

Abstract
Aromatic polyketides are a class of natural products that include many pharmaceutically important aromatic compounds. Understanding the structure and function of PKS will provide clues to the molecular basis of polyketide biosynthesis specificity. Polyketide chain reduction by ketoreductase (KR) provides regio- and stereochemical diversity. Two cocrystal structures of actinorhodin polyketide ketoreductase (act KR) were solved to 2.3 Å with either the cofactor NADP+ or NADPH bound. The monomer fold is a highly conserved Rossmann fold. Subtle differences between structures of act KR and fatty acid KRs fine-tune the tetramer interface and substrate binding pocket. Comparisons of the NADP+- and NADPH-bound structures indicate that the α6–α7 loop region is highly flexible. The intricate proton-relay network in the active site leads to the proposed catalytic mechanism involving four waters, NADPH, and the active site tetrad Asn114-Ser144-Tyr157-Lys161. Acyl carrier protein and substrate docking models shed light on the molecular basis of KR regio- and stereoselectivity, as well as the differences between aromatic polyketide and fatty acid biosyntheses. Sequence comparison indicates that the above features are highly conserved among aromatic polyketide KRs. The structures of act KR provide an important step toward understanding aromatic PKS and will enhance our ability to design novel aromatic polyketide natural products with different reduction patterns.
Nature creates a huge array of natural products that are diverse in their chemical structures and bioactivity. One such example are the polyketides, a large family of natural products that are an extremely rich source of bioactive molecules (1, 2). Representative compounds include cholesterol-lowering drugs (such as lovastatin) (3), antibiotics (such as tetracyclines and actinorhodin), and anticancer agents (such as doxorubicin, Figure 1A) (1, 2). The biosynthesis of these medically important polyketides is achieved by polyketide synthase (PKS),1 which synthesizes polyketides in high quantity and yields. Similar to fatty acid synthase (FAS), the PKSs are multifunctional enzymes that catalyze repeated chain elongations followed by optional chain modifications (4). The variation in chain length, choice of chain-building units, and chain modifications leads to the huge diversity among naturally occurring polyketides. Over the past decade, PKSs have been targets of intensive manipulation and analysis via genetic engineering (2, 5). These studies have given rise to >100 “unnatural” natural products as well as new technologies for manipulating natural product biosynthesis (6, 7). However, this endeavor has been severely hampered by the lack of molecular information about PKS subunits. Structural analyses of PKS subunits will help to answer important questions about polyketide biosynthesis, such as the molecular basis of regio- and stereospecificity of each enzyme, as well as the influence of protein–protein interactions on substrate specificity.
FIGURE 1.
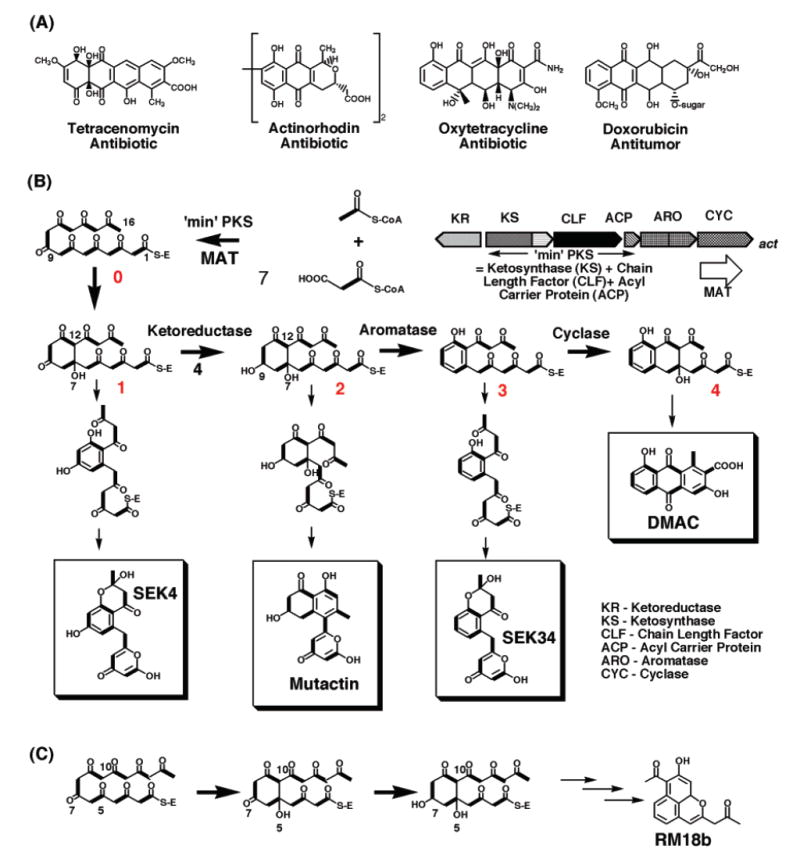
(A) Examples of aromatic polyketide compounds that are therapeutically important. (B) Biosynthesis of actinorhodin. (C) Alternative cyclization between C5–C10 leads to the reduction at C7 position and the eventual product RM18b.
There are at least three architecturally different types of PKSs (8-10), and the focus of this paper is on type II or “aromatic” PKSs. The type II PKSs synthesize aromatic polyketides such as actinorhodin and tetracycline. They are comprised of 5–10 distinct enzymes whose active sites are used iteratively in the chain elongation cycle (11, 12). The polyketide chain is covalently linked to acyl carrier protein (ACP). Following iterative chain elongation by the ketosynthase (KS)/chain length factor (CLF) heterodimer, the first ring is formed uncatalyzed either in the active site of KS/CLF or in the active site of KR, leading to intermediate 1 (Figure 1B). The polyketide chain is then reduced at the C9 position by ketoreductase (KR) to form intermediate 2, followed by subsequent aromatic ring formations catalyzed by aromatase and cyclase (12). Past studies has provided proposals for the function of each type II PKS subunit, as well as the origin of chain length control (11, 12). However, many key events are not well understood, such as the first ring formation and the molecular basis of the polyketide reduction.
The first polyketide chain modification reaction by the ketoreductase (KR) (Figure 1B) is chemically identical to the corresponding fatty acid ketoreduction, in which NADPH reduces a ketone to an alcohol. However, the regiospecificity is very different. Whereas fatty acid KR reduces every carbonyl group on the elongating chain, the aromatic polyketide KR has a high specificity for the C9-carbonyl group, except in special examples where an unusual cyclization pattern can lead to the reduction of other carbonyl groups (Figure 1C) (13, 14). Subsequently, the regiospecificity of the dehydration/cyclization reaction (catalyzed by ARO/CYC) is closely related to the C9 reduction (12). How KR achieves such accurate regiospecificity is not well understood. Similarly, very little is known about the molecular basis of KR stereospecificity. To expand polyketide biosynthesis beyond the current scope, it is essential to understand the molecular basis of KR regio- and stereospecificity.
