the abundance of ISGs, and the unique effect of HIV on B cells (9), as shown by the genes associated with terminal differentiation, including various surface markers (see next paragraph), proteins of secretory pathways, and immunoglobulin-related genes. Of the 17 ISGs found to be up-regulated in B cells of HIV-viremic patients, 9 were identical to genes induced by treatment of cells with IFNs (33) and 5 were identical to those found to be up-regulated in systemic lupus erythematosus (SLE; reference 34), an inflammatory disease that has similarities with HIV infection with regard to perturbations of B cells. Of note, five of the ISGs are also associated with lupus inclusions, intracellular formations found in B cells after treatment with type I IFNs (35), and in lymphoid tissues of HIV-infected patients (36). Several genes up-regulated in B cells of HIV-viremic patients were also found to be up-regulated in multiple myeloma, including BCMA, CXCR3, and annexin A2 (37). Considering that multiple myeloma is a B cell neoplasm characterized by expansion of plasma cells, these similarities in gene expression profiles further implicate terminal differentiation as a mechanism of B cell perturbation in HIV-viremic patients.
Figure 1.

Genes up-regulated greater than twofold in B cells of 10 HIV-viremic patients compared with 10 HIV-aviremic patients and 10 HIV-negative donors. The results depict all genes whose profiles were significantly different between HIV-viremic patients and the other groups of individuals (grouped together), as evaluated by T-statistics. The genes are ordered by the ratio of mean values between these two groups and represent one out of two independent hybridizations. A cut-off of twofold was used to produce this gene set. Relative levels of gene expression (median-centered normalized expression values) are shown in color according to the scale at the bottom, where red indicates up-regulation and green indicates down-regulation. Data points with CV ≥0.8 are depicted in gray. The categories of B and B-TD refer to genes associated with B cell activities and B cell terminal differentiation, respectively. The values in the column on the left represent median signal intensities obtained after linear normalization and averaging of duplicate spot signals for each clone.
Up-regulated Genes for B Cell Surface Markers Are Correlated with CD21low Population.
Several of the genes shown in Fig. 1 encode cell surface proteins, including BCMA, CD95, CD38, CXCR3, and CD11b. Increases in expression of BCMA, CD38, and CXCR3 have been associated with terminal differentiation of B cells (38–40), whereas CD95, also known as Fas, has been associated with IFN stimulation (33). To determine whether the up-regulation of genes of these B cell surface markers was linked to our previous observations regarding expression of CD21 (9), we evaluated the expression of each B cell marker relative to CD21 expression. Although it was impossible to perform the gene expression analyses on B cells fractionated into CD21low and CD21high subpopulations due to the paucity and fragility of cells after cell sorting, we took advantage of multicolor flow cytometry to analyze the expression of the various B cell markers of interest relative to CD21. As illustrated in Fig. 2 by analyses of a representative patient in each group, expression of BCMA, CD95, and CD38 was increased on B cells of HIV-viremic patients when compared with the representative HIV-aviremic patient and HIV-uninfected donor. Furthermore, the increased expression of these receptors was predominantly associated with the CD21low population, a population that was marginally present in B cells of HIV-aviremic and HIV-uninfected patients. Although not as dramatic, similar patterns were observed for CXCR3 and, to a lesser extent, CD11b (unpublished data), observations that were consistent with the gene expression profiles in Fig. 1.
Figure 2.
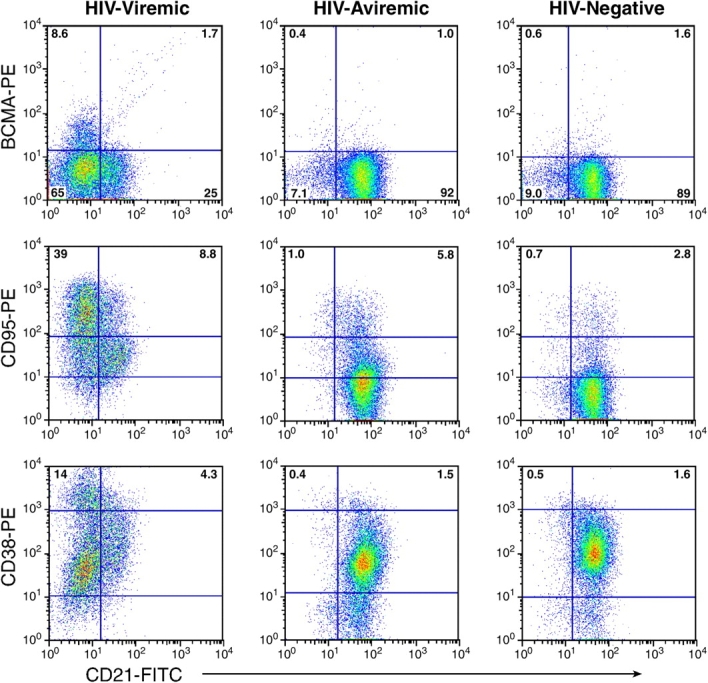
Phenotyping of B cell markers found to be up-regulated in HIV-viremic patients by microarray analyses. A gate was set on CD19-positive B cells, and cells were analyzed for expression of BCMA, CD95, and CD38 relative to CD21. Representative profiles from an HIV-viremic patient, an HIV-aviremic patient, and an HIV-negative individual are shown. Inset values shown for CD95 and CD38 represent percentage of B cells expressing high-intensity receptors delineated by the top horizontal bar.
