

Abstract
Both nicotinic and muscarinic cholinergic receptors are present in the optic tectum. To begin to understand how the activation of these receptors affects visual activity patterns, we have determined the types of physiological responses induced by their activation. Using tectal brain slices from the leopard frog, we found that application of nicotine (100 μM) evoked long-lasting responses in 60% of patch-clamped tectal cells. Thirty percent of these responses consisted of an increase in spontaneous postsynaptic currents (sPSCs) and had both a glutamatergic and GABAergic component as determined by the use of 6-cyano-7-nitroquinoxaline-2,3-dione (50 μM) and bicuculline (25 μM), respectively. Remaining response types consisted of an inward membrane current (16%) and an increase in sPSCs combined with an inward membrane current (14%). All responses could be elicited in the presence of tetrodotoxin (0.5 μM). Muscarinic receptor-mediated responses, induced by carbachol (100 μM) application after nicotinic receptor desensitization, produced responses in 70% of tectal cells. In contrast to responses elicited by nicotine, carbachol-induced responses could be evoked multiple times without significant decrement. Responses consisted of either an outward current (57%), a decrease in sPSCs (5%) or an increase in sPSCs, with (almost 6%) or without (almost 3%) an outward current. The response elicited by carbachol was not predicted by the response of the cell to nicotine.
Our results suggest that nicotinic receptors are found predominantly at presynaptic locations in the optic tectum while muscarinic receptors are most often present at postsynaptic sites. We conclude that both of these receptor types could substantially modulate visual activity by changing either the input to tectal neurons or the level of their response to that input.
Keywords: frog, retinotectal, acetylcholine, visual system, cholinergic, superior colliculus
Acetylcholine (ACh) is present in many regions throughout the visual system appearing in the retina, superior colliculus, lateral geniculate nucleus, suprachiasmatic nuclei and the visual cortex (Groos et al., 1983; Sastry, 1985; Hohmann and Berger-Sweeney, 1998; Bickford et al., 2000). Contributed by both intrinsic and extrinsic sources (Sherman and Koch, 1986; Nobili and Sannita, 1997; Binns, 1999), cholinergic activity influences system function, plasticity and development (Greuel et al., 1988; Hohmann and Berger-Sweeney, 1998; Lauder and Schambra, 1999). Disturbances of this activity can result in such things as delayed neuronal development, changes in cytoarchitecture (Hohmann and Berger-Sweeney, 1998; Lauder and Schambra, 1999) and impaired ocular dominance shifts (Bear and Singer, 1986; Gordon et al., 1990). However, how ACh affects system activity and contributes to the mechanisms responsible for normal brain organization is not understood.
The optic tectum of the frog provides an attractive model system for examining the ways in which cholinergic activity modulates visual system function and plasticity. Almost all of the ACh found in the amphibian tectum is supplied by a projection from the nucleus isthmi, a midbrain structure with which it is reciprocally connected (Desan et al., 1987; Marín and González, 1999). The importance of this input to system functioning is demonstrated by experiments in which the nucleus isthmi is lesioned. This manipulation results in a visual scotoma or an apparent blindness in the region of the visual field that maps to the tectal area that is no longer receiving isthmi input (Caine and Gruberg, 1985; Gruberg et al., 1991). Since the retinotectal projection remains entirely intact, these experiments indicate that the isthmotectal projection has a strong facilitating effect on retinotectal transmission. Such a facilitatory effect, dependent upon cholinergic activity, has been directly demonstrated in the goldfish (King and Schmidt, 1991). Effects on visual plasticity have also been demonstrated: cholinergic activity both maintains the topographic map of visual space created in the tectum and regulates substance P expression in a population of tectal cells (Tu et al., 2000).
The optic tectum of the frog contains both nicotinic and muscarinic cholinergic receptors (Sargent et al., 1989; Birdsall et al., 1980; Butt et al., 2000, 2001) but knowledge of the physiological consequences of their activation is incomplete. Assessments of the effects of nicotinic receptor activation have focused on a facilitation of glutamate release from retinal ganglion cell terminals (Titmus et al., 1999; Kuras and Gutmaniene, 2001) while muscarinic receptors have been associated with tectal cell membranes following an enhancement of single unit recordings in response to ACh microiontophoresis (Fite and Wang, 1986). Using patch-clamp techniques, we have examined the responses elicited in tectal neurons by nicotine and carbachol in order to begin to understand how nicotinic and muscarinic receptor activity, respectively, might modulate tectal activity patterns through presynaptic and postsynaptic means.