The aromatic polyketide KRs are highly homologous, with a sequence identity of 39–80% (Figure 2). They belong to the short-chain dehydrogenase/reductase (SDR) family (15), a large family of proteins that use NADPH or NADH as the cofactor and have an active site tyrosine in the center of the Rossmann fold (16). Among the 33 SDRs whose structures are available, the fatty acid KRs share the highest sequence identity to the polyketide KRs. Currently, four fatty acid KRs (also known as FabG) have been solved from Escherichia coli (17, 18), Thermatoga maritima (unpublished results), Mycobacterium tuberculosis (19), and Brassica napus (20). These fatty acid KRs share 30–43% sequence identity with the aromatic polyketide KRs. Past studies on these KRs have identified many residues that are important for enzyme catalysis. Further, the act KR–NADP+ complex crystallized in a condition similar to many SDR crystallization conditions and native gels indicate that the act KR–NADP+ complex is a tetramer, similar to many other SDRs (21). However, one important question remains unanswered: if enzymes in FAS and PKS pathways are similar, why does FAS make aliphatic acids while PKS generates such diverse products?
FIGURE 2.
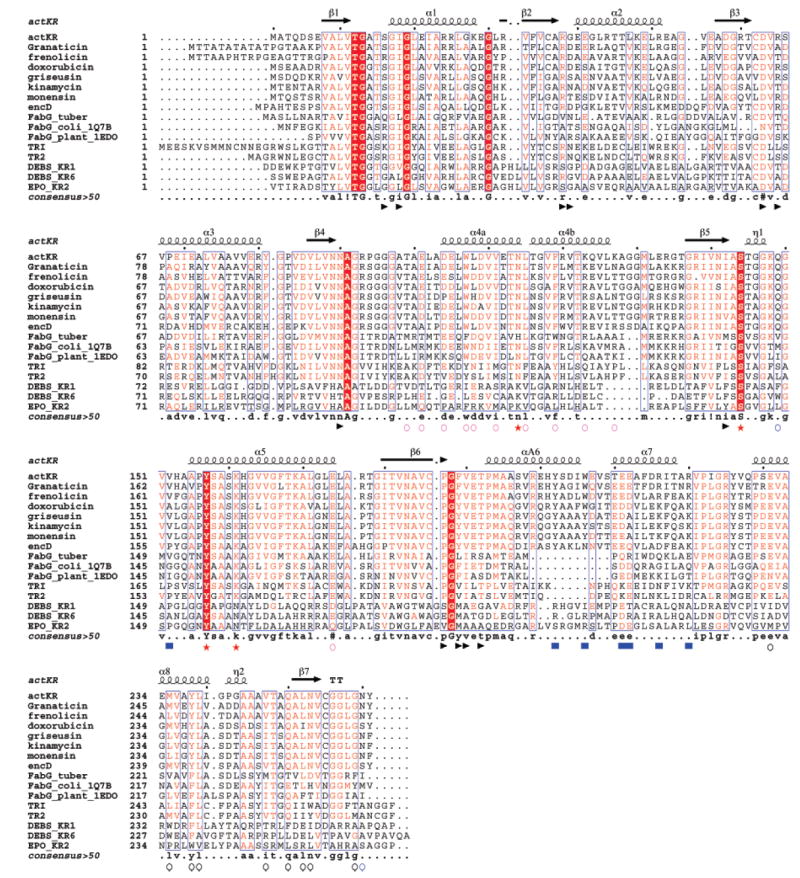
Sequence alignment among different SDRs. ActKR, granaticin, frenolicin, doxorubicin, griseusin, kinamycin, monesin, and encD are representative aromatic polyketide KRs. FabG_tuber, FabG_coli_1Q7B, and FabG_plnat_1EDO are the fatty acid KRs of M. tuberculosis, E. coli, and B. napus. TRI and TR2 are tropinone reductase I and II. DEBS_KR1, DEBS_KR6, and EPO_KR2 are type I (modular) polyketide KRs from DEBS module 1, module 6, and the second KR of epothilone synthase, respectively. Key: red star, catalytic tetrad; blue square, ACP docking site; blue triangle, cofactor binding; black circle, interface of monomers A and D; purple circle, interface of monomers A and B; blue circle, interface of monomers A and C; #, conserved hydrophilic residues; ! and $, conserved hydrophobic residues; %, conserved aromatic residues.
Here, we report the cocrystal structures of the actinorhodin polyketide KR (act KR) bound to NADP+ or NADPH. No polyketide KR structure has been previously reported. These structures allow extensive structural comparisons between act KR and other SDR enzymes. Combined with docking analysis of act KR with either the polyketide substrate or ACP, we discuss possible mechanisms leading to the reduced polyketide product with respect to selective binding motifs and protein–protein interactions.
MATERIALS AND METHODS
Expression and Purification
Strain BL21 λ(DE3) is an E. coli B strain lysogenized with λDE3, a prophage that expresses the T7 RNA polymerase from the IPTG-inducible lacUV5 promoter. Recombinant act KR was cloned into the pET28c vector (Novagen) by overnight restriction digestion of pRZ153 (22) with NdeI and EcoRI, followed by overnight ligation using T4 ligase, resulting in the plasmid pYT238. E. coli BL21 transformed with pYT238 was inoculated into 1 L of LB culture at 37 °C for 4 h followed by the induction of IPTG (1 mM) at 18 °C overnight. The cells were harvested (5000 rpm × 30 min), resuspended in lysis buffer (50 mM Tris, pH 7.5, 0.5 mM DTT, 5 mM imidazole, 10% glycerol), sonicated (5 × 30 s, 15000 rpm × 1 h), and purified by Ni-NTA (10 mL gel volume, 80 mL wash with lysis buffer, 30 mL elution with 100 mM imidazole) to yield >95% pure protein. The buffer was exchanged to either 50 mM sodium phosphate (pH 7.0) or 50 mM Tris (pH 7.0) by overnight dialysis, and the protein was concentrated with the Centricon YM-10 apparatus to concentrations of 20 and 15 mg/mL, respectively.
Crystallization of Act KR plus Cofactors
Cocrystals of act KR and NADP+ or NADPH were grown in sitting drops at room temperature by vapor diffusion. The protein buffer was 50 mM sodium phosphate (pH 7.0) and 10% glycerol for act KR plus NADP+ and 10 mM Tris (pH 7.0) for act KR plus NADPH. Drops were generated by mixing 2 μL of the purified protein solution (10 mg/mL protein, 5 mM NADP+ or NADPH) with 2 μL of well buffer above a well solution of 500 μL. The crystals of KR–NADP+ and KR–NADPH grew in 1 week. The crystallization condition of KR–NADP+ is very similar to the one previously reported (21). The crystals of both KR–NADP+ and KR–NADPH yield the same space group and similar cell dimensions (Table 1).