A more extensive validation of the microarray data was sought by extending the flow cytometric analyses illustrated in Fig. 2 to include the majority of the patients whose gene expression profiles are shown in Fig. 1. We not only concentrated on the genes that were found to be elevated by microarray analysis but also on B cell markers that could help us better determine the state of differentiation of the B cells. BCMA, described previously as an intracellular protein in B cells (41), was consistently up-regulated on the CD21low fraction of B cells enriched in HIV-viremic patients yet consistently absent from the surface of CD21high B cells (Fig. 3 A and Table S1, available at http://www.jem.org/cgi/content/full/jem.20032236/DC1), and levels of BCMA expression on B cells of HIV-viremic patients were significantly higher than on B cells of HIV-aviremic patients (P < 0.001) and HIV-negative donors (P < 0.01). A slightly different approach was used to demonstrate differences in levels of expression of CD95 and CD38; it is the high-density surface expression of each of these receptors that appears to be important for susceptibility to apoptosis and association with terminal differentiation, respectively (42, 43). Although CD38 was expressed on a high percentage of B cells in all three categories of patients studied, expression of high-intensity CD38 was increased on B cells of viremic patients compared with the other two groups (P < 0.01), with CD21low B cells accounting for the majority of the increases (Fig. 3 B and Table S1). When CD95 levels were analyzed, a similar pattern of expression was observed (Fig. 3 C and Table S1). CD21low B cells of HIV-viremic patients were found to express very high levels of CD95 when compared with their CD21low counterpart (P < 0.001), and levels of CD95 expression on B cells of HIV-viremic patients were significantly higher than on B cells of HIV-aviremic patients (P < 0.05) and HIV-negative donors (P < 0.001). Other distinctive patterns relative to CD21 expression included significantly decreased levels of CD20, CD22, CD124 (IL-4 receptor), and CD25 on CD21low B cells and significantly increased levels of CD27, CD80, CD86, CXCR3, and high-intensity CD22 on CD21low B cells (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20032236/DC1, and Table S1). These patterns of expression were consistent with previous observations made on B cells of HIV-viremic patients, including increased activation (CD80 and CD86; reference 10), decreased function (CD25; references 10, 16), and induction of terminal differentiation (CD20, CD25, CD27, and CXCR3; references 38, 39). It should be noted that for CD20 and CD27, changes in expression were not as pronounced as reported for plasma cells, and often missed if the analyzed cells came from cryopreserved samples (9). However, these changes were always present to some degree on the CD21low fraction of B cells of HIV-viremic patients, and easier to establish with a weak fluorochrome such as FITC (Fig. 2 and Fig. S1) compared with a strong one such as PE (9). Collectively, these phenotypic data validate the microarray data and extend our previous findings by demonstrating that the CD21low B cells are largely responsible for the alterations observed in HIV-viremic patients.
Figure 3.



Flow cytometric analysis of BCMA (A), CD38 (B), and CD95 (C) expression relative to CD21 on B cells. Levels of expression measured by gating on CD19-positive B cells are shown for nine HIV-viremic patients, eight HIV-aviremic patients, and nine HIV-negative individuals.
Increased Expression of CD95 Correlates with Increased Susceptibility to Fas-mediated Apoptosis.
Having demonstrated that CD21low B cells of HIV-viremic patients express aberrantly high levels of CD95, we sought to determine whether this subset was more prone to Fas-mediated apoptosis when compared with its CD21high counterpart and B cells of HIV-aviremic patients and HIV-negative donors. To this end, B cells were incubated with or without soluble FasL for 2 h and stained with annexin V and anti-CD21 mAb. As demonstrated in Fig. 4 A by comparing cells incubated at 4 and 37°C, B cells of the HIV-viremic patient were far more prone to spontaneous apoptosis than were B cells of the other two categories of patients and, furthermore, most of the apoptotic cells belonged to the CD21low subset of B cells. The addition of FasL further increased the level of apoptosis in B cells of HIV-viremic patients, and again the apoptotic cells were largely restricted to the CD21low subset of B cells that was over-represented in HIV-viremic patients. In HIV-aviremic patients and HIV-negative donors, addition of FasL induced marginal levels of apoptosis (Fig. 4 A). When data were collected on several individuals in each category, a direct correlation was found between the level of CD95 expression and the level of apoptosis induced by FasL (P < 0.0001; Fig. 4 B). Furthermore, when levels of HIV plasma viremia for each HIV-infected patient were plotted against Fas-mediated apoptosis, these two parameters were directly correlated (P = 0.001; Fig. 4 C). Given that triggering of the BCR has been shown to reverse Fas-mediated apoptosis in B cells (44), we tested whether the triggering of HIV-specific B cells by autologous virus could lead to protection from Fas-mediated apoptosis. Of four viremic patients tested, evidence for protection from Fas-mediated apoptosis was observed in one patient and only when the concentration of HIV p24 antigen was >1 μg/ml (unpublished data), which is 10,000 times above the highest levels reported in plasma (45). Hence, it is highly unlikely that the presence of virus in vivo can protect B cells of HIV-viremic patients from Fas-mediated apoptosis. Collectively, these observations demonstrate that B cells of HIV-viremic patients, and in particular the CD21low population, are more prone to spontaneous and Fas-mediated apoptosis than are B cells of HIV-aviremic patients and HIV-negative donors.
Figure 4.
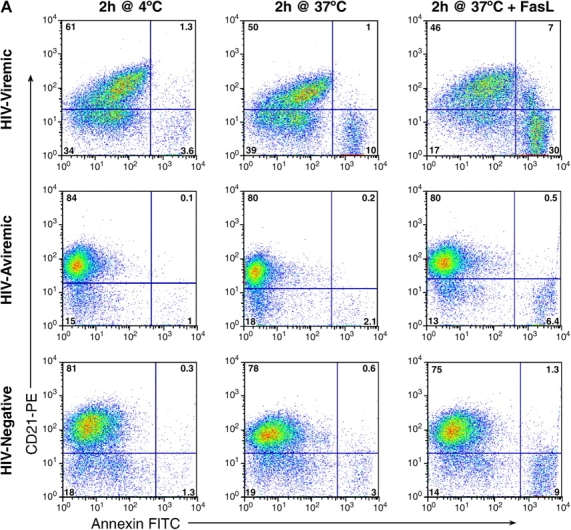

B cell responses to FasL. B cells were incubated at 4°C or at 37°C with and without FasL for 2 h and stained for CD21 and annexin V. (A) FACS® plots on live-gated cells are shown for a representative HIV-viremic patient, HIV-aviremic patient, and HIV-negative individual (see Table S1 for complete dataset). Correlation between percentage of annexin V staining in B cells where levels of staining without FasL were subtracted from levels with FasL and (B) percentage of CD95 expression on B cells or (C) levels of HIV plasma viremia in the HIV-infected patients (limit of detection was 50 copies/ml of plasma).
B Cells of HIV-viremic Patients Express Increased Levels of BCMA and TACI and Decreased Levels of BAFF-R.
