

Abstract
This review chronicles the synergistic growth of the fields of fatty acid and polyketide synthesis over the last century. In both animal fatty acid synthases and modular polyketide synthases, similar catalytic elements are covalently linked in the same order in megasynthases. Whereas in fatty acid synthases the basic elements of the design remain immutable, guaranteeing the faithful production of saturated fatty acids, in the modular polyketide synthases, the potential of the basic design has been exploited to the full for the elaboration of a wide range of secondary metabolites of extraordinary structural diversity.
1 Introduction
The enzymatic systems responsible for the de novo biosynthesis of saturated fatty acids and polyketide natural products share many similarities including the utilization of common precursors, similar chemistry, similar structures and overall architectural design. During the last decade, development of the two fields has proceeded in concert, with advances in each discipline often having immediate application to the other. The objective of this review is to examine how is it that, despite their similarities, the two systems are uniquely adapted for quite different functions: one involved universally in primary metabolism and dedicated to faithfully producing exclusively saturated fatty acids, the other endowed with remarkable malleability that allows adaptation to the task of producing a broad range of complex natural products.
Whereas the saturated fatty acids represent a relatively small group of simple molecules, polyketides constitute a large, structurally diverse class of natural products that includes plant flavonoids, fungal aflatoxins and a myriad of important pharmaceuticals that exhibit antibacterial, antifungal, immunosuppressive and antitumor properties. Currently, with the aid of nuclear magnetic resonance and mass spectroscopy, over 10 000 polyketides have been identified1 (Fig. 1). The enzyme systems responsible for the biosynthesis of fatty acids and polyketides have been classified according to their different architectural organization. In type I systems, the constituent catalytic components are covalently linked in multifunctional megasynthases, whereas in type II systems, the catalytic components are freestanding monofunctional polypeptides. The polyketide synthases (PKSs) are found in additional forms that include freestanding iteratively-acting condensing enzymes (type III) and an assortment of hybrid versions of the type I/type II systems and type I/nonribosomal peptide synthases.2 In the animal cytosolic type I FAS and the type I PKS systems that are found in bacteria, fungi, marine animals and plants, the catalytic domains are covalently linked in very long polypeptides and include a β-ketosynthase (KS), acyl transferase (AT), dehydratase (DH), enoylreductase (ER), β-ketoreductase (KR), acyl carrier protein (ACP) and thioesterase (TE). The animal type I FASs polypeptides contain a single copy of all seven functional domains, whereas the type I PKSs consist of multiple FAS-like ensembles, or ‘modules’, each containing a KS, AT and ACP with or without a full complement of the other catalytic domains. In the type I FAS system, the same set of enzymes is used iteratively for all steps in the chain elongation process, whereas in the PKS system adjacent modules are used sequentially in an assembly line manner. The type II FAS systems (found in bacteria, chloroplasts and mitochondria) and the type II PKS systems (found in prokaryotes) both utilize the component freestanding enzymes in an iterative manner (Fig. 2). This review will focus specifically on the type I FAS and PKS megasynthases, an area that has seen significant advances in recent years. Similarities persist from the global architectural level down to the molecular level, while differences provide clues as to the features important for the specific functional roles of the two systems. However, before reviewing these new exciting developments, it may be appropriate to examine the historical record that documents the fascinating co-evolution of these closely related subjects.
Fig. 1.
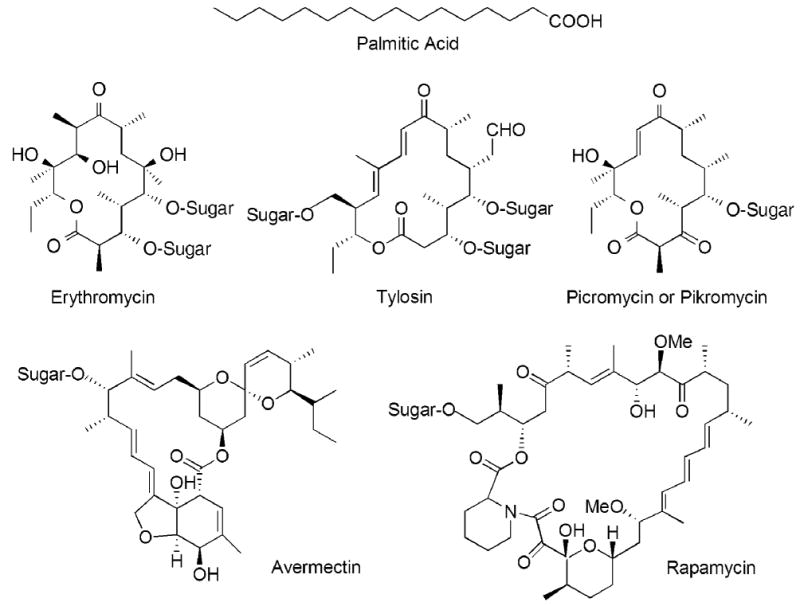
Chemical structures of some typical products of the animal FAS and modular PKSs. Many polyketides subsequently undergo modification by the addition of sugar residues.
Fig. 2.

Domain organization of the animal FAS and the DEBS modular PKS. In the FAS, the AT domain is responsible for loading both the starter and chain-elongating substrates, whereas in modular PKSs, these substrates are loaded by separate ATs in the loading and chain-extending modules. The intermediates formed by each of the DEBS modules are shown together with the final 14-membered ring product. The KR domain in module 3 of DEBS is inactive.
2 Historical perspective: the 20th century
It is an extraordinary fact that the earliest publications addressing the origins of fatty acids and polyketides actually appeared in the same volume of the Journal of the Chemical Society in 1907. On page 1806, John Norman Collie, a remarkable individual renowned for his achievements both as a chemist and as an explorer,3 who earlier had discovered that orcinol could be synthesized chemically from dehydroacetic acid, likely via a polyketone intermediate, suggested that polyphenols could be biosynthesized from a C2 precursor.4 Then on page 1831, Henry Stanley Raper reported that fatty acids likely were derived from a C2 precursor, although he was unable to decide whether it was ethanol, acetaldehyde or acetic acid.5 Unfortunately, it was not until the middle of the century, as isotopically labeled compounds became available, that these hypotheses could be seriously tested. In 1944, Rittenberg and Bloch6 fed to rats acetate labeled in the carboxyl group with 13C and in the methyl group with deuterium and measured the accumulation of both isotopes in body fat, concluding that “fatty acids are synthesized by successive condensation of C2 units”. Nevertheless, it was suspected that the actual metabolic intermediate was probably not acetate as such but some activated form, and the discovery of CoA in Fritz Lipmann’s laboratory just a few years later7 ultimately led to the conclusion that the activated form of acetate was in fact acetyl-CoA.8 The introduction of radioisotope labeling was also exploited in the polyketide field by Arthur Birch who, in 1955, fed [1-14C] acetate to Penicillium patulum and showed that this organism synthesized the aromatic polyketide 6-methylsalicylic acid by the head-to-tail condensation of four acetyl moieties.9
Following a period of intense competition between David Green’s group in Wisconsin and Feodor Lynen’s group in Munich, the enzymological details of the mitochondrial pathway for β-oxidation of fatty acids were revealed at the 1953 meeting of the American Society for Biological Chemistry in Chicago.10,11 By then, these teams had been joined by others in a race to characterize the fatty acid biosynthetic route, based on the notion that it likely proceeded by reversal of the β-oxidation pathway. Indeed, the demonstration that the β-oxidation enzymes could synthesize short chain fatty acids from acetyl-CoA appeared to support this hypothesis.12 Doubts were raised however, when Brady and Gurin found that mitochondrial-free soluble extracts from liver could synthesize long-chain fatty acids from acetate,13 and Popjak and Tietz reported that cytosolic extracts from lactating mammary glands were able to synthesize prodigious quantities of saturated fatty acids containing 4–16 C atoms.14 Although several groups had observed that fatty acid synthesis in animal tissues was markedly reduced when the customary Krebs–Ringer bicarbonate buffer was replaced by phosphate, radioactive CO2 was not incorporated into fatty acids,15,16 so the significance of this observation was not fully appreciated until the June 1958 Gordon Conference on Lipid Metabolism held at Kimball Union Academy in New Hampshire. On the evening of the 11th June, Roscoe Brady and Feodor Lynen had privately discussed the possibility that the requirement for bicarbonate might indicate that a carboxylation reaction was involved. One can imagine their surprise when the following day, at the conclusion of Salih Wakil’s talk, Norman Radin, not considered a player in this field, astutely enquired whether Dr Wakil had considered that the CO2 stimulation might point to malonyl-CoA being formed as a carboxylation intermediate. Dr Wakil replied that he had not, but now the cat was clearly out of the bag and the race was on to test this new hypothesis. Within a very short time Brady and Wakil independently confirmed and published that indeed acetyl-CoA was first carboxylated to malonyl-CoA, which was subsequently decarboxylated during the condensation reaction.17,18 Confirmation that the cytosolic ‘malonyl-CoA pathway’ was the major route for fatty acid synthesis in animals essentially discredited the concept of a mitochondrial pathway.19 At that time, it is unlikely anyone foresaw that this idea would eventually be resurrected, as indeed it was, when more than 40 years later the existence of a separate mitochondrial de novo pathway was firmly established; the enzymes of this pathway are now recognized as discreet freestanding proteins, closely resembling their type II prokaryotic counterparts.20–22 Within a decade, all of the enzymes of the malonyl-CoA pathway had been identified and characterized, primarily from Escherichia coli. A landmark discovery was the identification of a small heat-stable protein, later named acyl carrier protein (ACP), which was post-translationally modified at a serine residue by a phosphopantetheinyl moiety that functioned as a carrier for the reaction intermediates.23,24 These were heady times for the field of lipid biochemistry, and in 1964 the pioneering work of Konrad Bloch and Feodor Lynen on the biosynthesis of fatty acids and cholesterol was recognized when they shared the Nobel Prize in Medicine. By the late 1960s several laboratories had purified FASs from yeast and animal tissues as high molecular-mass proteins that contained all of the catalytic components required for the biosynthetic process. It was at this point in time that a simple nomenclature was proposed to distinguish the high molecular mass eukaryotic FASs (type I) from their counterparts which exist as separate freestanding proteins in prokaryotes and chloroplasts (type II).25 However, there was disagreement as to whether the type I FASs were multienzyme complexes, composed of multiple subunits, or were composed of large, multifunctional polypeptides. Electrophoresis of the yeast26 and liver27 FASs under denaturing conditions was reported to reveal 7 or 8 individual polypeptide species, and a small molecular mass polypeptide corresponding to the ACP could be isolated from several different FASs.28,29 In contrast, genetic and biochemical studies in Eckhart Schweizer’s laboratory indicated that the yeast FAS was comprised of only two non-identical, multifunctional peptides in which the individual components were covalently linked.30 At about this time, John Porter’s group published a series of papers claiming that the animal FAS actually consisted of two large non-identical subunits that could be separated by affinity chromatography.31,32 However, agonizingly exhaustive attempts to replicate these experiments by K. N. Dileepan, a postdoc in Stuart Smith’s laboratory, were unsuccessful. This period of uncertainty was finally resolved when it was emphatically demonstrated that co-purification of trace amounts of protease in FAS preparations resulted in proteolysis on exposure to the SDS used in electrophoresis.33
Susceptibility of the large FAS to proteolysis was quickly appreciated as a potential tool to dissect the FAS into catalytically competent components and resulted in the first successful isolation of a catalytically active domain, the TE, by controlled trypsinization of the whole protein.34 Through a combination of limited proteolysis,35 active-site labeling and peptide mapping,36 and ultimately sequencing of entire animal FASs,37–39 the approximate locations of the constituent domains were eventually established. Surprisingly, identification of the sequence elements characteristic of the various functional domains of the animal FAS was later to have a profound influence on the identification of the genes encoding the enzymes involved in macrolide biosynthesis and formulation of a model that could predict the structure of a macrolide based solely on the DNA sequence of these genes. As a result of the various sequencing and domain mapping efforts, as well as yeast genetic studies, it became clear that, although animals and fungi both utilize type I systems for de novo fatty acid biosynthesis in the cytosolic compartment, evolution of the megasynthases has proceeded along two distinctly different architectural lines in these organisms. Whereas the 0.54 MDa animal FAS is composed of two identical polypeptides, the fungal FAS is comprised of six pairs of non-identical subunits with a collective mass of 2.6 MDa. Furthermore, the arrangement of domains within the two polypeptides of the fungal FASs is quite different from that of the animal FAS and the structurally related modular PKSs. For this reason, the fungal FAS will not be discussed further here. Interested readers are referred to an excellent review on the fungal FASs40 and recent papers that document the crystal structures of fungal FASs at 3–4 Å resolution.41,42
Although the period from the 1950s through the 1980s saw major advances in the FAS field, there were relatively few biochemical studies made in the area of polyketides. The main emphasis at that time was in the exploitation of new synthetic organic chemistry strategies, and several spectacular total syntheses of polyketides were accomplished, including those for erythromycin, toralactone, maytansine, rifamycin and fumagillin. In addition, the Harris group conducted extensive studies on the chemical synthesis and cyclization of polyketides that laid a solid foundation for understanding the cyclization process. The period did see the first successful isolation and characterization of a PKS in Lynen’s laboratory: the 6-methylsalicylic acid synthase from Penicillium patulum.43 As it turned out, this type PKS is rather unusual in that the constituent domains function iteratively as in the type I FASs and this type of PKS is now referred to as type I, iterative.
In 1984, with the advancement of genetic techniques, the first set of genes encoding for biosynthesis of a polyketide, the aromatic polyketide actinorhodin, were identified in David Hopwood’s laboratory.44 This seminal achievement ignited strong interest in the genetics of PKSs and led to subsequent cloning and identification of many PKS gene clusters. The most celebrated discovery was undoubtedly the cloning of the genes encoding the enzymes of erythromycin biosynthesis in Saccharopolyspora erythraea by the groups led by Peter Leadlay and Leonard Katz.45,46 Identification of these genes was facilitated by David Hopwood, who made freely available a plasmid containing the erythromycin resistance gene that was used as a probe for locating the PKS genes.47 The structural genes encoding the biosynthetic enzymes for the aglycone portion of this antibiotic were found in three long open reading frames, each coding for an enormous polypeptide of ~350 kDa. Remarkably, the amino acid sequence of the encoded proteins strikingly resembled that of the animal FAS. Thus the three polypeptides were predicted to contain six repeated units, or ‘modules’, encoding FAS-like activities in which the functional domains were organized in the same order as in the animal FAS. However, several of the FAS-like modules appeared to lack certain of the enzymes responsible for β-carbon processing, and only the last module encoded an enzyme resembling a chain-terminating TE (Fig. 2). This landmark series of papers from the Leadlay and Katz laboratories45,48–50 revealed that the apparent colinearity of the genetic organization and order of the modules suggested a simple but elegant model that accounted for the precise structure of the polyketide product. In this model, each module was responsible for one elongation cycle analogous to a single cycle catalyzed by the animal FAS. Furthermore, each module carried an AT domain with the appropriate specificity to select the correct extender substrate and contained the appropriate complement of KR, DH and ER enzymes required to ensure the proper level of β-carbon processing for that particular elongation cycle. Thus, the product of one module would be transferred from the ACP of that module to the KS of the module acting immediately downstream, and the process would be repeated until finally the product of the last module was released through the action of the resident TE domain. The validity of this model was supported by inactivating the KR of module 5 and correctly predicting the structure of the new product.48 A clear understanding of the rules determining the chemical structure of complex macrolides was dramatically revealed almost overnight! There followed in the next decade a virtual avalanche of gene sequencing projects that sought to identify the genes responsible for the synthesis of a staggering range of macrolides and decipher the mechanism of their synthesis.51 One cannot overemphasize that the remarkable success of these endeavors has been due entirely to recognition of coding sequences for FAS-like domains and modules in the targeted gene clusters, and to an appreciation of the simple colinear relationship between genetic and biochemical events. For a more detailed account of the historical developments in the polyketide field, the reader is referred to the superb review by Jim Staunton and Kira Weissman in an earlier issue of this journal.1
3 The overall biosynthetic pathways catalyzed by the animal FASs and modular PKSs
3.1 The reaction sequence
A generic reaction scheme, applicable to both the FAS and modular PKSs, is shown in Fig. 3. In the FAS system, a primer substrate, usually an acetyl, is condensed successively with extender molecules, typically 7 malonyl moieties, and the β-ketoacyl moiety formed in each condensation reaction is completely reduced to a saturated acyl moiety prior to the following round of chain extension. Surprisingly, both the primer acetyl and chain-extender malonyl moieties are loaded onto the ACP phosphopantetheine by the same AT, the dual-specificity malonyl/acetyl transferase. Thus, each substrate is actually a competitive inhibitor of the other.52 The choice of substrate loaded is entirely random so the system relies on a sorting process in which acetyl and malonyl moieties are rapidly exchanged between CoA and FAS. When the appropriate substrate is loaded, then a productive reaction can follow, otherwise the inappropriately loaded substrate is transferred back to CoA, which must be present at all times during the FAS reaction to ensure efficient substrate sorting.53 The extremely rapid exchange of acetyl and malonyl moieties between CoA and FAS, catalyzed by the AT, ensures that this substrate sorting process is not rate limiting overall. Transfer of the primer moiety and the various saturated intermediates, from the ACP phosphopantetheine to the active-site cysteine of the KS is catalyzed by an innate transferase function of the KS. The vacant phosphopantetheine site is then filled by a malonyl moiety and a decarboxylative condensation event ensues. The β-ketoacyl moiety formed on the phosphopantetheine is then completely reduced to a saturated acyl moiety through the successive action of a KR, DH and ER. Both reductases utilize NADPH as hydride donor. The saturated acyl intermediate is then recycled to the active-site cysteine in preparation for the next condensation. Typically, after 7 turns of the cycle, when the saturated acyl chain-length reaches 16 carbon atoms, the fatty acyl moiety is released from the ACP phosphopantetheine through the action of the chain-terminating TE domain.
Fig. 3.

Generic reaction scheme for FASs and PKSs. The starter substrate for FASs typically is an acetyl moiety and the chain extender a malonyl moiety, whereas for PKSs, the starter is typically either acetyl or propionyl and the extender malonyl or methylmalonyl. In the FASs the β-ketoacyl moiety produced following each chain-extension reaction is completely reduced to a saturated carbon prior to the next condensation. In the modular PKSs, the β-ketoacyl moiety may undergo complete or partial reduction, leading to the formation of products that retain keto, hydroxyl or enoyl groups along the acyl chain.
The overall series of reactions catalyzed by modular PKSs is very similar to the FAS system in that a primer substrate bound to the KS active-site cysteine is condensed with the chain-extender substrate, and the resulting β-ketoacyl product is subjected to varying degrees of β-carbon processing prior to the next elongation step. As in the FASs, the reductive steps utilize NADPH as the hydride donor. On completion of the final elongation and β-carbon processing steps, the product is released from the phosphopantetheine thiol through the action of a TE. In both megasynthases, all reaction intermediates remain covalently associated with the PKS. Nevertheless, the logistics of the process differ in several significant ways. First, in the modular PKSs, loading of the primer and chain-extender substrates is catalyzed by separate dedicated ATs located on different modules; thus there is no competition between these substrates for the same AT active site, and different extender substrates can be utilized at each elongation step. Although, like their counterparts in the FAS, the AT domains responsible for loading the starter substrate exhibit relaxed substrate specificity and can accept primers with 2, 3 or 4 C-atoms,54 the AT domains responsible for loading the chain-extender substrates exhibit high specificity for either malonyl or methylmalonyl-CoA.55 Second, each round of chain extension and β-carbon processing is catalyzed by successive modules linked in assembly-line fashion so that the extent of β-carbon processing can differ at each elongation cycle. Thus, in contrast to the FAS KSs, which accept only saturated acyl moieties for chain extension, the PKS KSs are relatively promiscuous and can utilize either β-keto, β-hydroxy, enoyl or saturated substrates in the condensation reaction. Third, since the FASs by nature function in an iterative manner, the same ACP transfers the substrate for elongation to the KS and carries the chain-extender substrate in the ensuing condensation reaction. However, in the assembly-line PKS systems, these roles are performed by ACPs associated with adjacent FAS-like modules, which may be associated with either the same or different polypeptide chains.
Thus, the ACP domains, in addition to translocating the various intermediates through the condensation and β-carbon processing reactions, also provide the functional link between modules as they hand off the end product to the KS domain of the module immediately downstream, where the next chain extension and β-carbon processing reactions are conducted with the cooperation of the resident ACP domain of that module. Since some modules that function sequentially are located on different polypeptides, specific module recognition signatures are essential to ensure that the appropriate upstream and downstream modules are functionally coupled in a highly specific manner. Finally, the terminal TE domain catalyzes the intramolecular cyclization and release of product rather than hydrolysis to a free acid.
3.2 Catalytic efficiency of the megasynthases
Despite the fact that more enzyme-catalyzed reactions are necessary to produce palmitic acid (7 condensations and 21 β-carbon processing steps) than to produce 6-deoxyerythronolide B (6 condensations and 7 β-carbon processing reactions), the iterative FAS gets the job done in less than a second whereas the DEBS system requires ~2 minutes, at least when the rates are measured in vitro.56,57 The basis for this large difference is not immediately evident, although there is reason to suspect that the difficulty may lie in the transfer of intermediates from upstream to downstream polypeptides. It remains unclear as to whether modular PKSs comprised of multiple polypeptides do form a stable structure in vivo, or whether they engage in dynamic interactions that could limit the overall catalytic efficiency of the system.
4 Domain organization of the megasynthases
Modular PKSs are constructed of polypeptides that, like the FASs, may be ‘unimodular’ or may contain multiple modules, as many as nine in the gigantic mycolactone PKS.58 Undoubtedly, the best studied modular PKS is the 6-deoxyerythronolide synthase (DEBS), which contains 29 domains that are organized into one loading-module (AT-ACP) and six chain-extending modules (Fig. 2). Of the six chain-extender modules, only module 4 possesses a full complement of β-carbon processing enzymes and fully reduces the new β-keto moiety. Module 3 has no functional β-carbon processing enzymes (the KR domain is inactive) and thus leaves the new keto group unmodified whereas modules 1, 2, 5 and 6 have only the KR function and process the intermediate to the hydroxyl stage. Finally, after completion of six rounds of elongation and variable β-carbon processing, the TE domain catalyzes the intramolecular cyclization of the polyketide chain releasing 6-deoxyerythronolide (6-dEB), the precursor of erythromycin.
5 Megasynthase models
5.1 FAS: the rise and fall of the FAS head-to-tail model
5.1.1 The head-to-tail, fully extended polypeptide model
Work in several laboratories in the 1970s established that the dimeric FASs isolated from a variety of tissues could undergo reversible dissociation into their component monomers when exposed to low ionic strength buffers at refrigerator temperatures.59,60 The FAS monomers are unable to synthesize fatty acids, but on reassociation, induced by exposure to high ionic strength media at room temperature, full activity is restored. At that time, it appeared that the only function lost on dissociation was the ability to catalyze the condensation reaction. Catalysis of this reaction requires close cooperation between the active-site cysteine of the KS and the phosphopantetheine moiety of the ACP, first in the transfer of a saturated acyl moiety from phosphopantetheine to cysteine and then in the condensation of S-acyl cysteine and S-malonyl phosphopantetheine moieties. The subsequent discovery, by Jim Stoops and Salih Wakil, that the KS active-site cysteine of one subunit could be cross-linked by 1,3-dibromopropanone to the ACP phosphopantetheine of the companion subunit immediately suggested that the condensation reaction was catalyzed between KS and ACP domains from opposite subunits, and appeared to provide a logical explanation for the inability of FAS monomers to synthesize fatty acids.61 This observation became the cornerstone of a model for the type I FAS in which two fully extended polypeptides are orientated head-to-tail, such that two sites for condensation are created at the subunit interface by direct juxtaposition of the KS active-site cysteine thiol of one subunit with the phosphopantethine of the second subunit (Fig. 4A). Confirmation that the FAS contains two sites, or reaction chambers, for fatty acid synthesis was provided by demonstrating that a FAS dimer in which the chain-terminating TE was blocked assembles and retains two long fatty acyl chains, one on each ACP.62,63 Later refinement of the FAS domain map revealed that, consistent with the head-to-tail subunit concept, the KS and ACP domains are located far apart in the linear sequence (Fig. 2) and so the model enjoyed wide acceptance for 20 years in the FAS field. Later, as it became apparent that the modular PKSs shared a similar domain organization with the animal FASs, the head-to-tail model became the default model for interpreting experimental data for this megasynthase family too.