Table 1.
Crystallization, Data Collection, and Refinement Statistics
| KR–NADP+ | KR–NADPH | |
|---|---|---|
| (A) crystallization | 2 M sodium formate, 100 mM sodium phosphate (pH 7.5) | 2 M ammonium sulfate, 100 mM Tris (pH 8.5) |
| (B) crystallographic data | ||
| space group | P3221 | P3221 |
| cell dimension (Å) | 104.61, 104.61, 124.25, α = β = 90, γ = 120 | 103.70, 103.70, 122.68, α = β = 90, γ = 120 |
| resolution (Å) | 2.3 | 2.2 |
| mosaicity (deg) | 0.4 | 0.4 |
| no. of observations | 943814 | 1361501 |
| no. of unique reflections | 35529 | 39267 |
| completeness (%) (last shell) | 99.8 (100.0) | 100.0 (99.8) |
| I/σ(I) (last shell) | 18.0 (2.9) | 16.0 (1.8) |
| Rmerge (%) (last shell) | 11.5 (54.5) | 9.2 (50.6) |
| (C) refinement | ||
| resolution (Å) | 2.3 | 2.3 |
| no. of reflections | 33832 | 32785 |
| no. of protein atoms | 3680 | 3739 |
| no. of cofactor atoms | 96 | 96 |
| no. of waters | 164 | 194 |
| Rfree (%) | 24.2 | 24.6 |
| Rcrys (%) | 21.0 | 21.9 |
| (D) geometry | ||
| RMS bonds (Å) | 0.006 | 0.006 |
| RMS angles (deg) | 1.31 | 1.29 |
| RMS B main chain | 1.35 | 1.27 |
| RMS B side chain | 2.49 | 2.27 |
| Ramachandran plot (%) | ||
| most favored | 91 | 88.7 |
| favored | 9 | 10.8 |
| generously allowed | 0 | 0.5 |
Data Collection
X-ray diffraction data of KR–NADP+ and KR–NADPH were collected at the Stanford Synchrotron Radiation Laboratory (SSRL) and Advanced Light Source (ALS) to 2.2–2.3 Å (Table 1). Crystals were frozen in 30% glycerol plus 70% well solution. Diffraction intensities were integrated and reduced using the program DENZO and scaled using SCALEPACK (23). A summary of the crystallographic data is shown in Table 1.
Molecular Replacement and Refinement
Initial phases were determined by molecular replacement using a homology model based on the crystal structures of E. coli FabG (PDB code 1Q7C) that was generated by Swiss Model (17). A cross-rotational search followed by a translational search was performed using the program CNS (24). The noncrystallo-graphically related monomers were treated as rigid bodies and were refined using CNS to give an initial Rcrys of 48%. After the structure was rebuilt using Quanta, further refinement was performed using CNS (24). A preliminary round of refinement, using torsion angle simulated annealing followed by energy minimization and positional and individual B-factor refinement, reduced Rcrys to 35%. Subsequent rounds of model building and refinement were carried out using the maximum likelihood based approach implemented within CNS using all data to the highest resolution. Refinement was continued to an Rcrys of <27% (Rfree < 30%). Table 1 lists the statistics for refinement and components of the final model. After the Rfree decreased below 0.30, water molecules were added using CNS followed by visual inspection of the water molecules and final refinement of the B factor. Due to the disordered loop region (residues 200–215), the best Rcrys value that we could attain was 0.210 (Rfree = 0.242). However, outside this loop region, the electron density maps of both the protein and cofactor are well defined.
Model Docking
Docking between act KR–NADPH and act ACP (PDB code 2AF8) (25) was performed using the 3D-Dock program suite (26). ACP and KR were defined as the mobile and static models, respectively. The binding surface of act ACP was exhaustively searched. Two different docking simulations were conducted, one with the KR dimer and the second with the KR tetramer as the static model. Orientations were rapidly scored using an energy grid with a 6–9 attractive–repulsive van der Waals potential with the electrostatic term weighted by 0.01. The energy minimization option was activated with default values. The resulting structures were then energy-minimized using CNS (24). The 10 best complex models, from either KR tetramer–ACP or KR dimer–ACP docking, identified the same region of KR for ACP docking that is near the substrate binding cleft opening. The substrate docking models were generated manually, based on the three docking points discussed in the text, followed by energy minimization using CNS (24).
RESULTS AND DISCUSSION
Overall Fold
Similar to all SDR structures, act KR contains a typical Rossman fold (16), which consists of two right-handed β–α–β–α–β motifs that are connected by α3 (β1–α1–β2–α2–β3, β4–α4–β5–α5–β6; Figures 2 and 3B). Seven parallel β strands, in the order of 3–2–1–4–5–6–7, make up the core region that is flanked by eight helices. The cofactor NADPH is bound at the junction of two β–α–β–α–β motifs (Figure 3B). The polyketide substrate binding pocket is a large cleft (15 Å in width, 19 Å in length, 17 Å in depth) formed by helices α6–α7 and the loops between α4 and α7. For aromatic polyketide KRs, all SDR sequence motifs important for fold maintenance are conserved (Figure 2 and Table 2 in ref 15), such as TGxxxGxG (12–19), D63, and NNAG (89–92). Other important motifs are also conserved, such as the active site tetrad N114, S144, Y157 and K161, the active site N183, the PG motif (187–188), and T192 (Figure 2) (15). Indeed, the act KR structure has an overall RMSD of 0.8–1.3 Å to over 35 known SDR structures with a sequence identity of 19–43%. The biggest difference between the aromatic polyketide KRs and other SDRs [such as FabG (17) and tropinone reductase (27)] is a 10-residue insertion (residues 199–209) between helices 6 and 7 (Figure 2). This region consists of half of the substrate binding pocket and is the least conserved region among SDRs (15). It accounts for the different substrate specificity among SDRs. The α6–α7 region also has the highest B factor in the act KR crystal structure. From sequence comparison, all aromatic polyketide KRs have a highly conserved overall SDR fold and differ from other SDRs by the large insertion region that is important for substrate recognition.
FIGURE 3.