The microarray (Fig. 1) and phenotypic (Figs. 2 A and 3 A) analyses indicated increased expression of BCMA, one of three known receptors for BLyS (also known as BAFF, TALL-1, THANK, zTNF4, or TNFSF13b; reference 41), in and on B cells of HIV-viremic patients. Although the genes for the two other receptors for BLyS, namely BAFF-R and TACI, were not on the array used in this hybridization, we analyzed each receptor by flow cytometry. When levels of BCMA, TACI, and BAFF-R were measured on B cells from the three groups of patients, a clear trend was observed that contrasted B cells of HIV-viremic patients with those of HIV-aviremic and HIV-negative donors (Fig. 5). Consistent with data shown in Figs. 2 A and 3 A, BCMA expression was restricted to the CD21low population of the HIV-viremic patient. Expression of TACI was found to be more variable. However, overall, the B cells of HIV-viremic patients expressed significantly more TACI than did B cells of HIV-aviremic and HIV-negative donors (Table S1), and CD21low B cells of HIV-viremic patients expressed significantly more TACI than did their CD21high counterparts. Finally, significantly lower levels of BAFF-R were observed on B cells of HIV-viremic patients when compared with B cells of HIV-aviremic and HIV-negative donors (Fig. 5 and Table S1). The loss in BAFF-R expression was largely seen among CD21low B cells of the HIV-viremic patients with a median of 76.3% of CD21low B cells expressing BAFF-R compared with 96.3% for CD21high B cells (Fig. 6 A and Table S1; P < 0.001). Collectively, these data suggest that B cells of HIV-viremic patients, and in particular their CD21low B cells, tend to express increased levels of BCMA and TACI and decreased levels of BAFF-R. These data are consistent with findings of a recent paper (40), where the same pattern of increased BCMA and TACI and decreased BAFF-R expression was observed when B cells were differentiated into plasma cells in vitro, a process that is thought to occur in vivo in HIV-infected viremic patients (9).
Figure 5.

Representative expression of BCMA, TACI, and BAFF-R, as well as BLyS-binding, on B cells of HIV-viremic, HIV-aviremic, and HIV-negative individuals. A gate was set on CD19-positive B cells, and cells were analyzed for expression of BCMA, TACI, and BAFF-R relative to CD21. Representative profiles from an HIV-viremic patient, an HIV-aviremic patient, and an HIV-negative donor are shown (see Table S1 for complete dataset).
Figure 6.

Levels of (A) BAFF-R expression and (B) BLyS-binding on CD21low versus CD21high B cells of HIV-viremic patients. Levels of BAFF-R expression and BLyS-binding were measured on CD19-gated B cells of 16 HIV-viremic patients and are expressed as the percentage of BAFF-R expression on the CD21low and CD21high populations for each patient. (C) Survival effect of BLyS measured on B cells of HIV-viremic patients fractionated into CD21low and CD21high subsets. The percent survival after 16 h incubation was calculated as the fraction of annexin-positive cells measured in the presence of BLyS over annexin-positive cells measured in the absence of BLyS multiplied by 100.
The significance of the modulations in the expression of BCMA, TACI, and BAFF-R on B cells of HIV-viremic patients was investigated relative to ligand binding and ligand-mediated functional effects. Although BLyS has been shown to bind all three receptors, BAFF-R is thought to be the receptor with the highest affinity (46). Consistent with this observation, we found that regardless of the profile of expression of BCMA and TACI, BLyS-binding on B cells mirrored BAFF-R expression (Fig. 5). The significant reduction of BAFF-R expression on CD21low B cells of viremic patients was also observed for BLyS-binding (Fig. 6 B and Table S1; P < 0.001) and, furthermore, there was a significant correlation between BAFF-R expression and BLyS-binding on B cells of HIV-viremic patients (r = 0.69, P < 0.005; not depicted). Finally, we sought to determine whether the decrease in BLyS-binding on CD21low relative to CD21high B cells had an impact on the responsiveness of these B cell fractions to BLyS. One of the major effects of BLyS on mature B cells is to promote cell survival by preventing apoptosis (47). Accordingly, B cells of HIV-viremic patients, known to be highly susceptible to spontaneous apoptosis (Fig. 4 A and references 17, 18), were fractionated into CD21low and CD21high subsets and incubated with and without BLyS for 16 h. As shown in Fig. 6 C, the survival effect of BLyS was significantly more pronounced on CD21high compared with CD21low B cell fractions (P = 0.01) and, furthermore, there was a significant correlation between the differences in BLyS-binding between the two fractions and their responsiveness to BLyS (r = 0.66, P < 0.05; not depicted). Together, these data demonstrate that loss of BLyS-binding on B cells of HIV-viremic patients is largely restricted to the CD21low B cell population that in turn translates into a loss in BLyS-mediated survival.
Discussion
In the present work, we extend our understanding of how HIV viremia leads to aberrant activation of B cells, as evidenced by hypergammaglobulinemia (5–7), increased expression of activation markers (8, 10), and increased susceptibility to apoptosis (17, 18), by demonstrating that B cells of HIV-viremic patients up-regulate numerous genes associated with IFN stimulation and terminal differentiation. We also provide evidence of a unique intersection between these two pathways and our previous findings showing reduced expression of CD21 on B cells of HIV-viremic patients (9). Although the previous paper suggested that loss of CD21 expression, both at the protein and transcription levels, and concomitant increases in immunoglobulin secretion, decrease in proliferative potential, and appearance of plasmacytoid features were associated with terminal differentiation, the current work adds additional dimensions of dysfunction. CD95, an apoptosis marker that is not strongly modulated during B cell terminal differentiation (38, 48), but is up-regulated as a result of IFN stimulation (33), was markedly up-regulated on CD21low B cells of HIV-viremic patients. Furthermore, by showing that CD21low B cells were considerably more prone to Fas-mediated apoptosis than were their CD21high counterpart and B cells of HIV-aviremic patients and HIV-negative donors, and by demonstrating that there was a direct correlation between Fas-mediated apoptosis and HIV plasma viremia, our findings also offer a firm basis to explain why B cells of HIV-viremic patients are dysfunctional. In this regard, with an abnormally high percentage of B cells that are destined to terminal differentiation and apoptosis, HIV-viremic patients are less likely to respond adequately to antigenic stimulation. Finally, our findings also suggest that B cells of HIV-viremic patients lose responsiveness to BlyS-mediated survival signals as a result of activation-induced down-regulation of the main BlyS receptor, BAFF-R.