Experimental Procedures
Adult Rana pipiens, purchased from Charles Sullivan (Nashville, TN, USA), were used in these experiments. The animals were housed at room temperature in 10-gallon glass tanks and fed mealworms. All procedures used were approved by the Institutional Animal Care and Use Committee at the University of Kentucky. Every effort was made to minimize the number of animals used in this study and their suffering.
Tectal brain slices were prepared from adult leopard frogs that had been anesthetized by submersion in a 0.1% solution of ethyl m-aminobenzoate and then decapitated. The skulls of the frogs were then opened and the brains were removed. Each isolated brain was put in a Petri dish filled with ice-cold bath solution (pH=7.4) composed of (in mM): 112 NaCl, 2 KCl, 17 NaHCO3, 3 CaCl2, 3 MgCl2, 24.2 glucose and saturated with 95% O2 and 5% CO2. This solution was also used as the bath solution in the recording chamber (see below). After removal of the dura, the tissue was embedded in a 2% solution of low-melting-point agarose (type VII agarose, Sigma, St. Louis, MO, USA). The block was then cooled and hardened (Hickmott and Constantine-Paton, 1993; Malayev and Debski, 1998), trimmed to obtain the desired tissue orientation and sectioned into transverse tectal slices (350 μm) using a Campden vibrotome (WPI, Sarasota, FL, USA). Slices from mid-tectum were chosen for recording and transferred to the recording chamber (Warner Instrument Corporation, Hamden, CT, USA) where they were held in position using a “grid” and an “anchor.” The recording chamber was then positioned on the stage of an inverted microscope and washed with normal bath solution for at least 1 h prior to the commencement of an experiment. All experiments were done at a room temperature of 20–22 °C.
Patch micropipettes were fabricated from borosilicate glass (WPI) using a vertical puller (Narashige, Sea Cliff, NY, USA). Their resistance was in the range of 5–7 MΩ when back-filled with the following solution (in mM): 100 KCl, 15 NaCl, 10 HEPES, 10 EGTA, 3 MgCl2, 1 CaCl2, 3 ATP-Mg, 0.3 GTP-Na (pH=7.4; Malayev and Debski, 1998). This solution depolarized the Cl− reversal potential with the result that Cl−-mediated events led to inward currents when cells were voltage-clamped at −65 mV (the potential at which they were held during our recordings). This filling solution was chosen so that Cl−-mediated inhibitory postsynaptic potentials would not interfere with our ability to detect excitatory postsynaptic events. The tectum contains a substantial amount of GABA which activates GABAA receptors (Hickmott and Constantine-Paton, 1993; Li and Fite, 1998).
The recording patch pipette was positioned close to the main cellular layer of the tectum (layer 6; Székely and Lázár, 1976), and was slowly advanced into the slice until the pipette encountered a cell surface. After tight seal formation, the whole-cell configuration was obtained by application of negative pressure to the contents of the patch pipette.
Drug applications were performed using an external perfusion system, which consisted of several 30 ml syringes connected to a valve with multiple input ports and one output port. The solution flow was gravity driven and the flow rate was 1.4–1.6 ml/min. It took solutions approximately 11 s to reach the recording chamber. The working volume of this chamber was 150 μl.
All drugs applied to the preparation were dissolved in normal bath solution. Nicotine was applied for 30 s at 5-min intervals until no response was elicited from the patch-clamped cell on two consecutive applications. Because of an apparent and long-lasting desensitization of nicotinic receptors upon exposure to nicotine (see Results), individual tectal slices were used only once for recording of the reported nicotinic responses. Following the desensitization of nicotinic receptors by repeated nicotine exposure, carbachol was used to evoke muscarinic responses employing the same application paradigm as used with nicotine.
Membrane currents were recorded using an Axopatch 200 amplifier (Axon Instruments, Foster City, CA, USA) in the “whole-cell” mode. The signal from the amplifier was filtered at 5 kHz and was acquired with the pCLAMP sofware package using a DigiData 1200 interface (Axon Instruments). Membrane current was sampled at a 5-kHz frequency. The data were saved in digital format and then processed with pCLAMP. For current responses, peak amplitudes, response durations and latencies (time from beginning of an agonist application to half the peak amplitude of the response) were determined from the original traces. For changes in spontaneous postsynaptic currents (sPSCs), event histograms were constructed using Prism software (GraphPad Software Inc., San Diego, CA, USA). Responses were analyzed in 8-s bin widths to obtain peak frequency values and response-duration measurements. The change in peak frequency was calculated using the following formula: (peak frequency with the agonist−peak frequency before the agonist)/(peak frequency with the agonist+peak frequency before the agonist) (Titmus et al., 1999). One-second bin widths were used to determine half-peak latencies. Given values are ±S.E.M. Except as noted, statistical significance was determined using the unpaired t-test for nicotinic receptor data and a paired comparison for muscarinic receptor data. The null hypothesis was rejected if P<0.05.
