

Abstract
G protein-coupled receptors (GPCRs) are regulated by multiple families of kinases including GPCR kinases (GRKs). GRK4 is constitutively active towards GPCRs, and polymorphisms of GRK4γ are linked to hypertension. We examined, through co-immunoprecipitation, the interactions between GRK4γ and the Gα and Gβ subunits of heterotrimeric G proteins. Because GRK4 has been shown to inhibit Gαs-coupled GPCR signaling and lacks a PH domain, we hypothesized that GRK4γ would interact with active Gαs, but not Gβ. Surprisingly, GRK4γ preferentially interacts with inactive Gαs and Gβ to a greater extent than active Gαs. GRK4γ also interacts with inactive Gα13 and Gβ. Functional studies demonstrate that wild-type GRK4γ, but not kinase-dead GRK4γ, ablates isoproterenol-mediated cAMP production indicating that the kinase domain is responsible for GPCR regulation. This evidence suggests that binding to inactive Gαs and Gβ may explain the constitutive activity of GRK4γ towards Gαs coupled receptors.
G protein-coupled receptor kinases (GRKs) comprise a family of seven proteins that possess an amino terminal regulator of G protein signaling homology (RH) domain and a serine/threonine kinase domain similar to protein kinase C [1]. GRKs initiate homologous desensitization through phosphorylation of the third cytoplasmic loop or carboxy terminus of ligand bound/activated target receptors [1]. This desensitization of the activated receptor is crucial in regulating signal transduction as well as ligand-mediated receptor endocytosis. The GRK1-like family, GRK1 and GRK7, are found in rod and cone cells and are associated with opsins [2]; the GRK2-like family, GRK2 and GRK3 (also known as β-adrenergic receptor kinase 1 and 2, respectively), are ubiquitously expressed and associate with Gαq/11-coupled receptors [1]; and finally, the GRK4-like family comprises GRK4, GRK5, and GRK6, which compared to the other GRKs little is known about their biology [3]. GRK5 and GRK6 are expressed in multiple tissues [1], whereas GRK4 is expressed in the testis [4], kidney tubular cells [5], brain [6], and myometrium [7]. In this study we focused on GRK4.
GRK4 was first identified during positional cloning of the Huntington’s disease locus [8]. Early in vitro studies demonstrated that GRK4 phosphorylates the β2-adrenergic receptor (β2AR) in a PIP2-dependent manner [4]. Additionally, both GRK4 and GRK2, significantly decrease luteinizing hormone/chorionic gonadotropin receptor-mediated cAMP production [4]. These data suggest that GRK4 is similar to GRK2 in its action; however, further studies demonstrate that in a comparable system GRK4, but not GRK2, phosphorylates the β2AR in an agonist-independent manner resulting in increased recycling of the β2AR [9]. Additionally, GRK4, and particularly GRK4γ, is the only GRK that appears to be constitutively active [9]. In support of this, recent studies demonstrate that GRK4α phosphorylates the D1 dopamine receptor (D1R) independently of D1R agonists [10].
There are four published splice variants of GRK4 [4]. GRK4α is the longest transcript with 16 exons encoding a protein of 578 amino acids. GRK4β is missing exon 2 and produces a protein of 546 amino acids; the 32 missing amino acids occur after Gln17 of GRK4α, which, based upon the GRK6 crystal structure [11], correspond to the first two alpha helixes α0 and α1 of the RH domain. GRK4γ, the first GRK4 reported [8], lacks exon 15 and is 532 amino acids in length; the 46 missing amino acids occur after Glu515 and correspond to less than half of alpha helix α10 and all of α11 of the RH domain as well as a portion of the unstructured region following the RH domain [11]. GRK4δ, a 500 amino acid protein, is missing both exons 2 and 15. None of the alternative splicing events eliminate GRK4 activity [4]; however, most studies of GRK4 are conducted with GRK4α. Polymorphisms in GRK4 (numbering based on GRK4α: R65L, A142V, and A486V) are associated with hypertension [5; 12; 13]. Two of the polymorphisms, R65L and A142V, lie within the RH domain and transgenic mice carrying the polymorphisms of GRK4γ are hypertensive compared to mice carrying human wild-type GRK4γ [5] (personal communication with P.A. Jose). The mechanism of action of GRK4γ, with regard to hypertension, is proposed to be hyper-phosphorylation/desensitization of the dopamine D1R [5]. This desensitization prevents excretion of salt and relieves the renal dopaminergic-mediated inhibition of the renin-angiotensin system [14].