The increased expression of the death receptor CD95 and decreased expression of the survival receptor BAFF-R would be predicted to cause a net decrease in B cell numbers in the setting of ongoing HIV replication. The current work did not address this issue directly because its cross-sectional nature and the large donor-to-donor variability in total B cell numbers observed both in HIV-infected and HIV-negative donors were unlikely to yield conclusive results. These limitations were further compounded by complex and controversial issues of increased cell turnover and tissue redistribution associated with HIV viremia, which have been reported mainly for CD4+ T cells, but also suggested for B cells (49, 50). Nonetheless, when total B cell numbers were analyzed from a well-controlled longitudinal study of HIV-infected patients who cycled off (4 wk) and on (8 wk) antiretroviral therapy, a significant inverse correlation was observed between total B cell numbers in peripheral blood and HIV plasma viremia in seven out of nine patients who were on study for a minimum of 60 wk (reference 51 and unpublished data). Hence, despite the increased B cell turnover associated with ongoing HIV replication, there exists clear evidence that it is offset by increased B cell death, possibly as a result of modulations in the expression of TNFSF receptors such as CD95 and BAFF-R.
The roles of recently identified members of the TNFSF of receptors BCMA, TACI, and BAFF-R in B cell survival and proliferation remain nebulous in humans and only partially defined in mice (52, 53). Although a prominent role for BAFF-R in the development of B cells in mice has been definitively shown with BAFF-R–deficient mice (54), similar targeted deficiencies in BCMA and TACI have failed to demonstrate a clear role for these two receptors in B cell development (55–58). Three of these studies found B cell hyperactivity in the TACI-deficient mice, suggesting that TACI is a negative regulator of B cell function (56–58). However, TACI-deficient mice have also been shown to be deficient in T-independent humoral responses (56, 57), suggesting that TACI may be necessary for certain functions. In human B cells, the triggering of TACI with agonistic antibodies (NF-κB inducing) has been shown to interfere with CD40-mediated B cell proliferation, suggesting TACI may also deliver inhibitory signals in human B cells (58).
In this work, we observed a pattern of expression of the BLyS receptors BCMA, TACI, and BAFF-R that was clearly modulated by HIV plasma viremia. B cells of HIV-viremic patients expressed increased levels of BCMA, variable levels of TACI, and decreased levels of BAFF-R when compared with HIV-aviremic patients and HIV-negative individuals. These observations are consistent with findings showing that when B cells undergo terminal differentiation in vitro, they undergo the same BLyS receptor modulations as those described in the HIV-viremic patients (40). Also, in agreement with a recent paper describing much stronger binding kinetics between BLyS and BAFF-R than between BLyS and BCMA (46), we found that levels of BAFF-R expression dictated levels of BLyS binding, regardless of increases in TACI and BCMA expression. Furthermore, the loss of BLyS-binding on CD21low B cells of HIV-viremic patients also translated into a loss in BLyS-mediated survival, as indicated by increased apoptosis of CD21low compared with CD21high B cells in response to BLyS. Current efforts are underway to further evaluate the consequences of modulations in the expression of BCMA, TACI, and BAFF-R relative to responsiveness to the BLyS-related ligand APRIL, which binds BCMA and TACI, but not BAFF-R (54, 59).
The extensive phenotyping performed to better characterize the differentiation stage of B cells of HIV-viremic patients further established a pattern of expression that was clearly delineated by CD21 expression. It should be noted that we found no evidence that the CD21low population of B cells in HIV-viremic patients was any different phenotypically than the CD21low B cells of HIV-aviremic patients and HIV-negative donors; this population was merely overrepresented in HIV-viremic patients compared with the two other groups. Irrespective of disease status, the CD21low population was always more susceptible to apoptosis than its CD21high counterpart (Fig. 4 A). The B cell surface markers that were found to be up-regulated in HIV-viremic patients at the gene expression level were also found to be up-regulated at the protein level on the CD21low B cells. Consistent with published literature on the phenotypic profile of plasma cells (38, 60, 61), B cells of HIV-viremic patients exhibited many of the same characteristic features, including reduced expression of CD20, CD21, CD22, and CD25, as well as increased expression of BCMA, CD38, CXCR3, CD27, and CD86, with the CD21low fraction accounting for most of these modulations. In contrast, certain B cell markers that are down-regulated on plasma cells were not found to be down-regulated on B cells of HIV-viremic patients, including HLA-DR and surface Ig, or in the case of CD20, were down-regulated on a subset of CD21low B cells. CD27 has been shown to be decreased on B cells of HIV-infected patients (62, 63). However, these studies did not focus on the patients with detectable HIV plasma viremia, and we did not focus on patients with very low CD4+ T cell counts who do have a paucity of CD27-expressing B cells (unpublished data). CD22 was also discordant in that, whereas a fraction of CD21low B cells of viremic patients expressed reduced levels of CD22, consistent with features of plasma cells, another fraction expressed higher than normal intensities of CD22 (Fig. S1). Preliminary observations also indicated that expression of CD138 and the transcription factor Blimp-1, hallmarks of plasma cell differentiation (64), was not substantially modulated in and on B cells of HIV-viremic patients (unpublished data). However, considering the variability in the expression of these factors in primary human B cells and the heterogeneity of B cells in the peripheral blood (38, 65, 66), further investigation is needed. Nonetheless, the differences clearly delineated in this work suggest that B cells of viremic patients have a unique differentiation pathway and/or become engaged in terminal differentiation, but do not complete the process. Finally, the levels of CD95 observed on B cells of HIV-viremic patients were higher than those reported for blood-derived plasma cells (38), suggesting that the IFN-stimulated component of responses to viruses that affects CD95 expression may be imparting certain nonplasma cell features in HIV-viremic patients.
The up-regulation of ISGs appears to be an observation common not only to viral infections but also to other pathologic conditions, including inflammatory diseases such as SLE (34) and malignancies associated with high cell turnover (67). Of note, the loss of CD21 expression has also been observed on B cells of patients with inflammatory diseases such as SLE and rheumatoid arthritis (68, 69), but not on B cells of patients with hepatitis C virus infection (70), a viral infection that is often compared with HIV with regard to chronically high viral loads. Hence, it is difficult to establish which of the three factors, namely viral, inflammatory, or terminal differentiation, had a predominant effect on the B cells of HIV-viremic patients. Of note, early histological studies on tissues from HIV-infected patients found evidence of lupus inclusions or cisternae in the lymphoid sections (36), and these same intracellular formations were later reproduced by treating B cell lines with type I IFNs (71), and more recently these inclusions were observed in B cells isolated from HIV-viremic patients (Orenstein, J.M., personal communication). Several components of these inclusions were found to be up-regulated in B cells of HIV-viremic patients, providing a potential explanation for observations made >15 yr ago and further associating ongoing viral replication with morphological abnormalities of B cells.