Results
Physiological effects of nicotinic receptor activation
Responses arising from nicotinic receptor activation were identified by bath-applying nicotine (100 μM) for 30 s to patch-clamped neurons in the main cellular layer of the tectum, layer 6. This produced responses in 60% of the cells from which we recorded (n=267). Responses fell into one of three different categories: an increase in the frequency and amplitude of sPSCs (30% of total recordings), an increase in the frequency and amplitude of sPSCs combined with an inward membrane current (14% of total recordings) and an inward membrane current alone (16% of total recordings) (Fig. 1). Each of these responses could only be elicited upon the first exposure to nicotine; subsequent 30-s exposures at 5-min intervals did not produce any response. This was also true for 50 μM nicotine exposure. However, nicotine responses could again be obtained if the interval between applications was 55.7±5.7 min (n=20), although these responses were much smaller than the originals.
Fig. 1.

Responses produced in tectal cells by nicotinic receptor activation. One of three responses was evoked in tectal cells by application of nicotine (nic; 100 μM): an increase in the frequency and amplitude of sPSCs (A1, 30% of total recordings), an increase in the frequency and amplitude of sPSCs and an inward membrane current (B1, 14% of total recordings) or just an inward membrane current (C1, 16% of total recordings). Nicotine did not evoke any response in 40% of the cells. Arrows in A1, B1 and C1 indicate areas of traces displayed at a faster time scale prior to drug application (A2, B2 and C2) and during the peak period of the nicotine response (A3, B3 and C3).
The characteristics of the different nicotine-evoked responses are given in Table 1. The durations, latencies and amplitudes of each of the elements in the response class that combined both of these responses were not different from the characteristics of those elements found in isolation. Consequently, subsequent to this point, the response consisting of an increase in the frequency and amplitude of sPSCs combined with an inward membrane current is analyzed as if it were a simple combination of each of its two apparent components.
Table 1.
Characteristics of the different nicotine-evoked responses
| Nicotinic receptor responses | ||||
|---|---|---|---|---|
| ↑sPSCs | ↑sPSCs+Iin | Iin | ||
| ↑sPSCs | Peak frequency change (%) | 52.5±4.1 (n=25) | 56.9±5.0 (n=20) | – |
| Durationa (s) | 149.6±15.2 (n=13) | 148.6±18.5 (n=14) | – | |
| Latency (s) | 26.7±1.9 (n=21) | 22.5±1.9 (n=20) | – | |
| Iin | Peak amplitude (pA) | – | 19.4±3.3 (n=19) | 25.3±4.5 (n=20) |
| Durationa (s) | – | 167.0±23.1 (n=4) | 177.2±15.4 (n=6) | |
| Latency (s) | – | 24.3±1.8 (n=19) | 26.9±2.6 (n=20) | |
Calculated from cells recovering within a 5-min period after agonist exposure.
Abbreviations: Iin, inward current; sPSCs, spontaneous postsynaptic currents; –, not applicable.
A prominent feature of the nicotine-evoked responses was their long duration. Within the 5-min period usually allotted for recovery, 66% of the responses involving an increase in sPSCs returned to baseline levels of activity in 149.1±16.9 s (n=27). The remaining responses were still elevated over these levels by 20.6±4.7% (n=6) at the period's end. Inward-current responses returning to baseline within the recovery period had a mean duration of 173.1±18.5 s (n=10). For the vast majority of cells (74%), however, current persisted after 5 min and averaged 28.3±4.0% (n=29) of peak. The true duration of nicotine-evoked responses was measured in a subset of cells in which nicotine was applied only once and the response recorded until it had returned to pre-exposure levels. For responses consisting of increased sPSCs, the mean duration of the response was 577.8±97.0 s (range: 393.0–935.0 s; n=5). The true duration of inward-current responses was 464.5±83.0 s (n=16).
In order to determine if the nicotine-evoked responses were dependent upon the propagation of action potentials, preparations were exposed to tetrodotoxin (TTX). TTX (0.5 μM) application preceded nicotine exposure by 30 s and was maintained in the bath solution during that exposure and the subsequent nicotinic response. For the nicotine-evoked responses that consisted of increased sPSCs, TTX exposure significantly decreased the duration of the response (290.3±70.0 s; n=8; P<0.05). However, the half-peak latency and change in peak frequency of the response in TTX (26.5±2.7 s and 61.3±5.8%, respectively) were not significantly different from those obtained in its absence. TTX also significantly reduced the duration of the nicotine-evoked inward-current response (143.6±31.0 s; n=5; P<0.05; df=19) while having no effect on response mean amplitude and latency (27.4±10.1 pA and 25.8±2.1 s, respectively, n=6).