Fig. 4.
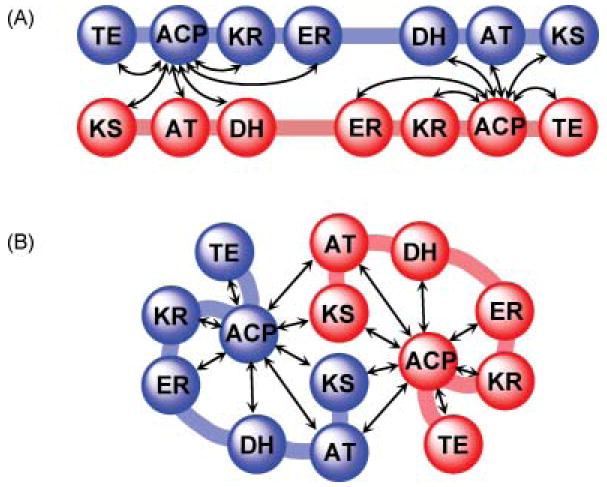
Cartoon representations of alternative models for the FAS. (A) The fully extended, head-to-tail orientated subunit model. (B) The head-to–head, coiled subunit model. In both models, arrows indicate the functional interactions occurring between domains, inter- and intrasubunit.
5.1.2 The head-to-tail model is challenged
5.1.2.1 Mapping of interdomain communications by mutant complementation
In the 1990s, development of a system for the expression of an appropriately post-translationally modified, fully active, recombinant FAS in insect Sf 9 cells64 offered the first real opportunity to test the validity of the head-to-tail model, initially by exploiting a mutant complementation approach.65 Pairs of inactive homodimeric FASs containing knockout mutations in different domains were combined together and subjected to dissociation and reassociation to form a mixed population of hetero- and homodimers. Thus the ability of any compromised domain to complement mutations in any other domain could be evaluated, since all homodimers would be inactive and any activity detected would be attributable only to the heterodimers. In this strategy, mutations on different subunits in domains that normally cooperate in the same reaction chamber will complement each other to produce a 50% active heterodimer, since both mutated domains will be associated with the same reaction chamber and all domains in the second chamber will be normal. On the other hand, mutations on different subunits in domains that normally operate independently in different reaction chambers will not complement each other, since each reaction chamber will be compromised by a single mutated domain. The first results obtained using this approach were totally unexpected. They revealed that the substrate loading and condensation reactions can be catalyzed by interaction of the ACP domains with the AT and KS domains of either subunit66 and the DH domain actually cooperates functionally with the ACP domain on the same subunit, more than 1100 residues distant67 (Fig. 5). The head-to-tail model had predicted that the ACP domain associated with one subunit should make functional contacts with the AT, KS and DH domains of the opposing subunit, but not with those of the same subunit. Most importantly, these findings suggested that the two subunits of FAS cannot be orientated in a fully extended head-to-tail arrangement, but are likely coiled in such a way as to permit functional interactions between domains distantly located on the same subunit.
Fig. 5.
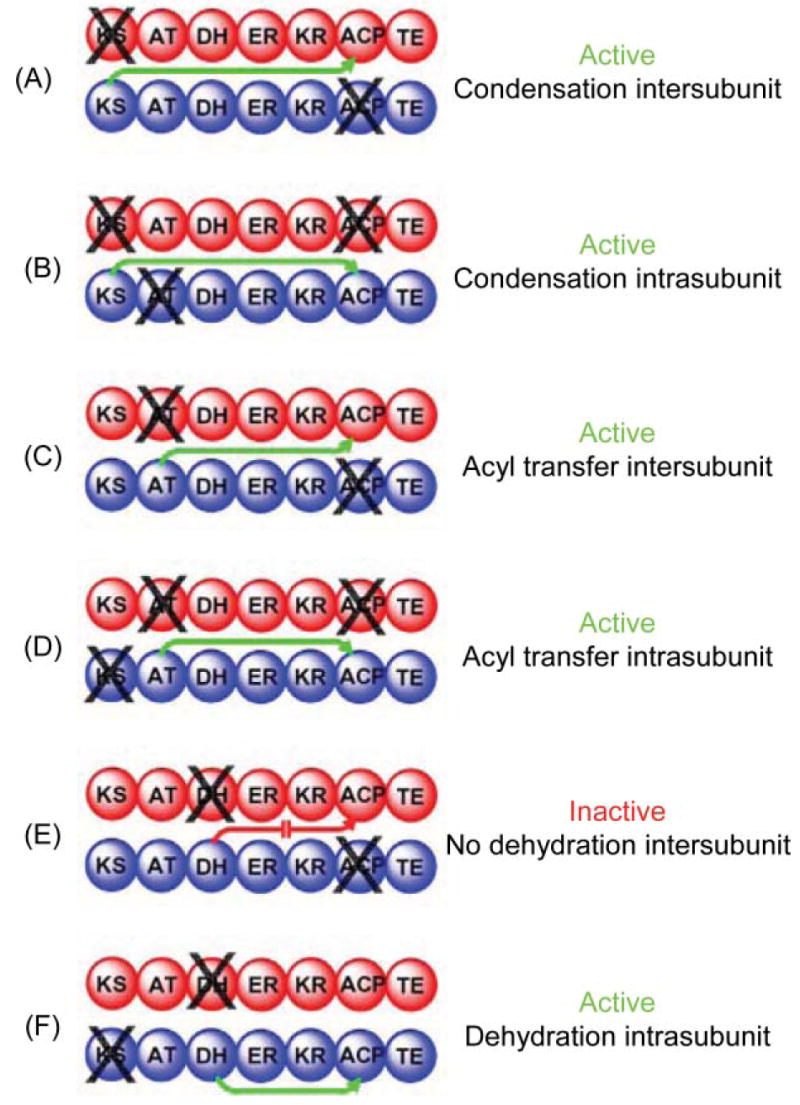
Mapping domain interactions in the FAS by mutant complementation analysis. The two individually mutated FAS subunits forming each heterodimer are colored red and blue and the site of the knockout mutation is indicated by X. Green arrows indicate which catalytic domains engage in functional interactions with an ACP domain. A red arrow indicates the blocked interaction responsible for an inactive heterodimer.
Subsequently, the mutant complementation strategy was refined by the introduction of a double tagging procedure that facilitated isolation of doubly tagged heterodimers (e.g. subunit A His6-tagged and subunit B FLAG-tagged) completely free of the parental homodimers (two His6-tagged A subunits or two FLAG-tagged B subunits) by sequential metal ion and anti-FLAG affinity chromatography.68 By eliminating any ambiguity as to the content of hetero- and homodimers in the preparation, this strategy permitted precise assessment of the specific activity of the heterodimers. A series of tagged knockout mutants was engineered, each mutant compromised in one of the seven functional domains, and a panel of more than 40 affinity-purified heterodimers was generated by hybridizing all possible combinations of the mutated subunits. The results confirmed and extended the earlier findings.69 Heterodimers comprised of a subunit containing either a KS or AT mutant, paired with a subunit containing mutations in any one of the other five domains, were active in fatty acid synthesis. On the other hand, heterodimers in which both subunits carried a knockout mutation in either the DH, ER, KR, ACP or TE domains were inactive (Table 1). Importantly, complementation was never observed simply by mixing any pair of mutated, catalytically inactive homodimers. Only after the subunits were randomized was complementation observed. Thus, no subunit exchange occurred under normal storage conditions, and complementation occurred exclusively intradimer and not interdimer. The catalytic activity of heterodimers containing complementary mutations never reached the theoretically expected value of 50% of the wild-type FAS activity. Depending on the particular mutant pair, activities were as low as 19% and never greater than 40%. These lower values were attributed to two factors. First, several of the heterodimeric mutants with low activity were found to be significantly less stable than the wild-type FAS. The second explanation related to the redundancy found in the mechanisms for substrate delivery and condensation, in that these processes can involve cooperation of domains either inter- or intrasubunit. Thus all heterodimers comprised of complementary mutants lack one of the two options for either substrate delivery or condensation, since either the KS or MAT domain of one subunit is compromised by mutation. Based on this evidence, the Smith group speculated that the availability of alternative routes for substrate delivery and condensation could increase the efficiency of the enzyme by approximately 20%.
Table 1.
Summary of mutant complementation analysisa
| KS0 | MAT0 | DH0 | ER0 | KR0 | ACP0 | TE0 | |
|---|---|---|---|---|---|---|---|
| KS0 | ○ | + | + | + | + | + | + |
| MAT0 | ○ | + | + | + | + | + | |
| DH0 | ○ | ○ | ○ | ○ | ○ | ||
| ER0 | ○ | ○ | ○ | ○ | |||
| KR0 | ○ | ○ | ○ | ||||
| ACP0 | ○ | ○ | |||||
| TE0 | ○ |
The matrix identifies the combinations of FAS subunits with knockout mutations in domains indicated by the superscript 0 that are strongly complementary (+) or non-complementary (○).
In summary, these results revealed that the substrate loading and condensation reactions are catalyzed by cooperation of an ACP domain of one subunit with the AT or KS domains, respectively, of either subunit. The β-carbon-processing reactions, responsible for the complete reduction of the β-ketoacyl moiety following each condensation step, and the chain termination reaction, are catalyzed by cooperation of an ACP domain with the KR, DH, ER and TE domains associated with the same subunit. These findings were inconsistent with the head-to-tail model and suggested that the two subunits must be coiled to allow functional interactions between domains distantly located on the same polypeptide (Fig. 4B).
5.1.2.2 Cross-linking of KS and ACP domains
An obvious implication of the mutant complementation analysis was that, if the ACP domain is able to interact functionally with the KS domain of either subunit, then cross-linking of the ACP should not be limited to the KS of the opposite subunit. This realization prompted re-examination of the specificity of dibromopropanone cross-linking, taking advantage of the ability to construct FASs lacking either or both the KS active site cysteine thiol (Cys161) and phosphopantetheine thiol (at Ser2151) in one or both subunits. The original study had shown that three molecular species with retarded electrophoretic mobilities were formed on treatment of the FAS with dibromopropanone. However, if cross-linking occurred only between KS active site cysteine and phosphopantetheine thiols on opposing subunits, only two species would be anticipated, i.e. dimers that are doubly or singly cross-linked interpolypeptide. How could a third cross-linked species be accounted for? The new study provided a clear answer to this conundrum, revealing that dibromopropanone indeed reacted with the FAS dimer to produce three discreet species: a doubly inter-subunit cross-linked species, a singly inter-subunit cross-linked species and, most significantly, an intra-subunit cross-linked species that accounted for 35% of the total FAS70 (Fig. 6). Thus the results of the mutant complementation and cross-linking experiments were in complete agreement and in support of the alternative model.
Fig. 6.
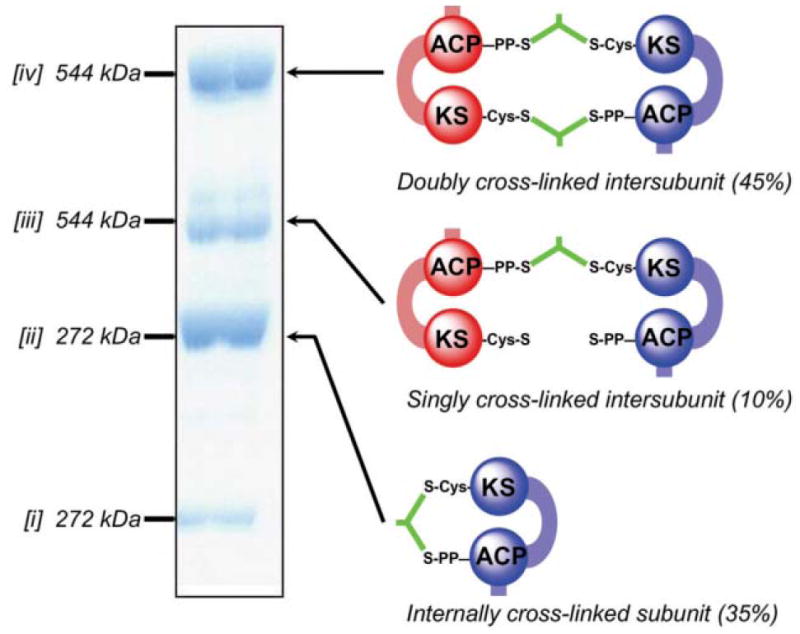
Cross-linking of FAS by dibromopropanone. The SDS–poly-acrylamide gel shows the three new cross-linked species [ii–iv] formed by treatment of the wild-type FAS with dibromopropanone. Species [i] represent residual subunits that have not undergone cross-linking. Molecular masses were determined by mass spectrometry. The identities of the cross-linked species were determined by performing cross-linking on a panel of mutants lacking either the ACP 4-phosphopantetheine or the KS active site cysteine on one or both subunits. The cartoons on the right illustrate the mode of cross-linking, either inter- or intra subunit.
5.1.2.3 The seven-KO punch
A second corollary to the alternative hypothesis was that, if the ACP domain is able to cooperate functionally with all catalytic domains located on the same subunit then, in the context of the dimer, it should be possible for one of the two subunits to catalyze all of the reactions leading to palmitate synthesis—a heretical notion for adherents to the old dogma! Astonishingly, this was indeed found to be the case. A heterodimer engineered from a wild-type subunit and a ‘seven knockout’ (KO) subunit, compromised by mutation in every one of the seven functional domains, was found to synthesize palmitate at about one third of the rate of a single reaction chamber in the wild-type homodimer.71 The possibility that the activity attributed to the 7KO/WT heterodimer could have resulted from subunit exchange and reformation of WT homodimers was discounted based on several independent lines of experimental evidence.
Clearly, the inability of the old head-to-tail model to explain either the cross-linking or mutant complementation data required that a new model be visualized that allowed for close contact and functional communication between domains distantly located on the same polypeptide (KS, AT and DH with ACP) as well as between domains located on opposite subunits (KS and AT with ACP) (Fig. 4B).
5.1.3 Cross-linking of FAS subunits via KS domains: the new head-to-head model
A key feature of the classical model illustrated in cartoon form in Fig. 4A was the clear spatial separation of the two KS domains that lie at opposite poles of the dimer. However, the KSs associated with the type II FASs are universally dimeric proteins in which the substrate-binding pocket is comprised of residues from both subunits.72–74 In the proposed alternative model, the KS domains are visualized as being positioned close to the center of the dimer, where they could possibly engage in homodimeric interactions (Fig. 4B). An experimental approach undertaken in the Smith laboratory to evaluate the oligomeric status of the KS domains in the context of the FAS dimer ultimately provided the final blow in the demise of the head-to-tail model.75
The first indication that the KS domains likely played an important role in maintaining the dimeric status of the FAS came from the observation that subunits lacking the KS domain do not form dimers. A survey of the crystal structures of homologous KSs from the type II FAS world revealed that the N-termini of the two subunits are located on the surface of the protein, typically only about 10–18 Å apart. A modeled three-dimensional structure for the FAS KS domain, based on the crystal structures of four type II KSs that exhibited 37–43% similarity with the type I KS domain, supported the view that it should be possible to cross-link the two N-termini of the FAS dimer with a relatively short spacer, provided an appropriate residue was present. Indeed, when a cysteine residue was engineered near the N-terminus of each subunit, up to 98% of the two polypeptides could be cross-linked by low concentrations of bismaleimido cross-linking reagents. Supportive evidence that the two subunits were cross-linked via the introduced cysteine residues was provided by the observation that heterodimers in which only one subunit carried the engineered cysteine did not undergo cross-linking. Final proof was provided by proteolytic digestion of the cross-linked FAS, isolation of the cross-linked peptide and sequencing by mass spectrometry. These experiments provided irrefutable evidence that the FAS subunits are arranged head-to-head and not head-to-tail as envisioned in the former model (Fig. 4A and B).
5.2 The architecture of modular PKSs
Throughout the period of upheaval in the FAS world during the 1990s, major strides were being made in deciphering the functional organization of the modular PKSs, mainly through collaborations between the Leadlay and Staunton laboratories in the UK and between the Khosla and Cane laboratories in the USA. Just as the development of a suitable expression system ushered in a new era in exploration of structure–function relationships in the animal FAS, so the identification of suitable host strains for production of modular PKSs played a crucial role in the engineering and characterization of mutant modular PKSs. Among the earliest strains to be utilized were derivatives of Streptomyces coelicolor and Streptomyces lividans from which the majority of the endogenous genes for polyketide synthesis had been deleted.76,77 In both strains endogenous phosphopantetheinyl transferases were present and able to perform the essential post-translational modification of ACP domains. More recently, the identification of phosphopantetheinyl transferases that can be coexpressed with PKSs has enabled the use of E. coli host strains that do not normally produce polyketides for expression of competent individual PKS modules and mutants thereof.78
5.2.1 Mapping of interdomain communications by mutant complementation
One of the first strategies exploited for probing details of the functionality of the modular PKSs was the same mutant complementation method used for the FASs, and here again, the strategy provided valuable information concerning the functional communications that take place both intra- and inter-polypeptide, as well as inter-module. In the first series of experiments, Chaitan Khosla and co-workers, using the DEBS1 + TE system, generated three different heterodimers from parental inactive mutated homodimers (Fig. 7). Heterodimers containing a KS knockout in module 2 of one polypeptide paired with a polypeptide containing either a KS knockout in module 1 or an ACP knockout in module 2 were capable of synthesizing the triketide lactone (panels A and B). However, a heterodimer comprised of a KS knockout in module 1 with an ACP knockout in module 2 was inactive (panel C). These results could only be satisfactorily explained by a model in which the condensation reaction is catalyzed by cooperation of the KS and ACP domains of complementary subunits and the transfer of the product of one module to the next occurs by a handover from the ACP to the KS domain immediately downstream on the same polypeptide. In a second series of experiments, again using the bimodular DEBS1-TE model system, heterodimers were generated from an inactive homodimer containing a knockout mutation in the AT domain of module 2 and an inactive homodimer containing either a knockout mutation in the KS domain of either module 1 or module 2 (panels D and E). Both heterodimers supported triketide synthesis at identical rates, indicating that the AT domains can service either of the two ACP domains associated with that module.79,80
Fig. 7.
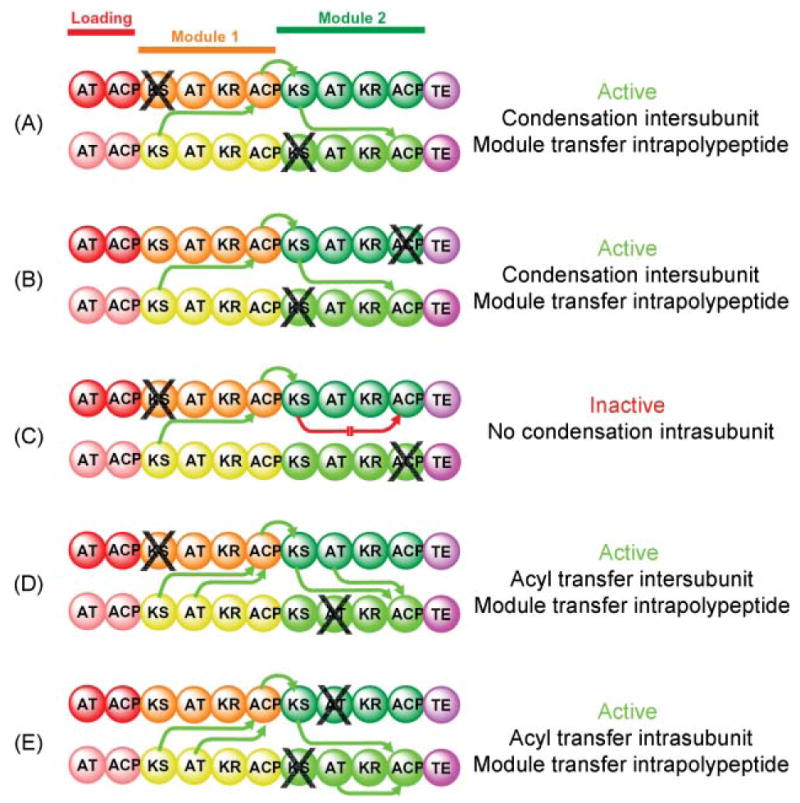
Mapping domain interactions in the DEBS PKS by mutant complementation analysis. The subunits used in this study were constructed by fusing the TE domain from module 6 to the C-terminus of the ACP domain in module 2. The site of a knockout mutation is indicated by X. The parental mutant homodimers were mixed together, the subunits were randomized to give a preparation consisting of hetero-and homodimers, then assayed for activity. Green arrows indicate which catalytic domains engage in functional interactions with an ACP domain and the directionality of intermodular substrate transfer. A red arrow indicates the blocked interaction responsible for an inactive heterodimer.
The results of these mutant complementation studies revealed yet another similarity in the modular PKSs and their FAS counterparts in that both systems incorporate an element of redundancy, since the AT domains can deliver the chain-extender substrate effectively to the ACP domain associated with the same subunit or the companion subunit. Nevertheless, whereas in the FASs this redundancy extends to the cooperation of the KS and ACP domains in catalysis of the condensation reaction, in the modular PKSs cooperation of KS and ACP domains appears to be exclusively an inter-subunit affair.
The early efforts to develop a model for the type I modular PKSs were based on the assumption that these proteins were likely constructed along similar architectural lines to the related type I FASs. Not surprising therefore, the mutant complementation results were initially rationalized according to the classical fully extended, head-to-tail orientated subunit theme.79 However, the validity of the head-to-tail model, as it might apply to the modular PKSs, was beginning to be questioned, and in 1996 the Cambridge group led by Staunton and Leadlay offered a strikingly different model.81
5.2.2 Challenging the head-to-tail model for modular PKSs: the Cambridge double helical model
The experimental foundation for what became known as the ‘Cambridge model’ was a comprehensive analysis of the fragments produced by limited proteolysis of the three multifunctional DEBS polypeptides, DEBS 1, 2 and 3. The Cambridge team subjected these proteins to limited proteolysis, by trypsin, elastase, endoproteinase Glu-C and endoproteinase Arg-C, identified the various fragments by N-terminal sequencing and assessed their oligomeric status by gel filtration. All fragments that contained whole chain-extender modules were dimeric, as was the individual TE domain. All of the smaller fragments were monomeric, including the AT-ACP from the loading module, the KS-AT from module 1, the individual KR domains from modules 1, 3, 5 and 6, and the individual ER domain from module 4.81,82
The limited proteolysis strategy had been previously exploited primarily as a tool for mapping of the domain organization of the FASs,35,36 but had also resulted in the isolation of two individual domains, the TE and AT domains, both of which were found to be monomeric.83,84 The discovery that the TE domain cleaved from DEBS3 was dimeric was surprising and impossible to reconcile with a head-to-tail arrangement of the DEBS subunits. As part of the same study, the authors found that a DEBS fragment containing all of module 5, when treated with dibromopropanone, yielded a cross-linked species of lower electrophoretic mobility, which they assumed to represent an interpolypeptide cross-linked dimer, as had originally been deduced for the FAS. However, the actual site of cross-linking by dibromopropanone was not verified experimentally. On the basis of these observations, Staunton and colleagues concluded that each module pair forms a dimer with the polypeptides orientated head-to-head, rather than head-to-tail.81 Furthermore, they speculated that identical pairs of subunits are twisted together to form a helix in which the KS, AT and ACP domains are positioned at the core of the helix, with the optional β-carbon processing domains forming loops that protrude out from the axis of the helix. With this arrangement, the twist in the helix was proposed to facilitate the interaction between KS and ACP domains across the subunit interface, thus accounting for the mutant complementation data obtained by the Khosla and Cane group. At the C-terminus of the last module the helical arrangement brings the two TE domains into close proximity, consistent with their observed dimeric structure (Fig. 8).1
Fig. 8.