In conclusion, we have combined DNA microarray data with phenotypic and functional analyses to establish a clear pattern of B cell dysregulation in HIV-viremic patients. This work supports and considerably extends our previous findings that HIV-mediated hyperactivation induces B cell terminal differentiation that is characterized by loss of CD21 expression, reduced proliferative potential, increased immunoglobulin secretion, and appearance of plasmacytoid features. The gene expression profiles described herein are consistent with the notion that HIV-mediated hyperactivation engages B cells along a pathway related to terminal differentiation. Furthermore, in validating the microarray data with phenotypic data, we find that the majority of aberrantly expressed cell surface markers are associated with the CD21low population of B cells that is enriched in HIV-viremic patients, diminished in HIV-aviremic patients, and mostly absent in HIV-negative individuals. Finally, we find that CD21low B cells of HIV-viremic patients, through shifts in expression of TNFSF receptors CD95, BCMA, and BAFF-R, may become more susceptible to cell death and less responsive to survival signals. Increased CD95 expression and correspondingly increased Fas-mediated apoptosis are unique features of the B cell activation profile in HIV-viremic patients as this death receptor is an ISG and not a receptor associated with terminal differentiation (33, 38). Together, the combination of gene expression profiling with phenotypic and functional analyses have enabled us to gain a clear understanding of the pathways that drive B cell dysfunction in HIV-viremic patients and offer new information on avenues that could be considered to reverse these pathways.
Acknowledgments
We thank J.A. Metcalf and C.A. Rehm for sample patient coordination. We are indebted to the patients for their participation and commitment to our research efforts.
This work was performed at the National Institute of Allergy and Infectious Diseases, National Institutes of Health.
The authors have no conflicting financial interests.
S. Moir and A. Malaspina contributed equally to this work.
Abbreviations used in this paper: BLyS, B lymphocyte stimulator; FasL, Fas ligand; ISG, IFN-stimulated gene; SLE, systemic lupus erythematosus; TNFSF, TNF superfamily.
References
- 1.Grossman, Z., M. Meier-Schellersheim, A.E. Sousa, R.M. Victorino, and W.E. Paul. 2002. CD4+ T-cell depletion in HIV infection: are we closer to understanding the cause? Nat. Med. 8:319–323. [DOI] [PubMed] [Google Scholar]
- 2.Hazenberg, M.D., D. Hamann, H. Schuitemaker, and F. Miedema. 2000. T cell depletion in HIV-1 infection: how CD4+ T cells go out of stock. Nat. Immunol. 1:285–289. [DOI] [PubMed] [Google Scholar]
- 3.Pantaleo, G., and A.S. Fauci. 1995. New concepts in the immunopathogenesis of HIV infection. Annu. Rev. Immunol. 13:487–512. [DOI] [PubMed] [Google Scholar]
- 4.Moir, S., A. Malaspina, Y. Li, T.W. Chun, T. Lowe, J. Adelsberger, M. Baseler, L.A. Ehler, S. Liu, R.T. Davey, et al. 2000. B cells of HIV-1–infected patients bind virions through CD21-complement interactions and transmit infectious virus to activated T cells. J. Exp. Med. 192:637–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pahwa, R., R.A. Good, and S. Pahwa. 1987. Prematurity, hypogammaglobulinemia, and neuropathology with human immunodeficiency virus (HIV) infection. Proc. Natl. Acad. Sci. USA. 84:3826–3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morris, L., J.M. Binley, B.A. Clas, S. Bonhoeffer, T.P. Astill, R. Kost, A. Hurley, Y. Cao, M. Markowitz, D.D. Ho, and J.P. Moore. 1998. HIV-1 antigen-specific and -nonspecific B cell responses are sensitive to combination antiretroviral therapy. J. Exp. Med. 188:233–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shirai, A., M. Cosentino, S.F. Leitman-Klinman, and D.M. Klinman. 1992. Human immunodeficiency virus infection induces both polyclonal and virus-specific B cell activation. J. Clin. Invest. 89:561–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Forster, R., G. Schweigard, S. Johann, T. Emrich, E. Kremmer, C. Nerl, and M. Lipp. 1997. Abnormal expression of the B-cell homing chemokine receptor BLR1 during the progression of acquired immunodeficiency syndrome. Blood. 90:520–525. [PubMed] [Google Scholar]
- 9.Moir, S., A. Malaspina, K.M. Ogwaro, E.T. Donoghue, C.W. Hallahan, L.A. Ehler, S. Liu, J. Adelsberger, R. Lapointe, P. Hwu, et al. 2001. HIV-1 induces phenotypic and functional perturbations of B cells in chronically infected individuals. Proc. Natl. Acad. Sci. USA. 98:10362–10367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Malaspina, A., S. Moir, S. Kottilil, C.W. Hallahan, L.A. Ehler, S. Liu, M.A. Planta, T.W. Chun, and A.S. Fauci. 2003. Deleterious effect of HIV-1 plasma viremia on B cell costimulatory function. J. Immunol. 170:5965–5972. [DOI] [PubMed] [Google Scholar]
- 11.Amadori, A., A. De Rossi, G.P. Faulkner-Valle, and L. Chieco-Bianchi. 1988. Spontaneous in vitro production of virus-specific antibody by lymphocytes from HIV-infected subjects. Clin. Immunol. Immunopathol. 46:342–351. [DOI] [PubMed] [Google Scholar]