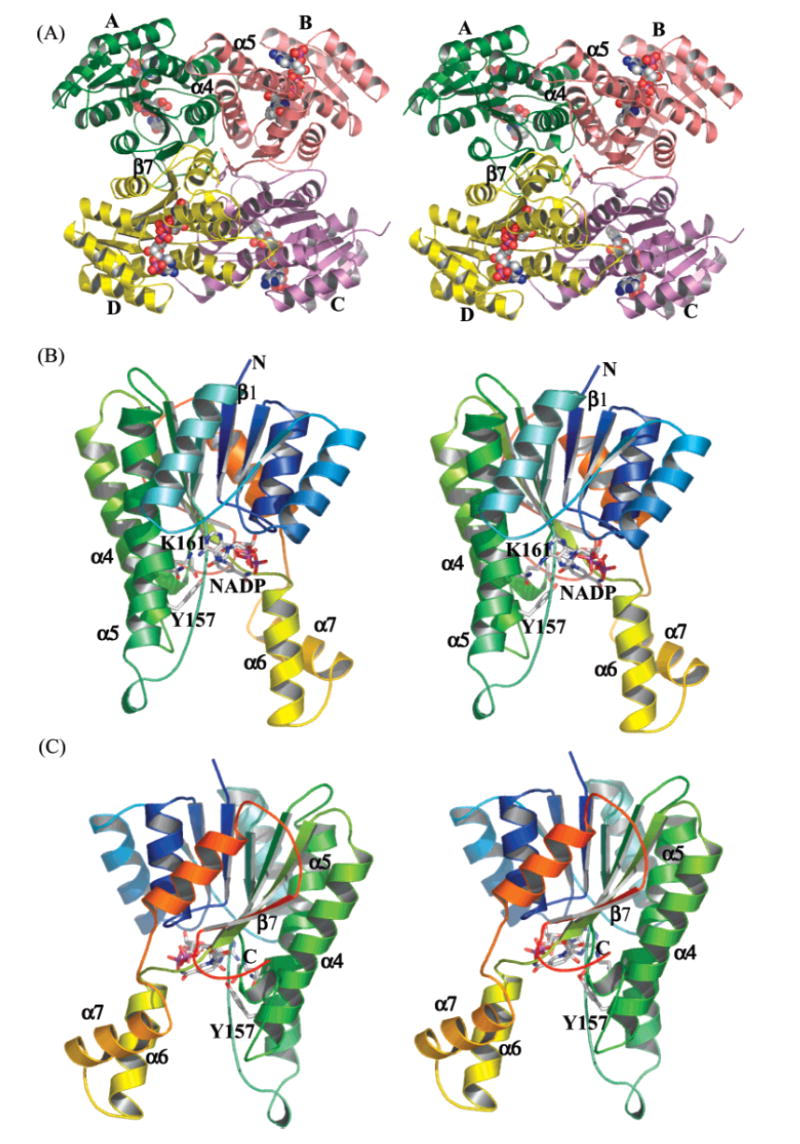
(A) Tetramer structure of act KR. Monomers A, B, C, and D are in yellow, green, red, and purple, respectively. (B) Front side and (C) back side (relative to NADPH binding) of the act KR monomer. The protein is colored from the N to C terminus in blue to red. The cofactor NADPH is shown in spheres in panel A and sticks in panels B and C.
Despite conservation of the conserved overall fold, the monomer–monomer interactions of act KR have subtle yet distinct differences from other SDRs. The biological act KR tetramer is related by the crystallographic 2-fold axis between monomers A–B and monomers C–D (Figure 3A). Monomers A and B, with an overall RMSD of 0.5 Å, adopt the same fold with minor differences due to crystal contacts. Compared to monomer A, monomer B has an additional five N-terminal amino acids and a less ordered loop (residues 201–213) between helices 6 and 7, the most flexible region. Each monomer binds the cofactor (NADP+ or NADPH, Figure 4A) in an identical motif. Three different monomer–monomer interactions maintain the tetramer structure: (1) between monomers A and B (Figure 3A), helical interactions between α4–α4′ are a mixture of hydrophobic (W107:F119′, L108:L115′) and hydrophilic (T99:E174′, E101:K127′, D104:R120′) interactions, while the interactions between α5–α5′ are van der Waals in nature (consists of V152, A159, A170 and L173); (2) monomers A and D form a tight, 14-strand β sheet (Figure 3A), in which the two β7 strands run antiparallel and side chain–side chain interactions are both hydrophobic (V247, M235, Y238, L239, L252) and hydrophilic (E231, Q250:N253′) in nature; (3) finally, the diagonal monomers A and C have no contacting secondary structures; however, at the junction of four monomers, N260:Q149′, R220:R220′, and Y261:Y261′ provide further interactions reenforcing the tetramer structure (Figure 3A). Y229, important for the FabG tetramer interaction (18), is not conserved in aromatic polyketide KRs. Among the interface residues, few (W107, L115, and Y238) are universally conserved among fatty acid KR, type I and type II (aromatic) polyketide KRs (Figure 2). For the β-strand interactions between monomers A and D, most residues are conserved between fatty acid and aromatic polyketide KRs. However, in the α-helix interactions between monomers A and B, many of the residues are only conserved in aromatic polyketide KRs but not in fatty acid KRs. The interaction between monomers A and C is also conserved only in aromatic polyketide KRs (Figure 2). As a result, the 2-fold symmetric axis between monomers A and B in act KR is tilted ~3° from the axis in E. coli FabG (18). Because of the close proximity of these quaternary interactions to the substrate binding pocket, the binding of NADPH to E. coli FabG exhibits negative cooperativity (18), indicating extensive communication between the active sites of the four monomers. Therefore, the differences at the quaternary structural level between fatty acid and polyketide KRs should have a substantial influence on the substrate specificity and allosteric behavior of aromatic polyketide KRs.
FIGURE 4.