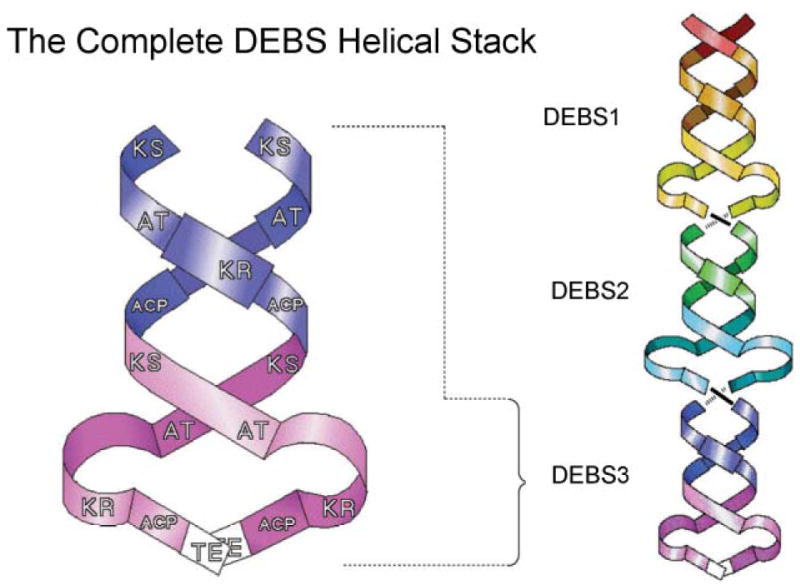
The Cambridge double helix model for modular PKSs. The core of the interwound helical dimer is formed by the KS, AT and ACP domains and the optional β-carbon processing enzymes loop away from the helix. Reproduced from ref. 1 with permission.
This bold departure from the head-to-tail model first proposed for the FASs was appealing in that it appeared to satisfactorily accommodate the results of the complementation analyses as well as the dibromopropanone cross-linking experiments. Probably an additional contributing factor was the new objections being raised against the head-to-tail model for FAS that clearly opened the door to speculation on possible alternatives. One ambiguity in the Cambridge model, however, was that although whole modules were found to be dimeric, as in the FASs, neither the KS–AT fragment, nor any of the β-carbon processing domains, were found to be dimeric, so that it was unclear how the dimeric modules were stabilized.
6 Structural analysis of the megasynthases
6.1 Electron microscopy of megasynthases
The earliest attempts to analyze the structure of animal FASs by electron microscopy, as well as small-angle neutron scattering, generated images that were interpreted as supporting the head-to-tail, fully extended subunit model.85–87 However, as evidence mounted challenging the old model, it became clear that higher resolution structures would be required to provide a structural basis for interpretation of the new experimental data. A collaboration between the laboratories led by Francisco Asturias and Stuart Smith was initiated to address this issue using electron microscopy. A major problem in analysis of proteins such as FAS is the inherent conformational variability, which can seriously limit the fidelity and resolution of a three-dimensional reconstruction that requires classification and averaging of images of many molecules. How could this limitation be overcome? Asturias and Smith reasoned that perhaps mutant FASs, when imaged in the presence of substrates, would display a more limited range of conformations that reflected their inability to catalyze a specific step in the reaction sequence. For example, an FAS unable to catalyze the condensation reaction, in the presence of substrates, was viewed as less likely to adopt conformations required for catalysis of the subsequent β-carbon processing or chain termination reactions. Indeed, when a dimeric FAS containing a mutated KS (Cys161Gln) was incubated with substrates, immediately preserved in negative stain and the images subjected to reference-free alignment and classification, substantially less conformational variability was observed. Similar results were obtained when KR and TE mutants were imaged in the presence of substrates and the images subjected to the same statistical analysis. The strategy worked! A preliminary three-dimensional reconstruction of the Cys161Gln mutant FAS was calculated to approximately 30 Å resolution by the random conical tilt method and used as a starting model for calculation of a reconstruction from images of unstained Cys161Gln FAS molecules preserved in amorphous ice (Fig. 9). Both the 30 Å and 15 Å resolution reconstructions revealed clearly that the symmetry of the FAS dimer was inconsistent with the head-to-tail model, which had predicted two identical cylindrical subunit structures lying side-by-side. Instead the structures exhibited pseudosymmetry about an axis running through the body of the dimer at right angles to that proposed in the head-to-tail model. Surprisingly, the two halves of the structure were not completely symmetrical. One side of the structure appeared to contain significantly more electron density than the other, suggesting that structural elements on one side of the dimer might be inherently more mobile and thus lost during the image averaging process. If this were the case then the implication was that the two subunits do not simultaneously adopt the same conformation. Reconstructions of FAS monomers were also calculated from molecules preserved in negative stain and found to resemble coiled structures rather than the cylindrical structures predicted by the head-to-tail model. Furthermore, when a structure was solved for a N-terminally His6-tagged FAS dimer that had been labeled with a nanogold Ni2+–nitriloacetic acid complex, two gold clusters were located near the center of the structure, not at opposite poles as would be predicted by the head-to-tail model. These findings were interpreted to indicate that the two FAS monomers are coiled in an overlapping arrangement consistent with the head-to-head model deduced earlier from the mutant complementation and cross-linking studies. However, the reconstructions did not allow distinction between back-to-back or crossed-over subunit arrangements (Fig. 9C).
Fig. 9.
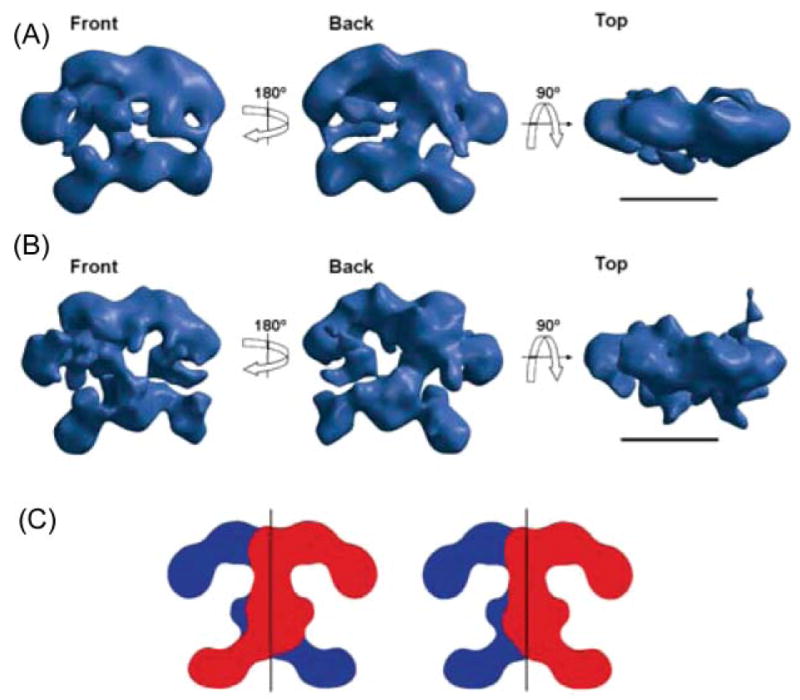
Structure of FAS determined by electron microscopy. (A) Calculated using the random conical tilt method from images of single particles of the Cys161Gln rat FAS mutant imaged under turnover conditions and preserved in negative stain; resolution ~30 Å. (B) Reconstruction of the same mutant, also imaged under turnover conditions, but preserved in amorphous ice; resolution ~16 Å. Scale bar 100 Å. (C) Alternative arrangements of the two subunits that are consistent with the structure of the dimer. Reproduced with permission from ref. 100.
Given the recent success in the application of electron microscopy to analyze the structure of the FAS, it is perhaps surprising that the technique has not been exploited successfully as a tool for probing the structural organization of the modular PKSs. One would have thought that their massive size would make these multimodular structures particularly well suited for single particle analysis, which might provide some clues as to how individual modules are docked together, both intra- and interpolypeptide. Indeed, Grant Jensen, an electron microscopist at Caltech, working in collaboration with Khosla’s group at Stanford, has made a major effort to image the DEBS3 structure by cryoelectron microscopy. Unfortunately the number of ordered structures observed was too few to perform alignment and classification, and it is unclear whether the limiting factor was sample quality, instability of the DEBS3 dimers, or inherent flexibility of the structures.
6.2 Crystallographic analysis of the megasynthases
In the last year, three landmark publications have provided unique insights into the structural organization of both the FASs and modular PKSs. The first groundbreaking study described the crystallization of a complete animal FAS while the others reported high-resolution crystal structures for partial PKS modules. Collectively, these publications categorically resolve many of the lingering controversial issues regarding the structural organization of these megasynthases, they provide invaluable data for appraising the various models proposed to explain the functional organization of these complexes, and challenge traditional ideas about domain boundaries and interdomain linkers.
6.2.1 The crystal structure of full-length FAS
Any enduring support for the antiparallel subunit model was finally crushed with the remarkable publication of a structure for the porcine FAS derived by crystallographic analysis.88 Nenad Ban and colleagues succeeded in obtaining crystals of the full-length FAS that diffracted to 4.5 Å but, unfortunately, this level of resolution was insufficient to identify individual side chains or to trace the complete backbone of the two subunits. The overall shape of the molecule agreed well with the 15 Å resolution structure derived by electron microscopy and clearly revealed, as had been suspected earlier from the electron microscopy study, a large dimerization interface running across the body of the molecule and perpendicular to the interface implicit in the head-to-tail model (Fig. 10A).88–90 Resolution of the structure was not improved by the inclusion of substrates or inhibitors, and so the authors adopted the innovative approach of fitting high-resolution crystal structures of individual homologous type II bacterial proteins into their electron density maps to reveal the location of the functional domains. The templates used for locating the catalytic domains were the KS II (FabB),73 the DH (FabA)91 and the KR (FabG) from E. coli92 and the malonyltransferase (FabD) from Streptomyces coelicolor;93 the best structural match for an ER was the zinc-free quinone reductase from T. thermophilus.94 The crystal structure confirmed the dimeric nature of the KS domains and their position in the central body of the structure. However, there were two quite unexpected findings, not predicted by any previous biochemical studies. The ER domains are dimeric and the DH domains appear pseudodimeric, with each of the pseudosubunits derived from adjacent regions of the same polypeptide. The second of the two pseudosubunits of the DH domain occupied ~200 residues of a ~650-residue region of the FAS previously referred to as the central core, the function of which was unclear. The AT and KR domains are both apparently monomeric. Thus, the oligomeric status of the KS, AT, DH, and ER domains turned out to be identical to that of their type II counterparts. The only exception appeared to be the monomeric KR domain, whose type II counterparts are tetramers95,96 (more about this later!). Ban and colleagues characterized the FAS dimer as consisting of a central body composed of the ER, DH and KS dimers flanked by a pair of arms and legs contributed by the monomeric KR domains and AT domains, respectively. Conspicuous by their absence from the structure were the ACP and TE domains, for which no appropriate electron density could be identified. Ironically these were the only two domains for which high resolution structures were available.97–99 The authors concluded that perhaps inherent mobility of these domains was responsible for the lack of any clearly attributable electron density and suggested that the most likely location of the ACP-TE region would be at the extremity of the arms, since in the linear sequence the ACP is positioned directly following the KR domain. Indeed additional blurred electron density is evident at the end of one of the arms (Fig. 10A), as was observed in the 15 Å electron density map derived from electron microscopy studies. It is possible then that this density could be attributable to the ACP-TE region of the FAS. Indeed, earlier unprocessed images of FAS dimers decorated with anti-TE antibodies revealed additional electron density at the ends of the long axis, consistent with the positioning of the TE domains at the extremities of the two arms of the crystal structure.86 Unfortunately, because the complete backbone could not be traced through the interdomain connecting regions, it is still unclear whether the arms and legs on the same side of the structure are associated with the same subunit. It remains a possibility that the two subunits actually cross over in the central body region, as suggested by the electron micrographic analyses (Fig. 9C).100
Fig. 10.

Crystallographic analysis of the megasynthases. (A) Structural overview of the porcine FAS. Fitted homologous domains are shown with a semitransparent surface representation of the experimental electron density. Left, front view; right, top and bottom views. Reproduced with permission from ref. 88. (B) Modeled structure of the section of a hypothetical PKS containing DH, ER, KR and ACP domains. Polyglycine linkers, in orange, were built to connect the domains. The molecule bound to DH is N,N-dimethyldodecylamineoxide (detergent bound in the TEII template structure) and to the reductases, NADPH. The phosphopantetheine of ACP is also displayed. Left, front view; right, top view. Reproduced with permission from ref. 89. (C) X-Ray structure of the KS–AT didomain from DEBS module 5, The three linker regions of the DEBS KS–AT include the N-terminal helices, the KS–AT linker and the C-terminal linker, colored in yellow in monomer A and purple in monomer B. In the porcine FAS model, these three linker regions contain electron density, but models of these regions were not built. Left, front view; right, bottom view. Adapted from ref. 90.
Based on the orientation of the type II structures fitted into the electron density map, Ban and colleagues deduced that the active sites of the two sets of domains are oriented facing each of the two lateral clefts in the structure that constitute two discrete reaction chambers. However, the two reaction chambers do not appear in identical conformations; the distances between active sites associated with the arms and legs are different on the two sides of the structure. This lateral asymmetry had also been observed in electron microscopic image reconstructions of the animal FAS and interpreted as suggesting that fatty acid synthesis at the two sites may proceed asynchronously, perhaps with one reaction chamber engaged in chain elongation while the other was engaged in β-carbon processing. By superimposing the two sides of the structure on each other, Ban and colleagues shrewdly exploited this lateral asymmetry to identify hinge regions that could facilitate the adoption of different conformations by the FAS (Fig. 11). Two hinges were located on each side of the structure, one between the KS and AT domains, and the other between the KR domains and the central body. These hinges could conceivably permit slight up-and-down movements of the arms and legs, resulting in alternative narrowing and widening of the lateral clefts. A third hinge was located in the narrow waist between the KS and DH domains.
Fig. 11.
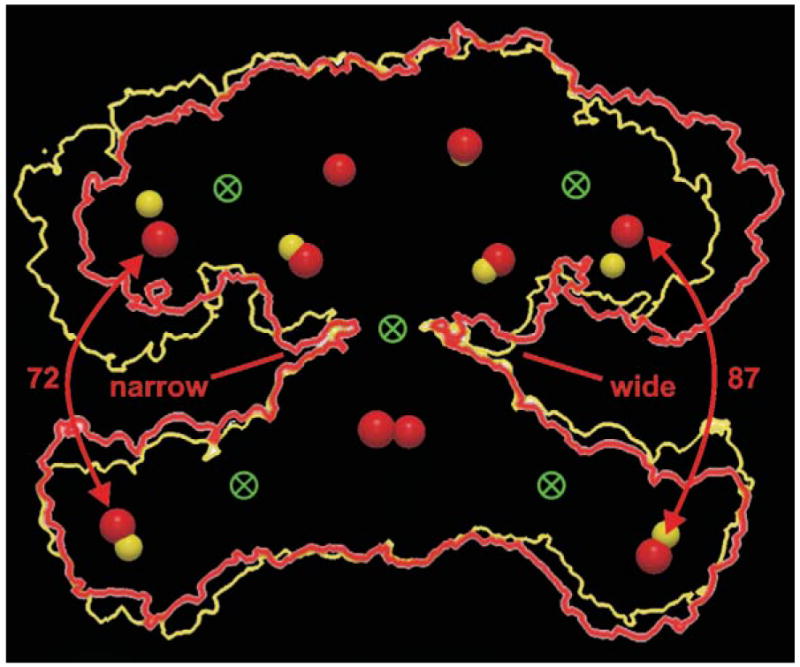
Interdomain hinges and conformational flexibility. An outline of the FAS dimer structure (red) was rotated 180° and the resulting outline (yellow) superimposed on the original. The active sites of the two structures are shown as red and yellow spheres and the possible location of hinges that could facilitate the conformational changes required to produce the two structures are shown as crossed circles. Reproduced with permission from ref. 88.
The mutant complementation and cross-linking studies revealed that the ACP domains can make contacts with the KS and AT domains of either subunit. Unfortunately, it is not yet clear from either the electron microscopy or crystallography analyses how the substantial conformational flexibility required for this feat is achieved.
6.2.2 The crystal structure of the KS–AT didomain from DEBS module 5
Not to be outdone, almost immediately following the publication of the FAS crystallographic study from Ban’s laboratory, the PKS community responded with their own startling revelations on the architectural organization of the modular PKSs. Khosla and co-workers solved the crystal structure of a 194 kDa fragment from DEBS module 5 that encompassed the KS and AT domains, revealing remarkable similarity to the equivalent domains of the FAS located in the leg region.90 However, the higher resolution (2.73 Å) provided considerably more detail than was evident from the FAS structure, and revealed surprising features in the linker regions flanking the catalytic domains. The three flanking regions are: a coiled-coil preceding the KS domain, a highly structured linker consisting of three β-strands and two helices that connects the KS and AT domains, and a 30-residue appendage to the C-terminus of the AT domain that wraps back over the AT domain and the KS-to-AT linker to make intimate contact with the KS domain (Fig. 10C). The coiled-coil feature is structurally and functionally independent of the KS domain and functions primarily as a polypeptide recognition element (see Sections 10.3.1 and 12.3). At the center of the structure is a dimeric KS domain flanked by a pair of monomeric AT domains. The structure exhibits an extensive (2828 Å2) dimer interface comprised of the KS catalytic domains and the N-terminal coiled-coil structure that protrude outwards away from the catalytic KS domains. The overall structures of both the KS dimer, characterized by a αβαβα fold, and the AT monomers, with a typical α,β-hydrolase-like core,101 are very similar to those of their prokaryotic type II and mitochondrial counterparts; the only notable difference is the extended C-terminal helix of the DEBS AT domain, which interacts closely with the KS-to-AT interdomain linker.
More remarkable is the striking similarity of the overall topology of the DEBS KS–AT didomain with its FAS counterpart, in which the KS domains forms a tightly interacting homodimer centered around the 2-fold axis flanked on either side by the two monomeric AT domains. In fact, when the structures of DEBS KS–AT and FAS are overlapped with the KS domains aligned, the positions of the two DEBS AT and FAS AT domains are almost identical, albeit with a twist of ~10° from the 2-fold axis. The only major difference in the two structures is the absence of the N-terminal coiled-coil docking element in the FAS. Because of the limited resolution of the FAS crystal structure, it was not possible to discern any details in the post-AT linker region. However, comparison of the amino acid sequences in this region reveals that the hydrophobic residues involved in domain–domain interactions and structurally constrained proline residues are conserved in animal FASs. It is highly likely then that in both types of megasynthase this region, previously assumed to represent merely a connecting region to downstream domains, in fact makes multiple contacts with the two upstream domains and plays an important role in stabilizing the relative position of the KS and AT domains. The remarkable similarity in the architectural details of the N-terminal region of the FAS and PKSs implies a close structural, functional and evolutionary relationship between these megasynthases.
6.2.3 The crystal structure of the pseudodimeric KR didomain of DEBS: the split-domain hypothesis
The location of KR domains in FASs and modular PKSs has historically been estimated by the recognition, and verification by mutagenesis, of a pyridine-nucleotide binding-site motif (GxG/AxxG/AxxxG/A) as part of a typical Rossmann αβ fold,92 predicted from the amino acid sequence. In modular PKSs that lack DH and ER domains, the KR domain is located adjacent to a core region (residues 1440–1665 in DEBS module 1), the function of which was unknown. This core region is also present in all FASs and in PKS modules that include both DH and ER domains, but in these situations the ER domain is inserted between the core and the KR domain. Attempts to engineer hybrid PKSs by KR replacement were successful only when part of the core region was included, and furthermore, mutagenesis of this core region in the context of the FASs had surprisingly eliminated binding of NADPH to the KR domain located ~400 residues downstream. Although these findings suggested that there might be structural elements in the core region that were important for KR function, no one could have anticipated the remarkable finding revealed by the crystallographic study recently reported by Keatinge-Clay and Stroud.89 These authors chose to crystallize an engineered protein equivalent to a large polypeptide region previously derived from DEBS1 by limited proteolysis that included both the KR domain and upstream core sequence. The structure of this 51 kDa protein with bound NADPH was solved to 1.79 Å resolution, revealing two subdomains each similar to a short-chain dehydrogenase/reductase (SDR) monomer. The first subdomain, derived from the core region, has a truncated Rossmann fold, lacking a pyridine nucleotide binding-site, and appears to perform a strictly structural role in stabilizing the adjacent catalytic subdomain, which contained bound NADPH. The structure of module 1 KR is presumably representative of the KR domains associated with modules 2, 3, 5 and 6, none of which contain DH or ER domains, and suggests that in all of these domains the previously unassigned core region actually represents the structural subdomain of the KR. But what about module 4, which contains a full complement of β-carbon-processing enzymes, where the core region is separated from the KR by the ER domain? By mapping their structure onto the predicted secondary structure for the core region of module 4, the authors concluded that this region contains a KR structural subdomain equivalent to that defined in their crystal structure. The astonishing implication of this finding is that the KR coding region can be interrupted by insertion of a ER coding sequence in modules that contain a full complement of β-carbon processing enzymes! Secondary structure prediction also suggested that the region downstream of the AT domain in module 4 adopts a double hotdog fold with the first hotdog fold corresponding to the sequence previously assigned to the DH domain. Thus the authors concluded that the DH domain was likely a pseudodimer and that part of the core region actually represents the second of the DH pseudosubunits. This deduction is in complete agreement with the finding that the DH domain of the FAS also adopts a pseudodimeric double hotdog fold, twice as long as was originally suspected. Thus in a complete PKS module, the unassigned sequence previously characterized as the core region actually may represent the second subdomain of the DH domain followed by the structural subdomain of the KR.
Using the structure of an E. coli TE II to represent the double hotdog structure of the DH domain102 and a dimeric quinone oxidoreductase to represent the medium chain dehydrogenase fold of the dimeric ER domain, the authors simulated a structure for the entire β-carbon processing region of DEBS module 4 that is remarkably similar to that deduced for the equivalent region of the FAS (Fig 10B). In support of their model, which requires that the ER domain be dimeric, the authors cloned and expressed the entire DH–ER–KR region from DEBS module 4 and found it to be dimeric. Based on this analysis, Keatinge-Clay and Stroud hypothesized that FASs too have split KR domains flanking the ER domain, supporting their argument by citing the disruptive effect of specific mutations in the core region on NADPH binding to the KR domain.
As appealing as this idea may be, one should bear in mind that the hypothesis relies heavily on secondary structure prediction and protein–protein docking simulations. In the structure shown in Fig. 10B, only that of the KR domain represents an actual crystal structure derived from a PKS. When we submitted the core region of DEBS module 4 to six different threading servers, including the 3D-PSSM used by Keatinge-Clay and Stroud, all predicted a short-chain dehydrogenase/reductase fold for the putative KR structural subdomain with >90% probability.92 In contrast, when we submitted the complete rat FAS core region (530 residues), an SAM-dependent methyltransferase fold103 was predicted with a 90–100% probability by all servers except 3D-PSSM, which found no clear solution. Furthermore, in their proposed three dimensional structure, Keatinge-Clay and Stroud placed the structural subdomain on the distal region of the ‘arm’; this assignation would appear somewhat arbitrary as no clear electron density in the 4.5 Å FAS structure could be attributed to a second region exhibiting characteristics of a truncated Rossmann fold. Clearly the true test of the model must ultimately come from real, rather than virtual, structural information.
7 Re-evaluation of the current status of the models for FAS and PKS
The availability of new provocative structural information for the animal FASs and modular PKSs make this an appropriate time to step back and reassess the status of the various models that have been proposed. A remarkable outcome is that, as additional structural data are assembled, it is becoming more and more apparent that the two megasynthases share a remarkably similar architecture. Clearly, it now has been established without doubt that in both types of megasynthase the companion subunits are orientated head-to-head, anchored by the dimeric N-terminal KS domains. But head-to-head does not necessarily imply tail-to-tail. And here there are some apparently unresolved differences between the two systems. The TE domains of FAS, whether released from the complex by limited proteolysis or expressed as recombinant proteins, are catalytically active monomers.83,104 On the other hand, the PKS TEs are dimeric regardless of how they are generated.81,97 Unfortunately, neither type of TE has yet been imaged in its native context, at the C-terminus of a FAS or a PKS module, although electron microscopy images of the animal FAS decorated with anti-TE antibodies appear consistent, with the two TE domains positioned at opposite ends of the structure.86 The structural information available appears consistent with the TE domains linked, via the ACP domains, to the monomeric KR domains located distantly on the two arms of the dimer in FASs and C-terminal PKS modules. For the two TE domains to dimerize would require a looping back of the ACP-TE domains into the central axis of the structure. Although both TEs crystallize as dimers,97,98 at least in the case of the FAS-TE, this represents a crystallization artifact of no functional significance (see Section 10.7).