The nicotine-induced increase in sPSCs had both a glutamatergic and GABAergic component. Application of the glutamatergic, non-NMDA receptor antagonist, 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX; 50 μM) reduced the magnitude of the nicotine-evoked response (Fig. 2). This reduction resulted in a significant decrease in response duration (155.6±30.3; n=6; P<0.01) and a significant increase in response latency (33.6±3.2 s; P<0.05). The peak frequency change (52.1±5.7%) was not significantly different from that obtained with nicotine alone. Although bicuculline (25 μM), the GABAA receptor antagonist, also appeared to reduce the duration of the nicotine-induced response (289.7±113.6 s; n=4), this reduction did not reach significance. The response latency (35.6±4.7) was significantly increased over that recorded with nicotine alone (P<0.05). Peak frequency (78.7±6.2%) was also significantly increased (P<0.05) although inspection of recordings showed that bicuculline very much reduced the amplitude of the nicotine-induced response overall (Fig. 2). Application of bicuculline in the presence of CNQX completely eliminated ongoing nicotine-induced responses (n=3) suggesting that glutamatergic and GABAergic activity accounted for the vast majority of the response.
Fig. 2.

The increased sPSC responses induced by nicotine consist of glutamatergic and GABAergic activity. Substantial portions of the nicotine-induced increases in sPSCs were blocked by the non-NMDA receptor antagonist CNQX (50 μM; middle trace) and the GABAA receptor antagonist bicuculline (25 μM; bottom trace). The solid line indicates the time during which nicotine was applied; the dotted line the time during which either CNQX or bicuculline was present. CNQX or bicuculline were introduced 30 s before the agonist (not shown). All recordings are from different cells. Abbreviations: nic, nicotine; bicu, bicuculline.
Physiological effects of muscarinic receptor activation
Muscarinic responses were evoked by application of carbachol (100 μM) after nicotinic receptor desensitization. Four different responses were observed (Fig. 3): an outward membrane current (57% of total recordings), an increase in the frequency and amplitude of sPSCs with an outward membrane current (6% of cells), a decrease in the frequency and amplitude of sPSCs (5% of cells), and an increase in the frequency and amplitude of sPSCs (3% of cells). The remaining 30% of cells had no response to carbachol exposure. The type of nicotinic response elicited in a given cell was not predictive of the type of muscarinic response that was evoked when that same cell was exposed to carbachol (Table 2). The observed distribution of responses was quite similar to the distribution calculated from the frequencies of the individual responses. However, cells that had a response to nicotine were more likely to have a response to carbachol than the general tectal cell population while those that did not, were less likely to respond to a subsequent application of carbachol (χ2 test; P<0.05).
Fig. 3.
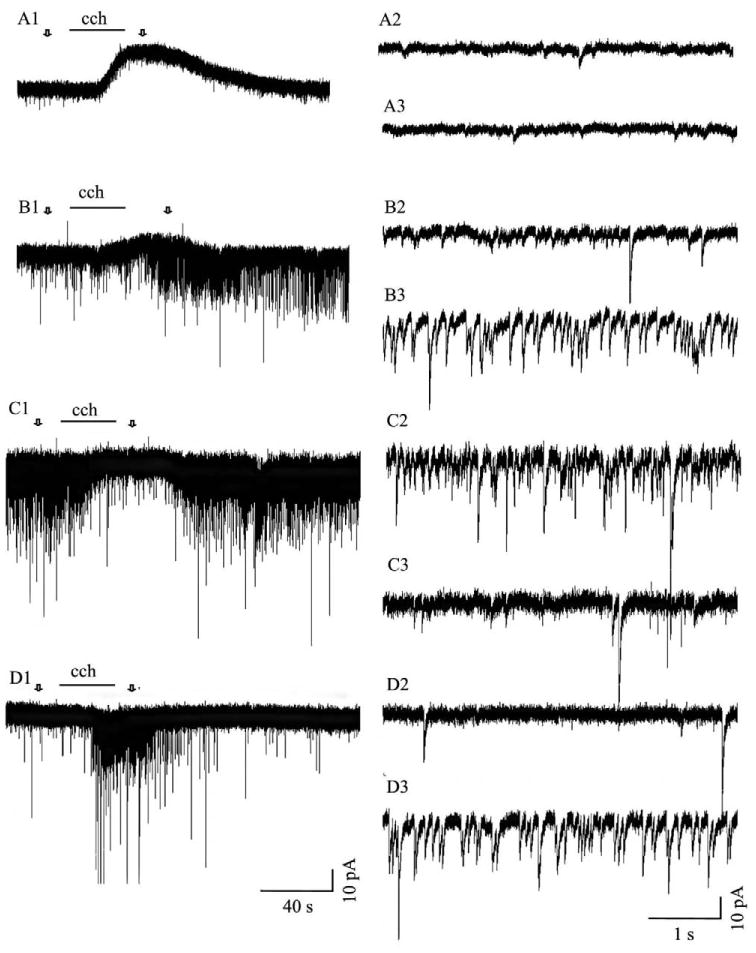