We examined which of the Gα subunits interact with GRK4γ in an effort to understand the biochemistry of GRK4γ and eventually examine the polymorphisms in the RH domain associated with hypertension. We hypothesized that GRK4γ would interact with active/GTP-bound Gαs but not inactive/GDP-bound Gαs, and also possibly interact with other G proteins in a GTP-dependent manner. Counter-intuitively, the data indicate that GRK4γ interacts preferentially with inactive/GDP-bound Gαs.
Materials and Methods
Materials
All agonists and drugs were obtained from Sigma-Aldrich (St. Louis, MO). The AU1 antibody was obtained from Covance (Berkley, CA). All other antibodies and materials for immunoprecipitation were obtained from Santa Cruz (Santa Cruz, CA). The following lysis buffers were used: TX-100 based buffer (1% Triton X-100, 100mM NaCl, 20mM Tris pH 7.5, 2mM EDTA, 10mM MgCl2, 10mM NaF, 40mM β-glycerol phosphate, and 1mM PMSF), RIPA buffer (TX-100 based buffer plus 0.1% SDS and 0.5% deoxycholate), and detergent-free buffer (TX-100 buffer without Triton X-100) with cells passed through a 27 gauge needle 10 times. The cAMP EIA Kit was obtained from Cayman Chemical (Ann Arbor, MI) and used per the manufactures specifications.
cDNA Constructs
GRK2 cDNA was purchased from ATCC (Manassas, VA). Human wild-type GRK4γ and GRK2 were subcloned into the expression vector pcDNA4/TO (Invitrogen). Kinase dead (K216M, K217M) GRK4γ [15] was created with the Stratagene (La Jolla, CA) quick change mutagenesis kit. The wild-type and constitutively active QL mutant G proteins were gifts of Dr. J.S. Gutkind. Wild-type G proteins were subcloned into pEF-AU1, a derivative of pCEFL, obtained from Dr. J.S. Gutkind.
Cell Culture and Transfection
HEK293T cells were grown in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% (v/v) FBS and 100 units/ml of penicillin/streptomycin (Invitrogen, Carlsbad, CA) in 60mm culture dishes coated with poly-D-Lysine (Sigma). HEK293T cells were transiently transfected using Qiagen’s PolyFect Transfection Reagent (Germantown, MD). Functional assays were conducted using immortalized renal proximal tubule cells (RPTCs) isolated from Wistar-Kyoto (WKY) rats (a gift from Dr. U. Hopfer). PTCs were cultured in DMEM/F12 containing 5% (v/v) FBS, 10 ng/ml epithelial growth factor (EGF), 4μg/ml dexamethasone, ITS+ diluted at 4 ml per bottle of media, and 100 units/ml of penicillin/streptomycin. EGF and ITS+ premix were obtained from BD Biosciences (Bedford, MA). PTCs were grown in 60mm culture dishes for the cAMP assay and transfected utilizing the FuGene 6 protocol (Roche, Indianapolis, IN).
Preparation of Cellular Extracts and Co-IP
Transfected HEK293T cells were serum starved overnight (at least 8 hours) prior to all experiments. For experiments with the AU1-tagged G proteins, all samples and buffers were supplemented with 10μM GDP. To activate Gα proteins, samples were treated with 100μM AlCl3 and 10mM NaF for 15 minutes at 37°C to generate AlF4−,. All buffers used with the AlF4− treated samples contained 100μM AlCl3 and 10mM NaF. For experiments with the wild-type and QL mutant Gα subunits, GDP and AlF4− were not used. Medium was removed and cells were lysed in one of the three lysis buffers, and clarified by centrifugation for 5 minutes at 4°C. An aliquot of the supernatant was reserved for analysis of the total cell lysate and the remaining sample was used for co-immunoprecipitation with an antibody directed against the AU1 tag, Gα subunit, or Gβ common for one hour at 4°C. Protein A/G PLUS-Agarose beads (Santa Cruz) were added for 30 additional minutes. Immune complexes were washed with PBS containing 1% NP-40 and 1mM PMSF, followed by SDS-PAGE on a 9% gel and western blot analysis.
