

Abstract
3-Isopropylmalate dehydrogenase (IPMDH, E.C. 1.1.1.85) from the thermophilic bacterium Thermus thermophilus HB8 is homologous to IPMDH from the mesophilic Escherichia coli, but has an approximately 17°C higher melting temperature. Its temperature optimum is 22–25°C higher than that of the E. coli enzyme; however, it is hardly active at room temperature. The increased conformational rigidity required to stabilize the thermophilic enzyme against heat denaturation might explain its different temperature-activity profile. Hydrogen/deuterium exchange studies were performed on this thermophilic-mesophilic enzyme pair to compare their conformational flexibilities. It was found that Th. thermophilus IPMDH is significantly more rigid at room temperature than E. coli IPMDH, whereas the enzymes have nearly identical flexibilities under their respective optimal working conditions, suggesting that evolutionary adaptation tends to maintain a “corresponding state” regarding conformational flexibility. These observations confirm that conformational fluctuations necessary for catalytic function are restricted at room temperature in the thermophilic enzyme, suggesting a close relationship between conformational flexibility and enzyme function.
Proteins and particularly enzymes generally are believed to be quite vulnerable structures sensitive to environmental changes, e.g., elevated temperatures. However, there are some exceptions. Extreme thermophilic microorganisms have optimum growth temperatures above 70°C. They have attracted the attention of the biotechnological research community because enzymes isolated from such bacteria are very resistant to heat denaturation. It seems likely that most enzymes, particularly the allosteric ones, work optimally at the edge of their stabilities. One reason for this might be the dynamic nature of the molecular events associated with enzymatic catalysis.
Protein molecules undergo relatively large fluctuations of internal energy, which also is reflected in conformational fluctuations (1, 2). The existence and significance of this flexibility already has been noticed by Linderström-Lang and Schellman (3), and later the “fluctuation fit” concept of enzyme function was developed by Straub (4) in terms of conformational fluctuations. The overwhelming first wave of protein structure data derived from x-ray crystallography overshadowed the dynamic approach by presenting impressive, easy-to-comprehend rigid models at atomic resolution. X-ray crystallographers were always aware of molecular motions, and they recognized the importance of temperature factors. However, all of this was not explained in terms of conformational dynamics for quite a number of years. Hydrogen isotope exchange (5–9) and fluorescence quenching experiments (10–13) maintained the molecular dynamics concept until the advent of molecular dynamics simulations and high-resolution proton NMR measurements on proteins. It was postulated that conformational fluctuations in a highly cooperative structure are correlated and can even be concerted (1). However, observations of the relation between enzyme function and conformational flexibility are sparse (14, 15).
A number of enzymes have been isolated from thermophilic and hyperthermophilic microorganisms (Thermus thermophilus, Thermotoga maritima, etc.; refs. 16–18). These thermophilic enzymes are stable and fully active at elevated temperatures. They are homologous with their mesophilic counterparts, the active sites of the homologous pairs, and most of their physico-chemical characteristics are very similar (19). Based on these similarities and on Arrhenius theory, the thermophilic enzymes are expected to be as active as their mesophilic counterparts at room temperature. However, this is not the case (17, 20). Cold denaturation cannot explain this behavior, because these enzymes do not unfold at room temperature (19).
Protein stability can be expressed in terms of the free energy difference associated with the macro- and micro-unfolding of the conformation (21). The macrostability is characterized by ΔGN→D (calculated per mol of the cooperative unit), i.e., by the work required to transfer the protein from the folded to the unfolded macroscopic state. On the other hand, microstability is characteristic of the rigidity of a structure, with ΔGmic as the Gibbs free energy associated with the local, reversible, noncooperative “unfolding” reactions within the folded state. Macrostability maintains the integrity of the native folded conformation, whereas microstability determines the flexibility of the protein, which is presumably responsible for optimum function.
There is a delicate balance between very large contributions of diverse stabilizing and destabilizing interactions involved in the formation of the folded, native three-dimensional structure of a protein, giving rise to a marginal free energy difference of stabilization. It is likely that this balance can be adjusted to substantial environmental changes just by one or a few changes in the amino acid sequence, restoring marginal stability and keeping flexibility at optimum level.