That two of the β-carbon processing enzymes, ER and DH, lie on the central axis of the FAS, and possibly in the PKSs too, and appear to be dimeric was not anticipated in any of the proposed models for either FAS or modular PKSs. The dimeric nature of the KS–AT structure and the likely dimeric nature of the ER domains in modular PKSs reveals one of the weaknesses in the Cambridge model, which relied heavily on gel filtration as the method to assess the oligomeric status of proteolytic fragments and judged the ER and KS–AT fragments to be monomeric. Indeed, these findings present a problem for the Cambridge double helical model, since a key feature of the model is that the β-carbon processing enzymes associated with the two subunits of each module loop out from opposite sides of the central axis and therefore are monomeric. Furthermore, the Cambridge model also requires that the AT domains be positioned along the central axis of the structure, whereas in reality they are located at the ends of the legs. Clearly, a revised model is required to meet these newly discovered structural constraints.
The available structural data does not yet reveal how a series of PKS modules is stacked. Many PKS modules typically lack the DH and ER domains which, at least in the FAS structure, lie on the central axis and appear to contribute to the dimerization interface. Clarification of this matter may well require a crystal structure that includes structural elements from adjacent modules or high quality electron microscopy reconstructions of a multimodular assembly. Whereas the head-to-head coiled sububunit model proposed for the FAS is generally consistent with the crystal structure derived in the Ban laboratory, certain features of the model are not readily explained. Thus, in the Smith model the ACP domains are positioned such that they are able to make functional contact with the KS and AT domains of either subunit (Fig. 4B). This feature of the model was based on the mutant complementation and cross-linking experiments (Fig. 5 and Fig. 6) that also indicated the intersubunit ACP contacts with KS and AT domains were favored over intrasubunit contacts. The redundancy in ACP-to-AT contacts was also confirmed for the modular PKSs (Fig. 7), and in this case the rates of acyl transfer were identical inter-and intrasubunit. As the ACP domains have yet to be imaged in the context of either megasynthase, and their range of movement has not been fully assessed, it is difficult to envision how these features will be accommodated in a structural framework. Ban and colleagues speculate that in their structure it would be very difficult for an ACP to reach the KS and AT domains of both subunits, in part because of the long distances that may be involved, but also because the KS and DH domains protrude sideways and would likely restrict access of the ACP domain to one side of the dimer. It is possible, of course, that the conformation captured in the crystal structure may represent only one of several different conformations that are visited by the megasynthase, and conceivably twisting of the upper and lower halves of the structure at the narrow waist region could bring the KS and AT domains of both subunits within range of either ACP. Again, this issue can likely only be resolved by examination of more complete structures and determining the range of motion that is enjoyed by the various domains, the ACP domain in particular.
8 Product specificity of FASs and modular PKSs
The main distinction between the two types of megasynthase is that whereas the FASs consist of a single module that is used iteratively and the limitation in growth of the acyl chain is a direct result of the substrate specificity of component enzymes, the PKSs are constructed of modules that are utilized sequentially so that the number of elongation cycles is strictly determined by the number of modules. Nevertheless, there are exceptions in both systems: FASs in which the intrinsic regulation of product chain length is overridden, and PKSs that break the rule of colinearity, so that the number of elongation steps does not correspond to the number of modules present.
8.1 Chain termination in FASs
Fidelity of the FAS system in producing only the fully saturated long chain fatty acid product is assured by three features. First, the KS can accept only saturated acyl intermediates for subsequent elongation; β-ketoacyl, β-hydroxyacyl and enoyl moieties are very poor substrates for this enzyme.105 Secondly, whereas saturated acyl chains containing 2–14 C atoms are good substrates for the KS, longer acyl chains are not readily translocated from the phosphopantetheine to the KS active-site cysteine thiol.56,105 So, when the TE activity of FAS is compromised, elongation to the 16 C atom stage takes less than 1 second, but further elongation to 18, 20 and 22 C atoms requires several minutes.106 Thirdly, the chain-terminating TE has very limited ability to hydrolyze short and medium chain-length acyl thioesters.83 Thus the specificities of the chain extension and chain termination steps complement each other perfectly to ensure that the predominant product released from the FAS is the 16 C atom fatty acid (Fig. 12). In contrast, in polyketide synthesis, product chain-length is controlled primarily by the total number of FAS-like modules associated with the particular PKS. Thus, when presented with the natural starter and chain-extender substrates, modular PKSs are ‘hard-wired’ to perform the appropriate number of condensation and β-C-processing reactions. Indeed, in the specific case of the DEBS PKS, all six KS domains are capable of extending unnatural substrates of various chain lengths from diketide to decaketide.54 Nevertheless, it is possible that in other PKSs, substrate specificity of the KS domains may also play a role in ensuring the forward flow of intermediates through the assembly line.
Fig. 12.
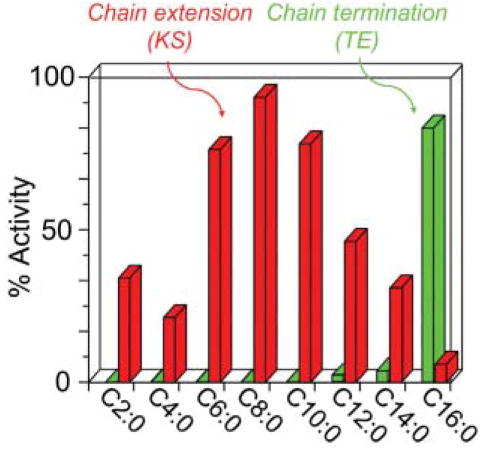
Substrate specificities of the chain-extending and chain-termination enzymes of the animal FAS. Chain-extension activity represents the activity of the KS domain interthiol acyltransferase and chain termination activity, the activity of the TE domain, both assayed using acyl-CoAs as model substrates.
8.2 Exceptions to the normal control of product chain length
8.2.1 The role of type II thioesterases
The strict control of product chain-length specificity intrinsic to FASs operates in most lipogenic tissues throughout the animal kingdom. However, there are exceptions. The best characterized examples are found in specialized tissues such as the avian preen glands and lactating mammary glands, which synthesize appreciable quantities of shorter chain-length fatty acids (C6–C14). In mammals, these short and medium chain-length fatty acids are important components of milk fat triglycerides, and in aquatic birds, as components of preen oil, they play a critical role in waterproofing feathers. These unusual fatty acids result, not from the presence of unique FASs in these glands, but from the presence of tissue-specific chain-terminating TE enzymes.107,108 The TEs are freestanding type II enzymes that exhibit broad acyl chain-length specificity and have the remarkable ability to interact with the FAS and hydrolyze intermediate chain-length acyl moieties from the phosphopantetheine of the ACP domain without slowing the overall rate of fatty acid synthesis.108
Type II TEs are also found encoded proximal to gene clusters for modular PKSs. The first of these TEs to be characterized, PikAV, is encoded immediately downstream of the gene cluster for the modular PKSs responsible for the production of both hexa-and heptaketides in Streptomyces venezuelae.109,110 This organism produces three hexaketides derived from 10-deoxymethynolide and two heptaketides derived from narbonolide (Fig. 13). The gene cluster contains a loading module and six chain-extending modules, the last two being encoded on two separate polypeptides PikAIII (module 5) and PikAIV (module 6).111 The hexaketides are produced by bypassing the chain-extension reaction normally catalyzed by module 6. Initially, it was hypothesized that the type II TE encoded by pikAV might function as an alternative chain-terminating enzyme by interacting directly with module 5 to release the hexaketide product. Indeed, consistent with this concept, the pikAV TE appears to be closely related to the animal TE II enzymes (>60% sequence similarity) that function as alternative chain-terminating enzymes for fatty acid synthesis.109,110 However, the subsequent finding that inactivation of the pikAV TE II led to reduced production of both the hexa- and heptaketide products was inconsistent with this viewpoint, and an alternative role for type II TEs as editing or proof-reading enzymes has been proposed.110 According to this hypothesis, premature decarboxylation of extender substrate moieties, without accompanying condensation, can generate acyl-ACP species that, if not removed, can block elongation sites on the PKS. The pikAV TE II has a broad specificity toward acyl-ACP substrates and is therefore capable of unblocking the ACP sites and restoring functionality to the PKS. A similar TE II has been found associated with the DEBS system.112 Thus these structurally related TEs associated with FAS and PKS systems perform very similar functions: both have broad acyl chain-length specificity and interact with type I synthase systems to catalyze hydrolysis of acyl-ACP thioester intermediates. The crucial difference is that in the FAS context, the released fatty acids themselves have an important role in the physiology of the tissue, whereas in the context of the PKS they represent the products of an editing system that functions to clear the ACP domains of non-productive intermediates.
Fig. 13.

The modular architecture of the ‘Pik’ PKS. An acyl chain released from module 5 and cyclized forms 10-deoxymethynolide, whereas cyclization of the product released from module 6 forms narbonolide.
8.2.2 Module skipping
Elimination of pikAV TE II as a player in the production of hexaketides by the six-module PKS required an alternative explanation for the bypassing of module 6, a process that has been termed ‘module skipping’. Further experimentation quite unexpectedly revealed that, under certain culture conditions, translation of module 6 was initiated from an alternative start site, resulting in the production of an N-terminally truncated polypeptide that lacked part of the KS domain. Production of the truncated module 6 transcript correlated with the production of hexaketide. Apparently, the truncated KS domain, which retains the cysteine nucleophile, cannot carry out a condensation reaction but is able to accept the hexaketide product from the module 5 ACP and transfer it directly to the module 6 ACP, where it is removed by the resident TE domain.
8.2.3 Programmed iteration by PKS modules
The colinearity rule is also broken in some modular PKSs that use a module more than once during polyketide synthesis in a phenomenon commonly referred to as ‘stuttering’. One of the first examples documented was in Saccharopolyspora erythraea, when it was shown that two octaketides are synthesized as minor products by the use of module 4 twice in successive elongation cycles.113 The two octaketides differ only in that either an acetyl or propionyl moiety was utilized as the starter substrate. Since DEBS normally rigorously abides by the rule of colinearity, this phenomenon was referred to as aberrant stuttering. Subsequently however, several additional examples of stuttering have been described that appear to play a vital role in the biosynthesis of the major polyketide product and therefore might more appropriately be termed programmed iteration.114 These include the stigmatellin PKS of Stigmatella aurantiaca that caries out ten elongation cycles with only nine modules,115 the borrelidin PKS from Streptomyces parvulus Tü4055 that carries out eight elongation cycles with only six modules116 and the aureothin PKS of Streptomyces thioluteus that carries out five elongation cycles with only four modules.117 The possibility that programmed iteration may result from the presence of additional modules distantly located in the chromosome, or from non-stoichiometric association of constituent modules, appears to have been discounted. In each of these examples, the product formed in the first elongation cycle by the iterating module is transferred back to the active-site cysteine of the KS domain of the same module, rather than to the KS of the downstream module. This, of course, is exactly as occurs normally in the animal FAS at the conclusion of every elongation cycle. Indeed, in a recent comprehensive review on exceptions to the colinearity rule, Moss et al. have suggested that programmed iteration occurs most commonly in modules that catalyze full β-carbon processing and are not covalently connected to a downstream module; that is, in modules that closely resemble a FAS.114 Thus, if the interaction between non-covalently associated modules is dynamic rather than stable, the product of the first elongation cycle might be more likely to be recycled back through the same module. As appealing as the hypothesis may seem, the aureothin PKS does not fit neatly into this concept (Fig. 14).
Fig. 14.
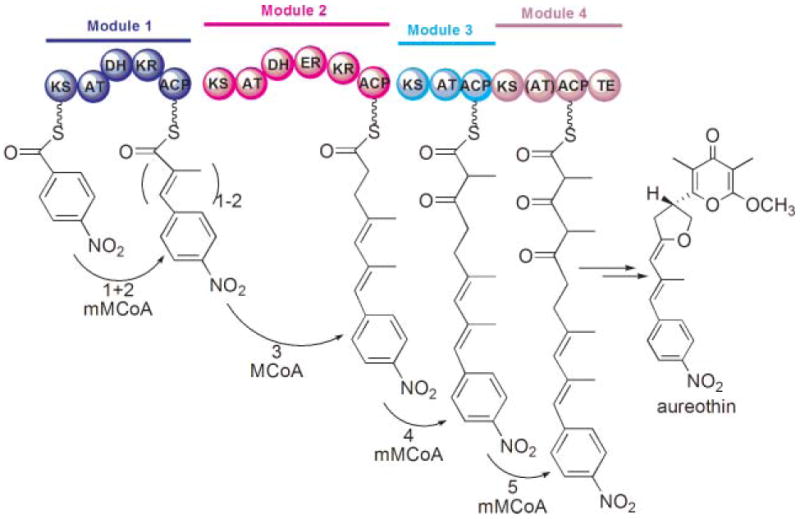
Programmed iteration by the aureothin modular PKS. An unusual starter moiety, p-nitrobenzoyl, is subjected to two rounds of chain extension in module 1 by methylmalonyl-CoA (mMCoA) followed by a normal chain-extension process successively in modules 2–4.
In this system, the iterating module 1, which resides on a unimodular polypeptide, does not have a full complement of β-C-processing enzymes and, lacking an ER function, results in the retention of a diene in the final product. Furthermore, covalent fusion of modules 1 and 2 does not eliminate the stuttering, and the same product is formed by the iterative use of module 1. Thus, the absence of a covalent linkage between the iterating and downstream modules does not appear to be responsible for the stuttering. Furthermore, in each of the examples of programmed iteration studied thus far, the number of iterations employed appears to be highly regulated. For example, in the borrelidin PKS, module 5, the only unimodular polypeptide housing a full complement of β-C-processing enzymes, appears to be utilized three times, and in the aureothin PKS, module 1 is utilized twice. So how is the number of iterations so tightly controlled? One possibility perhaps is that specificity of acyl transfer between modules is a key factor. In this scenario, the product of the iterating module may not be a good substrate, either for transfer to, or condensation by, the downstream KS domain until the required number of chain-extension reactions has occurred. Although in general PKS modules usually have a broad tolerance toward incoming ketide chains,118 hybrid PKSs have been engineered where the substrate tolerance of KS domains limits the efficiency of intermodule coupling.119 Regardless of the actual mechanism governing programmed iteration, the phenomenon clearly illustrates that, under some circumstances, modular PKSs can function just like a FAS with the product of one round of elongation being recycled through the same set of enzymes more than once. Indeed, in type I modular PKSs the transfer step from donor ACP to downstream acceptor KS appears to be a freely reversible reaction, and it is unclear at present why reverse transfer from downstream to upstream modules occurs so rarely.120
9 Stereospecificity in FASs and PKSs
Most of the details concerning the stereochemistry of fatty acid biosynthesis in eukaryotes were deciphered in the laboratories of Gordon Hammes and John Cornforth, who had earlier shared the 1975 Chemistry Nobel Prize (with Vladimir Prelog) for his work on the stereochemistry of enzyme-catalyzed reactions. Their studies established that the KS-catalyzed condensation reaction proceeds with inversion of configuration at the malonyl C2 position, followed by KR-catalyzed reduction of the 3-keto moiety to the 3R alcohol by transfer of the pro-4S hydride from NADPH, and DH-catalyzed dehydration to a trans-enoyl moiety by the syn elimination of the 2S hydrogen and the 3R hydroxyl as water. In the animal FAS, the final reduction to a fully saturated acyl moiety by the ER domain proceeds by transfer of the pro-4R hydride of NADPH to the pro-3R position with simultaneous addition of a solvent proton to the pro-2S position, a syn addition,121 as in prokaryotes.122 Surprisingly, the yeast FAS appears to be an exception, since this ER takes the pro-4S hydride of NADPH into the pro-3S position and the solvent proton is incorporated at the pro-2S position in an anti addition.123 Although these elegant studies may seem rather academic for a pathway that results ultimately in the loss of all of the chiral centers and formation of an unbranched saturated acyl moiety, this is certainly not the case for polyketide synthesis where the final products typically retain stereochemical features that are a direct consequence of the specificity of the component enzymes (Fig. 15).
Fig. 15.

The stereochemistry of FAS and PKS.
One of the commonest types of PKS module elongates the acyl chain with a methylmalonyl moiety and reduces the condensation product only as far as the alcohol. Under these circumstances, the 2-methyl-3-hydroxyacyl moiety produced has two new chiral centers and all four combinations of methyl and alcohol stereochemistry are theoretically possible: (2R,3S), (2S,3R), (2R,3R) and (2S,3S). In 1965, Walter Celmer noted that, despite the variation within a given polyketide structure, there are strong position-specific structural and stereochemical homologies among the hundreds of known polyketides.124 His inference that these patterns were indicative of a common genetic origin for stereochemical control at these centers is referred to as Celmer’s rule. So how is stereochemical control exercised and how do the stereochemical preferences of the component enzymes compare to their FAS counterparts?
Stereochemistry at the methyl-branched centers clearly is not controlled by a choice of isomer by the AT domains, since they all exhibit absolute specificity for the 2S isomer of methylmalonyl-CoA. As with the FASs, in the modular PKSs the condensation reactions always proceed with inversion of configuration, resulting in the 2R configuration at the methyl center. However, in some PKS modules the methyl center is epimerized, resulting in the formation of the 2S isomer.125 Domain swapping experiments have implicated the KS domain as being responsible for epimerization.126 However, the KR domain has also been implicated based on the observation that no module lacking a KR domain has been found to catalyze epimerization.89 In the case of DEBS modules 1, 3 and 4, the methyl groups are of the S configuration and must therefore undergo epimerization following the condensation and accompanying inversion of configuration, whereas in modules 2, 5 and 6, the methyl groups are of the R configuration and have not undergone epimerization (Fig. 2). In those rare instances where animal FASs use methylmalonyl moieties as chain extenders, for example in avian preen glands, the methyl branched-chain products are always of the R configuration at the methyl centers.127 Yet, unlike their counterparts in modular PKSs, the AT domains associated with the animal FASs are actually capable of utilizing the 2R and 2S isomers of methylmalonyl-CoA equally well. The explanation, once again, appears to be substrate availability, since the methylmalonyl-CoA produced in these tissues by cytosolic acetyl-CoA carboxylase is exclusively the 2S isomer.127 Given that propionyl-CoA carboxylase and methylmalonyl-CoA epimerase are both mitochondrial enzymes, the 2R isomer is unlikely to be available for fatty acid synthesis in the cytosol.
In modular PKSs, as in the FASs, the hydride utilized in reduction of the ketoacyl moiety by KR is derived from the 4S face of NADPH.128 However, in contrast to FAS, which always produces the 3R hydroxyl, the KRs associated with DEBS produce hydroxyls of both the 3R configuration, in module 1, and the 3S configuration, in modules 2, 5 and 6. Since chirality at the 3-hydroxyl is determined by the direction of hydride addition to the keto group (re or si face), the stereochemical outcome must depend on different orientations of the β-ketoacyl moieties in the KR active site relative to the NADPH cofactor (see Section 10.4).
Surprisingly, KR domains associated with modular PKSs can also influence the stereochemical outcome at methyl chiral centers. In the modules where the epimerization takes place at the 2-methyl center, both 2R and 2S isomers are potentially available for reduction at C3. However, it appears that the KR domain in such modules selects specifically the 2S isomer for reduction, resulting in formation of a 2S-methyl-3R-hydroxyacyl moiety.129
The vast majority of polyketides contain least one double bond (the much-studied DEBS PKS is atypical in this regard) generated through the action of a DH domain. These double bonds are of the trans (E) type, as are the cryptic unsaturated intermediates of fatty acid synthesis. In exceptional cases, such as the epothilone PKS, the DH appears to catalyze formation of a cis double bond, although details of the mechanism have yet to be deciphered.130
David Cane and colleagues131 have examined the amino acid sequences of KR domains associated with PKS modules that also have functional DH domains and noticed that all possessed a conserved aspartate residue (in the ‘LDD’ motif—see the KR discussion in Section 10) that typically serves as an indicator for 3R stereochemistry.132 Using the picromycin/methymycin PKS as a model, Cane’s team confirmed experimentally that in module 2, which has an active DH domain, the associated KR domain generates specifically a hydroxy intermediate with 3R stereochemistry.131 The implication of this finding is that the DH domains of FAS and the picromycin PKSs appear to have the same stereospecificity and generate the trans (E) unsaturated moieties from 3R hydroxyacyl substrates through the syn elimination of water. An important consequence is that engineering novel PKSs by the introduction of this picromycin DH domain likely will only succeed when paired with a KR domain that specifically produces a hydroxyacyl moiety with 3R stereochemistry. However, module 2 of the picromycin PKS uses malonyl-CoA as the extender, where either of the C2 hydrogens are potentially available for elimination. Does the dehydration reaction follow the same stereochemical course in modules that use a methylmalonyl moiety, where only one hydrogen is potentially available, either 2R or 2S depending on the configuration of the methyl moiety? In DEBS module 4, the methyl moiety at C2 is likely to be in the 2S configuration, based on the stereochemical structure of the product 6-deoxyerythronolide B (Fig. 15). However, the KR domain (with an ‘LAD’ motif in DEBS KR4), based on the sequence and the Cane result, is predicted to produce the 3R hydroxyl intermediate. As in the FASs, the product of the dehydration is again in the trans configuration, based on evidence from ER4 deletion experiments.133 So, there are two possible scenarios during ER reduction: either NADPH transfers a pro-4R hydride to the pro-3R position, followed by an anti addition of the solvent proton at the 2R position (similar to the yeast FAS but from the mirror direction), or NADPH transfers a pro-4S hydride to the pro-3S position, followed by a syn addition of the solvent proton at the 2R position (similar to the animal FAS but from the mirror direction). Clearly further experimentation is required to resolve this ambiguity.
10 The structures, catalytic mechanisms and substrate specificity of individual domains
In the following section, the catalytic domains in type I modular PKS and FAS are compared in terms of structure, mechanism and substrate specificity. For FAS, experimentally determined structures are available only for the ACP and TE domains. The 4.5 Å Ban structure was deposited to PDB as a C-alpha trace, based on which we have generated structures for other domains using secondary structure prediction (six different threading servers) and homology modeling (using SwissModel, Geno3d and EsyPred3D). For the DEBS PKS, high resolution crystal structures are available for, KS, AT, KR and TE, and only the structures for DH and ER domains are produced by modeling. Despite these limitations, a comprehensive picture does emerge that reveals remarkable similarity between equivalent domains of type I FAS and PKS, in terms of overall fold and catalytic mechanism. Nevertheless, several of the domains exhibit different substrate specificities in the context of the two megasynthase types, and these unique features are highlighted in the following text. For type I modular PKSs, the readers are referred to excellent reviews by O’Hagan, Rawlings and Hill in this journal.134–138 For a detailed analysis of the individual type II FAS enzymes, readers are referred to an outstanding recent review by Stephen White, Charles Rock and colleagues.139
10.1 ACP
The mammalian FAS apo ACP structures have been solved by NMR and X-ray crystallography and consist of a four-helix bundle stabilized by multiple inter-helical hydrophobic interactions99,140 (Fig. 16A). The predicted structures of the DEBS ACP domains also exhibit a similar fold.141 The conserved serine residue that is the site of post-translational modification lies at the N-terminal end of helix-2. Thus, the type I ACP structures are very similar to that of a typical type II ACP, in which helix 2 is characterized by a highly conserved cluster of negatively charged residues and is regarded as a universal ‘recognition helix’ involved in interactions with other proteins99,142–145 (Fig. 16B). Although the amino acid sequences of the ACP domains of type I FASs are less well conserved and do not generally retain the strong negatively-charged character, there is evidence indicating that helix 2 also plays an important role in the docking of ACP with other proteins. For example, the crystal structure of the human FAS ACP domain bound to its cognate phosphopantetheinyl transferase reveals that helix 2 makes direct contact with multiple residues on the transferase.140 Similarly, sequence analysis and homology modeling studies on the ACP domains of modular PKSs are also consistent with the role of helix 2 as a universal interaction motif.141 Functional assays have revealed that KS3-AT3 and KS6-AT6 didomain constructs exhibit much higher affinities for their cognate ACPs than for ACPs from other modules, suggesting that the ACPs may have module-specific binding sites on their surface.146 Consistent with this idea, the predicted DEBS ACP1–6 structures display distinct differences in surface charge distribution.
Fig. 16.

The ACP structure. (A) Ribbon diagram of the rat FAS ACP (2PNG), colored blue to red, corresponding to N to C termini.99,140 The post-translationally modified serine residue on helix 2 is shown. (B) The ACP surface electrostatic potential colored from red (negative) to blue (positive). (C) The crystal structure of the decanoyl thioester of E. coli ACP. The phosphopantetheine moiety is shown in red and the decanoyl moiety in green. Adapted from ref. 147.