Results and Discussion
Examination of common Gα subunits
Before conducting any co-immunoprecipitation (co-IP) studies, the levels of endogenous and transfected GRK4γ were examined in 293T cells. No endogenous GRK4γ was detected by western blot in 293T cells. Initial co-IP studies were conducted with a panel of the commonly used heterotrimeric G protein alpha subunits: s, i1-3, q, 11, 12, and 13 (Fig. 1). The Gα subunits were subcloned into pEF-AU1 in order to conduct the analyses in parallel using a single antibody for detection. Each Gα subunit was examined in the active, AlF4− treated, conformation (Fig. 1a and 1b) and inactive conformation (Fig. 1c and 1d). After stimulation with AlF4−, GRK4γ co-immunoprecipitates with AU1-Gαs and to a lesser extent AU1-Gα13 (Fig. 1a). Surprisingly, when the experiment is conducted without AlF4−, representing the inactive Gα subunits, GRK4γ also co-immunoprecipitates with AU1-Gαs and to a lesser extent AU1-Gα13 (Fig. 1c). These results were opposite to our initial hypothesis.
Figure 1.

GRK4γ interacts with G-proteins in the active and inactive state. 293T cells were transfected with an AU1-tagged Gα subunit, and GRK4γ (a & c) or GRK2 (b & d). The G-proteins were stimulated as indicated and immunoprecipitated with AU1 antibody. The first of the three blots in each set is the co-immunoprecipitation, and the lower two blots are the controls to indicate that the GRK and Gα subunits were expressed. Note, Gαq and Gα11 are not well expressed, which is why the IP blot was used instead of total cell lysate (TCL) in panels a, b & d. Each experiment was conducted a minimum of two times.
To confirm that the conditions represent active and inactive Gα subunits, we conducted identical studies with GRK2 (Fig. 1b and d). As shown in Figure 1b GRK2 interacts with AlF4− treated (active) AU1-Gαq. Moreover, in samples lacking AlF4− (inactive) GRK2 is not co-immunoprecipitated with any of the AU1-Gα subunits (Fig 1d). These data partially fit what is known about GRK2 and G protein interactions[16; 17]; however, GRK2 should also interact with Gα11. Most likely this interaction is not observed due to low expression of AU1-Gα11. The expression levels of the various G proteins are significantly different, and Gαq and Gα11 are expressed the least (Figure 1). Therefore, we utilized wild-type Gαq and Gα11 and constitutively active Q209L Gαq and Q209L Gα11 in co-IP experiments with GRK2. As previously reported [16; 17], GRK2 interacted with the active form of Gαq and Gα11 (data not shown). Therefore, IP is an accurate measure of GRK4γ interactions with active and inactive G proteins.
Interaction of active and inactive Gαs with GRK4γ
Due to the experimental setup of the Gα subunit panel, direct comparison of the inactive and AlF4− activated blots is not appropriate. To directly compare each Gα subunit in the active and inactive conformation, two series of experiments were conducted. First, experiments similar to those shown in figure 1 were conducted; each Gα subunit in the active and inactive conformation was run on the same gel for direct comparison of their interaction with GRK4γ. As shown in figure 2a, GRK4γ co-immunoprecipitates with inactive AU1-Gαs to a greater extent than with AlF4− activated AU1-Gαs. Second, in order to avoid artifacts that may arise from using tagged constructs and AlF4−, untagged wild-type and constitutively active Q213L Gαs subunits were expressed to obtain the inactive and active state of the G protein, respectively. GRK4γ robustly co-immunoprecipitated with inactive Gαs, but little interaction was found with Q213L Gαs (Fig. 2b). These data confirm the AU1-tagged/AlF4− treatment results. Thus indicating that the observed interaction between GRK4γ and inactive Gαs is not an artifact of the tagged Gαs, AlF4−, or the antibodies used. However, in all experiments presented thus far, the co-IP was conducted in Triton X-100 based lysis buffer. To determine if the interaction of inactive Gαs with GRK4γ is an artifact of the lysis buffer, we repeated the studies shown previously with three different lysis buffers (Fig. 2c). GRK4γ interacts with wild-type Gαs to a much greater extent than Q213L Gαs in all detergents examined, including detergent-free lysis. Overexposure of the IP blot indicates that native Gαs will also pull down the overexpressed GRK4γ. Similar experiments were conducted with AU1-Gαs with and without AlF4−, and showed GRK4γ interaction with inactive Gαs (data not shown). Therefore, the data are not an artifact of the lysis buffer. GRK4γ preferentially interacts with inactive Gαs.