Responses in tectal cells evoked by muscarinic receptor activation. Application of carbachol (cch; 100 μM), after desensitization of nicotinic ACL receptors, produced four different responses: an outward membrane current (A1, 57% of the cells), an increase in the frequency and amplitude of sPSCs superimposed upon an outward current (B1, 6% of total recordings), a decrease in the frequency and amplitude of sPSCs (C1, 5% of total recordings) or an increase in the frequency and amplitude of sPSCs (D1, 3% of total recordings). Carbachol did not induce any response in 30% of the cells. Arrows in A1, B1 and C1 indicate areas of traces displayed at a faster time scale prior to drug application (A2, B2 and C2) and during the peak period of the carbachol response (A3, B3 and C3).
Table 2.
The different responses recorded in tectal cells upon nicotinic receptor activation were randomly were associated with those produced by muscarinic receptor activation
| Nicotinic receptor responses (#) | ||||||
|---|---|---|---|---|---|---|
| ↑sPSCs | ↑sPSCs+Iin | Iin | No response | Total (%) | ||
| Iout | 49 | 23 | 27 | 52 | 57 | |
| Muscarinic | Iout+↑sPSCs | 6 | 1 | 0 | 8 | 6 |
| Receptor | ↑sPSCs | 3 | 0 | 1 | 3 | 3 |
| Responses | ↓sPSCs | 7 | 2 | 3 | 2 | 5 |
| (#) | No response | 15 | 12 | 12 | 41 | 30 |
| Total (%) | 30 | 14 | 16 | 40 | 100a | |
Data are only from cells which were sequentially exposed to both nicotine and carbachol.
Approximate value because of rounding off.
Abbreviations: Iin, inward current; Iout, outward current; sPSCs, spontaneous postsynaptic currents.
As with the nicotinic responses, the carbachol-induced response consisting of both outward current and an increase in sPSCs, appeared to result from combining the properties of the two single response classes. The properties of the outward current response in combination with the increase in sPSCs did not differ from its properties when it occurred alone (Table 3). The latency and duration of the increase in sPSCs in combination with the outward current were statistically identical to the values for those properties associated with only an increase in sPSCs. However, in the presence of an outward current, the change in peak frequency attained by an increase in sPSCs (43.0±4.6%; n=11) was significantly lower from that attained in the absence of this current (64.3±9.5%; n=6; P<0.05).
Table 3.
Characteristics of the different carbachol-evoked responses
| Iout | Muscarinic receptor reponse | ||||
|---|---|---|---|---|---|
| Iout+↑ sPSCs | ↑ sPSCs | ↓sPSCs | |||
| Iout | Peak amplitude (pA) | 7.2±1.1 (n=20) | 7.0±0.8 (n=18) | – | – |
| Duration (s) | 81.3±7.3 (n=20) | 88.8±7.3 (n=18) | – | – | |
| Latency (s) | 22.7 ± 1.5 (n=20) | 22.6±1.5 (n=17) | – | – | |
| ↑sPSCs | Peak frequency change (%) | – | 43.0±4.6 (n=11) | 64.3±9.5 (n=6) | – |
| Duration (s) | – | 81.5±7.8 (n=7) | 85.6±14.8 (n=6) | – | |
| Latency (s) | – | 28.5±2.6 (n=9) | 28.1±2.3 (n=6) | – | |
| ↓sPSCs | Peak frequency change (%) | – | – | – | −45.5±11.8 (n=5) |
| Duration (s) | – | – | – | 67.0±15.5 (n=5) | |
| Latency (s) | – | – | – | 27.4±2.3 (n=5) | |
Abbreviations: Iout, outward current; sPSCs, spontaneous postsynaptic currents. –, not applicable.