One explanation of the loss of activity of thermophilic enzymes at room temperature, where most related enzymes are fully active, might lie in their increased conformational rigidity required to stabilize the protein against heat denaturation. To check this hypothesis, comparative H/D exchange studies on a thermophilic-mesophilic enzyme pair were carried out. Recording and analysis of the time course of H/D exchange is a powerful tool to compare the conformational flexibilities and microstabilities of protein molecules, whereas CD and differential scanning calorimetry (DSC) measurements can report on the macrostability of the protein molecule.
We have devised an experimental setup to check this hypothesis and find a relationship between enzyme activity and conformational fluctuations. 3-Isopropylmalate dehydrogenase (IPMDH, E.C. 1.1.1.85), a thermophilic enzyme isolated from Th. thermophilus (22) and characterized in detail (16), was chosen for our studies. Its three-dimensional structure is known from x-ray crystallography to a resolution of 2.2 Å (23). Mesophilic counterparts of this enzyme have been isolated from several sources (24–26).
The three-dimensional structure of the E. coli IPMDH was obtained initially by homology modeling (27), and the atomic coordinates are now also available from x-ray crystallography (28). We have chosen this enzyme as a mesophilic counterpart of the Th. thermophilus IPMDH. The overall topology of E. coli IPMDH is very similar to that of Th. thermophilus IPMDH: the rms deviation between equivalent α-carbon atoms of the two structures is 1.58 Å (28). The active site residues are fully conserved (29). A slight conformational difference is present next to the active site, caused by an insertion in position 92 in the E. coli sequence (28), although superimposition of the two structures reflects an extremely high degree of structural similarity (30).
In this work, the macrostabilities of Th. thermophilus and E. coli IPMDH were compared by thermal unfolding experiments using DSC and CD spectroscopy. Activity measurements versus pH, temperature, and salt concentrations were performed to determine the optimal working conditions for the enzymes. The microstabilities were measured by the H/D exchange method using Fourier-transformed infrared (FT-IR) spectroscopy. The main goal of this paper was to compare the microstabilities, i.e., the flexibilities of the mesophilic and the thermophilic IPMDHs, to see whether they converge under “physiologically equivalent” conditions.
MATERIALS AND METHODS
Reagents.
Threo-DL-3-isopropylmalic acid was purchased from Wako Biochemicals (Osaka) and NAD from Boehringer Mannheim. Chromatography media were obtained from Pharmacia. D2O (99.95% purity) was obtained from Merck. Other chemicals (high purity grade) were products of Merck and Sigma.
Enzyme Preparation.
Th. thermophilus IPMDH was produced in E. coli BMH 71–18 cells, using a recombinant plasmid pUTL118 (31) carrying the leuB gene from Th. thermophilus. Cells were grown in Luria-Bertani medium at 37°C in the presence of 100 μg/ml of ampicillin and harvested in the stationary phase. Mesophilic IPMDH was expressed in the E. coli strain RDK1782 (32) transformed with pWally, a derivative of pBluescript KS− carrying the leuB gene from E. coli. Plasmids pWally and pUTL118 were a kind gift from Gerlind Wallon (Brandeis University, Waltham, MA). Cells were grown at 30°C in the presence of 100 μg/ml of ampicillin and 50 μg/ml of kanamycin. After induction during the midlogarithmic phase (temperature was increased to 42°C for 1 h) cells were further grown for 3 h at 37°C.
IPMDHs were purified by using a similar procedure to that described in ref. 16. The final products were observed as a single band on SDS/PAGE gel stained with Coomassie brilliant blue.
Enzyme Activity Measurements.
The activity of IPMDH was measured in 20 mM potassium phosphate buffer, pH 7.6, containing 25 μl of enzyme solution, 0.4 mM DL-3-isopropylmalate, 0.8 mM NAD, 0.2 mM MnCl2, and 0.3 M KCl in a final volume of 700 μl. In the case of the Th. thermophilus enzyme, the KCl concentration was increased to 1.0 M (16). Initial velocities were determined by monitoring the absorbance of NADH formed at 340 nm on a Jasco (Tokyo) V-500 spectrophotometer equipped with a Grant [Grant Instruments (Cambridge, U.K.)] Y6 thermostat.
Measurement of pH.
Extreme care was taken with pH measurements. The pDs of the samples were measured after the spectroscopic (CD and FT-IR) measurements with a Pharmacia pH monitor using a Radelkis (Budapest, Hungary) combined glass electrode. pD was calculated as pD = pDread + 0.4, where pDread is the pH meter reading (33). Measuring in D2O, pDread was found 0.15 pH units higher than the pH of the same buffer composition measured in H2O.