Figure 2.
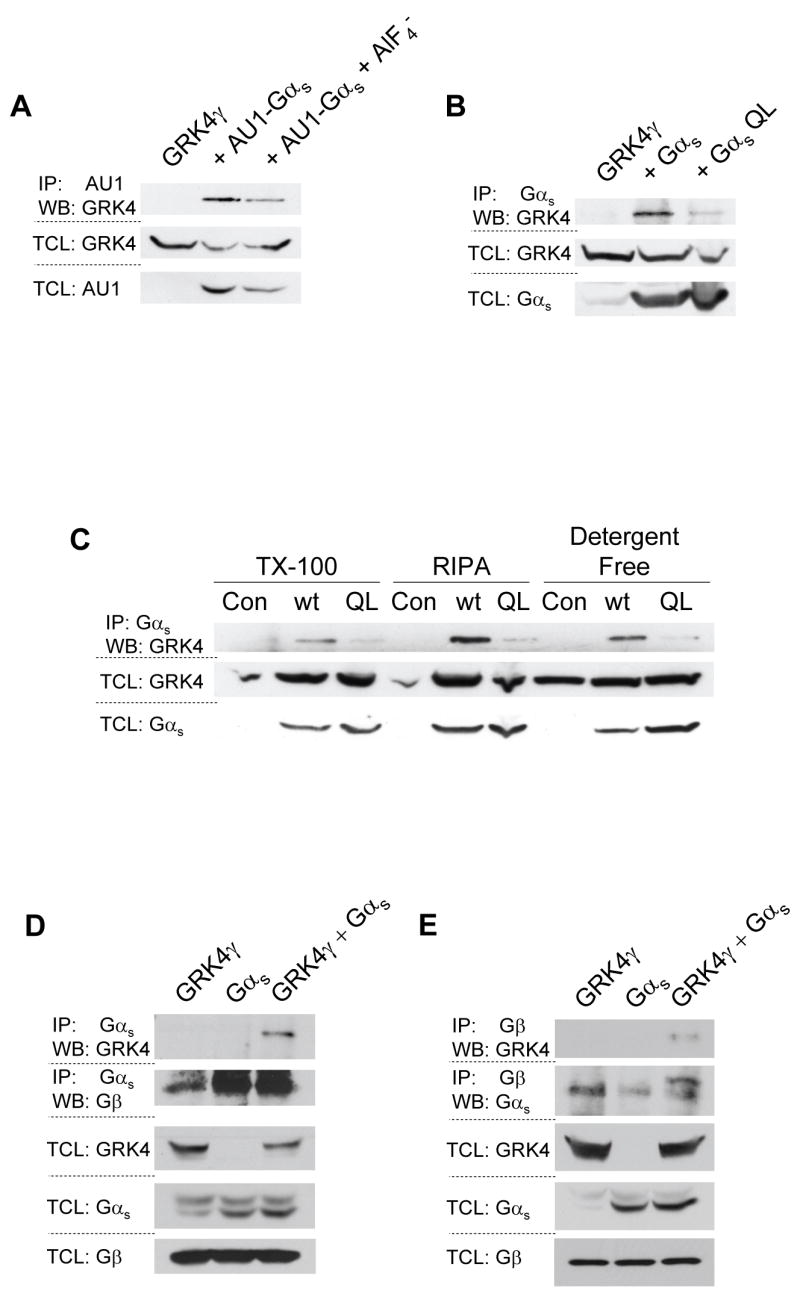
GRKγ interacts preferentially with inactive Gαs and Gβ. 293T cells were transfected with GRK4γ and either AU1-tagged Gαs (a), or wild-type and constitutively active Gαs (b & c). AU1 tagged Gαs was stimulated with AlF4−. 293T cells were lysed with either a triton X-100 (TX-100) based lysis buffer, a RIPA buffer, or a detergent-free buffer to examine the effects of the lysis buffer (c). Endogenous Gβ is co-immunoprecipitated with GRK4γ by Gαs (d), conversely endogenous Gβ co-immunoprecipitates GRK4γ (e). In all panels the upper blots are the co-immunoprecipitation and the lower blots are the control total cell lysates (TCL). Each experiment was conducted a minimum of two times
Interaction of an RH-domain containing protein with an inactive Gα subunit has never been described. As stated, these results run counter to our hypothesis. Given that this is an overexpression system, and 293T cells are known to generate copious amounts of protein, we investigated the interaction of GRK4γ, Gαs, and Gβ subunits. Gβ subunits are bound to inactive Gα subunits. If GRK4γ displaces Gβ from Gαs, then it is likely that the binding is an artifact of overexpression. Using endogenous Gβ, overexpressed GRK4γ, and overexpressed wild-type Gαs, we conducted co-IP studies with antibodies to Gαs/olf (Fig. 2d) and Gβ common (Fig. 2e). In both cases GRK4γ was pulled down only in the presence of overexpressed Gαs. Gβ was pulled down with the antibody to Gαs/olf (Fig. 2d). Similarly, Gαs was pulled down with the antibody to Gβ common, and GRK4γ appears to increase the pull down of Gαs (Fig. 2e). Because there is no apparent displacement of Gβ by GRK4γ, the interaction is most likely specific. The Gβ data are difficult to interpret because the antibody is interacting with four of the five Gβ variants, which will pull down all the Gα subunits. Alternatively, it is unknown if Gβ binds to GRK4γ. Gβ binds to the plextrin homology (PH) domain of GRK2 [17], however the GRK4 family members do not contain a PH domain [11]. Although it is unlikely that Gβ directly binds to GRK4γ, the experiments conducted here cannot discriminate between direct binding and indirect binding through Gαs or Gα13. However, it is clear that GRK4γ, Gαs, and Gβ are bound together in one complex.