- 12.Lane, H.C., H. Masur, L.C. Edgar, G. Whalen, A.H. Rook, and A.S. Fauci. 1983. Abnormalities of B-cell activation and immunoregulation in patients with the acquired immunodeficiency syndrome. N. Engl. J. Med. 309:453–458. [DOI] [PubMed] [Google Scholar]
- 13.Miedema, F., A.J. Petit, F.G. Terpstra, J.K. Schattenkerk, F. de Wolf, B.J. Al, M. Roos, J.M. Lange, S.A. Danner, J. Goudsmit, et al. 1988. Immunological abnormalities in human immunodeficiency virus (HIV)-infected asymptomatic homosexual men. HIV affects the immune system before CD4+ T helper cell depletion occurs. J. Clin. Invest. 82:1908–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ballet, J.J., G. Sulcebe, L.J. Couderc, F. Danon, C. Rabian, M. Lathrop, J.P. Clauvel, and M. Seligmann. 1987. Impaired anti-pneumococcal antibody response in patients with AIDS-related persistent generalized lymphadenopathy. Clin. Exp. Immunol. 68:479–487. [PMC free article] [PubMed] [Google Scholar]
- 15.Conge, A.M., K. Tarte, J. Reynes, M. Segondy, J. Gerfaux, M. Zembala, and J.P. Vendrell. 1998. Impairment of B-lymphocyte differentiation induced by dual triggering of the B-cell antigen receptor and CD40 in advanced HIV-1-disease. AIDS. 12:1437–1449. [DOI] [PubMed] [Google Scholar]
- 16.Moir, S., K.M. Ogwaro, A. Malaspina, J. Vasquez, E.T. Donoghue, C.W. Hallahan, S. Liu, L.A. Ehler, M.A. Planta, S. Kottilil, et al. 2003. Perturbations in B cell responsiveness to CD4+ T cell help in HIV-infected individuals. Proc. Natl. Acad. Sci. USA. 100:6057–6062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Muro-Cacho, C.A., G. Pantaleo, and A.S. Fauci. 1995. Analysis of apoptosis in lymph nodes of HIV-infected persons. Intensity of apoptosis correlates with the general state of activation of the lymphoid tissue and not with stage of disease or viral burden. J. Immunol. 154:5555–5566. [PubMed] [Google Scholar]
- 18.Samuelsson, A., A. Sonnerborg, N. Heuts, J. Coster, and F. Chiodi. 1997. Progressive B cell apoptosis and expression of Fas ligand during human immunodeficiency virus type 1 infection. AIDS Res. Hum. Retroviruses. 13:1031–1038. [DOI] [PubMed] [Google Scholar]
- 19.Jacobson, M.A., H. Khayam-Bashi, J.N. Martin, D. Black, and V. Ng. 2002. Effect of long-term highly active antiretroviral therapy in restoring HIV-induced abnormal B-lymphocyte function. J. Acquir. Immune Defic. Syndr. 31:472–477. [DOI] [PubMed] [Google Scholar]
- 20.Horvath, A., D. Banhegyi, A. Biro, E. Ujhelyi, A. Veres, L. Horvath, Z. Prohaszka, A. Bacsi, V. Tarjan, L. Romics, et al. 2001. High level of anticholesterol antibodies (ACHA) in HIV patients. Normalization of serum ACHA concentration after introduction of HAART. Immunobiology. 203:756–768. [DOI] [PubMed] [Google Scholar]
- 21.Notermans, D.W., J.J. de Jong, J. Goudsmit, M. Bakker, M.T. Roos, L. Nijholt, J. Cremers, J.A. Hellings, S.A. Danner, and A. de Ronde. 2001. Potent antiretroviral therapy initiates normalization of hypergammaglobulinemia and a decline in HIV type 1-specific antibody responses. AIDS Res. Hum. Retroviruses. 17:1003–1008. [DOI] [PubMed] [Google Scholar]
- 22.Taoufik, Y., I. Peguillet, M.G. de Goer, M. Lambert, B. Gubler, A. Trylesinski, J.F. Delfraissy, and O. Lantz. 2001. Effect of highly active antiretroviral therapy on expression of interleukin-10 and interleukin-12 in HIV-infected patients. J. Acquir. Immune Defic. Syndr. 26:303–304. [DOI] [PubMed] [Google Scholar]
- 23.De Milito, A., A. Nilsson, K. Titanji, R. Thorstensson, E. Reizenstein, M. Narita, S. Grutzmeier, A. Sonnerborg, and F. Chiodi. 2003. Mechanisms of hypergammaglobulinemia and impaired antigen-specific humoral immunity in HIV-1 infection. Blood. 103:2180–2186. [DOI] [PubMed] [Google Scholar]
- 24.Tedder, T.F., L.T. Clement, and M.D. Cooper. 1984. Expression of C3d receptors during human B cell differentiation: immunofluorescence analysis with the HB-5 monoclonal antibody. J. Immunol. 133:678–683. [PubMed] [Google Scholar]
- 25.Makar, K.W., C.T. Pham, M.H. Dehoff, S.M. O'Connor, S.M. Jacobi, and V.M. Holers. 1998. An intronic silencer regulates B lymphocyte cell- and stage-specific expression of the human complement receptor type 2 (CR2, CD21) gene. J. Immunol. 160:1268–1278. [PubMed] [Google Scholar]
- 26.Sung, C., B. Nardelli, D.W. LaFleur, E. Blatter, M. Corcoran, H.S. Olsen, C.E. Birse, O.K. Pickeral, J. Zhang, D. Shah, et al. 2003. An IFN-beta-albumin fusion protein that displays improved pharmacokinetic and pharmacodynamic properties in nonhuman primates. J. Interferon Cytokine Res. 23:25–36. [DOI] [PubMed] [Google Scholar]
- 27.Neale, G.A., and G.R. Kitchingman. 1991. mRNA transcripts initiating within the human immunoglobulin mu heavy chain enhancer region contain a non-translatable exon and are extremely heterogeneous at the 5′ end. Nucleic Acids Res. 19:2427–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klein, U., Y. Tu, G.A. Stolovitzky, J.L. Keller, J. Haddad Jr., V. Miljkovic, G. Cattoretti, A. Califano, and R. Dalla-Favera. 2003. Transcriptional analysis of the B cell germinal center reaction. Proc. Natl. Acad. Sci. USA. 100:2639–2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meffre, E., F. Papavasiliou, P. Cohen, O. de Bouteiller, D. Bell, H. Karasuyama, C. Schiff, J. Banchereau, Y.J. Liu, and M.C. Nussenzweig. 1998. Antigen receptor engagement turns off the V(D)J recombination machinery in human tonsil B cells. J. Exp. Med. 188:765–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li, S.C., P.B. Rothman, J. Zhang, C. Chan, D. Hirsh, and F.W. Alt. 1994. Expression of I mu-C gamma hybrid germline transcripts subsequent to immunoglobulin heavy chain class switching. Int. Immunol. 6:491–497. [DOI] [PubMed] [Google Scholar]