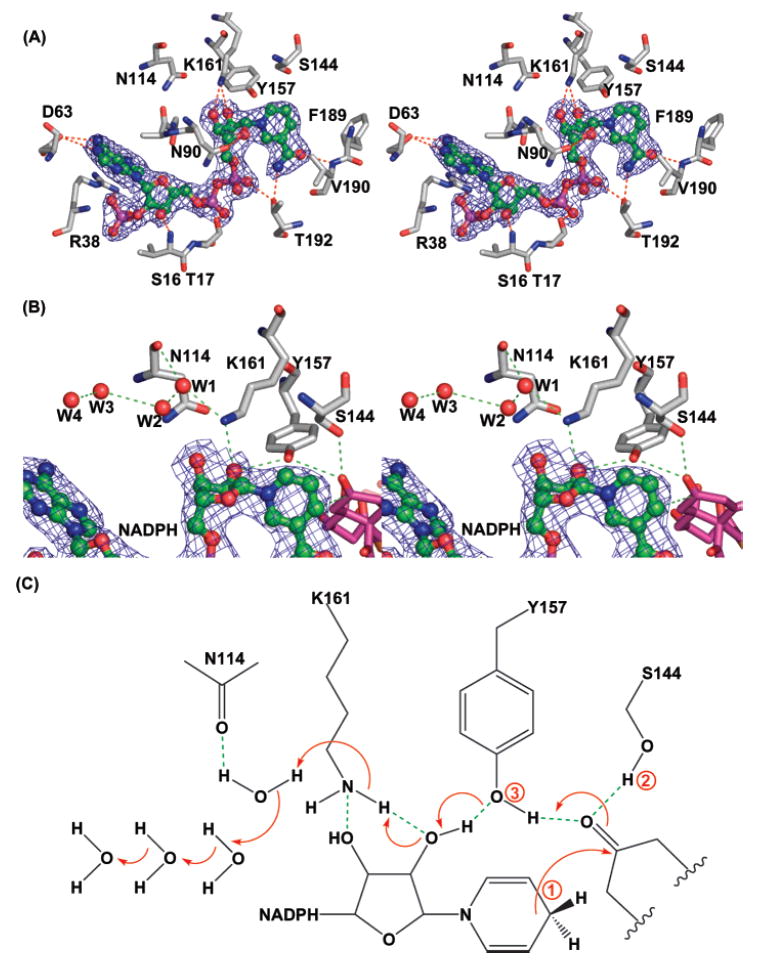
(A) The cofactor-binding pocket is highly conserved. NADPH (in green) is shown in the ∣2Fo − Fc∣ map; the hydrogen bonds are shown in red. (B) The active site consists of four crystalline waters, NADPH, and the catalytic tetrad N114-S144-Y157-K161. Hydrogen bonds are in red, NADPH is in green with the electron density map, and a three-point docked intermediate 1 is in purple. (C) Cartoon representation of the proton-relay mechanism. The three docking points for intermediate 1 are labeled in the order of the proton-relay event.
Cofactor Binding
Nearly all residues important for binding the cofactor NADPH are highly conserved among SDRs (Figure 2) (15), including I18, G39, D63, I142, Y157, K161, and F189 (Figure 4A). This indicates that the cofactor-binding mode should be very similar among type I, type II, and fatty acid KRs. Indeed, the cofactor position is almost identical when the structures of act KR and FabG (17) are superimposed. There is a minor deviation in the adenine ring position; however, residues that interact with the adenine are also less conserved, including S16, R38, and R65 (Figures 2 and 4A). As a result, the high conservation of the cofactor-binding site provides an excellent opportunity to pinpoint the location where hydride transfer takes place.
The structures of act KR–NADP+ and act KR–NADPH are virtually identical, especially surrounding the cofactor-binding pocket (Figure 4A). Except for minor differences in the flexible α6–α7 region, presumably due to differences in crystal packing, the two structures have an overall RMSD of 0.4 Å, and even the crystalline water molecules are conserved. Our results are consistent with the cocrystal structures of FabG and tropinone reductase II (17, 27), in which no major conformational change is observed between enzyme–NADP+ and enzyme–NADPH complexes (28). The two structures reported here represent an important step toward dissecting the catalytic mechanism of KRs.
The Active Site
The active site of act KR is located near the nicotinamide ring of NADPH. The active site N114 is located on α4 while S144, Y157, and K161are located at the β5–α5 loop (Figures 3B and 4B). Contrary to the E. coli FabG structure (18), we did not observe any loop structure variation among different monomers. Key residues Y157 and K161 form hydrogen bonds with the ribose and nicotinamide ring of NADPH, while four crystalline water molecules that form extensive hydrogen bonds with N114 and K161 are observed in both enzyme–NADP+ and enzyme–NADPH complexes. This extensive proton-relay water network is very similar to the one observed in E. coli FabG–NADP+ (17). In fact, the four water molecules are located at nearly identical positions. The proton-relay water network has been proposed to be an important catalytic mechanism for bacterial 3β/17β-hydroxysteroid dehydrogenase (PDB code 1HXH) and E. coli FabG (PDB code 1Q7B) (Figure 4C) (17, 29). Furthermore, the “kink” of α4 near N114, proposed to be important for the FabG water network and catalysis, is also observed in act KR (Figure 2) (17). Due to the similarity between the active sites of act KR and E. coli FabG, the proposed water-relay mechanism should also apply to act KR. Briefly, the ketone substrate is hydrogen bonded to both S144 and Y157 that constitute the oxyanion hole. Following hydride transfer from NADPH to the ketone substrate, the alkoxide is stabilized by the oxyanion hole while the tyrosyl proton is transferred to the alkoxide. An extensive proton relay then takes place to replenish the proton extracted from the tyrosyl OH, sequentially including the 2-OH of NADPH ribose and lysine NH, followed by the four water molecules (Figure 4C). In the type I polyketide KRs, the positions of the active site lysine and asparagine are swapped (Figure 2), although this should not hamper the proton-relay event. After further literature search and structure comparisons, we found that many previously determined SDR structures have similar proton-relay water networks that have not been noted before. For example, the same proton-relay water network is observed in the plant fatty acid KR from B. napus (PDB code 1EDO) (20), which has an identical NADP+ binding motif and water molecule positions as in E. coli FabG. Many other SDR enzymes also have similar proton-relay networks, such as glucose dehydrogenase (PDB code 1GCO, with NAD as the cofactor) (30) and trihydroxynaphthalene reductase (PDB code 1GOO) (31). On the other hand, the water network is not observed in apo E. coli FabG and in the fatty acid KR from T. maritima (PDB code 1O5I; unpublished results) that has an arginine instead of a lysine at K161. This indicates the importance of cofactor binding and the active site lysine/asparagine to maintain the water network. From these structural and sequence comparisons, the proton-relay network is a conserved catalytic mechanism in fatty acid and polyketide KRs that maintain similar cofactor-binding motifs and active site lysines and asparagines.
The ACP Docking Site
The interdomain communication between ACP and KR is crucial for polyketide chain reduction. We used docking simulations between the act KR–NADPH tetramer and the averaged NMR structure of act ACP (PDB code 2AF8) (25) to probe for the ACP binding site of KR. Out of 9300 possible docking solutions, all of the 10 most energetically favored solutions mapped to the same KR site near the entrance of the substrate binding channel. The KR docking site consists of α6–α7 from both monomers A and C (Figure 5A), the most flexible region of act KR. Except for minor hydrophobic interactions (L33: V152′, L43:W206′), the KR–ACP interactions are mostly hydrophilic in nature due to the highly electronegative surface of ACP (D32:E221′, D41:R177′, D62:R216″, D63:R220″; Figure 5B). Similar docking simulations have been performed for E. coli FabG, where two FabG arginines (R149 and R172) near α4 and α5 are identified to be important for FabG–ACP interactions (32). These two arginines are not conserved in aromatic polyketide KRs; therefore, the distinct docking results do not come as a surprise. Our results are plausible for two reasons: (1) by docking at the substrate pocket opening, ACP can induce a conformational change of α6–α7 in monomers A and C that should be important for subsequent substrate binding; (2) this binding motif also allows ACP to interact with two monomers at the same time and therefore induce allosteric interactions between the active sites of KR tetramers. Finally, several key residues important in the docking solution of KR, such as V152, Y202, W206, E212, and R220, are conserved among aromatic polyketide KRs, implying that the interactions between KR and ACP may be conserved for the aromatic polyketide KRs while unconserved residues may be necessary for distinguishing the different KR–ACP pairs.