Interaction of active and inactive Gα13 with GRK4γ
Experiments similar to those presented in figures 2a and 2b were conducted with all Gα subunits examined in figure 1. Initially weak interactions between GRK4γ and many of the Gα subunits were observed, but only the AU1-Gα13 interaction with GRK4γ proved to be specific when using the wild-type and QL mutants. This supports previous findings that GRK4 does not interact with Gαq or Gα11 [18]. In accordance with figure 1, GRK4γ is co-immunoprecipitated with inactive AU1-Gα13 and to a lesser extent when treated with AlF4− (Fig. 3a). Repetition with wild-type and constitutively active Q226L Gα13 indicated that inactive Gα13 binds to GRK4γ significantly more than active Gα13 (Fig. 3b). To confirm these results, the wild-type and Q226L Gα13 co-IP was repeated under detergent-free conditions. As shown in figure 3c, inactive Gα13 co-immunoprecipitates with GRK4γ, whereas constitutively active Gα13 does not interact with GRK4γ. Furthermore, GRK4γ is co-immunoprecipitated with Gβ only in the presence of overexpressed Gα13 (Fig. 3d) similar to what is seen with Gαs (Fig 2e). However, Gβ pulled down an equal amount of Gα13 in all conditions suggesting that the system was saturated. As noted previously, the binding order cannot be determined in such an assay; however, the data indicate that GRK4γ, Gα13, and Gβ are in a complex. However, the interaction of GRK4γ with Gα13 is weaker than that of GRK4γ with Gαs (Fig. 1a and 1c).
Figure 3.

GRK4γ interacts with inactive Gα13. 293T cells were transfected with GRK4γ and either AU1 tagged Gα13 (a), or wild-type and constitutively active Gα13 (b & c). The AU1 tagged Gα13 was stimulated with AlF4−. To test if the lysis buffer had any effects, 293T cells were lysed in detergent-free buffer (c). Endogenous Gβ co-immunoprecipitates GRK4γ and Gα13 (d). In all panels the upper blots are the co-immunoprecipitation and the lower blots are the control total cell lysates. Each experiment was conducted twice.