CD.
CD measurements were carried out on a Jasco J-720 spectropolarimeter equipped with a Neslab Instruments (Portsmouth, NH) RTE-100 computer-controlled thermostat. All cells were cylindrical, water-jacketed quartz cells. For recording far-UV spectra, cells of 0.1 cm and 0.01 cm were used, and the protein concentrations were set to 0.4 mg/ml.
For heat denaturation studies, a protein concentration of 1 mg/ml was used. For ordinary and heavy water experiments, samples were prepared in H2O, lyophilized, and dissolved in the appropriate amount of H2O and D2O, respectively. Unfolding was monitored at 221 nm with a heating rate of 50°C/h. Transition temperatures were determined by using the first derivatives of the transition curves.
DSC Measurements.
DSC measurements were performed on a DASM-4 instrument (Institute of Protein Research, Russian Academy of Science, Pushchino). Denaturation curves were recorded between 10°C and 100°C using a scan rate of 1°C/min. Samples were accurately dialyzed against the buffers used in the activity measurements, and the dialysis buffer was used as a reference. Protein concentrations of 1.0–1.5 mg/ml were used. Heat capacities were calculated as outlined by Privalov (34).
FT-IR Measurements.
The kinetics of H/D exchange in D2O were measured by FT-IR spectroscopy on a Bruker IFS 28 FT-IR photometer using the procedure described earlier (5, 7). CaF2 cells with a pathlength of 110 μm were used both for the sample and background measurements. The temperature was measured with a sensor attached directly to the cell windows. Measurements were carried out at 25.0 ± 0.1°C and near the activity optimum of each enzyme, i.e., 48.0 ± 0.1°C for the E. coli and 70.0 ± 0.1°C for the Th. thermophilus enzyme.
The samples were dialyzed in 0.02 M potassium phosphate buffer solutions, pH 6.6 and pH 7.6, containing various concentrations of KCl (0, 0.3, and 1.0 M), and were lyophilized above liquid nitrogen for 6 h. The loss of activity was negligible on lyophilization. Aliquots of buffers also were lyophilized and used for background measurements.
Lyophilized samples (1 mg of the proteins) were dissolved in D2O. The time of the addition of D2O was taken as the start of exchange. A series of IR spectra (400–4,000 cm−1 region) was recorded starting 30–40 s after initiating the H/D exchange. A resolution of 2 cm−1 was used. The number of scans used for recording a spectrum was adjusted to the speed of the exchange (four scan measurements every 10 sec at the beginning and 128 scans every 10 min toward the end).
H/D Exchange.
Absorbances of the amide I and amide II bands were evaluated from the spectra at the wavenumbers of their maxima, 1,650 cm−1 and 1,547.5 cm−1, corrected with the baseline absorbance measured at 1,789 cm−1.
The fraction of the unexchanged peptide hydrogen atoms was determined as
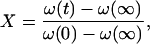 |
1 |
where ω(t) is the ratio of the amide II and amide I absorbances, corrected with the absorbance of the baseline, at time t:
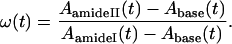 |
2 |
ω(0) is the amide II/amide I ratio of the undeuterated proteins and ω(∞) is the ratio for the fully deuterated proteins.
To obtain ω(0), IR spectra of the undeuterated protein samples were measured in KBr pastille, in nujol, and in H2O. A value of ω(0) = 0.65 ± 0.02 was obtained for both enzymes. When calculating ω(0) from the H2O measurement, only the value of AamideII was used, and the value of AamideI measured in D2O was substituted into Eq. 2 to obtain ω(0). The rationale behind this calculation was that the value of AamideI in H2O could not be determined with high accuracy because of errors appearing during the correction of the spectrum for the H2O excluded by the protein.
The value of ω(∞) was determined from samples incubated in D2O for 14 days at high temperatures (45°C for E. coli IPMDH and 65°C for Th. thermophilus IPMDH). Approximately half of the protein was precipitated during the incubation. Measurements were carried out after clearing the samples by centrifugation (12,000 g, 15 min, room temperature). A value of ω(∞) = 0.10 ± 0.01 was obtained for both enzymes.