FIGURE 5.
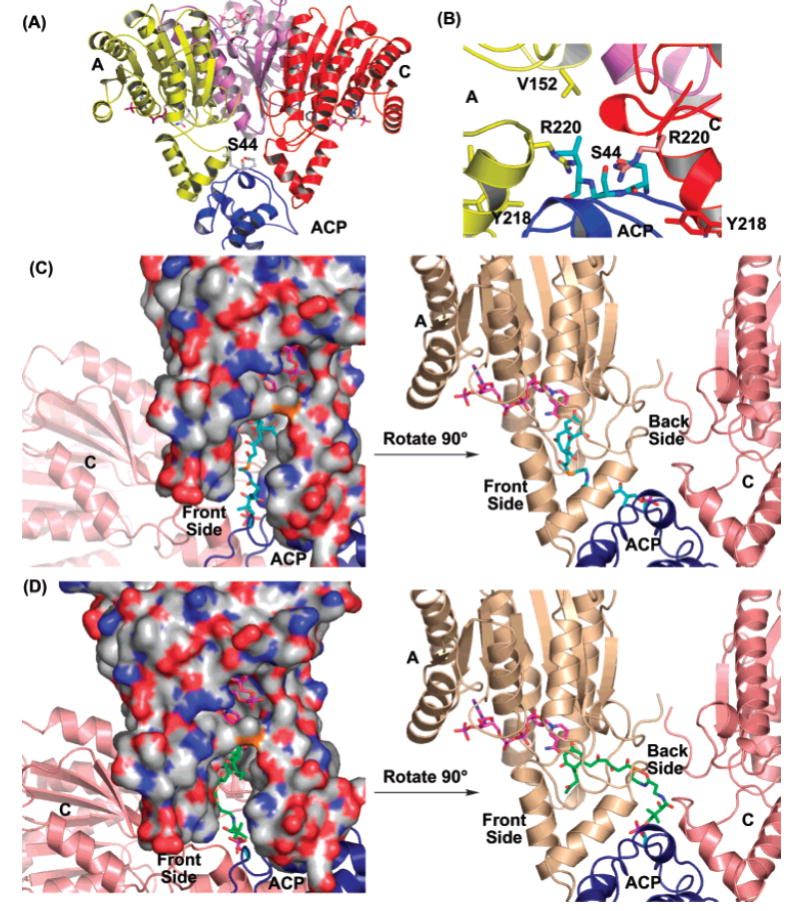
(A) Overview of the ACP and KR docking result. (B) Details of the ACP–KR interaction indicate a mixture of hydrophilic and hydrophobic interactions. (C) The front-side binding motif of intermediate 1 (in blue), leading to the R stereomer. (D) The back-side binding motif of intermediate 1 (in green), leading to the S stereomer. In (A)–(D), ACP is in blue, while the act KR monomers A and C are in yellow and red, respectively.
Substrate Binding
The binding motif of the polyketide substrate is closely associated to its regio- and stereospecificity. Understanding substrate binding is therefore of great importance for combinatorial polyketide biosynthesis. Toward this goal, we generated the coordinates of the cyclic polyketide substrate 1 (with phosphopantetheine attached) and docked this intermediate into the binding pocket of act KR. Due to the intricate hydrogen-bonding network of the KR active site, this docking is well-defined by three docking points in the KR active site: the side chains of S144 and Y157 (the oxyanion hole) should be within hydrogen-bonding distance of the C9-carbonyl oxygen of 1, and the C4′ of the NADPH nicotinamide ring should be within hydride transfer distance to C9 of substrate 1. As a result of this three-point docking, only two binding motifs are possible: the polyketide substrate 1 can bind either from the same face as the cofactor-binding pocket (referred to as the front-side motif) (Figures 3B and 5C) or from the face opposite the cofactor-binding pocket (referred to as the back-side motif) (Figures 3C and 5D). The front-side motif will result in the R stereomer of the reduced product 2, while the back-side motif will result in the S stereomer of 2. Manual docking of 1 followed by CNS energy minimization indicates that 1 can bind act KR with either motif without evoking extensive protein conformational change [note that past attempts to determine the stereochemistry of mutactin at the C9 position have not been successful (33)]. Furthermore, the phosphate of 1 in both motifs is physically within binding distance of S44 of the docked ACP where the phosphopantetheine arm is attached in vivo (Figure 5C,D). Finally, efforts to dock the carbonyl groups of 1 at positions other than C9-carbonyl (such as C3, C5, C7, C11, or C13) at the reaction center resulted in extensive steric clashes between 1 and the enzyme. Therefore, the docking of 1 revealed that, among the eight carbonyl groups that are possible for ketoreduction, the C9-carbonyl is in an optimum position for enzyme-mediated reduction without evoking extensive conformational change.
Molecular Basis of Regio- and Stereospecificity
The stereoselective signature motifs for the modular polyketide KRs have been proposed to be LDD (93–95) and PxxxN (141–148) (34, 35). These sequence motifs are proposed to control the orientation of the substrate, so that the presence of these motifs results in the formation of the R stereomer while the absence of these motifs results in the S stereomer. For act KR, or the aromatic polyketide KRs in general, none of these motifs are conserved (Figure 2). This does not imply that aromatic KRs favor the S stereomer; rather, aromatic polyketide KRs may have a very different binding motif from that of modular polyketide KRs. As previously discussed, the stereospecificity of KR should arise from different binding orientations of the substrate 1, similar to the results observed for tropinone reductases I and II (27). Finally, the docking of ACP at the substrate entrance of two KR monomers indicates that allosteric interactions can be introduced upon ACP binding, which is observed in the case of FabG (18).