Unlike the responses elicited by nicotine application, carbachol-evoked responses could be induced multiple times in the same cell. There was a tendency for the amplitude of the response to decrease with repeated exposure and for the latency to increase. Quantitative analysis of the outward current responses evoked by four successive exposures of carbachol did not reveal any significant changes in either latency, peak current or response duration (Fig. 4). Carbachol-elicited increases in sPSCs also did not show any significant decrement with repeated exposures.
Fig. 4.
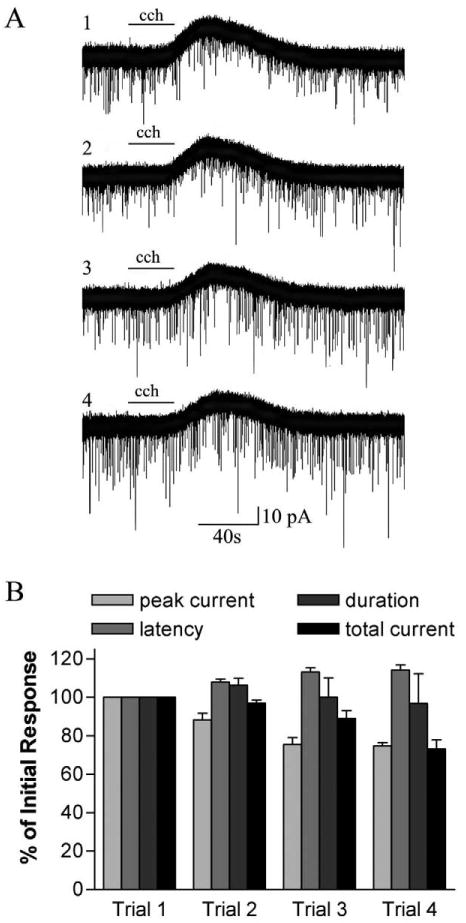
Repeatability of carbachol-induced outward-current responses. (A) Sequential 30-s applications of carbachol (cch; 100 μM) produced repeated outward-current responses. Top trace is the response produced upon the initial application of carbachol; bottom trace, the response on the fourth application. The interval between carbachol applications is 5 min. (B) Amplitude and/or frequency, latency and duration of sequential responses to carbachol are plotted as a percent of the response to the initial application (trial 1). Carbachol-induced outward-current response features are relatively stable but have a non-significant tendency to decrease in amplitude and total current, and increase in latency with increasing trial number (n=3).
We used atropine, a general muscarinic receptor antagonist, to confirm that the responses that we were inducing with carbachol were in fact mediated by muscarinic receptors. Application of atropine (2 μM) 30 s prior to carbachol, eliminated 94.7±5.3% of the peak outward current response (n=3; Fig. 5A). Carbachol-induced increases in sPSCs were similarly blocked by atropine (n=2; Fig. 5B). We were unable to test the effectiveness of atropine in preventing carbachol-induced reductions in sPSCs.
Fig. 5.

Atropine blocks carbachol-induced responses. (A) Carbachol (100 μM) application induced outward current in a tectal cell (top trace). A second application of carbachol in the presence of atropine (2 μM) elicited a minimal response in this same cell. (B) A carbachol-induced increase in sPSCs (top trace) was prevented by the presence of atropine (bottom trace). Solid line indicates the time during which carbachol (cch) was applied; dotted line, the time during atropine was present. The interval between the beginning of the first and second applications of carbachol in both A and B was 5 min.
Exposure to TTX did not affect carbachol-induced outward current responses. Peak outward current (9.5±2.9 versus 12.36±4.3 pA), latency (35.8±3.3 versus 29.3±1.9 s) or duration (106.1±11.05 versus 110.2±12.6 s) were not significantly changed by the presence or absence, respectively, of TTX (n=6). Consistent with the induced outward current being due to an increase in K+ conductance, a known consequence of M2-like muscarinic receptor activation (Felder, 1995), the carbachol-activated conductance had a linear I–V curve with a reversal potential of −72.4±3.5 mV (n=4).
Discussion
Nicotinic receptor location in the optic tectum
Nicotinic receptors have been generally regarded as having a presynaptic location in the optic tectum following their immunocytochemical identification on retinal ganglion cell terminals (Sargent et al., 1989). More recent work in Xenopus has provided physiological evidence for this terminal location by showing that activation of nicotinic receptors increases sPSCs recorded in patch-clamped tectal cells (Titmus et al., 1999). These sPSCs are thought to reflect an increase in neurotransmitter release from glutamatergic retinal ganglion cells brought about by increased terminal calcium levels (Edwards and Cline, 1999). As shown in extracellular recordings in Rana temporaria, endogenous ACh facilitates the responses evoked by the stimulation of individual retinal ganglion cell axons (Kuras and Gutmaniene, 2001). Receptor binding confirms the presence of nicotinic binding sites in retinorecipient tectal layers while indicating their presence at lower levels in other tectal layers (Butt et al., 2000).