X, the ratio of unexchanged peptide hydrogen atoms, is a function of time, pDread, and temperature. The results were interpreted in terms of the EX2 mechanism (5), i.e., under the assumption that the fluctuations exposing buried H atoms are fast compared with the rate of exchange of the solvent-exposed peptide groups. In this case, the exchange is supposed to proceed as a series of simultaneous first-order reactions:
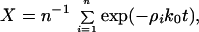 |
3 |
where n is the number of peptide hydrogens in the protein molecule and ρi is the probability of finding the ith peptide group exposed to the solvent. The chemical exchange rate constant, k0, is a function of pD and temperature (T in°C). According to NMR studies performed on model peptides, the value of k0 is slightly dependent on the neighboring residues of the individual peptide groups (35, 36). When using the FT-IR technique, however, individual peptide groups cannot be distinguished. Therefore, an empirical value of
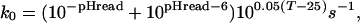 |
4 |
obtained with poly-dl-alanine (5) was used in the calculations as a good approximation. Because the present work is a comparative study of homologous proteins, the actual value of k0, in fact does not influence the results. Kinetic data of the exchange were plotted as the ratio of unexchanged protons, X vs. log(k0t), in the form of relaxation spectra (7).
RESULTS AND DISCUSSION
Catalytic Properties and Experimental Conditions.
The aim of this study was to compare the stabilities, enzymatic activities, and bulk flexibilities of a thermophilic and a mesophilic IPMDH at room temperature (25°C) and under the conditions of optimum function. IPMDH is a good choice because the structures and the active sites between the E. coli and the Th. thermophilus enzymes are highly conserved (28). All micro- and macrostability measurements were carried out under the respective optimum conditions for both enzymes. The enzyme activity of IPMDH depends on temperature, pH, and the ions added to the buffer. K+ is the most effective in activating these enzymes (16), whereas Na+ does not activate them at all. We followed the oxidative decarboxylation reaction at various pHs, temperatures, and KCl concentrations. The latter cannot be ignored because the Th. thermophilus enzyme is not fully active at the 0.3 M KCl concentration optimal for the activity of the E. coli IPMDH. The temperature, pH, and KCl optima for the enzymes are listed in Table 1. The pH dependence (data not shown) has a broad plateau for both enzymes. The temperature dependence of the specific activities at pH 7.6 and at optimum KCl concentrations are presented in Fig. 1. The difference in optimum temperature of these enzyme variants is more than 20°C, and the catalytic activity of the thermophilic enzyme is negligible at room temperature. “Cold denaturation” cannot account for this decrease according to our CD and DSC measurements.
Table 1.
Optimal working conditions for E. coli and Th. thermophilus IPMDH
| pHopt (25°C) | Topt | [KCl]opt | |
|---|---|---|---|
| E. coli | pH 7.5–7.9 | 48–52°C | 0.3–0.6 M |
| Th. thermophilus | pH 7.3–7.7 | 70–77°C | 1–2 M |
The quantities were measured as described in Materials and Methods.
Figure 1.

Specific activity-temperature profiles of E. coli (x) and Th. thermophilus (+) IPMDHs, in 0.3 M and 1.0 M KCl, respectively, in 20 mM potassium phosphate buffer pH 7.6 containing 0.2 mM MnCl2.
Macrostability.
The thermal unfolding properties of both enzymes were characterized by DSC and temperature-dependent CD measurements. The DSC melting profiles were recorded in the temperature range of 10–100°C and are presented in Fig. 2. Both IPMDHs undergo a temperature-induced cooperative transition from the native state to the unfolded state, with melting temperatures Tm = 73.5°C for the E. coli enzyme and Tm = 90°C for the Th. thermophilus enzyme. The heat absorption curves showed sharp single peaks, reflecting the high cooperativity of the unfolding transition. Sequential heating experiments showed that the unfolding of IPMDH was not completely reversible, therefore no other thermodynamic parameters were calculated.
Figure 2.

Calorimetric melting profile of E. coli (thin line) and Th. thermophilus (thick line) IPMDHs measured with a heating rate of 1°C/min, in 20 mM potassium phosphate, pH 7.6. KCl concentrations were set to 0.3 M and 1.0 M, respectively. Melting temperatures are 73.5°C for the E. coli and 90.0°C for the Th. thermophilus enzymes.