An important question for aromatic polyketide biosynthesis is the timing of the first ring cyclization. The cyclization of the polyketide chain [producing intermediate 1 (Figure 1A)] can be formed uncatalyzed either in the active site of KS/CLF or in the active site of KR. From substrate docking simulation of the highly flexible linear polyketide chain (intermediate 0), it is physically possible to reduce many carbonyl groups of intermediate 0 other than the C9-carbonyl group without evoking protein conformational changes. Due to the flexibility of intermediate 0, this will result in the loss of the C9 regiospecificity, contradicting results from previous studies that aromatic polyketide KRs are highly specific for C9 reduction. Rather, it is more reasonable to cyclize 0 to 1 prior to the binding of 1 to KR. Under the dual constraints imposed by the ring structure of 1 and the three-point docking of the KR active site, the C9-carbonyl group is optimally positioned for ketoreduction when the C7–C12 cyclization takes place (Figure 5C,D). Similarly, in the case of C5–C10 cyclization (Figure 1C), the C7 position (para and meta to the two bulky ring substituents) is selectively reduced, resulting in the formation of RM18b. Docking simulation of the C5–C10 cyclization product indeed placed the C7 position favorably for ketoreduction. Therefore, the cyclization event that leads to 1 is likely to happen before its binding to KR, and the active site of both KS/CLF (36) and KR may be important for the C9 regiospecificity.
We believe these factors help to define the fundamental differences between fatty acid and aromatic polyketide biosynthesis. It is physically possible for KR to reduce every carbonyl group of a growing linear polyketide chain, as is observed for fatty acid biosynthesis. Because the helix 6–7 region is very flexible, the polyketide binding pocket should also be highly adaptive to substrates with different polyketide chain lengths. However, for aromatic polyketide biosynthesis that involves a cyclized polyketide intermediate, it is not energetically favorable for KR to reduce carbonyl groups that have an energy penalty imposed by constraints of the active site and substrate geometry. Thus, this key cyclization event differentiates aromatic PKS from FAS by the control of substrate geometry and protein–protein interactions during ketoreduction. In conclusion, the two cocrystal structures of act KR presented here provide important clues about the control of aromatic polyketide regio- and stereospecificity and suggest future directions for engineering PKS to obtain polyketides with altered chemical structures.
Acknowledgments
We thank Prof. Yi Tang for providing the pYT238 plasmid and Prof. Chaitan Khosla for providing the pRZ153 plasmid. We express deep gratitude to Prof. Yi Tang, Prof. Naomi Morrissette, and Mr. Brian Ames for reading the manuscript and for insightful suggestions.
Footnotes
The atomic coordinates have been deposited in the Protein Data Bank (accession codes 1X7G and 1X7H).
Abbreviations: KR, ketoreductase; FabG, β-ketoacyl (acyl carrier protein) reductase; act, actinorhodin; PKS, polyketide synthase; NADP, nicotinamide adenine dinucleotide diphosphate; NADPH, reduced nicotinamide adenine dinucleotide diphosphate; SDR, short-chain dehydrogenase/reductase; ACP, acyl carrier protein.
References
- 1.Hopwood DA. Genetic contributions to understanding polyketide synthases. Chem Rev. 1997;97:2465–2498. doi: 10.1021/cr960034i. [DOI] [PubMed] [Google Scholar]
- 2.Staunton J, Weissman KJ. Polyketide biosynthesis: a millennium review. Nat Prod Rep. 2001;18:380–416. doi: 10.1039/a909079g. [DOI] [PubMed] [Google Scholar]
- 3.Tobert JA. Lovastatin and beyond: the history of the HMG-CoA reductase inhibitors. Nat Rev Drug Discov. 2003;2:517–526. doi: 10.1038/nrd1112. [DOI] [PubMed] [Google Scholar]
- 4.O’Hagan D. Biosynthesis of fatty acid and polyketide metabolites. Nat Prod Rep. 1993;10:593–624. doi: 10.1039/np9931000593. [DOI] [PubMed] [Google Scholar]
- 5.Khosla C, Keasling JD. Metabolic engineering for drug discovery and development. Nat Rev Drug Discov. 2003;2:1019–1025. doi: 10.1038/nrd1256. [DOI] [PubMed] [Google Scholar]
- 6.Walsh CT. Polyketide and nonribosomal peptide antibiotics: modularity and versatility. Science. 2004;303:1805–1810. doi: 10.1126/science.1094318. [DOI] [PubMed] [Google Scholar]
- 7.Nicholson TP, Winfield C, Westcott J, Crosby J, Simpson TJ, Cox RJ. First in vitro directed biosynthesis of new compounds by a minimal type II polyketide synthase: evidence for the mechanism of chain length determination. Chem Commun (Cambridge) 2003:686–687. doi: 10.1039/b300847a. [DOI] [PubMed] [Google Scholar]
- 8.Shen B. Polyketide biosynthesis beyond the type I, II and III polyketide synthase paradigms. Curr Opin Chem Biol. 2003;7:285–295. doi: 10.1016/s1367-5931(03)00020-6. [DOI] [PubMed] [Google Scholar]
- 9.Leadlay PF. Combinatorial approaches to polyketide biosynthesis. Curr Opin Chem Biol. 1997;1:162–168. doi: 10.1016/s1367-5931(97)80005-1. [DOI] [PubMed] [Google Scholar]
- 10.Rawlings BJ. Type I polyketide biosynthesis in bacteria (Part A–erythromycin biosynthesis) Nat Prod Rep. 2001;18:190–227. doi: 10.1039/b009329g. [DOI] [PubMed] [Google Scholar]
- 11.Axford LC, Simpson TJ, Willis CL. Synthesis and incorporation of the first polyketide synthase free intermediate in monocerin biosynthesis. Angew Chem Int Ed Engl. 2004;43:727–730. doi: 10.1002/anie.200352652. [DOI] [PubMed] [Google Scholar]
- 12.McDaniel R, Ebert-Khosla S, Hopwood DA, Khosla C. Rational design of aromatic polyketide natural products by recombinant assembly of enzymatic subunits. Nature. 1995;375:549–554. doi: 10.1038/375549a0. [DOI] [PubMed] [Google Scholar]