Our work supports the idea that some nicotinic receptors have a terminal location but adds to previous observations in several important ways. Although inward current responses induced by nicotinic receptor agonists had been reported in the tectum (Titmus et al., 1999), these responses had not been characterized. Our finding that such currents can be recorded in 30% of cells makes it clear that nicotinic receptors, in addition to their presynaptic locations, have a substantial postsynaptic presence within the tectum. Our work also underscores the idea that nicotinic receptors are not present on all retinal ganglion cell terminals. There are a number of different retinal ganglion cell classes in the frog and their terminals end in different tectal layers. Not all classes of retinal ganglion cells project to every tectal cell although it is thought that all layer 6 cells do receive some direct retinal input (Hughes, 1990; Nakagawa et al., 1994). Therefore, if all retinal ganglion cell terminals had nicotinic receptors on them, we would have expected to have seen a nicotine-evoked increase in sPSCs in every tectal cell from which we recorded. Yet such an increase was seen in only 44% of the tectal cells. We cannot rule out the possibility that the soma of cells in which no nicotine-induced increase in sPSCs was recorded were electrically isolated from their retinal input. However, the evoking of this response in only a subset of cells is consistent with previous observations that antibodies against nicotinic receptors bind to some but not all of the tectal layers that contain retinal ganglion cell terminals (Sargent et al., 1989; Titmus et al., 1999).
Another contribution of our work to the understanding of nicotinic receptor function in the optic tectum is our characterization of the nicotine-induced sPSC components. In addition to its excitatory effects and in contrast to what has been concluded for Xenopus (Titmus et al., 1999), nicotinic receptor activation led to some inhibitory activity. Along with the expected glutamatergic response, the increase in sPSCs produced by nicotine also includes GABAergic activity. There are several possible explanations for this result. One is that GABAergic retinal ganglion cell terminals have presynaptic nicotinic receptors. Although the retinal projection to the tectum is overwhelmingly glutamatergic (Nistri et al., 1990; Hickmott and Constantine-Paton, 1993; Xiao et al., 1999; Kuras and Gutmaniene, 2001), GABA-containing retinal ganglion cells do in fact project to the tectum (Li and Fite, 2001). These terminals may or may not also release glutamate. There is precedence for modulation of both GABA and glutamate release by nicotinic receptors in embryonic spinal cord (Fucile et al., 2002). Another possibility is that some nicotinic receptors are located on GABAergic terminals from other sources. Sources of GABA in the frog optic tectum include tectal interneurons as well as nucleus isthmi terminals (Li and Fite, 1998).
Our nicotine-evoked responses had long-lasting durations. Polysynaptic, action potential-mediated activity contributed to the prolonged nature of these responses as indicated by the ability of TTX to reduce response durations. However, responses greatly outlasted agonist application even in the presence of TTX. A slow rate of clearance of the agonist from our slice preparation and non-spike-mediated local circuit interactions are some of the many factors that might be responsible for this prolongation.
Nicotinic receptors have also been investigated in the superior colliculus, the mammalian homologue of the optic tectum. In the superior colliculus, nicotinic receptors are concentrated in the superficial gray layers where information from the retina is inputted and integrated (Clarke et al., 1985; Perry and Kellar, 1995; Marks et al., 1998; Whiteaker et al., 2000; Yeh et al., 2001). Optic nerve lesion significantly reduces the density of nicotinic binding sites suggesting an association with the glutamatergic retinal ganglion cell terminals (Prusky and Cynader, 1988). Activation of these receptors results in the release of glutamate, further strengthening the idea that they are located predominantly on presynaptic terminals (Binns and Salt, 2000).
Muscarinic receptor location in the tectum
Muscarinic receptors, present in the superior colliculus (Aubert et al., 1992; Zubieta and Frey, 1993; Levey et al., 1994), are also found throughout the optic tectum of the leopard frog (Butt et al., 2001). Previous extracellular recordings had suggested that these receptors are on tectal cell membranes (Fite and Wang, 1986).