Analogous results were obtained by far-UV CD spectroscopy. The temperature-dependent unfolding of the secondary structure elements was followed at 221 nm in the temperature range of 25–97°C (data not shown). Sharp cooperative transitions were observed with midpoint temperatures of 73.6 and 90.1°C for the mesophilic and the thermophilic enzymes, respectively. The difference in the unfolding temperatures was 16.5°C, somewhat smaller than the difference between the enzyme activity maxima (cca. 20°C).
The Effect of D2O on the Stability and Enzyme Activity.
Because our H/D exchange experiments were done in heavy water, the effect of this solvent on both the activity and the stability of the enzymes also was checked. Enzymatic activities of the samples were measured after the H/D exchange experiments in H2O and proved to be unchanged within the limits of experimental error. The far-UV CD spectra measured in heavy water did not show any difference in the secondary structure of the enzymes when compared with spectra in ordinary water. The temperature-dependent unfolding was followed by CD spectroscopy at 221 nm in the temperature range of 25–98°C (Fig. 3 A and B). The same buffer was used as for the H/D exchange experiments. Unfolding temperatures are listed in Table 2. Both enzymes are stabilized to a similar extent by D2O in the full pH range used in our experiments, as reflected by the slight increase in transition temperatures.
Figure 3.

Heat denaturation curves of E. coli (thin line) and Th. thermophilus (thick line) IPMDHs in D2O followed by CD with a heating rate of 50°C/h at 221 nm in 20 mM potassium phosphate buffer containing 0.3 M or 1.0 M KCl. Measurements were carried out at pD 7.15 (A) and pD 8.15 (B). Corresponding melting temperatures are 77.4°C for the E. coli and 92.2°C for the Th. thermophilus enzyme (pD 7.15) and 74.5°C and 90.9°C (pD 8.15).
Table 2.
Melting temperatures of IPMDHs measured by CD spectroscopy in D2O in 20 mM potassium phosphate buffer at a KCl concentration of 0.3 M (E. coli) and 1.0 M (Th. thermophilus)
|
Tm, °C
|
||
|---|---|---|
| pD 7.15 | pD 8.15 | |
| E. coli | 77.4 | 74.5 |
| Th. thermophilus | 90.9 | 92.2 |
pD = pDread + 0.4.
The Effect of Salt on H/D Exchange.
The optimal KCl concentrations for the two enzymes are different (see Table 1). To check the effect of the difference in salt concentrations on H/D exchange, measurements were performed with the E. coli IPMDH using a buffer containing 1.0 M KCl. The exchange kinetics were the same as in the presence of 0.3 M KCl within the limits of experimental error (data not shown). According to Bai et al. (36), KCl does not influence H/D exchange significantly at higher pHs. Our results also indicate that the shift in KCl concentration from 0.3 M to 1.0 M has a negligible effect.
Microstability and Flexibility.
The flexibility of protein molecules is reflected in perpetual conformational fluctuations. These reversible, noncooperative local rearrangements expose buried segments of the polypeptide chain to the solvent. In D2O, H/D exchange occurs during such exposure. The probability distribution of the accessibility of peptide hydrogens can be determined by H/D exchange experiments. This distribution function is the so-called exchange relaxation spectrum (7, 37). By the FT-IR technique, only those protons can be followed that are exposed to the solvent with a probability smaller than 10−1–10−2; faster ones are surface protons, which are irrelevant from our point of view. Because the halftime of exchange for the full distribution of amide groups in a protein covers 8–10 orders of magnitude, and because H/D exchange in the pH range used is a OH− catalyzed reaction, our measurements were carried out at two pD values (7.15 and 8.15) around the flat pH optima of the enzymes to expand the fraction of peptide hydrogens covered by the measurements.