Studies on type II ACPs using NMR indicate that the bound acyl moieties can adopt two different states; one involving direct interaction with the ACP through the cleft flanked by helices α2 and α3, the other allowing direct exposure to solvent.142 This two-state equilibrium may facilitate functional interaction of the bound acyl moieties with other catalytic domains. Recently, the crystal structures of the several E. coli type II acyl-ACPs (hexanoyl, heptanoyl and decanoyl) have been solved;147 the decanoyl-ACP structure is shown (Fig. 16C). In all of these structures the saturated acyl chain, the thioester bond and the β-mercaptoethylamine section of the phosphopantetheine moiety are sequestered in the hydrophobic cavity within the four-helix bundle. The size of the cavity appears to expand as the length of the bound acyl chain increases, although the overall fold is not altered. Prior to each round of chain extension, the growing acyl chain relinquishes its connection with the ACP and is transferred to the active-site cysteine of the KS. This transfer requires the phosphpantetheinyl moiety to uncoil from its position at the entrance to the helical core of the ACP and thread the acyl chain into the KS active site. Interaction with the β-carbon processing enzymes does not necessitate removal of the acyl chain from the hydrophobic core of the ACP but presumably, when the more polar intermediates are present on the ACP (β-keto, β-hydroxy and enoyl), the thioester and β-carbon atoms must be more exposed to allow access to the appropriate enzymes. Thus there appears to be some inherent plasticity within the four-helix structure and the degree of exposure of intermediates to the surface may depend on the structure of the particular intermediate.
In a type I system, exactly how an ACP domain bearing a particular acyl intermediate locates the domain appropriate for catalysis of the next reaction is unknown. However, the new structural data for type II acyl-ACPs suggests that the degree of exposure of the acyl thioester bound to the phosphopantetheine and/or the degree of expansion of the four-helix bundle may provide cues that facilitate recognition by the appropriate enzyme. In the absence of any obvious ‘road map’ in the reaction chambers of the megasynthases, one can only speculate that perhaps a random search is concluded when the correct match of substrate and enzyme is made and a productive interaction can follow.
10.2 The AT domains responsible for substrate recruitment and loading
An important difference between the animal FASs and modular PKSs is that in the FASs, a single AT is responsible for loading both the acetyl starter and malonyl chain-extender substrates, whereas in the PKSs these two functions are carried out by specialized ATs: in the loading module, an AT primes the ACP typically with an acetyl or propionyl moiety and in the chain-extending modules, the ATs load the ACPs with either a malonyl or methylmalonyl moiety.
10.2.1 AT architecture
The crystal structures for the DEBS AT5 domain and the porcine FAS AT are characterized by an α/β-hydrolase-like core domain and an appended smaller subdomain with a ferredoxin-like structure, consisting of a 4-stranded antiparallel β-sheet capped by two helices (Fig. 17A). The active site lies in a gorge between the two subdomains. This architecture is very similar to that of the type II malonyl-CoA:ACP transacylases of E. coli and Streptomyces coelicolor.93,148 A novel feature of the type I AT revealed by the DEBS AT5 crystal structure, however, is the greatly extended C-terminal helix (residues 857–867) that interacts closely with the KS-to-AT linker.
Fig. 17.

(A) The DEBS AT5 domain, showing the overall fold with the active site Ser in purple, and the substrate-binding pocket under the Ser in the cleft formed by domains A and B. The four structural elements discussed in the text (items 1–4) that are important for specificity are highlighted in red, purple, green and blue, respectively. (B) Proposed catalytic mechanism of the AT domain. R = rest of acyl chain.
10.2.2 AT catalytic mechanism
Both type I and II ATs catalyze the transfer of acyl moieties between CoA and ACP via a ping-pong bi-bi catalytic mechanism using a Ser-His catalytic dyad.149 The catalytic serine (S581 in rat FAS and S642 in DEBS AT5) is positioned in a highly conserved GHSXG motif that lies at a sharp turn between a β-sheet and α-helix; a structural feature commonly referred to as the ‘nucleophilic elbow’.101 The nucleophilicity of the serine is enhanced by hydrogen bonding to a conserved histidine (H745 in AT5 and H683 in rat FAS). For extender ATs, the sequestration of the substrate (malonyl- or methylmalonyl-CoA) in the cleft between the two subdomains is facilitated by direct interaction of the 3-carboxylate anion with the positively charged side-chain of a conserved arginine (R667 in DEBS AT5, R606 in rat FAS).93,148,150 The active site serine then undergoes nucleophilic attack on the thioester carbonyl group, resulting in the formation of a tetrahedral intermediate, whose negative charge is stabilized by an oxyanion hole, formed most likely by two backbone amides (Fig. 17B). The exiting CoA moiety is protonated by the active-site histidine and replaced by ACP. The ACP phosphopantetheine thiol mounts a nucleophilic attack on the ester carbonyl, the second tetrahedral intermediate is formed and resolved again by the active site histidine which, by reprotonation of the catalytic serine, releases the acyl-ACP product. A remarkable feature of the ATs, both type I and type II, is that unlike the structurally-related α/β hydrolases, the acyl–enzyme complex is stable and deacylation occurs only in the presence of specific thiol acceptors. Zygmunt Derewenda and colleagues have suggested that it is the unique location of the oxyanion hole in the ATs that prohibits the positioning of a potentially hydrolytic water molecule such that it can be activated by the catalytic histidine.148
10.2.3 Substrate specificity of AT
10.2.3.1 Substrate availability or substrate choice?
The FAS AT domain can effectively transfer acetyl, propionyl, butyryl, acetoacetyl, β-hydroxybutyryl and crotonyl starter substrates with similar efficiency in vitro; even a phenylacetyl moiety can function as a starter, although there appears to be no physiological importance to this phenomenon.151 The most commonly used starter in vivo appears to be the acetyl moiety. The same AT domain is also utilized by FASs to load the chain extender, which is almost always a malonyl moiety. Nevertheless, in certain specialized tissues, such as the preen glands of waterfowl and the harderian glands of mammals, the FASs appear to use propionyl-CoA as primer and methylmalonyl-CoA as the chain extender, so that odd-carbon-number methyl-branched fatty acids are synthesized.152,153 However, this atypical choice of substrates is not attributable to the presence of FASs with unusual substrate specificity, since FASs isolated from tissues that do not normally generate these products in vivo are capable of producing them in vitro, when presented with propionyl-CoA and methylmalonyl-CoA as substrates.152 Rather, the production of odd-carbon-number methyl-branched fatty acids appears to depend entirely on the availability of propionyl-CoA and methylmalonyl-CoA in vivo.154 Remarkably then, although FASs can utilize malonyl-CoA ~2 orders of magnitude more efficiently than methylmalonyl-CoA, the major products of the preen gland are branched-chain fatty acids.
The AT domain associated with the loading module of type I modular PKSs, like that of its counterpart in FASs, exhibits remarkable substrate tolerance and can utilize acetyl-, propionyl-, isopropionyl-, isobutyryl-, crotonyl-, phenylacetyl-, hydroxybutyryl- and isopentyl-CoA, both in vivo and in vitro.55,155–159 In comparison, the AT domains that load the chain extender in DEBS modules 1–6 are extremely fastidious, exhibiting a strict structural and stereochemical specificity for 2S-methylmalonyl-CoA and the 2S-methylmalonyl thioester of N-acetylcysteamine (NAC).155,157,160 The AT domains in the chain-extending modules of DEBS therefore act as one of the key gatekeepers of type I polyketide biosynthesis.54
In summary, clearly the nature of the primer and extender utilized by the FAS is governed primarily by substrate availability in specific tissues. The primer used by modular PKSs also appears to be influenced by substrate availability but the extender is selected by highly specific extender AT domains.
10.2.3.2 Molecular basis of AT specificity
Past studies indicate that there are several possible structural and sequence elements important for AT substrate specificity. The new crystal structure and in silico substrate docking of the DEBS AT5 domain now sheds light on the molecular basis for its specificity for the 2S-methylmalonyl-CoA in the following elements:
The ‘RVDVVQ’ motif. This motif, 30 residues upstream of the active site serine, was first recognized in the rapamycin synthase.161 Later, in an analysis of over 200 AT domains, Gokhale and co-workers derived more precise motifs [RQSED]V[DE]VVQ for methylmalonyl-AT and ZTX$[AT][QE] for malonyl-AT, in which Z is a hydrophilic residue and $ is an aromatic residue.162 The structures of DEBS AT5 and modeled FAS AT revealed that the motif is located in a 35 Å-long amphipathic helix (residues 613–630 of AT5 and 550–567 of rat AT) that defines the substrate-binding pocket. Although it is not certain whether this helix makes contact with the substrate, subtle residue changes in this helix may alter its interaction with neighboring structural elements, leading to a change in the substrate pocket size/shape. This consensus sequence has reliable predictive value for the assignment of substrate specificity to AT domains associated with other PKSs.163 Consistent with the observed substrate specificity, according to this motif, AT5 is predicted to be specific for methylmalonyl-CoA, while the FAS ATs display a mixed sequence character and accept both malonyl- and methylmalonyl-CoA (albeit 100 times slower than malonyl-CoA). Further, replacement of the RVDVLQ motif with DTLYAQ in DEBS AT4 produced an active AT that accepted both malonyl- and methylmalonyl-CoA as substrate.164
The GHSXG motif. Sequence alignments and crystal structures indicate that residue X adjacent to the catalytic serine is a branched hydrophobic amino acid (valine or isoleucine) in malonyl-specific ATs and a less bulky residue (glutamine or methionine) in all other ATs.161 Based on the DEBS AT5 structure, a branched hydrophobic residue at this position would result in a steric clash with the methyl group of methylmalonyl-CoA.90 Consistent with the motif prediction, the ‘X’ residue is Q643 in AT5 (a methylmalonyl-specific AT) and either leucine or valine in FAS ATs that exhibit a preference for malonyl over methylmalonyl.
The YASH motif. At the position 100 residues downstream of the active site serine, the methylmalonyl-specific ATs (such as the DEBS extender ATs) have a ‘YASH’ motif, while malonyl-specific AT has a ‘HAFH’ motif.161,164 Gokhale and co-workers further refined these motifs as [YVW]ASH for methylmalonyl-specific ATs and [HTVY]AFH for malonyl-specific ATs.162 Based on a mutational analysis on DEBS, this motif should be the dominant structural feature among motifs #1, #2 and #3.164 In DEBS AT4, mutations of ‘YASH’ to ‘HAFH’ did result in the relaxation of substrate specificity. However, in S. coelicolor AT, mutations of F200 to serine (the ‘GAFH’ motif to ‘GASH’) did not change the malonyl preference.165 In FAS, the corresponding residues are ‘[MILV]AFH’, consistent with a general preference for malonyl over methylmalonyl. Nevertheless, the AT5 structure clearly indicates the importance of hydrophobic pocket formation (which requires the tyrosine in ‘YASH’) for methylmalonyl-CoA binding. Further, the 2S sterespecificity is also clarified: in addition to the predicted hydrophobic interaction of Tyr742 (of ‘YASH’) with the α-methyl group of the substrate, Gln643 (the ‘X’ in GHSXG) can orient the bound methylmalonyl through a hydrogen bond with the substrate carboxylate. In contrast, the side chain of His745 would cause a steric clash with the methyl group of 2R-methylmalonyl-CoA, thereby accounting for the observed stereospecificity.
The C-terminal region. Swapping a short ~30-residue C-terminal segment between rapamycin synthase AT2 and DEBS AT2/AT6 indicates that this region is important for substrate specificity.155 The DEBS AT5 and modeled FAS-AT structures indicate that this region corresponds to an α–β–α motif, in which one α-helix forms part of the substrate pocket entrance. The success of the domain swapping can be explained by the location of the α–β–α motif, which is separated from the remaining protein fold by a loop, and the α–β–α lies near the protein surface. Therefore this region can be exchanged without disruption of the protein fold and may be considered as a possible target for re-engineering of substrate specificity.
In the DEBS starter-loading AT domain the motifs described above are: (1) RVEVVQ instead of RVDVVQ, (2) GHSIG instead of GHSQG, (3) MAAH instead of YASH; the C-terminal 30 residues (motif 4) of ATs associated with the loading and extender modules are well conserved. These findings indicate that the main difference between the loading and extender ATs is in motif #3. Furthermore, the conserved active-site arginine, important for docking of the malonate C3-carboxylate and thus a signature residue for malonyl or methylmalonyl-AT, is usually absent in loading AT domains. Mutagenesis studies with the AT domain of the animal FAS established clearly that the conserved active-site arginine is essential for binding of malonyl moieties. Replacement of this residue actually enhanced activity toward acetyl and longer chain-length starter substrates. In contrast, the AT counterparts in the chain-extender modules of PKSs are inactive toward acetyl moieties.54 It remains unclear as to how the FAS AT domain is able to transfer both malonyl and acetyl moieties with equal efficiency.
Although the DEBS AT5 crystal structure explains its stereo-preference, the specificity between malonyl- versus methylmalonyl-CoA (or methylmalonyl- versus propionyl-CoA) is likely to be a combinatorial result of different structural elements that interact throughout the entire protein fold, rather than the influence of a limited number of residues. A molecular phylogenetic approach such as that applied to chalcone synthase, in which up to 30 active site residues were mutated to engineer substrate specificity, is likely necessary in order to control the AT specificity.
10.3 KS
10.3.1 The dimeric thiolase fold
The crystal structure of the KS domain from DEBS module 5 (KS5)90 and the porcine FAS KS model reveal an overall fold characteristic of the thiolase-superfamily (Fig. 18A). These KSs are homodimers, each subunit consisting of two subdomains that exhibit the same βαβαβαββ topology, suggesting they have evolved through an ancestral gene duplication event. In the KS domains associated with both PKSs and FASs, the extensive dimer interface is stabilized by a pair of hydrogen-bonded antiparallel β-strands. However, uniquely in the KS domains located at the N-termini of PKS polypeptides, a coiled-coil structure projecting from the N-termini also makes a significant contribution to the dimer interface. The role of the coiled-coil may be to facilitate protein dimerization and to act as a docking domain with the complementary upstream C-terminal docking domain to ensure appropriate positioning of modules within the assembly line (see Section 12.3).
Fig. 18.

The KS domain. (A) Overall fold comparison: left, DEBS KS; right, FAS KS; monomers are shown in green and blue and the dimer interface helix-loop in red. (B) Molecular basis of substrate specificity. The FAS KS channel is narrower with limited flexibility, reflecting its limited substrate tolerance for branched acyl chains, while the DEBS KS5 channel is much wider, reflecting its substrate tolerance; the substrate-binding pockets are identified by arrows. The active site Cys is shown as yellow or red spheres in both panels. (C) Proposed mechanism.
The dimeric nature of the KS domains appears to be crucial in promoting dimerization of the entire polypeptide chains. Although, in the FAS, the ER and DH domains also contribute significantly to the extensive subunit interface running through the central body of the structure (Fig. 10A), in a FAS engineered without KS domains, intersubunit contacts between ER and DH domains are insufficient to promote dimerization and this mutant FAS is entirely monomeric.75 Conversely, since many PKS modules lack both ER and DH domains, the KS subunit interface and their N-terminal coiled-coil appendage are apparently sufficient to promote dimerization of the full-length polypeptide (see Section 12.3 for discussion of interpolypeptide coiled-coil interactions).
10.3.2 KS reaction mechanism
Similar to the well-studied FabF and FabB KSs of E. coli, the KSs associated with type I FASs and modular PKSs employ a Cys-His-His triad at the active center. Although the KSs are probably the most widely studied of any of the type I and type II FAS enzymes, and crystal structures are available for many of them, controversy has raged for many years concerning the precise role of the conserved residues in the reaction mechanism.166,167 Only recently does a consensus appear to have been reached that envisions essentially identical mechanisms for the type I and type II KSs.168,169 The KSs form a new carbon–carbon bond by a Claisen condensation reaction that involves three distinct steps: acyl transfer, decarboxylation and condensation (Fig. 18C). Nucleophilicity of the active-site cysteine (C161 in rat FAS and C199 in DEBS KS5), located at the N-terminus of an α-helix, benefits from the positive charge generated by the helix-dipole, effectively lowering its thiol pKa. The oxyanion hole formed by the backbone amides of the active-site cysteine and a residue located near the C-terminus neutralizes the negative charge on the thioester carbonyl, stabilizing the tetrahedral intermediate. The free ACP is released and the chain extender, malonyl- or methylmalonyl-ACP, binds to the enzyme. In the second step, an active-site water molecule, likely activated by a conserved histidine, attacks the malonyl C3 carboxylate, releasing bicarbonate. Formation of the carbanion at C2 is likely facilitated by the second conserved histidine, which stabilizes the enol intermediate. In the final step, the carbanion attacks the acylthioester to form the tetrahedral intermediate, which is stabilized initially by the oxyanion hole then breaks down to form the β-ketoacyl product. In the crystal structure for DEBS KS5 and the modeled rat KS structure, the positions of the catalytic triad and the oxyanion hole can be overlapped completely, consistent with identical catalytic mechanisms for the two types of enzyme.
Some type I modular PKSs, such as those responsible for synthesis of niddamycin, tylactone and 10-deoxymethynolide/narbonolide, contain an unusual KS domain at the N-terminus of their loading module in which the active-site cysteine residue is replaced by glutamine. Surprisingly, the role of these ‘KSQ’ domains was first clarified by engineering a cysteine-to-glutamine mutation in the KS domain of the animal FAS.170 The residue replacement eliminated condensation activity but increased the uncoupled rate of malonyl decarboxylation by more than 2 orders of magnitude. On the basis of this observation, the Smith group suggested that the function of KSQ domains associated with PKS loading modules was to optimize availability of the starter units by catalyzing the decarboxylation of malonyl or methylmalonyl moieties to acetyl or propionyl moieties, respectively. This inference was supported by the observation that the AT domains adjacent to the KSQ domains contained the signature sequence motifs associated with specificity for either malonyl (niddamycin PKS) or methylmalonyl (tylactone and 10-deoxymethynolide/narbonolide PKSs) and contained the conserved arginine residue required for interacting with the 3-carboxylate of these substrates. As indicated in Section 10.2.3.2, this arginine residue is absent from loading AT domains that translocate acetyl or propionyl moieties. The proposed role for KSQ domains in generating the starter units for polyketide synthesis was confirmed by the Leadlay group,171 affording another nice example of the synergism that has characterized development of the FAS and PKS fields in recent years.
10.3.3 KS substrate specificity
In general, the KS domains associated with modular PKSs are quite tolerant of substrate chain length, and all six KS domains of DEBS can accept substrates that vary in length from a diketide to decaketide.54 The animal FAS also is able to accommodate substrates of varied chain-length that can include methyl-branches, derived from methylmalonyl extenders,127 as well as aromatic moieties introduced from phenylacetyl primers.151 The DEBS KS5 structure provides a possible explanation for this broad specificity. A unique feature of the structure is the dimer interface loop region (marked red in Fig. 18A) that replaces a helix in type II KSs and appears to contribute to a larger substrate-binding cavity (Fig. 18B). Homology models of the DEBS KS1, KS2, KS3, KS4 and KS6.54 as well as the KS domain of the rat FAS, indicate that they all likely possess this generous substrate-binding cavity.
Nevertheless, the FASs are highly selective in that they will only accept saturated acyl chains for elongation, and there is evidence that some PKS KSs can discriminate on the basis of the β-carbon atom status, when presented with unnatural substrates. For example, DEBS KS2 will accept diketide substrates with 3-hydroxyl or 3-keto groups but not 3-deoxy diketide substrates. Similar limited substrate tolerance has been reported for other KS domains. A possible explanation for this phenomenon may be found in the structural organization of the KS substrate-binding channel. As in the KSs associated with type II FASs and PKSs, the substrate channel of the type I enzymes can be divided into two halves, corresponding to the substrate-binding and phosphopantetheine-binding regions. The substrate-binding region, which extends from the active site Cys toward the dimer interface, is varied in amino acid sequence between different KS domains of DEBS, and may be important for defining substrate specificity and specific KS dimer formation. However, high-resolution structures of additional DEBS KS domains are needed in order to evaluate the relationship between sequence and substrate specificity. On the other hand, the phosphopantetheine-binding region, which stretches from the enzyme surface to the active site cysteine, is relatively well conserved in all six DEBS and FAS KS domains, reflecting its universal role in binding the phosphopantetheine moiety.
10.4 KR
10.4.1 The KR short-chain dehydrogenase fold
Both FAS and PKS KRs belong to the short-chain dehydrogenase family.92 In the porcine FAS structure, only one KR domain was built into the model. However, as indicated earlier, the DEBS KR1 structure actually contains two KR subdomains, one catalytic, the other structural and exhibiting a truncated Rossmann fold. The β1 and β8 of the structural subdomain bridge both the structural and catalytic subdomains and appear to contribute to stabilization of the pseudodimeric structure of the entire KR domain (Fig. 19A).
Fig. 19.
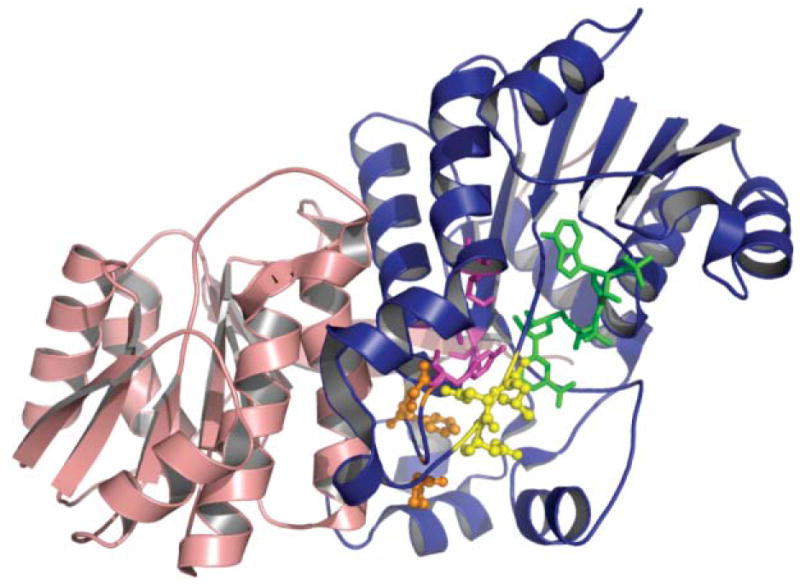
The KR domain. Crystal structure of the DEBS1 KR domain. The catalytic subdomain is shown in blue, the structural subdomain in red, the active site residues in purple, the phenylalanine, proline and glycine residues important for substrate binding in orange, the ‘LDD’ loop in yellow and NADP in green. Adapted from ref. 89.
10.4.2 KR active site and catalytic mechanism
The cofactor NADPH binds DEBS KR1 in a long groove. The catalytic tyrosine, Y1813, and serine, S1800, are positioned similarly as in other short-chain dehydrogenases. Although the positions of the conserved lysine, K1776, and asparagine, N1817, are swapped, the K1776 amine engages in the same interaction observed in other SDR enzymes. In view of the highly conserved nature of the catalytic residues, the KR domains of both PKSs and FASs likely utilize the same proton-relay mechanism proposed for the E. coli FabG.142,172,173 Thus in the reduction of the ketone to the alcohol, the hydride is derived from NADPH and the proton derived from the conserved active-site tyrosine is replenished through solvent, via the active-site lysine.
10.4.3 The molecular basis of KR substrate specificity
Based on sequence alignment, the stereoselective signature motifs for the modular PKS KRs have been proposed to be ‘LDD’ and PxxxN,132,174 and the presence of these motifs predicts formation of the 3R stereomer (or ‘D stereomer’) whereas the absence of these motifs predicts the 3S stereomer (or ‘L-stereomer’, Fig. 15). The crystal structure of KR1 has shed light on the proposed motifs, and shows that F141, P144, and G148 are part of a loop directly adjacent to the catalytic tyrosine, whereas the 93–95 ‘LDD’ motif is located in a loop adjacent to the active site. In KR2, residues W141 and 93–95 ‘PQQ’ are the counterparts of F141 and ‘LDD’ in KR1. The importance of these motifs was demonstrated by mutational analyses of KR1 and KR2, in which the double mutation of KR1 (F141W, P144G) resulted in a switch of the alcohol stereochemistry.175 The identified structural motifs have great potential for future stereospecific biosynthesis of new macrolides.176 Finally, although a high resolution FAS KR structure is not yet available, sequence alignment indicates that the ‘LDD’ motif corresponds to ‘LRD’, and is therefore predicted to produce the R stereochemistry. Consistent with this prediction, the mammalian FAS does produce a 3R hydroxyacyl moiety.