- 13.McDaniel R, Ebert-Khosla S, Fu H, Hopwood DA, Khosla C. Engineered biosynthesis of novel polyketides: influence of a downstream enzyme on the catalytic specificity of a minimal aromatic polyketide synthase. Proc Natl Acad Sci USA. 1994;91:11542–11546. doi: 10.1073/pnas.91.24.11542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hertweck C, Xiang L, Kalaitzis JA, Cheng Q, Palzer M, Moore BS. Context-dependent behavior of the enterocin iterative polyketide synthase; a new model for ketoreduction. Chem Biol. 2004;11:461–468. doi: 10.1016/j.chembiol.2004.03.018. [DOI] [PubMed] [Google Scholar]
- 15.Oppermann U, Filling C, Hult M, Shafqat N, Wu X, Lindh M, Shafqat J, Nordling E, Kallberg Y, Persson B, Jornvall H. Short-chain dehydrogenases/reductases (SDR): the 2002 update. Chem–Biol Interact. 2003;143–144:247–253. doi: 10.1016/s0009-2797(02)00164-3. [DOI] [PubMed] [Google Scholar]
- 16.Rossmann MG, Argos P. Protein folding. Annu Rev Biochem. 1981;50:497–532. doi: 10.1146/annurev.bi.50.070181.002433. [DOI] [PubMed] [Google Scholar]
- 17.Price AC, Zhang YM, Rock CO, White SW. Cofactor-induced conformational rearrangements establish a catalytically competent active site and a proton relay conduit in FabG. Structure (Cambridge) 2004;12:417–428. doi: 10.1016/j.str.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 18.Price AC, Zhang YM, Rock CO, White SW. Structure of beta-ketoacyl-[acyl carrier protein] reductase from Escherichia coli: negative cooperativity and its structural basis. Biochemistry. 2001;40:12772–12781. doi: 10.1021/bi010737g. [DOI] [PubMed] [Google Scholar]
- 19.Cohen-Gonsaud M, Ducasse S, Hoh F, Zerbib D, Labesse G, Quemard A. Crystal structure of MabA from Mycobacterium tuberculosis, a reductase involved in long-chain fatty acid biosynthesis. J Mol Biol. 2002;320:249–261. doi: 10.1016/S0022-2836(02)00463-1. [DOI] [PubMed] [Google Scholar]
- 20.Fisher M, Kroon JT, Martindale W, Stuitje AR, Slabas AR, Rafferty JB. The X-ray structure of Brassica napus beta-keto acyl carrier protein reductase and its implications for substrate binding and catalysis. Struct Folding Des. 2000;8:339–347. doi: 10.1016/s0969-2126(00)00115-5. [DOI] [PubMed] [Google Scholar]
- 21.Teartasin W, Limpkin C, Glod F, Spencer J, Cox RJ, Simpson TJ, Crosby J, Crump MP, Hadfield AT. Expression, purification and preliminary X-ray diffraction analysis of a ketoreductase from a type II polyketide synthase. Acta Crystallogr D: Biol Crystallogr. 2004;60:1137–1138. doi: 10.1107/S0907444904007905. [DOI] [PubMed] [Google Scholar]
- 22.Zawada RJ, Khosla C. Heterologous expression, purification, reconstitution and kinetic analysis of an extended type II polyketide synthase. Chem Biol. 1999;6:607–615. doi: 10.1016/s1074-5521(99)80112-4. [DOI] [PubMed] [Google Scholar]
- 23.Otwinoswski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 24.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D: Biol Crystallogr. 1998;54(Part 5):905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 25.Crump MP, Crosby J, Dempsey CE, Parkinson JA, Murray M, Hopwood DA, Simpson TJ. Solution structure of the actinorhodin polyketide synthase acyl carrier protein from Streptomyces coelicolor A3(2) Biochemistry. 1997;36:6000–6008. doi: 10.1021/bi970006+. [DOI] [PubMed] [Google Scholar]
- 26.Smith GR, Sternberg MJ. Evaluation of the 3D-Dock protein docking suite in rounds 1 and 2 of the CAPRI blind trial. Proteins. 2003;52:74–79. doi: 10.1002/prot.10396. [DOI] [PubMed] [Google Scholar]
- 27.Nakajima K, Yamashita A, Akama H, Nakatsu T, Kato H, Hashimoto T, Oda J, Yamada Y. Crystal structures of two tropinone reductases: different reaction stereospecificities in the same protein fold. Proc Natl Acad Sci USA. 1998;95:4876–4881. doi: 10.1073/pnas.95.9.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu YJ, Zhang YM, Rock CO. Product diversity and regulation of type II fatty acid synthases. Biochem Cell Biol. 2004;82:145–155. doi: 10.1139/o03-076. [DOI] [PubMed] [Google Scholar]
- 29.Filling C, Berndt KD, Benach J, Knapp S, Prozorovski T, Nordling E, Ladenstein R, Jornvall H, Oppermann U. Critical residues for structure and catalysis in short-chain dehydrogenases/reductases. J Biol Chem. 2002;277:25677–25684. doi: 10.1074/jbc.M202160200. [DOI] [PubMed] [Google Scholar]
- 30.Yamamoto K, Kurisu G, Kusunoki M, Tabata S, Urabe I, Osaki S. Crystal structure of glucose dehydrogenase from Bacillus megaterium IWG3 at 1.7 Å resolution. J Biochem (Tokyo) 2001;129:303–312. doi: 10.1093/oxfordjournals.jbchem.a002858. [DOI] [PubMed] [Google Scholar]
- 31.Liao D, Basarab GS, Gatenby AA, Valent B, Jordan DB. Structures of trihydroxynaphthalene reductase-fungicide complexes: implications for structure-based design and catalysis. Structure (Cambridge) 2001;9:19–27. doi: 10.1016/s0969-2126(00)00548-7. [DOI] [PubMed] [Google Scholar]
- 32.Zhang YM, Wu B, Zheng J, Rock CO. Key residues responsible for acyl carrier protein and beta-ketoacyl-acyl carrier protein reductase (FabG) interaction. J Biol Chem. 2003;278:52935–52943. doi: 10.1074/jbc.M309874200. [DOI] [PubMed] [Google Scholar]
- 33.Zhang H-I, He X-g, Adefaranti A, Galluci J, Cole SP, Beale JM, Keller PJ, Chang C-j, Floss HG. Mutactin, a novel polyketide from Streptomyces coelicolor. Structure and biosynthetic relationship to actinorhodin. J Org Chem. 1990;55:1682–1684. [Google Scholar]
- 34.Caffrey P. Conserved amino acid residues correlating with ketoreductase stereospecificity in modular polyketide synthases. ChemBioChem. 2003;4:654–657. doi: 10.1002/cbic.200300581. [DOI] [PubMed] [Google Scholar]
- 35.Reid R, Piagentini M, Rodriguez E, Ashley G, Viswanathan N, Carney J, Santi DV, Hutchinson CR, McDaniel R. A model of structure and catalysis for ketoreductase domains in modular polyketide synthases. Biochemistry. 2003;42:72–79. doi: 10.1021/bi0268706. [DOI] [PubMed] [Google Scholar]
- 36.Keatinge-Clay AT, Maltby DA, Medzihradszky KF, Khosla C, Stroud RM. An antibiotic factory caught in action. Nat Struct Mol Biol. 2004;11:888–893. doi: 10.1038/nsmb808. [DOI] [PubMed] [Google Scholar]