The time course of the H/D exchange of both IPMDHs was followed by FT-IR spectroscopy. Fig. 4 shows a set of spectra for a typical experiment together with the spectra of the unexchanged and totally exchanged enzymes. The absorbance bands around 1,650 cm−1 and 1,550 cm−1 correspond to the amide I band and amide II band, respectively. The amide I band arises from the in-plane C=O stretching vibration weakly coupled with C-N stretching and in-plane N-H bending. The amide II band is associated with in-plane N-H bending strongly coupled with C-N stretching (38). H/D exchange of amide protons decreases the absorbance of the amide II band. The N-D band can be found around 1,450 cm−1. The ratio of unexchanged peptide hydrogen atoms was calculated as a function of time, pDread, and temperature. The results were interpreted in terms of the EX2 mechanism (5), i.e., under the assumption that the fluctuations exposing buried H atoms are fast compared with the exchange rate of the solvent-exposed peptide groups. The evaluation of data and the basis of the representation are described in Materials and Methods. The relaxation spectra reflect probability distributions. The thin lines in Figs. 5 and 6 represent the H/D exchange curves of hypothetical polypeptides exposing their peptide hydrogens in a cooperative way with probability ρi. Compared with this set of curves, the relaxation spectra indicate that the conformational fluctuations exposing the buried peptide hydrogens to the solvent are noncooperative, overlapping, local movements with varying probabilities for various parts of the protein. The shift of the relaxation spectrum toward the right upper corner, i.e. toward smaller ρ values, reflects an increase in the conformational stability.
Figure 4.

A typical H/D exchange experiment on E. coli IPMDH at 25°C, pD 7.15 in the time range from 30 sec to 24 h. The amide II band (at approximately 1,550 cm−1) shows the decreasing number of amide protons. The broad band at 1,450 cm−1 reflects the increasing number of ND groups and HDO molecules. Arrows show the direction of changes. Spectra of undeuterated and totally deuterated proteins (thin lines) were measured as described in Materials and Methods.
Figure 5.

H/D exchange data, summarized in the form of relaxation spectra for both E. coli and Th. thermophilus IPMDHs at 25°C. E. coli, pD 7.15 (▵), pD 8.15 (□); Th. thermophilus, pD 7.15 (▿), pD 8.15 (○). X is the fraction of unexchanged peptide hydrogens, t is the time. k0, the chemical exchange rate constant, was calculated according to Eq. 4. The solid lines represent the exchange rate curves for hypothetical polypeptides characterized by the ρ values indicated in the figure. ρ is the probability of solvent exposure of the peptide groups (see Materials and Methods). Curves indicate a more rigid structure for the thermophilic enzyme.
Figure 6.

H/D exchange data of IPMDHs, at two different pD values at their temperature optima. E. coli, 48°C, pD 7.15 (▵), pD 8.15 (□); Th. thermophilus, 70°C, pD 7.15 (▿), pD 8.15 (○). Curves obtained for the two enzymes show very similar flexibilities.
Fig. 5 shows the relaxation spectra of both enzymes at 25°C, at pD 7.15 and 8.15. The E. coli IPMDH is characterized by a continuous spectrum, i.e. the curves recorded at the two pHs overlap, whereas the Th. thermophilus IPMDH shows a slight flexibility change as a function of pH, as reflected by the shift of the curve when the pH is changed. The major conclusion from this set of experiments is that at 25°C the thermophilic IPMDH is significantly less flexible than the mesophilic one, irrespective of the pH.
As shown in Fig. 6, at temperatures near the activity optima of the enzymes, the flexibility of the thermophilic IPMDH is strongly increased, becoming similar to the conformational flexibility of its mesophilic counterpart. The curves recorded at the respective optimum temperatures are close to each other in this case. The slight upward bend at the initial part of the curves measured at 70°C might be caused by imperfect temperature adjustment during the first 30–60 sec of the experiment.
To sum up, at 25°C, the relaxation spectra of thermophilic and mesophilic IPMDHs are far apart from each other, whereas they almost coincide at the respective optimum temperatures of the enzymes. The relaxation spectra are similar in shape and position, reflecting similar distributions of the conformational fluctuations. This observation points to the significance of conformational flexibility in enzyme function and demonstrates how conformational flexibility is “adjusted” to the optimum working temperatures of the enzymes. We hypothesize that the increased rigidity of the thermophilic enzyme results, at 25°C, in restricting the conformational fluctuations required for catalytic function. These experiments suggest a direct relationship among conformational stability, flexibility, and enzyme function in the case of IPMDH.
CONCLUSION
In this work, we have examined the question of whether the low activity of certain thermophilic enzymes at room temperature may be caused by restricted conformational movements of the protein molecule, in other words, whether the activity of such an enzyme is related to its flexibility. As a model system we chose an enzyme, IPMDH, having two variants with significantly different heat stabilities, isolated from a mesophilic and a thermophilic organism. H/D exchange as followed by the FT-IR method was used to compare enzyme flexibilities. All measurements were carried out at the optimal pHs and salt concentrations. The analysis of the obtained relaxation spectra clearly showed that the thermophilic IPMDH is significantly more rigid at room temperature than its mesophilic counterpart. However, the two enzyme variants, which are adapted to different environmental conditions, showed very similar flexibility at temperatures near their activity optima. Our measurements strongly support the idea that enzyme activity and conformational flexibility are closely correlated, and evolutionary adaptation of proteins to different physiological temperatures tends to maintain “corresponding states” regarding conformational flexibility among others (39).