10.5 DH
10.5.1 The DH double hotdog fold
One of the surprises revealed by the 4.5 Å electron density map of the porcine FAS88 was that the DH domain appeared to be comprised of two subdomains that, despite no obvious sequence similarity, exhibit the same ‘hotdog’ fold.177 DHs are notorious for their lack of sequence similarity, a factor that hindered the original locating of the domain in the animal FAS. Nevertheless, the expanded double hotdog DH domain is very similar in three-dimensional structure to the dimeric bacterial DHs, exemplified by the bacterial FabA and FabZ enzymes,91,178 and related ‘hotdog fold’ enzymes, such as the E. coli and human TE II;179 parenthetically, these ‘TE II’ enzymes do not have any role in fatty acid synthesis. In this class of protein fold, the long, non-polar central helix—the ‘sausage’—nestles in a seven-stranded β-sheet that forms the ‘bun’. The two hotdog subdomains of the type I DHs interact extensively by forming a 14-strand β-sheet and by the interactions between the ‘hotdog’ helices from each subdomain. In the homodimeric type II DHs two active sites are formed at the subunit interface, each involving a catalytic histidine and aspartate (or glutamate) from opposite subunits (Fig. 20).91,178 However, the type I pseudodimeric DHs likely have only a single active site, since mutation of a single active-site histidine residue (His878 in the rat FAS) completely eliminates all DH activity.180 The location of the active-site acidic residue, presumably in the second of the two pseudosubunits, has not yet been identified. However, modeling and multiple sequence alignments indicate that a likely candidate is an aspartate residue located ~150 residues downstream from the active-site histidine (Asp1032 in rat FAS, Asp2571 in DEBS2) as part of a DxxxQ motif that is positionally conserved in many DHs. The conserved glutamine residue hydrogen bonds to the active site residue Asp84 or Glu63, respectively in Pseudomonas aeruginosa FabZ and E. coli FabA.178 We should emphasize that, as yet, no high resolution structure has been solved for a DH from a type I FAS or modular PKS, and the conclusions drawn above rely heavily on modeled structures fitted to the C-alpha trace for the porcine FAS and a homology model of the DEBS module 4 DH domain.
Fig. 20.
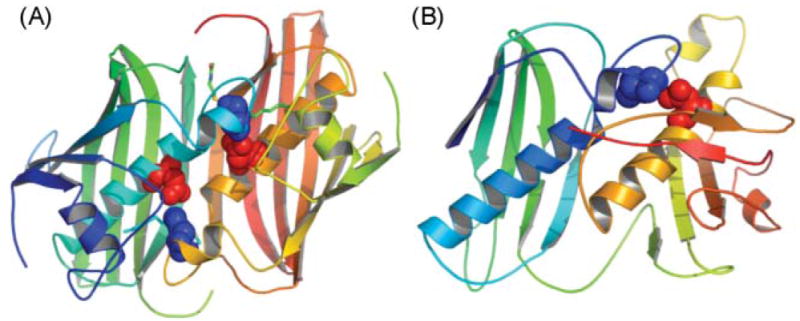
The DH domain. The E. coli DH, FabA (A) and modeled DH domain of FAS (B) exhibit two hotdog folds. The structures are colored from blue to red, from N- to C-terminus. The active site histidine (blue) and aspartate (red) residues are shown.
10.5.2 DH catalytic dyad mechanism
The PKS and FAS DH domains are members of a large family of non-metal dehydratases. The type II counterpart from E. coli, FabA, enjoys a special place in the history of enzymology as the first enzyme for which a mechanism-based ‘suicide’ inactivation was recognized, using 3-decynoyl-N-acetylcysteamine, an analogue of cis-3-decenoyl-ACP.181 The reaction catalyzed by type I and type II DHs is freely reversible and the equilibrium actually favors hydration.182,183 A two-base mechanism has been proposed for FabA with His70 and Asp84 of opposing subunits centered in the largely hydrophobic substrate-binding pocket, and both residues may participate in substrate protonation and deprotonation.91 In the absence of a crystal structure for the type I DHs, it is not yet clear whether the megasynthase DHs employ a similar mechanism.
10.5.3 DH substrate specificity
Compared to other domains, fewer studies have been conducted on the specificity of the DH domains of the type I FASs and modular PKSs. Much work has been done in the E. coli DH (FabA and FabZ), in which motions of substrate pocket side chains may help accommodate the substrates.184 Similar conformational changes will also be necessary for the mammalian FAS DH domain (which is structurally similar to FabA) in order to accommodate substrates longer than 10 carbons. Recently, in the first in vitro biochemical study of a DH-containing PKS module, the DH specificity has been examined in the picromycin/methymycin synthase (PICS),131 and this study has been discussed in Section 9.
10.6 ER
10.6.1 The ER medium-chain dehydrogenase fold
Although no high resolution crystal structures are available for ERs from type I FAS, an ER domain from the M. tuberculosis type I PKS ppsC (part of a six-module complex responsible for mycocerosic acid biosynthesis) has been solved (PDB code 1pqw). Compared to the FAS or DEBS ERs, which were predicted to contain two domains, the solved ppsC ER structure contains only one domain with a Rossman fold for cofactor (NADP or NAD) binding and shares 39% and 37% sequence identity with the predicted Rossman-fold region of rat FAS ER and DEBS ER4, respectively. The structure of the porcine FAS revealed that the ER consists of a typical medium-chain dehydrogenase fold with a catalytic and a NADP-binding domain (the Rossman fold)185 and that the ER domains are actually dimeric. Predicted structures for the animal FAS and DEBS module 4 ER domains indicate that they likely share a very similar architecture in which the dimeric structure is stabilized through extensive interactions at the subunit interface across the Rossman-fold within a 10-strand β sheet (Fig. 21).
Fig. 21.
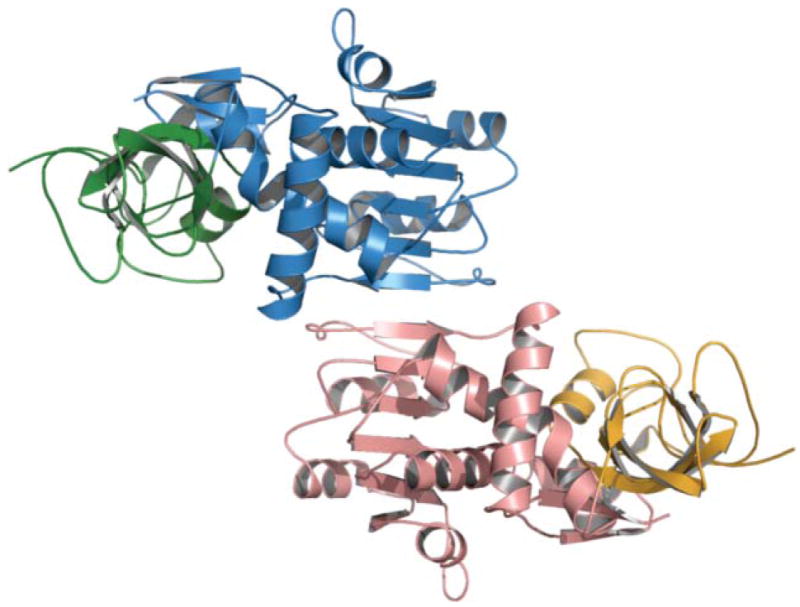
The rat FAS ER homology model (built using the C-alpha trace of the porcine FAS model) shows a tightly interacting dimer. The nucleotide-binding and catalytic domains are colored blue and green in monomer A and red and orange in monomer B. The dimer interface spans 10 β-strands located in the nucleotide-binding domain.
10.6.2 ER active site and catalytic mechanism
As in all members of the medium chain dehydrogenase family, the ER active site is predicted to reside in a cleft that includes the NADPH binding site and other active site residues. The location of the NADPH binding site in the animal FAS, identifiable by the characteristic GxGxxG/AxxxG/A motif, has been confirmed by mutagenesis.182 In both ER and KR, the active site hydrophilic residues are thought to stabilize cofactor binding through hydrogen bond interactions with the hydroxyl groups on the nicotinamide ribose. However, the ER and KR reaction mechanisms differ in the relative substrate positions during the hydride transfer step. Presumably, the active site tyrosine and lysine should be important for the cofactor binding, as well as catalytic mechanism, similar to the one proposed for type II FAS ER domain. However, the active site of ER for either FAS or PKS remains to be determined by a high resolution structure and accompanying mutational analyses.
10.6.3 ER substrate specificity
The FAS ER exhibits broad substrate chain-length specificity and can reduce trans-2 unsaturated fatty acids having 4 to at least 20 carbon atoms.182 This relaxed chain length specificity may be attributable to presence of an open hydrophobic substrate-binding cleft on the enzyme surface, rather than a deep, closed-end binding pocket, That ER-containing PKS modules can usually be freely swapped in environments that require handling of different chain-length substrates suggests that these enzymes also exhibit relatively relaxed substrate specificity.54
10.7 TE
The TE domain is the only catalytic domain for which crystal structures have been derived for both types of megasynthase. The human FAS TE, the DEBS TE, and the PICS TE all exhibit the classic features of the α/β hydrolase fold (Fig. 22), which consists of a central seven-stranded β-sheet with the second strand (β2) antiparallel to the remaining strands.97,98,186,187 However, the FAS TE structure differs from that of the PKS TE in that it contains an additional small all-helical subdomain. Residues in each secondary structural element are highly conserved, especially in regions surrounding the catalytic triad. Many of these conserved residues are presumably important for proper protein folding. Consistent with the sequence alignment, when the three-dimensional structures of FAS TE, DEBS TE and PICS TE are overlapped, the major core region (β-sheet) can be superimposed near-perfectly, with an RMSD of 1.2–1.45 Å. The positions of most α-helices (two on one side and three on the other) can also be overlapped well between the FAS and PKS TE domains. The DEBS and PICS TEs are dimers in solution, and indeed their crystal structures were also solved as dimers with an extensive subunit interface (1700–2350 Å2). The FAS TEs, however, are catalytically active as monomers, and dimerization has not been observed in solution.
Fig. 22.
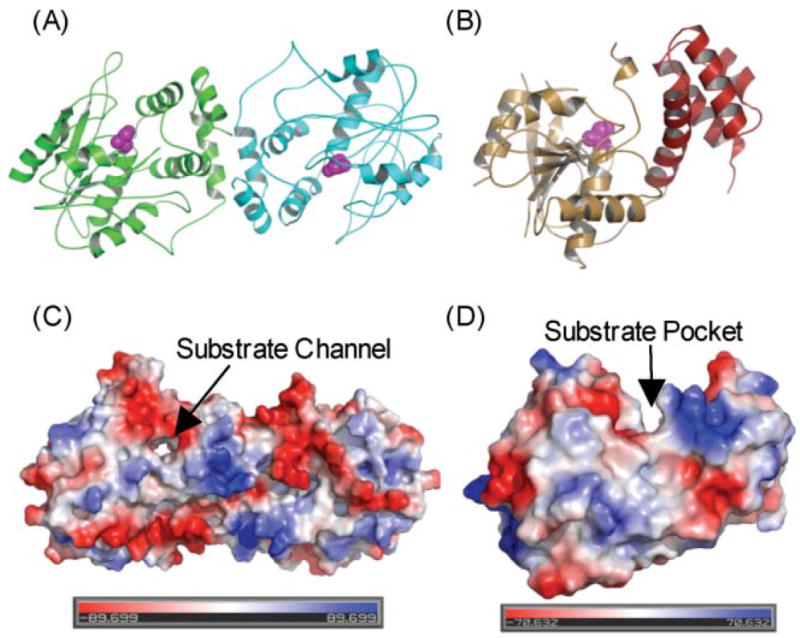
The TE domain. (A) Overall fold of DEBS TE dimer; monomer A in green and monomer B in blue. (B) Overall fold of FAS TE monomer; domain A in orange, domain B in red. The active-site serine is shown in purple. (C) Molecular surface of DEBS TE and FAS TE, colored in red (negative) and blue (positive) to reflect electrostatic potential.
10.7.1 The substrate channel shape
The major difference between FAS and PKS TE lies in its substrate channel shape. In DEBS and PICS TE, an unusual, 20 Å-long substrate channel (containing the active site) passes through the entire protein, suggesting the passage of the substrate through the protein (Fig. 22A,B). In comparison, the FAS TE comprised of two dissimilar subdomains, A and B. While the A subdomain (the α/β hydrolase fold) overlaps well with DEBS TE, the smaller subdomain B, inserted between α5 and α6, is uniquely composed entirely of α-helices and arranged in an atypical fold. As a result, the substrate pocket of the FAS TE is not a channel, but a hydrophobic groove at the interface of the two subdomains. The length and largely hydrophobic nature of the groove and pocket are consistent with the high selectivity of the FAS TE for palmitoyl acyl substrate, while the size, shape and residue properties of the PKS TE is also consistent with its corresponding specificity. The active site of both types of TE is comprised of a Ser-His-Asp triad and, typical of an α/β-hydrolase, the active-site serine is located at a nucleophilic elbow, a sharp turn between a helix and a β strand. Despite the large difference in their substrate pocket shape, the active site triads of FAS TE and DEBS TE (S2308-H2481-D2338 of FAS TE and S142-H258-D169 of DEBS TE) and the backbone amides of the proposed oxyanion hole residues can be completely superimposed, consistent with a common catalytic mechanism.
Moreover, a comparison of the structures of PICS TE at different pH values also revealed that the size of the substrate channel increases with increasing pH. The pH effect of the substrate channel geometry appears to be caused by an overall increase in the number of negative charges developed in the substrate channel as pH increases.186 The variable channel geometry may also influence the region-selectivity between the hydrolysis and macrocyclization activites of TE (see Section 10.7.3 for details).
TEs catalyzing cyclization reactions are also found in related non-ribosomal peptide synthetases, where the assembled peptide chain is cyclized by a terminal TE domain. Comparison of the TE domains of FAS, PKS and non-ribosomal peptide synthetases (surfactin TE and fengycin TE188,189) reveals that whereas the α/β-hydrolase fold and the active site triad are highly conserved, the substrate pockets (size, shape, residue distributions) vary greatly, even between surfactin and fengycin TE domains. Therefore, substrate pocket shape appears to be a key factor in determining the substrate specificity of the TE domains associated with all of these systems.
10.7.3 The TE mechanism
Facilitated by the direct covalent linkage between the two domains, the acyl-ACP docks to the TE. In DEBS TE, there is an arginine-rich groove that may also serve as the ACP docking site. The polyketide (or fatty-acyl) chain is transacylated from the phosphopantetheine of ACP to the nucleophilic active site Ser. The accurate orientation of the polyketide or fatty-acyl chain involves close interactions between the substrates and the active site residues. In the case of FAS TE, it is the hydrophobic interaction and the size/shape of the hydrophobic binding groove that determines its substrate specificity, and hydrolysis then takes place and the fatty-acyl chain is released from the FAS. In the case of DEBS and PICS TE, several functional groups on the polyketide chain can anchor the polyketide chain via hydrogen bonds, such that the C13-OH is placed next to the acyl-O-serine linkage, resulting in cyclization between C=O and C13-OH.186 Recently, the crystal structure of affinity-labeled picromycin TE was also solved, shedding light on the identity of the oxyanion hole and substrate recognition, although specific active site hydrogen bonds were not identified from this structure.187
10.7.4 The molecular basis of substrate specificity of TEs
In vitro, the FAS TE exhibits specificity for C16-acyl substrates, with a sharp decline in activity for chain lengths longer than C18 or shorter than C16 (see Section 8.1). The substrate specificity of the FAS TE is consistent with the FAS TE crystal structure docked with a C16 hexadecyl sulfonyl fluoride (HDSF) in the substrate-binding pocket.98 The modeled HDSF-TE structure revealed that the C16 palmitoyl chain fits well within the groove, with the first 12 carbon atoms exposed to the solvent and the remaining 4 carbon atoms held in place in the distal pocket. As a result, a fatty-acyl chain shorter than 14 carbons will result in nonproductive binding, while a substrate longer than 18 carbons will not be accommodated by the groove.
The crystal structures of DEBS and PICS TE also aid in rationalization of the observed substrate specificity of the modular PKS TE.97,186,187 Modeling exercises indicate that the active site can orient the 6-deoxyerythronolide B precursor uniquely, while at the same time shielding the active site from external water and promote cyclization. Therefore, the substituents on the polyketide substrates are as important as the TE active site for controlling the cyclization versus hydrolysis activity. Consistent with the above observation, simple C4–C10 alkyl chains that lack the functional substituents of a natural polyketide substrate can be hydrolyzed but not cyclized by both DEBS and PICS TE domains,118,190 presumably because of the lack of hydrogen bonds to steer the substrate in an orientation favorable for cyclization. On the other hand, recent studies on the epothilone191 and picromycin TEs have shown that synthetic S-acyl N-acetyl cysteamine analogs with substituents that mimic the natural polyketide stereochemistry can be cyclized by the TE domains.187,192 The application of the TE domains in modular PKS and NRPS should provide a novel chemoenzymatic avenue for the synthesis of macrolactones and macrolactams.
11 Swinging arms or swinging domains?
The idea that covalently attached mobile prosthetic groups play an important role in the coupling of sequential reactions in a pathway was introduced several decades ago and has been discussed in the context of various multifunctional enzyme systems. They include the lipoyllysines that shuttle electrons and acetyl moieties around the 2-oxo acid dehydrogenases and the biotinyl-lysines that serve as CO2 carriers in carboxylases.193 The term ‘swinging arm’ (actually schwingarm) appears to have been first used in the context of the FAS by Lynen194 in characterizing the role of the phosphopantetheine moiety of the ACP domain that carries the various acyl intermediates as they visit the different catalytic sites of the complex. The distances between the various catalytic centers of the FAS have been measured by cross-linking, fluorescence techniques and, more recently, by crystallographic analysis. For example, the phosphopantetheine thiol and KS active site thiols clearly can approach within 5 Å of each other to allow cross-linking by dibromopropanone. However, distances between the phosphopantetheine thiol and the active sites of TE, KR and ER have been estimated, in the resting enzyme, to be at least 40 Å by fluorescence resonance energy transfer, and distances between the active sites of the KS, AT and KR domains appear to be >70 Å (Fig. 11). The crystal structure of the DEBS module 5 KS–AT also reveals that the distance between the KS and AT active centers (~80 Å ) is too great to be spanned by a 20 Å-long phosphopantetheine moiety.90 Clearly then, there must be a requirement for some mobility in the system to allow the phosphopantetheine to reach all of these distant locations within the two catalytic chambers. So does the phosphopantetheine arm indeed swing? The problem with this idea was graphically illustrated by Ban and colleagues in their crystallographic analysis of the porcine FAS when they pointed out that a 20 Å-long phosphopantetheine arm is sufficient only to reach from the perimeter of the functional domains into the active site. This arrangement clearly would require that the entire ACP domain make close contact sequentially with each functional domain, and so it likely that it is in fact the entire ACP domain that swings. This idea is consistent with the absence of any clear electron density in FAS crystals that could be assigned unambiguously to an ACP domain. It seems likely that other domains may also be capable of significant motion, as evidenced by the structural asymmetry observed in both the electron microscope and crystallographic studies. Presumably, this domain mobility could result from a combination of internal domain elasticity and flexibility in the linker regions between functional domains. An important role of the flexible phosphopantetheine moiety that is often overlooked is that it also facilitates insertion and movement of the acyl intermediates within the hydrophobic core of the ACP itself, as is graphically illustrated in the structure of the decanoyl-ACP (Fig. 16C and accompanying text).
12 Linkers
A key feature distinguishing the type I FASs and modular PKSs from their type II counterparts is the presence of discrete connecting regions between the component catalytic elements. In both the type I FASs and in individual PKS modules, adjacent domains are covalently linked. Uniquely in the PKSs, covalent linkers also connect individual modules and non-covalent linkers facilitate the docking of unimodular and multimodular polypeptides with their appropriate partners.
12.1 Interdomain linkers
Several of the interdomain linker regions of the FASs are relatively rich in alanine and proline residues, as are the interdomain segments of the dihydrolipoyl transacetylase polypeptide chains of pyruvate dehydrogenase, where they appear to be well adapted for allowing a degree of flexibility while avoiding completely collapsed conformations.193 To date, the only FAS linker that has been studied in any detail is that connecting the ACP and TE domains. Covalent linkage of the TE and ACP domains is essential for efficient product release in both the FAS and PKS systems; when separated from their cognate ACP domains, these TEs are at least two orders of magnitude less efficient in hydrolysis of their acyl-ACP substrates.104,195 Flexibility of this linker in the FAS system was assessed by measuring the rotational correlation time of a fluorophore bound to the active-site serine of the TE domain.196 This linker does appear to be inherently flexible, and the TE domain enjoys considerably more conformational mobility than the rest of the of the FAS protein, no doubt a contributing factor to the absence of any clear electron density corresponding to this domain in FAS crystals. Increasing the length of the linker by 13 residues resulted in even greater mobility of the TE domain without affecting the activity of the FAS to any great extent. Surprisingly, although removal of the entire 22-residue linker resulted in a doubling of the rotational correlation time (i.e. decreased mobility of the TE domain), the modified FAS retained the ability to synthesize fatty acids, albeit at a reduced rate, ~28% of that observed for the wt FAS. Clearly, in this Δ22 mutant, the ability of the TE domain to access the growing acyl chain was rate-limiting and the product chain length shifted from predominantly 16 to 18 carbon atoms. This study confirmed the inherent mobility of the TE domain, but also revealed that integrity of the FAS is quite tolerant to changes in length and flexibility of the linker region. The retention of activity in a mutant lacking the entire linker suggests that additional factors, perhaps the inherent mobility of the entire ACP domain, must also contribute to the successful docking of the long-chain acyl moiety into the TE active site. Other linker regions in the megasynthases appear to be more conserved and structured. For example, a region between the KS and AT domains of the DEBS PKS adopts a novel αβ-fold, and the region immediately following the C-terminus of the AT domain wraps back over both the AT domain, the KS-AT linker and the KS domain in a manner which would appear to restrict any movement of the KS and AT domains relative to one another.90 Clearly, the role of each of the linkers will have to be evaluated on a case-by-case basis. Some, such as the ACP-TE linker, may function as simple flexible tethers, while others may function more like structured hinges, allowing limited movement in a manner that facilitates efficient communication between cooperating domains.
12.2 Intermodule linkers
Type I PKS polypeptides may be uni- or multimodular. The sequences separating covalently connected modules are typically ~20 residues in length and are poorly conserved except for the presence of proline residues near the middle of the linker.118 No structural analyses have been reported for these linkers, but most likely they function primarily as ‘non-specific’ tethers that hold adjacent modules in close proximity. For example, DEBS module 6 can be fused to module 1 with the linker that normally connects modules 1 and 2 to generate a construct (module 1+ module 6 + TE) that exhibits comparable activity to a module 1+ module 2 + TE construct that includes its natural intermodule linker.120
12.3 Interpolypeptide linkers
One of the structural features unique to the modular PKSs are the N- and C-terminal linkers, or docking domains, that facilitate interaction between adjacent polypeptides in the polyketide assembly line. These short complementary N- and C-terminal sequence elements interact in a specific manner to ensure that each polypeptide interacts only with its appropriate partner in the assembly line. They function independently of the catalytic domains to which they are attached and complementary docking domains can be transferred to facilitate intermodular chain transfer between unnatural polypeptide partners. On the other hand, polypeptides coupled to mismatched N- and C-terminal docking domains have extremely limited ability to effect chain transfer.197
Insight into the recognition mechanism, by which polypeptides dock with appropriate neighbors, was provided by an NMR analysis of a 120-residue fusion protein consisting of the C-terminal docking domain of DEBS2 and the N-terminal docking domain of DEBS3, referred to as Dock 2–3.198 The NMR structure revealed that Dock 2–3 adopts a stable dimeric structure in which residues 1–80 (mimicking the DEBS2 C-terminus) adopt a three α-helical formation and residues 83–120 (mimicking the DEBS3 N-terminus) contain a single longer α-helix, consistent with the coiled-coil structure that was subsequently identified at the N-terminus of the DEBS KS–AT crystal structure (Fig. 10C). Interactions between helices 1 and 2 and between 3 and 4 divide the structure into two subdomains, A and B, which are connected by flexible tethers and do not appear to interact. Subdomain A (Fig. 23A) contains an unusual intertwined four α-helix bundle formed by helices 1, 2, 1′, and 2′ that may also serve to stabilize the entire DEBS2 dimer at its C-terminus. Subdomain B (Fig. 23B) is comprised of helices 3 and 3′, 4, and 4′ as a parallel coiled-coil dimer, which also may stabilize the DEBS3 dimer at its N-terminus. Because of the flexible connection between subdomains A and B, their relative orientations could not be determined with certainty. These findings indicate that the docking domains may serve two functions: docking of PKS subunits and stabilization of the PKS homodimers.