Function of GRK4γ
Recent structural studies indicate that the interaction of Gαq with the RH domain of GRK2 occurs in a manner distinct from Gαq/RGS interactions in that the interaction is more like an interaction with an effector not a RGS [17]. GRK2 does not increase the intrinsic catalytic rate of Gαq, which is proposed to be a general method of action of GRKs. The RH domain of GRK6, a member of the GRK4 subfamily, structurally resembles GRK2 suggesting that the mechanisms of action and areas of Gα subunit binding are similar [11]. However, the area of GRK2 that interacts with Gαq, α5-α6 helixes of the bundle sub-domain of the RH domain, is highly diverged in GRK6 (3.1% identity and 34.4% similarity). Furthermore, the predicted α5-α6 helixes of GRK4 has a 15.5% identity and 28.1% similarity with GRK2, and GRK4 and GRK6 only share 25% identity and 59.4% similarity over the same sequence. Although the α5-α6 region of GRK4 and GRK6 may not be the region that interacts with Gα subunits, the absence of significant similarity suggests that the interaction of the Gα subunit with its respective GRK does not necessarily have one unified mechanism.
In order to examine the physiological consequence of the interactions with inactive Gα subunits, functional assays were conducted to test the effects of GRK4γ and kinase dead K216M, K217M GRK4γ (KD GRK4γ) on Gαs- and Gα13-mediated cellular signaling. We generated KD GRK4γ as previously reported [15] (Fig 4a). As a control, we examined the interaction of KD GRK4γ with wild-type Gαs (Fig. 4b). KD GRK4γ interacts identically to wild-type GRK4γ (Fig. 2d). Similar studies were conducted with Q213L Gαs, wild-type Gα13, and Q226L Gα13 that demonstrated KD GRK4γ interacts with the inactive Gα subunits (data not shown). Thus, the dual lysine to methionine mutation can be used to discriminate between kinase activity driven effects of GRK4γ and interactions with Gα subunit driven events.
Figure 4.

Functional properties of GRK4γ. Kinase dead (KD) GRK4γ was generated through oligonucleotide-directed mutagenesis (a). This mutation does not abrogate binding to inactive Gαs (b). cAMP production was measured in WKY RPTCs transfected with cDNA expression vectors, indicated at the base of the histogram, and stimulated for 30 min with Isoproterenol (n = 4). Bars with different letters are significantly different (p < 0.05) via ANOVA with Tukey-Kramer multiple-comparison post hoc test.
To examine Gαs-mediated signaling, we utilized WKY RPTCs; one of the few cells to express endogenous GRK4 [4–7]. WKY RPTCs were transfected with empty vector, GRK4γ, KD GRK4γ, or GRK2 and then stimulated with vehicle, or 30 nM or 10 μM Isoproterenol for 30 min in the presence of 500 μM IBMX, a phosphodiesterase inhibitor. Intracellular cAMP measurements indicated that 30 nM Isoproterenol is saturating, thus, only this data is shown (Fig. 4c). All data were normalized to the control empty vector transfection. GRK2 served as a positive control. No effects were observed in unstimulated RPTCs. Stimulation doubled cAMP levels in KD GRK4γ and vector transfected RPTCs, but had no significant effect in GRK4γ and GRK2 transfected RPTCs. An independently conducted experiment utilizing 1 μM fenoldopam, a partial D1-like receptor agonist [19], and Isoproterenol confirmed GRK4γ’s inhibitory action, and revealed that KD GRK4γ can enhance fenoldopam-mediated cAMP accumulation (data not shown). Fenoldopam had a marginal effect on the RPTCs except when KD GRK4γ was present, suggesting that KD GRK4γ acts in a classic dominant negative fashion. Experiments were conducted to examine Gα13-mediated events; however, these studies were inconclusive.
Summary and Conclusion
A caveat of these studies is that they are conducted with GRK4γ. The gamma isoform is missing a portion of the carboxy terminus of full-length GRK4. Based on the co-crystallization of GRK2, Gαq, and Gβ, the missing portion of GRK4γ is most likely not directly involved in the interaction with the Gα subunit [17]. However, this does not preclude a variation in protein folding that alters GRK4γ-Gα interactions. Therefore, these studies should not be extended to all GRK4 variants or the other GRK4 family members.