Acknowledgments
We thank Dr. G. Wallon from Brandeis University for providing us with the plasmids pWally, pUTL118, and the strain RDK1782. We are grateful to Dr. F. Vonderviszt and A. Szilágyi for critical reading of the manuscript. This work was supported by grants of the Hungarian Science Foundation (OTKA Grants No. T022370 and F20874), the grant of The Ministry of Education (FKFP 0166/1997), and the U.S.-Hungarian Joint Fund (Grant No. 155/91).
ABBREVIATIONS
- IPMDH
3-isopropylmalate dehydrogenase
- DSC
differential scanning calorimetry
- FT-IR
Fourier-transformed infrared
References
- 1.Careri G, Fasella P, Gratton E. CRC Crit Rev Biochem. 1975;3:141–164. doi: 10.3109/10409237509102555. [DOI] [PubMed] [Google Scholar]
- 2.Cooper A. Prog Biophys Mol Biol. 1984;34:181–214. doi: 10.1016/0079-6107(84)90008-7. [DOI] [PubMed] [Google Scholar]
- 3.Linderström-Lang K M, Schellman J A. In: The Enzymes. Boyer P, editor. New York: Academic; 1959. pp. 443–510. [Google Scholar]
- 4.Straub F B. Adv Enzymol. 1964;2:89–114. doi: 10.1002/9780470122716.ch3. [DOI] [PubMed] [Google Scholar]
- 5.Hvidt A, Nielsen S O. Adv Protein Chem. 1966;21:287–386. doi: 10.1016/s0065-3233(08)60129-1. [DOI] [PubMed] [Google Scholar]
- 6.Rosenberg A, Woodward C K. J Biol Chem. 1970;245:4677–4683. [PubMed] [Google Scholar]
- 7.Závodszky P, Johansen J T, Hvidt A. Eur J Biochem. 1975;56:67–72. doi: 10.1111/j.1432-1033.1975.tb02207.x. [DOI] [PubMed] [Google Scholar]
- 8.Englander J J, Downer N W, Englander S W. J Biol Chem. 1982;257:7982–7986. [PubMed] [Google Scholar]
- 9.Woodward C, Simon I, Tüchsen E. Mol Cell Biochem. 1982;48:135–160. doi: 10.1007/BF00421225. [DOI] [PubMed] [Google Scholar]
- 10.Lehrer S S. Biochemistry. 1971;10:3254–3263. doi: 10.1021/bi00793a015. [DOI] [PubMed] [Google Scholar]
- 11.Eftink M R, Ghiron C A. Biochemistry. 1977;16:5546–5551. doi: 10.1021/bi00644a024. [DOI] [PubMed] [Google Scholar]
- 12.Somogyi B, Papp S, Rosenberg A, Seres I, Matko J, Welch G R, Nagy P. Biochemistry. 1985;24:6674–6679. doi: 10.1021/bi00344a056. [DOI] [PubMed] [Google Scholar]
- 13.Rosenberg A, Ng K, Punyiczki M. J Mol Liquids. 1989;42:31–43. [Google Scholar]
- 14.Vihinen M. Protein Eng. 1987;1:477–480. doi: 10.1093/protein/1.6.477. [DOI] [PubMed] [Google Scholar]
- 15.Varley P G, Pain R H. J Mol Biol. 1991;220:531–538. doi: 10.1016/0022-2836(91)90028-5. [DOI] [PubMed] [Google Scholar]
- 16.Yamada T, Akutsu N, Miyazaki K, Kakinuma K, Yoshida M, Oshima T. J Biochem. 1990;108:449–456. doi: 10.1093/oxfordjournals.jbchem.a123220. [DOI] [PubMed] [Google Scholar]
- 17.Wrba A, Schweiger A, Schultes V, Jaenicke R, Závodszky P. Biochemistry. 1990;29:7584–7592. doi: 10.1021/bi00485a007. [DOI] [PubMed] [Google Scholar]
- 18.Miyazaki K, Yaoi T, Oshima T. Eur J Biochem. 1994;221:899–903. doi: 10.1111/j.1432-1033.1994.tb18805.x. [DOI] [PubMed] [Google Scholar]