- 31.Porton, B., H.T. Kao, and P. Greengard. 1999. Characterization of transcripts from the synapsin III gene locus. J. Neurochem. 73:2266–2271. [DOI] [PubMed] [Google Scholar]
- 32.Biron, C.A., K.B. Nguyen, G.C. Pien, L.P. Cousens, and T.P. Salazar-Mather. 1999. Natural killer cells in antiviral defense: function and regulation by innate cytokines. Annu. Rev. Immunol. 17:189–220. [DOI] [PubMed] [Google Scholar]
- 33.Der, S.D., A. Zhou, B.R. Williams, and R.H. Silverman. 1998. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc. Natl. Acad. Sci. USA. 95:15623–15628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bennett, L., A.K. Palucka, E. Arce, V. Cantrell, J. Borvak, J. Banchereau, and V. Pascual. 2003. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J. Exp. Med. 197:711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rich, S.A., and T.R. Owens. 1984. Purified recombinant human leukocyte interferons IFLrA and IFLrD induce human lupus inclusions in Raji and Daudi cells. J. Interferon Res. 4:335–345. [DOI] [PubMed] [Google Scholar]
- 36.Orenstein, J.M., O.T. Preble, P. Kind, and R. Schulof. 1987. The relationship of serum alpha-interferon and ultrastructural markers in HIV-seropositive individuals. Ultrastruct. Pathol. 11:673–679. [DOI] [PubMed] [Google Scholar]
- 37.Claudio, J.O., E. Masih-Khan, H. Tang, J. Goncalves, M. Voralia, Z.H. Li, V. Nadeem, E. Cukerman, O. Francisco-Pabalan, C.C. Liew, et al. 2002. A molecular compendium of genes expressed in multiple myeloma. Blood. 100:2175–2186. [DOI] [PubMed] [Google Scholar]
- 38.Medina, F., C. Segundo, A. Campos-Caro, I. Gonzalez-Garcia, and J.A. Brieva. 2002. The heterogeneity shown by human plasma cells from tonsil, blood, and bone marrow reveals graded stages of increasing maturity, but local profiles of adhesion molecule expression. Blood. 99:2154–2161. [DOI] [PubMed] [Google Scholar]
- 39.Kunkel, E.J., and E.C. Butcher. 2003. Plasma-cell homing. Nat. Rev. Immunol. 3:822–829. [DOI] [PubMed] [Google Scholar]
- 40.Avery, D.T., S.L. Kalled, J.I. Ellyard, C. Ambrose, S.A. Bixler, M. Thien, R. Brink, F. Mackay, P.D. Hodgkin, and S.G. Tangye. 2003. BAFF selectively enhances the survival of plasmablasts generated from human memory B cells. J. Clin. Invest. 112:286–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mackay, F., P. Schneider, P. Rennert, and J. Browning. 2003. BAFF AND APRIL: a tutorial on B cell survival. Annu. Rev. Immunol. 21:231–264. [DOI] [PubMed] [Google Scholar]
- 42.Terstappen, L.W., S. Johnsen, I.M. Segers-Nolten, and M.R. Loken. 1990. Identification and characterization of plasma cells in normal human bone marrow by high-resolution flow cytometry. Blood. 76:1739–1747. [PubMed] [Google Scholar]
- 43.Krammer, P.H. 2000. CD95's deadly mission in the immune system. Nature. 407:789–795. [DOI] [PubMed] [Google Scholar]
- 44.Rothstein, T.L., J.K. Wang, D.J. Panka, L.C. Foote, Z. Wang, B. Stanger, H. Cui, S.T. Ju, and A. Marshak-Rothstein. 1995. Protection against Fas-dependent Th1-mediated apoptosis by antigen receptor engagement in B cells. Nature. 374:163–165. [DOI] [PubMed] [Google Scholar]
- 45.Prado, J.G., A. Shintani, M. Bofill, B. Clotet, L. Ruiz, and J. Martinez-Picado. 2004. Lack of longitudinal intrapatient correlation between p24 antigenemia and levels of human immunodeficiency virus (HIV) type 1 RNA in patients with chronic hiv infection during structured treatment interruptions. J. Clin. Microbiol. 42:1620–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pelletier, M., J.S. Thompson, F. Qian, S.A. Bixler, D. Gong, T. Cachero, K. Gilbride, E. Day, M. Zafari, C. Benjamin, et al. 2003. Comparison of soluble decoy IgG fusion proteins of BAFF-R and BCMA as antagonists for BAFF. J. Biol. Chem. 278:33127–33133. [DOI] [PubMed] [Google Scholar]
- 47.Do, R.K., E. Hatada, H. Lee, M.R. Tourigny, D. Hilbert, and S. Chen-Kiang. 2000. Attenuation of apoptosis underlies B lymphocyte stimulator enhancement of humoral immune response. J. Exp. Med. 192:953–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Defrance, T., M. Casamayor-Palleja, and P.H. Krammer. 2002. The life and death of a B cell. Adv. Cancer Res. 86:195–225. [DOI] [PubMed] [Google Scholar]
- 49.Kovacs, J.A., R.A. Lempicki, I.A. Sidorov, J.W. Adelsberger, B. Herpin, J.A. Metcalf, I. Sereti, M.A. Polis, R.T. Davey, J. Tavel, et al. 2001. Identification of dynamically distinct subpopulations of T lymphocytes that are differentially affected by HIV. J. Exp. Med. 194:1731–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.De Boer, R.J., H. Mohri, D.D. Ho, and A.S. Perelson. 2003. Turnover rates of B cells, T cells, and NK cells in simian immunodeficiency virus-infected and uninfected rhesus macaques. J. Immunol. 170:2479–2487. [DOI] [PubMed] [Google Scholar]