Fig. 23.
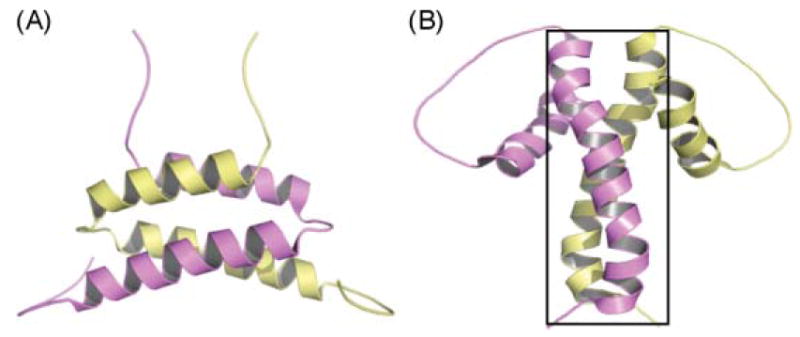
The A and B domains of Dock 2–3. (A) Domain A, (B) domain B. Monomer A is colored in yellow and monomer B in purple. The N-terminal coiled-coil observed in the crystal structure of DEBS module 5 KS–AT is enclosed in a box.
Correct pairing of subunits in modular PKSs comprised of several different polypeptides is obviously critical in ensuring the proper assembly of these megasynthases. In this regard, it may be significant that the residues involved in forming the coiled-coil structure of the KSs are not conserved among DEBS modules.90 It seems feasible that these critical structural features also may play an important role in guaranteeing specificity of homodimerization. This is not an issue for the FAS of course, since only one type of FAS polypeptide is produced in the cytosolic compartment.
13 Perspective
In the century that has elapsed since Collie and Raper first speculated on the origin of fatty acids and polyketides, most of the details of the pathways have been resolved, and finally in this last year we have a clearer understanding of the structural organization of the megasynthases responsible for their biosynthesis. Similarities in the FASs and modular PKSs appear to persist from the level of the individual reaction mechanisms and the structures of individual catalytic domains to the global architectural organization of the entire complexes. In the FASs the basic elements of the design are immutable: one functional copy of every domain per polypeptide, iterative use of the same set of enzymes and rigidly adhered-to specificities of chain elongation and chain termination enzymes that ensure the faithful production of a saturated long-chain fatty acid. On the other hand, the PKSs provide a remarkable example of the parsimony of nature in exploiting the full potential of a particular architectural design. Docking elements have been introduced that facilitate formation of multimodular structures, and certain domains can be omitted from the basic ensemble, allowing the formation of individually tailored intermediates at each station in the assembly line, retention of chiral centers, and ultimately the production of an astonishingly broad range of natural products. Nevertheless, examples are being uncovered where PKS modules have reverted to the archetypal iterative mode, further emphasizing the common origins of the FAS and PKS systems.
The recent revelations resulting from crystallographic analysis of FASs and PKSs have raised awareness of the extraordinary architecture of these megasynthases to a new level, and have provided a solid platform for addressing some of the unsolved questions concerning their functional operation. The enigmatic ‘central core’ region of the megasynthases has all but vanished, essentially being reassigned to important roles as subdomains of the DH and KR regions. Thus the domain maps of the FAS and modular PKSs likely can be revised to reflect this new information (Fig. 24). Nevertheless, full acceptance of this unexpected outcome will require higher resolution structures, particularly of the KR–ER region, and biochemical studies are needed to confirm the catalytic role of the second subdomain of the DH and the requirement for the structural KR subdomain for KR catalytic activity. The new structural data also necessitate a rethinking as to the nature and role of interdomain linkers. The original notion that the linkers acted merely as semiflexible tethers that held adjacent domains in convenient proximity is clearly not broadly applicable. Indeed, to date only the ACP-TE linker seems to fit this stereotype. Some, such as the KS–AT and post-AT linkers, are highly structured, make intimate contact with catalytic domains, and appear to contribute extensively to stabilization of the overall structure. The nature of the other linkers remains to be determined.
Fig. 24.
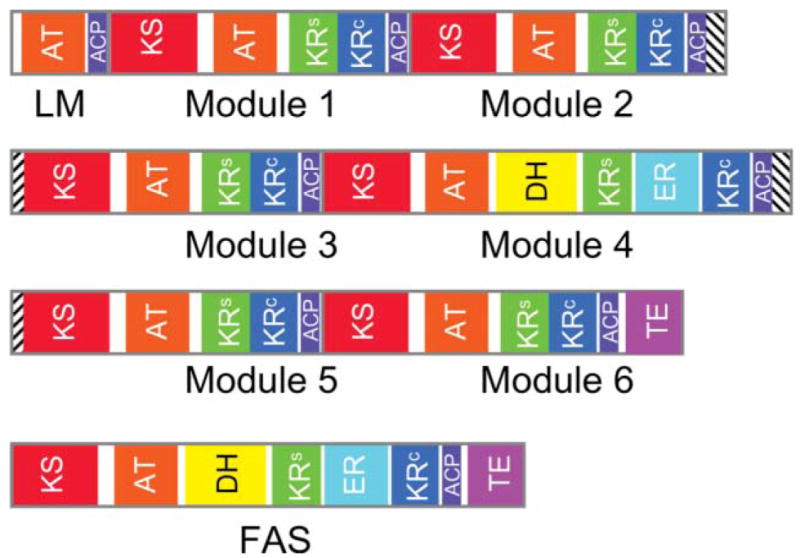
Revised domain maps for the animal FAS and a modular PKS. The domain maps for the loading module (LM) and six extender modules of DEBS include the interpolypeptide docking domains (cross-hatched). The KR domain of module 3 is inactive. In both the FAS and modular PKS, the region previously referred to as the central core is assigned to the KR structural subdomain (KRs) and, where present, the second subdomain of the DH. Thus the KR catalytic (KRc) and structural subdomains are contiguous in DEBS modules 1, 2, 3, 5 and 6, but are split by the ER domain in module 4 and in the FAS.
One of the most fascinating questions concerning the functional aspects of the megasynthases is how the ACPs gain access to each of the functional domains that appear, at least in the structures currently available, to be separated by formidable distances. Mapping the range of motion of the ACP domain, and perhaps of other domains within these flexible macromolecules, poses special difficulties for analysis by X-ray crystallography. Although electron microscopy cannot match crystallographic analysis in terms of resolution, it does have one significant advantage in that the shape of every individual molecule is recorded, regardless of conformation. The challenge is to develop robust procedures for the identification and classification of molecules representative of the various conformations occupied by the protein. Impressive progress has been made recently by Stan Burgess and colleagues in the UK in using electron microscopy to characterize the structure and flexibility of single molecules of the microtubule motor protein dynein.199 The trick employed was to align images based on features of only one part of the molecule and then group the images into classes based on the structural features of other parts of the molecule. The averaged images of each group then identifies consistent features of the class, and the series of class averages reveals the range of shapes adopted by the molecule. The early successful application of electron microscopy to analysis of FAS structure offers encouragement that this approach may provide even more insight into the functional operation of the megasynthases.
Acknowledgments
The authors are grateful to Drs Roscoe Brady and Norm Radin for sharing their recollections of the discovery of malonyl-CoA as an intermediate in fatty acid synthesis and to Grant Jensen for discussing his unpublished electron microscopy experiments. Our sincere thanks to Adrian Keatinge-Clay, Nenad Ban and Jim Staunton for their generosity in supplying us with high quality figures. We are particularly grateful to Andrzej Witkowski, Adrian Keatinge-Clay, Leonard Katz, Craig Townsend and David Hopwood for their helpful suggestions and critical reading of the manuscript. Stuart Smith is supported by grants DK16073 and GM069717 from the NIH, and Sheryl Tsai by the Pew Foundation and the American Heart Association (0665164Y).
Biographies
 Stuart Smith was born in Durham, England. He received his B.Sc. in 1962, and later his Ph.D. under the guidance of Ray Dils in Medical Biochemistry from the University of Birmingham, England. After a postdoctoral fellowship in Jerusalem with Shimon Gat, he moved to Children’s Hospital Oakland Research Institute in California, where his research has focused primarily on the structure, mechanism of action and regulation of lipogenic enzymes in animals. He was awarded a D.Sc. degree from his alma mater in 1980.
Stuart Smith was born in Durham, England. He received his B.Sc. in 1962, and later his Ph.D. under the guidance of Ray Dils in Medical Biochemistry from the University of Birmingham, England. After a postdoctoral fellowship in Jerusalem with Shimon Gat, he moved to Children’s Hospital Oakland Research Institute in California, where his research has focused primarily on the structure, mechanism of action and regulation of lipogenic enzymes in animals. He was awarded a D.Sc. degree from his alma mater in 1980.
 Shiou-Chuan (Sheryl) Tsai was born in 1969 in Taipei, Taiwan. She received her B.S. and M.S. in Chemistry from National Taiwan University, where she studied the syntheses and organometallic chemistry of tripodal ligands with Prof. Shiuh-Tzung Liu. She then received her Ph.D. in Chemistry from University of California, Berkeley in 1999, where she examined hydrogen tunneling phenomena in enzymes with Prof. Judith Klinman. As a postdoctoral fellow, Sheryl investigated the structural enzymology of polyketide synthase in the laboratories of Prof. Chaitan Khosla at Stanford University and Prof. Robert Stroud at the University of California, San Francisco. In 2003, Sheryl joined the University of California, Irvine, as an Assistant Professor. Her research interest includes enzyme structures and mechanisms of multi-domain enzyme complexes, such as acyl-CoA carboxylase, polyketide synthase, fatty acid synthase and sugar-synthesizing enzymes
Shiou-Chuan (Sheryl) Tsai was born in 1969 in Taipei, Taiwan. She received her B.S. and M.S. in Chemistry from National Taiwan University, where she studied the syntheses and organometallic chemistry of tripodal ligands with Prof. Shiuh-Tzung Liu. She then received her Ph.D. in Chemistry from University of California, Berkeley in 1999, where she examined hydrogen tunneling phenomena in enzymes with Prof. Judith Klinman. As a postdoctoral fellow, Sheryl investigated the structural enzymology of polyketide synthase in the laboratories of Prof. Chaitan Khosla at Stanford University and Prof. Robert Stroud at the University of California, San Francisco. In 2003, Sheryl joined the University of California, Irvine, as an Assistant Professor. Her research interest includes enzyme structures and mechanisms of multi-domain enzyme complexes, such as acyl-CoA carboxylase, polyketide synthase, fatty acid synthase and sugar-synthesizing enzymes
References
- 1.Staunton J, Weissman KJ. Nat Prod Rep. 2001;18:380. doi: 10.1039/a909079g. [DOI] [PubMed] [Google Scholar]
- 2.Shen B. Curr Opin Chem Biol. 2003;7:285. doi: 10.1016/s1367-5931(03)00020-6. [DOI] [PubMed] [Google Scholar]
- 3.Taylor WC. Snows of yesteryear: J Norman Collie, mountaineer. Holt, Rinehart and Winston; Toronto: 1973. [Google Scholar]
- 4.Collie J. J Chem Soc, Trans. 1907;91:1806. [Google Scholar]
- 5.Raper H. J Chem Soc, Trans. 1907;91:1831. [Google Scholar]
- 6.Rittenberg D, Bloch K. J Biol Chem. 1944;154:311. [Google Scholar]
- 7.Kaplan NO, Lipmann F. J Biol Chem. 1948;174:37. [PubMed] [Google Scholar]
- 8.Klein HP, Lipmann F. J Biol Chem. 1953;203:101. [PubMed] [Google Scholar]
- 9.Birch A, Massy-Westropp P, Moye C. Aust J Chem. 1955;8:539. [Google Scholar]
- 10.Mahler HR. Fed Proc. 1953;12:694. [PubMed] [Google Scholar]
- 11.Lynen F. Fed Proc. 1953;12:683. [PubMed] [Google Scholar]
- 12.Seubert W, Greull G, Lynen F. Angew Chem. 1957;69:359. [Google Scholar]
- 13.Brady RO, Gurin S. J Biol Chem. 1952;199:421. [PubMed] [Google Scholar]
- 14.Popjak G, Tietz A. Biochem J. 1955;60:147. doi: 10.1042/bj0600147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brady RO, Gurin S. J Biol Chem. 1950;186:461. [PubMed] [Google Scholar]
- 16.Klein HP. J Bacteriol. 1957;73:530. doi: 10.1128/jb.73.4.530-537.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brady RO. Proc Natl Acad Sci U S A. 1958;44:993. doi: 10.1073/pnas.44.10.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wakil SJ. J Am Chem Soc. 1958;80:6465. [Google Scholar]
- 19.Wakil SJ. J Lipid Res. 1961;2:1. [Google Scholar]
- 20.Miinalainen IJ, Chen ZJ, Torkko JM, Pirila PL, Sormunen RT, Bergmann U, Qin YM, Hiltunen JK. J Biol Chem. 2003;278:20154. doi: 10.1074/jbc.M302851200. [DOI] [PubMed] [Google Scholar]
- 21.Zhang L, Joshi AK, Smith S. J Biol Chem. 2003;278:40067. doi: 10.1074/jbc.M306121200. [DOI] [PubMed] [Google Scholar]
- 22.Zhang L, Joshi AK, Hofmann J, Schweizer E, Smith S. J Biol Chem. 2005;280:12422. doi: 10.1074/jbc.M413686200. [DOI] [PubMed] [Google Scholar]
- 23.Majerus PW, Alberts AA, Vagelos PR. Proc Natl Acad Sci U S A. 1964;51:1231. doi: 10.1073/pnas.51.6.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sauer F, Pugh EL, Wakil SJ, Delaney R, Hill RL. Proc Natl Acad Sci U S A. 1964;52:1360. doi: 10.1073/pnas.52.6.1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brindley DN, Matsumura S, Bloch K. Nature. 1969;224:666. [Google Scholar]
- 26.Willecke K, Ritter E, Lynen F. Eur J Biochem. 1969;8:503. doi: 10.1111/j.1432-1033.1969.tb00555.x. [DOI] [PubMed] [Google Scholar]
- 27.Yang PC, Butterworth PHW, Bock RM, Porter JW. J Biol Chem. 1967;242:3501. [Google Scholar]
- 28.Qureshi AA, Lornitzo FA, Hsu RY, Porter JW. Arch Biochem Biophys. 1976;177:379. doi: 10.1016/0003-9861(76)90451-3. [DOI] [PubMed] [Google Scholar]
- 29.Roncari DAK. J Biol Chem. 1974;249:7035. [PubMed] [Google Scholar]
- 30.Schweizer E, Kniep B, Castorph H, Holzner U. Eur J Biochem. 1973;39:353. doi: 10.1111/j.1432-1033.1973.tb03133.x. [DOI] [PubMed] [Google Scholar]
- 31.Lornitzo FA, Qureshi AA, Porter JW. J Biol Chem. 1974;249:1654. [PubMed] [Google Scholar]
- 32.Lornitzo FA, Qureshi AA, Porter JW. J Biol Chem. 1975;250:4520. [PubMed] [Google Scholar]
- 33.Stoops JK, Arslanian MJ, Oh YH, Aune KC, Vanaman TC, Wakil SJ. Proc Natl Acad Sci U S A. 1975;72:1940. doi: 10.1073/pnas.72.5.1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith S, Agradi E, Libertini L, Dileepan KN. Proc Natl Acad Sci U S A. 1976;73:1184. doi: 10.1073/pnas.73.4.1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsukamoto Y, Wong H, Mattick JS, Wakil SJ. J Biol Chem. 1983;258:15312. [PubMed] [Google Scholar]
- 36.Hardie DG, McCarthy AD. Evolution of fatty acid synthase systems by gene fusion. In: Hardie DG, Coggins JR, editors. Multidomain proteins—structure and evolution. Elsevier; Amsterdam: 1986. [Google Scholar]
- 37.Amy C, Witkowski A, Naggert J, Williams B, Randhawa Z, Smith S. Proc Natl Acad Sci U S A. 1989;86:3114. doi: 10.1073/pnas.86.9.3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schweizer M, Takabayashi K, Laux T, Beck KF, Schreglmann R. Nucleic Acids Res. 1989;17:567. doi: 10.1093/nar/17.2.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Holzer KP, Liu W, Hammes GG. Proc Natl Acad Sci U S A. 1989;86:4387. doi: 10.1073/pnas.86.12.4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schweizer E, Hofmann J. Microbiol Mol Biol Rev. 2004;68:501. doi: 10.1128/MMBR.68.3.501-517.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lomakin IB, Xiong Y, Steitz TA. Cell. 2007;129:319. doi: 10.1016/j.cell.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 42.Jenni S, Leibundgut M, Boehringer D, Frick C, Mikolasek B, Ban N. Science. 2007;316:254. doi: 10.1126/science.1138248. [DOI] [PubMed] [Google Scholar]
- 43.Dimroth P, Walter H, Lynen F. Eur J Biochem. 1970;13:98. doi: 10.1111/j.1432-1033.1970.tb00904.x. [DOI] [PubMed] [Google Scholar]
- 44.Malpartida F, Hopwood DA. Nature. 1984;309:462. doi: 10.1038/309462a0. [DOI] [PubMed] [Google Scholar]
- 45.Cortes J, Haydock SF, Roberts GA, Bevitt DJ, Leadlay PF. Nature. 1990;348:176. doi: 10.1038/348176a0. [DOI] [PubMed] [Google Scholar]
- 46.Tuan JS, Weber JM, Staver MJ, Leung JO, Donadio S, Katz L. Gene. 1990;90:21. doi: 10.1016/0378-1119(90)90435-t. [DOI] [PubMed] [Google Scholar]
- 47.Hopwood DA. Chem Rev. 1997;97:2465. doi: 10.1021/cr960034i. [DOI] [PubMed] [Google Scholar]
- 48.Donadio S, Staver MJ, McAlpine JB, Swanson SJ, Katz L. Science. 1991;252:675. doi: 10.1126/science.2024119. [DOI] [PubMed] [Google Scholar]
- 49.Donadio S, Katz L. Gene. 1992;111:51. doi: 10.1016/0378-1119(92)90602-l. [DOI] [PubMed] [Google Scholar]
- 50.Bevitt DJ, Cortes J, Haydock SF, Leadlay PF. Eur J Biochem. 1992;204:39. doi: 10.1111/j.1432-1033.1992.tb16603.x. [DOI] [PubMed] [Google Scholar]
- 51.Jacobsen JR, Hutchinson CR, Cane DE, Khosla C. Science. 1997;277:367. doi: 10.1126/science.277.5324.367. [DOI] [PubMed] [Google Scholar]
- 52.Plate CA, Joshi VC, Wakil SJ. J Biol Chem. 1970;245:2868. [PubMed] [Google Scholar]
- 53.Stern A, Sedgwick B, Smith S. J Biol Chem. 1982;257:799. [PubMed] [Google Scholar]
- 54.Khosla C, Gokhale RS, Jacobsen JR, Cane DE. Annu Rev Biochem. 1999;68:219. doi: 10.1146/annurev.biochem.68.1.219. [DOI] [PubMed] [Google Scholar]
- 55.Liou GF, Lau J, Cane DE, Khosla C. Biochemistry. 2003;42:200. doi: 10.1021/bi0268100. [DOI] [PubMed] [Google Scholar]
- 56.Chang SI, Hammes GG. Acc Chem Res. 1990;23:363. [Google Scholar]
- 57.Pieper R, Ebert-Khosla S, Cane D, Khosla C. Biochemistry. 1996;35:2054. doi: 10.1021/bi952860b. [DOI] [PubMed] [Google Scholar]
- 58.Stinear TP, Mve-Obiang A, Small PL, Frigui W, Pryor MJ, Brosch R, Jenkin GA, Johnson PD, Davies JK, Lee RE, Adusumilli S, Garnier T, Haydock SF, Leadlay PF, Cole ST. Proc Natl Acad Sci U S A. 2004;101:1345. doi: 10.1073/pnas.0305877101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kumar S, Dorsey JA, Muesing RA, Porter JW. J Biol Chem. 1970;245:4732. [PubMed] [Google Scholar]
- 60.Smith S, Abraham S. J Biol Chem. 1971;246:6428. [PubMed] [Google Scholar]
- 61.Wakil SJ, Stoops JK. Structure and Mechanism of Fatty Acid Synthetase. In: Boyer PD, editor. The Enzymes. XVI. Academic Press; New York: 1983. p. 3. [Google Scholar]
- 62.Singh N, Wakil SJ, Stoops JK. J Biol Chem. 1984;259:3605. [PubMed] [Google Scholar]
- 63.Smith S, Stern A, Randhawa ZI, Knudsen J. Eur J Biochem. 1985;152:547. doi: 10.1111/j.1432-1033.1985.tb09230.x. [DOI] [PubMed] [Google Scholar]
- 64.Joshi AK, Smith S. Biochem J. 1993;296:143. doi: 10.1042/bj2960143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Witkowski A, Joshi AK, Smith S. Biochemistry. 1996;35:10569. doi: 10.1021/bi960910m. [DOI] [PubMed] [Google Scholar]
- 66.Joshi AK, Witkowski A, Smith S. Biochemistry. 1998;37:2515. doi: 10.1021/bi971886v. [DOI] [PubMed] [Google Scholar]
- 67.Joshi AK, Witkowski A, Smith S. Biochemistry. 1997;36:2316. doi: 10.1021/bi9626968. [DOI] [PubMed] [Google Scholar]
- 68.Joshi AK, Rangan VS, Smith S. J Biol Chem. 1998;273:4937. doi: 10.1074/jbc.273.9.4937. [DOI] [PubMed] [Google Scholar]
- 69.Rangan VS, Joshi AK, Smith S. Biochemistry. 2001;40:10792. doi: 10.1021/bi015535z. [DOI] [PubMed] [Google Scholar]
- 70.Witkowski A, Joshi AK, Rangan VS, Falick AM, Witkowska HE, Smith S. J Biol Chem. 1999;274:11557. doi: 10.1074/jbc.274.17.11557. [DOI] [PubMed] [Google Scholar]
- 71.Joshi AK, Rangan VS, Witkowski A, Smith S. Chem Biol. 2003;10:169. doi: 10.1016/s1074-5521(03)00023-1. [DOI] [PubMed] [Google Scholar]
- 72.Moche M, Schneider G, Edwards P, Dehesh K, Lindqvist Y. J Biol Chem. 1999;274:6031. doi: 10.1074/jbc.274.10.6031. [DOI] [PubMed] [Google Scholar]