- 19.Jaenicke R. Eur J Biochem. 1991;202:715–728. doi: 10.1111/j.1432-1033.1991.tb16426.x. [DOI] [PubMed] [Google Scholar]
- 20.Hecht K, Wrba A, Jaenicke R. Eur J Biochem. 1989;183:69–74. doi: 10.1111/j.1432-1033.1989.tb14897.x. [DOI] [PubMed] [Google Scholar]
- 21.Privalov P L, Tsalkova T N. Nature (London) 1979;280:693–696. doi: 10.1038/280693a0. [DOI] [PubMed] [Google Scholar]
- 22.Tanaka T, Kawano N, Oshima T. Biochemistry. 1981;89:677–682. doi: 10.1093/oxfordjournals.jbchem.a133245. [DOI] [PubMed] [Google Scholar]
- 23.Imada K, Sato M, Tanaka M, Katsube Y, Matsuura Y, Oshima T. J Mol Biol. 1991;222:725–738. doi: 10.1016/0022-2836(91)90508-4. [DOI] [PubMed] [Google Scholar]
- 24.Parsons S J, Burns R O. Methods Enzymol. 1970;17:793–799. [Google Scholar]
- 25.Kohlhaw G B. Methods Enzymol. 1988;166:429–435. doi: 10.1016/s0076-6879(88)66056-3. [DOI] [PubMed] [Google Scholar]
- 26.Kawaguchi H, Inagaki K, Kuwata Y, Tanaka H, Tano T. J Biochem. 1993;114:370–377. doi: 10.1093/oxfordjournals.jbchem.a124183. [DOI] [PubMed] [Google Scholar]
- 27.Magyar C, Szilágyi A, Závodszky P. Protein Eng. 1996;9:663–670. doi: 10.1093/protein/9.8.663. [DOI] [PubMed] [Google Scholar]
- 28.Wallon G, Kryger G, Lovett S T, Oshima T, Ringe D, Petsko G A. J Mol Biol. 1997;266:1016–1031. doi: 10.1006/jmbi.1996.0797. [DOI] [PubMed] [Google Scholar]
- 29.Kadono S, Sakurai M, Moriyama H, Sato M, Hayashi Y, Oshima T, Tanaka N. J Biochem. 1995;118:745–752. doi: 10.1093/oxfordjournals.jbchem.a124975. [DOI] [PubMed] [Google Scholar]
- 30.Yura T, Mori H, Nagai H, Nagata T, Ishihama A, Fujita N, Ishono K, Mizobuchi K, Nakata A. Nucleic Acids Res. 1992;20:3305–3308. doi: 10.1093/nar/20.13.3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kagawa Y, Nojima H, Nukiwa N, Ishizuka M, Nakajima T, Yashuhara T, Tanaka T, Oshima T. J Biol Chem. 1984;259:2956–2960. [PubMed] [Google Scholar]
- 32.Luisi-DeLuca C, Kolodner R. Genetics. 1989;122:269–278. doi: 10.1093/genetics/122.2.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gary R, Bates R G, Robinson R E. J Phys Chem. 1964;68:3806–3809. [Google Scholar]
- 34.Privalov P L. Adv Protein Chem. 1979;33:167–241. doi: 10.1016/s0065-3233(08)60460-x. [DOI] [PubMed] [Google Scholar]
- 35.Molday R S, Kallen R G. J Am Chem Soc. 1972;94:6739–6745. [Google Scholar]
- 36.Bai Y, Milne J S, Mayne L, Englander S W. Proteins. 1993;17:75–86. doi: 10.1002/prot.340170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hvidt A, Wallevik K. J Biol Chem. 1972;247:1530–1535. [PubMed] [Google Scholar]
- 38.Susi H. Methods Enzymol. 1972;26:455–472. doi: 10.1016/s0076-6879(72)26024-4. [DOI] [PubMed] [Google Scholar]
- 39.Jaenicke R, Závodszky P. FEBS Lett. 1990;268:344–349. doi: 10.1016/0014-5793(90)81283-t. [DOI] [PubMed] [Google Scholar]