GRK4 is the most active GRK under basal conditions, and GRK4γ appears to have the lowest level of receptor-mediated activity [9]. This may, in part, be explained by our results indicating that GRK4γ binds preferentially to inactive Gαs. Binding of GRK4γ to the inactive receptor-Gα complex would allow GRK4γ greater access to the G protein-coupled receptor, and thus could result in increased phosphorylation of the inactive receptor. Moreover, the apparent lower affinity for active Gαs may also explain the aforementioned low level of receptor-mediated activity. GRK4γ and GRK2 functionally inhibit Gαs-mediated accumulation of cAMP (Fig. 4). Thus, in RPTCs, GRK4γ functions as hypothesized, but through a novel mechanism. This may be a cell type specific response since similar studies show little effect of wild-type GRK4γ in regulating dopamine-mediated cAMP production in CHO cells [5].
In conclusion, GRK4γ binds to inactive Gαs > inactive Gα13 ≫ active Gαs ≫ active Gα13, yet still appears to function normally by inhibiting receptor-mediated signaling in a physiologically relevant cell type. These phenomena may account for the previously reported increased basal activity of GRK4γ. The molecular mechanism mediating this interaction requires further elucidation. Once the mechanism of wild-type GRK4γ binding to Gαs is determined, the effects of the polymorphisms in the RH domain associated with hypertension can then be clearly examined.
Acknowledgments
We would like to thank Dr. J. Silvio Gutkind (National Institute of Dental and Craniofacial Research, NIH, Bethesda, MD) for providing the Gα proteins, and Dr. Pedro A. Jose for his funding support (1 P01 HL074940-04 and 2 R37 HL23081-26). Additionally, this work was directly supported by a grant from the American Health Assistance Foundation, National Heart Foundation to Dr. B.T. Andresen.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Krupnick JG, Benovic JL. The role of receptor kinases and arrestins in G protein-coupled receptor regulation. Annu Rev Pharmacol Toxicol. 1998;38:289–319. doi: 10.1146/annurev.pharmtox.38.1.289. [DOI] [PubMed] [Google Scholar]
- 2.Weiss ER, Raman D, Shirakawa S, Ducceschi MH, Bertram PT, Wong F, Kraft TW, Osawa S. The cloning of GRK7, a candidate cone opsin kinase, from cone- and rod-dominant mammalian retinas. Mol Vis. 1998;4:27. [PubMed] [Google Scholar]
- 3.Metaye T, Gibelin H, Perdrisot R, Kraimps JL. Pathophysiological roles of G-protein-coupled receptor kinases. Cell Signal. 2005;17:917–928. doi: 10.1016/j.cellsig.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 4.Premont RT, Macrae AD, Stoffel RH, Chung N, Pitcher JA, Ambrose C, Inglese J, MacDonald ME, Lefkowitz RJ. Characterization of the G protein-coupled receptor kinase GRK4. Identification of four splice variants. J Biol Chem. 1996;271:6403–6410. doi: 10.1074/jbc.271.11.6403. [DOI] [PubMed] [Google Scholar]
- 5.Felder RA, Sanada H, Xu J, Yu PY, Wang Z, Watanabe H, Asico LD, Wang W, Zheng S, Yamaguchi I, Williams SM, Gainer J, Brown NJ, Hazen-Martin D, Wong LJ, Robillard JE, Carey RM, Eisner GM, Jose PA. G protein-coupled receptor kinase 4 gene variants in human essential hypertension. Proc Natl Acad Sci USA. 2002;99:3872–3877. doi: 10.1073/pnas.062694599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sallese M, Lombardi MS, De Blasi A. Two isoforms of G protein-coupled receptor kinase 4 identified by molecular cloning. Biochem Biophys Res Commun. 1994;199:848–854. doi: 10.1006/bbrc.1994.1306. [DOI] [PubMed] [Google Scholar]