- 73.Olsen JG, Kadziola A, von Wettstein-Knowles P, Siggaard-Andersen M, Larsen S. Structure (Cambridge, MA, U S) 2001;9:233. doi: 10.1016/s0969-2126(01)00583-4. [DOI] [PubMed] [Google Scholar]
- 74.Price AC, Rock CO, White SW. J Bacteriol. 2003;185:4136. doi: 10.1128/JB.185.14.4136-4143.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Witkowski A, Ghosal A, Joshi AK, Witkowska HE, Asturias FJ, Smith S. Chem Biol. 2004;11:1667. doi: 10.1016/j.chembiol.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 76.Kao CM, Katz L, Khosla C. Science. 1994;265:509. doi: 10.1126/science.8036492. [DOI] [PubMed] [Google Scholar]
- 77.Xue Q, Ashley G, Hutchinson CR, Santi DV. Proc Natl Acad Sci U S A. 1999;96:11740. doi: 10.1073/pnas.96.21.11740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pfeifer BA, Admiraal SJ, Gramajo H, Cane DE, Khosla C. Science. 2001;291:1790. doi: 10.1126/science.1058092. [DOI] [PubMed] [Google Scholar]
- 79.Kao CM, Pieper R, Cane DE, Khosla C. Biochemistry. 1996;35:12365. doi: 10.1021/bi9616312. [DOI] [PubMed] [Google Scholar]
- 80.Gokhale RS, Lau J, Cane DE, Khosla C. Biochemistry. 1998;37:2524. doi: 10.1021/bi971887n. [DOI] [PubMed] [Google Scholar]
- 81.Staunton J, Caffrey P, Aparicio JF, Roberts GA, Bethell SS, Leadlay PF. Nat Struct Biol. 1996;3:188. doi: 10.1038/nsb0296-188. [DOI] [PubMed] [Google Scholar]
- 82.Aparicio JF, Caffrey P, Marsden AF, Staunton J, Leadlay PF. J Biol Chem. 1994;269:8524. [PubMed] [Google Scholar]
- 83.Lin CY, Smith S. J Biol Chem. 1978;253:1954. [PubMed] [Google Scholar]
- 84.Rangan VS, Witkowski A, Smith S. J Biol Chem. 1991;266:19180. [PubMed] [Google Scholar]
- 85.Stoops JK, Wakil SJ, Uberbacher EC, Bunick GJ. J Biol Chem. 1987;262:10246. [PubMed] [Google Scholar]
- 86.Kitamoto T, Nishigai M, Sasaki T, Ikai A. J Mol Biol. 1988;203:183. doi: 10.1016/0022-2836(88)90101-5. [DOI] [PubMed] [Google Scholar]
- 87.Brink J, Ludtke SJ, Yang CY, Gu ZW, Wakil SJ, Chiu W. Proc Natl Acad Sci U S A. 2002;99:138. doi: 10.1073/pnas.012589499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Maier T, Jenni S, Ban N. Science. 2006;311:1258. doi: 10.1126/science.1123248. [DOI] [PubMed] [Google Scholar]
- 89.Keatinge-Clay AT, Stroud RM. Structure. 2006;14:737. doi: 10.1016/j.str.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 90.Tang Y, Kim CY, Mathews II, Cane DE, Khosla C. Proc Natl Acad Sci U S A. 2006 [Google Scholar]
- 91.Leesong M, Henderson BS, Gillig JR, Schwab JM, Smith JL. Structure. 1996;4:253. doi: 10.1016/s0969-2126(96)00030-5. [DOI] [PubMed] [Google Scholar]
- 92.Persson B, Kallberg Y, Oppermann U, Jornvall H. Chem Biol Interact. 2003;143–144:271. doi: 10.1016/s0009-2797(02)00223-5. [DOI] [PubMed] [Google Scholar]
- 93.Keatinge-Clay AT, Shelat AA, Savage DF, Tsai SC, Miercke LJ, O’Connell JD, 3rd, Khosla C, Stroud RM. Structure (Cambridge, MA, U S) 2003;11:147. doi: 10.1016/s0969-2126(03)00004-2. [DOI] [PubMed] [Google Scholar]
- 94.Shimomura Y, Kakuta Y, Fukuyama K. J Bacteriol. 2003;185:4211. doi: 10.1128/JB.185.14.4211-4218.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Korman TP, Hill JA, Vu TN, Tsai SC. Biochemistry. 2004;43:14529. doi: 10.1021/bi048133a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hadfield AT, Limpkin C, Teartasin W, Simpson TJ, Crosby J, Crump MP. Structure. 2004;12:1865. doi: 10.1016/j.str.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 97.Tsai SC, Miercke LJ, Krucinski J, Gokhale R, Chen JC, Foster PG, Cane DE, Khosla C, Stroud RM. Proc Natl Acad Sci U S A. 2001;98:14808. doi: 10.1073/pnas.011399198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chakravarty B, Gu Z, Chirala SS, Wakil SJ, Quiocho FA. Proc Natl Acad Sci U S A. 2004;101:15567. doi: 10.1073/pnas.0406901101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Reed MA, Schweizer M, Szafranska AE, Arthur C, Nicholson TP, Cox RJ, Crosby J, Crump MP, Simpson TJ. Org Biomol Chem. 2003;1:463. doi: 10.1039/b208941f. [DOI] [PubMed] [Google Scholar]
- 100.Asturias FJ, Chadick JZ, Cheung IK, Stark H, Witkowski A, Joshi AK, Smith S. Nat Struct Mol Biol. 2005;12:225. doi: 10.1038/nsmb899. [DOI] [PubMed] [Google Scholar]
- 101.Holmquist M. Curr Protein Pept Sci. 2000;1:209. doi: 10.2174/1389203003381405. [DOI] [PubMed] [Google Scholar]
- 102.Cheng Z, Song F, Shan X, Wei Z, Wang Y, Dunaway-Mariano D, Gong W. Biochem Biophys Res Commun. 2006;349:172. doi: 10.1016/j.bbrc.2006.08.025. [DOI] [PubMed] [Google Scholar]
- 103.Martin JL, McMillan FM. Curr Opin Struct Biol. 2002;12:783. doi: 10.1016/s0959-440x(02)00391-3. [DOI] [PubMed] [Google Scholar]
- 104.Naggert J, Witkowski A, Wessa B, Smith S. Biochem J. 1991;273:787. doi: 10.1042/bj2730787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Witkowski A, Joshi A, Smith KS. Biochemistry. 1997;36:16338. doi: 10.1021/bi972242q. [DOI] [PubMed] [Google Scholar]
- 106.Libertini LJ, Smith S. Arch Biochem Biophys. 1979;192:47. doi: 10.1016/0003-9861(79)90070-5. [DOI] [PubMed] [Google Scholar]
- 107.Knudsen J, Clark S, Dils R. Biochem J. 1976;160:683. doi: 10.1042/bj1600683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Libertini LJ, Smith S. J Biol Chem. 1978;253:1393. [PubMed] [Google Scholar]
- 109.Chen S, Roberts JB, Xue Y, Sherman DH, Reynolds KA. Gene. 2001;263:255. doi: 10.1016/s0378-1119(00)00560-6. [DOI] [PubMed] [Google Scholar]
- 110.Kim BS, Cropp TA, Beck BJ, Sherman DH, Reynolds KA. J Biol Chem. 2002;277:48028. doi: 10.1074/jbc.M207770200. [DOI] [PubMed] [Google Scholar]
- 111.Xue Y, Zhao L, Liu HW, Sherman DH. Proc Natl Acad Sci U S A. 1998;95:12111. doi: 10.1073/pnas.95.21.12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hu Z, Pfeifer BA, Chao E, Murli S, Kealey J, Carney JR, Ashley G, Khosla C, Hutchinson CR. Microbiology. 2003;149:2213. doi: 10.1099/mic.0.26015-0. [DOI] [PubMed] [Google Scholar]
- 113.Wilkinson B, Foster G, Rudd BAM, Taylor NL, Blackaby AP, Sidebottom PJ, Cooper DJ, Dawson MJ, Buss AD, Gaisser S, Bohm IU, Rowe CJ, Cortés J, Leadlay PF, Staunton J. Chem Biol. 2000;7:111. doi: 10.1016/s1074-5521(00)00076-4. [DOI] [PubMed] [Google Scholar]
- 114.Moss SJ, Martin CJ, Wilkinson B. Nat Prod Rep. 2004;21:575. doi: 10.1039/b315020h. [DOI] [PubMed] [Google Scholar]
- 115.Gaitatzis N, Silakowski B, Kunze B, Nordsiek G, Blocker H, Hofle G, Muller R. J Biol Chem. 2002;277:13082. doi: 10.1074/jbc.M111738200. [DOI] [PubMed] [Google Scholar]
- 116.Olano C, Wilkinson B, Sanchez C, Moss SJ, Sheridan R, Math V, Weston AJ, Brana AF, Martin CJ, Oliynyk M, Mendez C, Leadlay PF, Salas JA. Chem Biol. 2004;11:87. doi: 10.1016/j.chembiol.2003.12.018. [DOI] [PubMed] [Google Scholar]
- 117.He J, Hertweck C. ChemBioChem. 2005;6:908. doi: 10.1002/cbic.200400333. [DOI] [PubMed] [Google Scholar]
- 118.Gokhale RS, Tsiji SY, Cane DE, Khosla C. Science. 1999;284:482. doi: 10.1126/science.284.5413.482. [DOI] [PubMed] [Google Scholar]
- 119.Watanabe K, Wang CC, Boddy CN, Cane DE, Khosla C. J Biol Chem. 2003;278:42020. doi: 10.1074/jbc.M305339200. [DOI] [PubMed] [Google Scholar]
- 120.Wu N, Tsuji SY, Cane DE, Khosla C. J Am Chem Soc. 2001;123:6465. doi: 10.1021/ja010219t. [DOI] [PubMed] [Google Scholar]
- 121.Anderson VE, Hammes GG. Biochemistry. 1984;23:2088. doi: 10.1021/bi00304a033. [DOI] [PubMed] [Google Scholar]
- 122.Saito K, Kawaguchi A, Seyama Y, Yamakawa T, Okuda S. Eur J Biochem. 1981;116:581. doi: 10.1111/j.1432-1033.1981.tb05375.x. [DOI] [PubMed] [Google Scholar]
- 123.Sedgwick B, Morris C. J Chem Soc, Chem Commun. 1980;96 [Google Scholar]
- 124.Celmer WD. J Am Chem Soc. 1965;87:1801. doi: 10.1021/ja01086a038. [DOI] [PubMed] [Google Scholar]
- 125.Weissman KJ, Timoney M, Bycroft M, Grice P, Hanefeld U, Staunton J, Leadlay PF. Biochemistry. 1997;36:13849. doi: 10.1021/bi971566b. [DOI] [PubMed] [Google Scholar]
- 126.Holzbaur IE, Ranganathan A, Thomas IP, Kearney DJ, Reather JA, Rudd BA, Staunton J, Leadlay PF. Chem Biol. 2001;8:329. doi: 10.1016/s1074-5521(01)00014-x. [DOI] [PubMed] [Google Scholar]
- 127.Kim YS, Kolattukudy PE. J Biol Chem. 1980;255:686. [PubMed] [Google Scholar]
- 128.McPherson M, Khosla C, Cane D. J Am Chem Soc. 1998;120:3267. [Google Scholar]
- 129.Holzbaur IE, Harris RC, Bycroft M, Cortes J, Bisang C, Staunton J, Rudd BA, Leadlay PF. Chem Biol. 1999;6:189. doi: 10.1016/S1074-5521(99)80035-0. [DOI] [PubMed] [Google Scholar]
- 130.Tang L, Ward S, Chung L, Carney JR, Li Y, Reid R, Katz L. J Am Chem Soc. 2004;126:46. doi: 10.1021/ja030503f. [DOI] [PubMed] [Google Scholar]
- 131.Wu J, Zaleski TJ, Valenzano C, Khosla C, Cane DE. J Am Chem Soc. 2005;127:17393. doi: 10.1021/ja055672+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Caffrey P. ChemBioChem. 2003;4:654. doi: 10.1002/cbic.200300581. [DOI] [PubMed] [Google Scholar]
- 133.Donadio S, McAlpine JB, Sheldon PJ, Jackson M, Katz L. Proc Natl Acad Sci U S A. 1993;90:7119. doi: 10.1073/pnas.90.15.7119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Rawlings BJ. Nat Prod Rep. 2001;18:190. doi: 10.1039/b009329g. [DOI] [PubMed] [Google Scholar]
- 135.Rawlings BJ. Nat Prod Rep. 2001;18:231. doi: 10.1039/b100191o. [DOI] [PubMed] [Google Scholar]
- 136.Hill AM. Nat Prod Rep. 2006;23:256. doi: 10.1039/b301028g. [DOI] [PubMed] [Google Scholar]
- 137.O’Hagan D. Nat Prod Rep. 1993;10:593. doi: 10.1039/np9931000593. [DOI] [PubMed] [Google Scholar]
- 138.O’Hagan D. Nat Prod Rep. 1992;9:447. doi: 10.1039/np9920900447. [DOI] [PubMed] [Google Scholar]
- 139.White SW, Zheng J, Zhang YM, Rock Annu Rev Biochem. 2005;74:791. doi: 10.1146/annurev.biochem.74.082803.133524. [DOI] [PubMed] [Google Scholar]
- 140.Bunkoczi G, Joshi A, Papagrigoriu E, Arrowsmith C, Edwards A, Sundstrom M, Weigelt J, Von Delft F, Smith S, Oppermann U. Structure of aminoadipate–semialdehyde dehydrogenase–phosphopantetheinyl transferase in complex with cytosolic acyl carrier protein and CoA, Structural Genomics website. 2006 ( http://www.sgc.ox.ac.uk)
- 141.Weissman KJ, Hong H, Popovic B, Meersman F. Chem Biol. 2006;13:625. doi: 10.1016/j.chembiol.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 142.Zhang YM, Wu B, Zheng J, Rock CO. J Biol Chem. 2003;278:52935. doi: 10.1074/jbc.M309874200. [DOI] [PubMed] [Google Scholar]
- 143.Crump MP, Crosby J, Dempsey CE, Parkinson JA, Murray M, Hopwood DA, Simpson TJ. Biochemistry. 1997;36:6000. doi: 10.1021/bi970006+. [DOI] [PubMed] [Google Scholar]
- 144.Findlow SC, Winsor C, Simpson TJ, Crosby J, Crump MP. Biochemistry. 2003;42:8423. doi: 10.1021/bi0342259. [DOI] [PubMed] [Google Scholar]
- 145.Li Q, Khosla C, Puglisi JD, Liu CW. Biochemistry. 2003;42:4648. doi: 10.1021/bi0274120. [DOI] [PubMed] [Google Scholar]
- 146.Chen AY, Schnarr NA, Kim CY, Cane DE, Khosla C. J Am Chem Soc. 2006;128:3067. doi: 10.1021/ja058093d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Roujeinikova A, Simon WJ, Gilroy J, Rice DW, Rafferty JB, Slabas AR. J Mol Biol. 2007;365:135. doi: 10.1016/j.jmb.2006.09.049. [DOI] [PubMed] [Google Scholar]
- 148.Serre L, Verbree EC, Dauter Z, Stuitje AR, Derewenda ZS. J Biol Chem. 1995;270:12961. doi: 10.1074/jbc.270.22.12961. [DOI] [PubMed] [Google Scholar]
- 149.Ruch FE, Vagelos PR. J Biol Chem. 1973;248:8086. [PubMed] [Google Scholar]
- 150.Rangan VS, Smith S. J Biol Chem. 1997;272:11975. doi: 10.1074/jbc.272.18.11975. [DOI] [PubMed] [Google Scholar]
- 151.Smith S, Stern A. Arch Biochem Biophys. 1983;222:259. doi: 10.1016/0003-9861(83)90523-4. [DOI] [PubMed] [Google Scholar]
- 152.Buckner JS, Kolattukudy PE, Rogers L. Arch Biochem Biophys. 1978;186:152. doi: 10.1016/0003-9861(78)90474-5. [DOI] [PubMed] [Google Scholar]
- 153.Seyama Y, Otsuka H, Kawaguchi A, Yamakawa T. J Biochem. 1981;90:789. doi: 10.1093/oxfordjournals.jbchem.a133535. [DOI] [PubMed] [Google Scholar]
- 154.Jang SH, Cheesbrough TM, Kolattukudy PE. J Biol Chem. 1989;264:3500. [PubMed] [Google Scholar]
- 155.Lau J, Fu H, Cane DE, Khosla C. Biochemistry. 1999;38:1643. doi: 10.1021/bi9820311. [DOI] [PubMed] [Google Scholar]
- 156.Lau J, Cane DE, Khosla C. Biochemistry. 2000;39:10514. doi: 10.1021/bi000602v. [DOI] [PubMed] [Google Scholar]
- 157.Liou GF, Khosla C. Curr Opin Chem Biol. 2003;7:279. doi: 10.1016/s1367-5931(03)00016-4. [DOI] [PubMed] [Google Scholar]
- 158.Del Vecchio F, Petkovic H, Kendrew SG, Low L, Wilkinson B, Lill R, Cortes J, Rudd BA, Staunton J, Leadlay PF. J Ind Microbiol Biotechnol. 2003;30:489. doi: 10.1007/s10295-003-0062-0. [DOI] [PubMed] [Google Scholar]
- 159.Hong H, Appleyard AN, Siskos AP, Garcia-Bernardo J, Staunton J, Leadlay PF. FEBS J. 2005;272:2373. doi: 10.1111/j.1742-4658.2005.04615.x. [DOI] [PubMed] [Google Scholar]
- 160.Marsden AF, Caffrey P, Aparicio JF, Loughran MS, Staunton J, Leadlay PF. Science. 1994;263:378. doi: 10.1126/science.8278811. [DOI] [PubMed] [Google Scholar]
- 161.Haydock S, Aparicio J, Molnar I, Khaw STL, Konig A, Marsden A, Galloway I, Staunton J, Leadlay P. FEBS Lett. 1995;374:246. doi: 10.1016/0014-5793(95)01119-y. [DOI] [PubMed] [Google Scholar]
- 162.Yadav G, Gokhale RS, Mohanty D. J Mol Biol. 2003;328:335. doi: 10.1016/s0022-2836(03)00232-8. [DOI] [PubMed] [Google Scholar]
- 163.Long PF, Wilkinson CJ, Bisang CP, Cortes J, Dunster N, Oliynyk M, McCormick E, McArthur H, Mendez C, Salas JA, Staunton J, Leadlay PF. Mol Microbiol. 2002;43:1215. doi: 10.1046/j.1365-2958.2002.02815.x. [DOI] [PubMed] [Google Scholar]
- 164.Reeves CD, Murli S, Ashley GW, Piagentini M, Hutchinson CR, McDaniel R. Biochemistry. 2001;40:15464. doi: 10.1021/bi015864r. [DOI] [PubMed] [Google Scholar]
- 165.Koppisch AT, Khosla C. Biochemistry. 2003;42:11057. doi: 10.1021/bi0349672. [DOI] [PubMed] [Google Scholar]
- 166.Heath RJ, Rock CO. Nat Prod Rep. 2002;19:581. doi: 10.1039/b110221b. [DOI] [PubMed] [Google Scholar]
- 167.Siggaard-Andersen M, Bangera G, Olsen J, von Wettstein-Knowles P. In: Defining the functions of highly conserved residues in β-ketoacyl synthases. Sanchez J, Cerda-Olmedo E, Martinez-Force E, editors. Secretariado de Publicaciones, Universidad de Sevilla; 1998. [Google Scholar]
- 168.Witkowski A, Joshi AK, Smith S. Biochemistry. 2002;41:10877. doi: 10.1021/bi0259047. [DOI] [PubMed] [Google Scholar]
- 169.Zhang YM, Hurlbert J, White SW, Rock CO. J Biol Chem. 2006;281:17390. doi: 10.1074/jbc.M513199200. [DOI] [PubMed] [Google Scholar]
- 170.Witkowski A, Joshi AK, Lindqvist Y, Smith S. Biochemistry. 1999;38:11643. doi: 10.1021/bi990993h. [DOI] [PubMed] [Google Scholar]
- 171.Bisang C, Long PF, Cortés J, Westcott J, Crosby J, Matharu AL, Cox RJ, Simpson TJ, Staunton J, Leadlay PF. Nature. 1999;401:502. doi: 10.1038/46829. [DOI] [PubMed] [Google Scholar]
- 172.Price AC, Zhang YM, Rock CO, White SW. Biochemistry. 2001;40:12772. doi: 10.1021/bi010737g. [DOI] [PubMed] [Google Scholar]
- 173.Price AC, Zhang YM, Rock CO, White SW. Structure. 2004;12:417. doi: 10.1016/j.str.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 174.Reid R, Piagentini M, Rodriguez E, Ashley G, Viswanathan N, Carney J, Santi DV, Hutchinson CR, McDaniel R. Biochemistry. 2003;42:72. doi: 10.1021/bi0268706. [DOI] [PubMed] [Google Scholar]
- 175.Baerga-Ortiz A, Popovic B, Siskos AP, O’Hare HM, Spiteller D, Williams MG, Campillo N, Spencer JB, Leadlay PF. Chem Biol. 2006;13:277. doi: 10.1016/j.chembiol.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 176.O’Hare HM, Baerga-Ortiz A, Popovic B, Spencer JB, Leadlay PF. Chem Biol. 2006;13:287. doi: 10.1016/j.chembiol.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 177.Dillon SC, Bateman A. BMC Bioinf. 2004;5:109. doi: 10.1186/1471-2105-5-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Kimber MS, Martin F, Lu Y, Houston S, Vedadi M, Dharamsi A, Fiebig KM, Schmid M, Rock CO. J Biol Chem. 2004;279:52593. doi: 10.1074/jbc.M408105200. [DOI] [PubMed] [Google Scholar]
- 179.Li J, Derewenda U, Dauter Z, Smith S, Derwenda Z. Nat Struct Biol. 2000;7:555. doi: 10.1038/76776. [DOI] [PubMed] [Google Scholar]
- 180.Joshi AK, Smith S. J Biol Chem. 1993;268:22508. [PubMed] [Google Scholar]
- 181.Helmkamp GM, Jr, Bloch K. J Biol Chem. 1969;243:6014. [PubMed] [Google Scholar]
- 182.Witkowski A, Joshi AK, Smith S. Biochemistry. 2004;43:10458. doi: 10.1021/bi048988n. [DOI] [PubMed] [Google Scholar]
- 183.Heath RJ, Rock CO. J Biol Chem. 1995;270:26538. doi: 10.1074/jbc.270.44.26538. [DOI] [PubMed] [Google Scholar]
- 184.Heath RJ, Rock CO. J Biol Chem. 1996;271:27795. doi: 10.1074/jbc.271.44.27795. [DOI] [PubMed] [Google Scholar]
- 185.Stankovich MT, Sabaj KM, Tonge PJ. Arch Biochem Biophys. 1999;370:16. doi: 10.1006/abbi.1999.1385. [DOI] [PubMed] [Google Scholar]
- 186.Tsai SC, Lu H, Cane DE, Khosla C, Stroud RM. Biochemistry. 2002;41:12598. doi: 10.1021/bi0260177. [DOI] [PubMed] [Google Scholar]
- 187.Giraldes JW, Akey DL, Kittendorf JD, Sherman DH, Smith JL, Fecik RA. Nat Chem Biol. 2006;2:531. doi: 10.1038/nchembio822. [DOI] [PubMed] [Google Scholar]
- 188.Bruner SD, Weber T, Kohli RM, Schwarzer D, Marahiel MA, Walsh CT, Stubbs MT. Structure (Cambridge, MA, U S) 2002;10:301. doi: 10.1016/s0969-2126(02)00716-5. [DOI] [PubMed] [Google Scholar]
- 189.Samel SA, Wagner B, Marahiel MA, Essen LO. J Mol Biol. 2006;359:876. doi: 10.1016/j.jmb.2006.03.062. [DOI] [PubMed] [Google Scholar]
- 190.Lu H, Tsai SC, Khosla C, Cane DE. Biochemistry. 2002;41:12590. doi: 10.1021/bi026006d. [DOI] [PubMed] [Google Scholar]
- 191.Boddy CN, Schneider TL, Hotta K, Walsh CT, Khosla C. J Am Chem Soc. 2003;125:3428. doi: 10.1021/ja0298646. [DOI] [PubMed] [Google Scholar]
- 192.He W, Wu J, Khosla C, Cane DE. Bioorg Med Chem Lett. 2006;16:391. doi: 10.1016/j.bmcl.2005.09.077. [DOI] [PubMed] [Google Scholar]
- 193.Perham RN. Annu Rev Biochem. 2000;69:961. doi: 10.1146/annurev.biochem.69.1.961. [DOI] [PubMed] [Google Scholar]
- 194.Sumper M, Oesterhelt D, Riepertinger C, Lynen F. Eur J Biochem. 1969;10:377. doi: 10.1111/j.1432-1033.1969.tb00701.x. [DOI] [PubMed] [Google Scholar]
- 195.Gokhale RS, Hunziker D, Cane DE, Khosla C. Chem Biol. 1998;6:117. doi: 10.1016/S1074-5521(99)80008-8. [DOI] [PubMed] [Google Scholar]
- 196.Joshi AK, Witkowski A, Berman HA, Zhang L, Smith S. Biochemistry. 2005;44:4100. doi: 10.1021/bi047856r. [DOI] [PubMed] [Google Scholar]
- 197.Tsuji SY, Cane DE, Khosla C. Biochemistry. 2001;40:2326. doi: 10.1021/bi002463n. [DOI] [PubMed] [Google Scholar]
- 198.Broadhurst RW, Nietlispach D, Wheatcroft MP, Leadlay PF, Weissman KJ. Chem Biol. 2003;10:723. doi: 10.1016/s1074-5521(03)00156-x. [DOI] [PubMed] [Google Scholar]
- 199.Burgess SA, Walker ML, Sakakibara H, Oiwa K, Knight PJ. J Struct Biol. 2004;146:205. doi: 10.1016/j.jsb.2003.10.005. [DOI] [PubMed] [Google Scholar]