- 51.Dybul, M., E. Nies-Kraske, M. Daucher, K. Hertogs, C.W. Hallahan, G. Csako, C. Yoder, L. Ehler, P.A. Sklar, M. Belson, et al. 2003. Long-cycle structured intermittent versus continuous highly active antiretroviral therapy for the treatment of chronic infection with human immunodeficiency virus: effects on drug toxicity and on immunologic and virologic parameters. J. Infect. Dis. 188:388–396. [DOI] [PubMed] [Google Scholar]
- 52.Mackay, F., and C. Ambrose. 2003. The TNF family members BAFF and APRIL: the growing complexity. Cytokine Growth Factor Rev. 14:311–324. [DOI] [PubMed] [Google Scholar]
- 53.Schneider, P., and J. Tschopp. 2003. BAFF and the regulation of B cell survival. Immunol. Lett. 88:57–62. [DOI] [PubMed] [Google Scholar]
- 54.Thompson, J.S., S.A. Bixler, F. Qian, K. Vora, M.L. Scott, T.G. Cachero, C. Hession, P. Schneider, I.D. Sizing, C. Mullen, et al. 2001. BAFF-R, a newly identified TNF receptor that specifically interacts with BAFF. Science. 293:2108–2111. [DOI] [PubMed] [Google Scholar]
- 55.Xu, S., and K.P. Lam. 2001. B-cell maturation protein, which binds the tumor necrosis factor family members BAFF and APRIL, is dispensable for humoral immune responses. Mol. Cell. Biol. 21:4067–4074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.von Bulow, G.U., J.M. van Deursen, and R.J. Bram. 2001. Regulation of the T-independent humoral response by TACI. Immunity. 14:573–582. [DOI] [PubMed] [Google Scholar]
- 57.Yan, M., H. Wang, B. Chan, M. Roose-Girma, S. Erickson, T. Baker, D. Tumas, I.S. Grewal, and V.M. Dixit. 2001. Activation and accumulation of B cells in TACI-deficient mice. Nat. Immunol. 2:638–643. [DOI] [PubMed] [Google Scholar]
- 58.Seshasayee, D., P. Valdez, M. Yan, V.M. Dixit, D. Tumas, and I.S. Grewal. 2003. Loss of TACI causes fatal lymphoproliferation and autoimmunity, establishing TACI as an inhibitory BLyS receptor. Immunity. 18:279–288. [DOI] [PubMed] [Google Scholar]
- 59.Yan, M., J.R. Brady, B. Chan, W.P. Lee, B. Hsu, S. Harless, M. Cancro, I.S. Grewal, and V.M. Dixit. 2001. Identification of a novel receptor for B lymphocyte stimulator that is mutated in a mouse strain with severe B cell deficiency. Curr. Biol. 11:1547–1552. [DOI] [PubMed] [Google Scholar]
- 60.Hauser, A.E., G.F. Debes, S. Arce, G. Cassese, A. Hamann, A. Radbruch, and R.A. Manz. 2002. Chemotactic responsiveness toward ligands for CXCR3 and CXCR4 is regulated on plasma blasts during the time course of a memory immune response. J. Immunol. 169:1277–1282. [DOI] [PubMed] [Google Scholar]
- 61.Horst, A., N. Hunzelmann, S. Arce, M. Herber, R.A. Manz, A. Radbruch, R. Nischt, J. Schmitz, and M. Assenmacher. 2002. Detection and characterization of plasma cells in peripheral blood: correlation of IgE+ plasma cell frequency with IgE serum titre. Clin. Exp. Immunol. 130:370–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nagase, H., K. Agematsu, K. Kitano, M. Takamoto, Y. Okubo, A. Komiyama, and K. Sugane. 2001. Mechanism of hypergammaglobulinemia by HIV infection: circulating memory B-cell reduction with plasmacytosis. Clin. Immunol. Immunopathol. 100:250–259. [DOI] [PubMed] [Google Scholar]
- 63.De Milito, A., C. Morch, A. Sonnerborg, and F. Chiodi. 2001. Loss of memory (CD27) B lymphocytes in HIV-1 infection. AIDS. 15:957–964. [DOI] [PubMed] [Google Scholar]
- 64.Calame, K.L., K.I. Lin, and C. Tunyaplin. 2003. Regulatory mechanisms that determine the development and function of plasma cells. Annu. Rev. Immunol. 21:205–230. [DOI] [PubMed] [Google Scholar]
- 65.Tarte, K., F. Zhan, J. De Vos, B. Klein, and J. Shaughnessy, Jr. 2003. Gene expression profiling of plasma cells and plasmablasts: toward a better understanding of the late stages of B-cell differentiation. Blood. 102:592–600. [DOI] [PubMed] [Google Scholar]
- 66.Zhan, F., E. Tian, K. Bumm, R. Smith, B. Barlogie, and J. Shaughnessy, Jr. 2003. Gene expression profiling of human plasma cell differentiation and classification of multiple myeloma based on similarities to distinct stages of late-stage B-cell development. Blood. 101:1128–1140. [DOI] [PubMed] [Google Scholar]
- 67.Shaffer, A.L., A. Rosenwald, E.M. Hurt, J.M. Giltnane, L.T. Lam, O.K. Pickeral, and L.M. Staudt. 2001. Signatures of the immune response. Immunity. 15:375–385. [DOI] [PubMed] [Google Scholar]
- 68.Marquart, H.V., A. Svendsen, J.M. Rasmussen, C.H. Nielsen, P. Junker, S.E. Svehag, and R.G. Leslie. 1995. Complement receptor expression and activation of the complement cascade on B lymphocytes from patients with systemic lupus erythematosus (SLE). Clin. Exp. Immunol. 101:60–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Illges, H., M. Braun, H.H. Peter, and I. Melchers. 2000. Reduced expression of the complement receptor type 2 (CR2, CD21) by synovial fluid B and T lymphocytes. Clin. Exp. Immunol. 122:270–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ni, J., E. Hembrador, A.M. Di Bisceglie, I.M. Jacobson, A.H. Talal, D. Butera, C.M. Rice, T.J. Chambers, and L.B. Dustin. 2003. Accumulation of B lymphocytes with a naive, resting phenotype in a subset of hepatitis C patients. J. Immunol. 170:3429–3439. [DOI] [PubMed] [Google Scholar]
- 71.Bockus, D., F. Remington, J.Y. Luu, M. Bean, and S. Hammar. 1988. Induction of cylindrical confronting cisternae (AIDS inclusions) in Daudi lymphoblastoid cells by recombinant alpha-interferon. Hum. Pathol. 19:78–82. [DOI] [PubMed] [Google Scholar]