- 7.Brenninkmeijer CB, Price SA, Lopez BA, Phaneuf S. Expression of G-protein-coupled receptor kinases in pregnant term and non-pregnant human myometrium. J Endocrinol. 1999;162:401–408. doi: 10.1677/joe.0.1620401. [DOI] [PubMed] [Google Scholar]
- 8.Ambrose C, James M, Barnes G, Lin C, Bates G, Altherr M, Duyao M, Groot N, Church D, Wasmuth JJ. A novel G protein-coupled receptor kinase gene cloned from 4p16.3. Hum Mol Genet. 1992;1:697–703. doi: 10.1093/hmg/1.9.697. [DOI] [PubMed] [Google Scholar]
- 9.Menard L, Ferguson SS, Barak LS, Bertrand L, Premont RT, Colapietro AM, Lefkowitz RJ, Caron MG. Members of the G protein-coupled receptor kinase family that phosphorylate the beta2-adrenergic receptor facilitate sequestration. Biochemistry. 1996;35:4155–4160. doi: 10.1021/bi952961+. [DOI] [PubMed] [Google Scholar]
- 10.Rankin ML, Marinec PS, Cabrera DM, Wang Z, Jose PA, Sibley DR. The D1 dopamine receptor is constitutively phosphorylated by G protein-coupled receptor kinase 4. Mol Pharmacol. 2006;69:759–769. doi: 10.1124/mol.105.019901. [DOI] [PubMed] [Google Scholar]
- 11.Lodowski DT, Tesmer VM, Benovic JL, Tesmer JJ. The structure of G protein-coupled receptor kinase (GRK)-6 defines a second lineage of GRKs. J Biol Chem. 2006;281:16785–16793. doi: 10.1074/jbc.M601327200. [DOI] [PubMed] [Google Scholar]
- 12.Speirs HJ, Katyk K, Kumar NN, Benjafield AV, Wang WY, Morris BJ. Association of G-protein-coupled receptor kinase 4 haplotypes, but not HSD3B1 or PTP1B polymorphisms, with essential hypertension. J Hypertens. 2004;22:931–936. doi: 10.1097/00004872-200405000-00014. [DOI] [PubMed] [Google Scholar]
- 13.Lohmueller KE, Wong LJ, Mauney MM, Jiang L, Felder RA, Jose PA, Williams SM. Patterns of genetic variation in the hypertension candidate gene GRK4: ethnic variation and haplotype structure. Ann Hum Genet. 2006;70:27–41. doi: 10.1111/j.1529-8817.2005.00197.x. [DOI] [PubMed] [Google Scholar]
- 14.Jose PA, Eisner GM, Felder RA. Dopamine and the kidney: a role in hypertension? Curr Opin Nephrol Hypertens. 2003;12:189–194. doi: 10.1097/00041552-200303000-00010. [DOI] [PubMed] [Google Scholar]
- 15.Sallese M, Salvatore L, D’Urbano E, Sala G, Storto M, Launey T, Nicoletti F, Knopfel T, De Blasi A. The G-protein-coupled receptor kinase GRK4 mediates homologous desensitization of metabotropic glutamate receptor 1. FASEB J. 2000;14:2569–2580. doi: 10.1096/fj.00-0072com. [DOI] [PubMed] [Google Scholar]
- 16.Carman CV, Parent JL, Day PW, Pronin AN, Sternweis PM, Wedegaertner PB, Gilman AG, Benovic JL, Kozasa T. Selective regulation of Galpha(q/11) by an RGS domain in the G protein-coupled receptor kinase, GRK2. J Biol Chem. 1999;274:34483–34492. doi: 10.1074/jbc.274.48.34483. [DOI] [PubMed] [Google Scholar]
- 17.Tesmer VM, Kawano T, Shankaranarayanan A, Kozasa T, Tesmer JJ. Snapshot of activated G proteins at the membrane: the Galphaq-GRK2-Gbetagamma complex. Science. 2005;310:1686–1690. doi: 10.1126/science.1118890. [DOI] [PubMed] [Google Scholar]
- 18.Iacovelli L, Salvatore L, Capobianco L, Picascia A, Barletta E, Storto M, Mariggio S, Sallese M, Porcellini A, Nicoletti F, De Blasi A. Role of G protein-coupled receptor kinase 4 and beta-arrestin 1 in agonist-stimulated metabotropic glutamate receptor 1 internalization and activation of mitogen-activated protein kinases. J Biol Chem. 2003;278:12433–12442. doi: 10.1074/jbc.M203992200. [DOI] [PubMed] [Google Scholar]
- 19.Grenader A, Healy DP. Fenoldopam is a partial agonist at dopamine-1 (DA1) receptors in LLC-PK1 cells. J Pharmacol Exp Ther. 1991;258:193–198. [PubMed] [Google Scholar]