In this study, we induced muscarinic responses by using carbachol. Nicotinic receptors, which can also be activated by carbachol, were desensitized with nicotine prior to carbachol exposure. We know of no evidence indicating that this desensitization would affect subsequently recorded muscarinic receptor responses. The carbachol-evoked responses that we obtained were blocked by the muscarinic receptor agonist, atropine, indicating that they were in fact mediated by muscarinic receptor activation. Further evidence that the carbachol used was selectively stimulating muscarinic receptors comes from the recorded responses themselves which had very little overlap with nicotine-evoked responses. Responses induced by carbachol application could roughly be classified as two types: outward current and/or changes in sPSC frequency. Two response classes, an outward current and a decrease in sPSCs, together accounted for 88% of the carbachol-evoked responses recorded. Neither of these two response types was ever seen to result from nicotine exposure. The remaining carbachol-evoked responses (12%) all included an increase in sPSC frequency. This response type was also observed with nicotine exposure but was present in 73.3% of responding cells.
The vast majority of muscarinic responses involved the induction of outward current, either alone (81%) or in combination with an increase in sPSCs in patch-clamped tectal cells (8%). These responses were not blocked by TTX suggesting that carbachol was having a direct effect on the recorded tectal cell membrane. Furthermore, the induced current had a reversal potential consistent with current flow through a K+ channel. Modification of K+ channels is a described consequence of muscarinic receptor activation (Felder, 1995). We are therefore confident in placing muscarinic receptors on tectal cell membranes where they could modulate cell activity postsynaptically.
Both increases and decreases in the frequencies of sPSCs could also be induced by carbachol. Because of the infrequency with which these responses were observed, we were not able to test whether they could be evoked in the presence of TTX. Nor were we able to characterize these sPSCs as to whether they were excitatory and/or inhibitory in nature. Nevertheless, it is clear that muscarinic receptor activation can change the level of synaptic input onto layer 6 cells in both a positive and negative fashion. The results of optic nerve lesion experiments make it unlikely that muscarinic receptors are located on retinal ganglion cell terminals (Butt et al., 2001) although terminal locations on extraretinal input to the tectum or on tectal cells themselves remain possible. A postsynaptic location could also achieve the reported changes in sPSCs: muscarinic receptors can directly elevate neuronal firing rates (Fujino and Oertel, 2001; Wenzel et al., 2002) and in particular, have been shown to do so for GABAergic neurons in the hippocampus (Vogt and Regehr, 2001). Although we did not observe such an excitation in response to carbachol application, large GABAergic interneurons are present outside of our recording area in the superficial tectal layers and could respond with such an excitation (Fite and Wang, 1986; Wang and Matsumoto, 1990).
Our observation that nicotinic responses did not correlate with a given type of muscarinic response is somewhat disappointing. This finding is, however, consistent with other observations that fail to find an association between particular cell morphologies and individual responses (Wang and Matsumoto, 1990; Hickmott and Constantine-Paton, 1993; Malayev and Debski, 1998; Titmus et al., 1999). Nevertheless, it is difficult to understand how the system could function if nicotinic responses were randomly associated with muscarinic ones. It is more pleasing to postulate that layer 6 cells fall into many different cholinergic subpopulations that need further fractionation before correlative patterns become evident.
Understanding the consequences of cholinergic activity
In the leopard frog, the interruption of cholinergic input to a tectal lobe creates a visual scotoma in the contralateral monocular visual field that results in an inability to respond to prey or threat stimuli (Gruberg et al., 1991). This scotoma may be caused, at least in part, by disruptions in the cholinergic-dependent mapping of the retinal projection onto the optic tectum (Tu et al., 2000). Such observations suggest that cholinergic activity plays a necessary and facilitating role in obtaining normal system function and organization.
Electrical stimulation of the nucleus isthmi, the major source of tectal ACh, inhibits 70% and excites 30% of responding tectal cells (Wang and Matsumoto, 1990). Although the nucleus isthmi projections contain other neurotransmitters that could also contribute to these effects (Liu and Debski, 1995; Li and Fite, 2001), in the present study we demonstrate that nicotinic and muscarinic receptor activation could account for both the excitatory and inhibitory responses of individual optic tectal cells. Linking these responses to the apparent facilitatory effects of cholinergic activity on the tectum as a whole is difficult at this point. Part of this difficulty comes from the fact that layer six contains many different populations of cells (Debski et al., 1995; Tostivint et al., 1996; Gabriel et al., 1998; Li and Fite, 1998; Lázár, 2001; Stuesse et al., 2001) and we do not know how any of these cell types function in generating the overall tectal response. Thus it is unclear, for example, how the muscarinic receptor-mediated inhibition of some layer 6 cells will affect overall tectal activity patterns. A second difficulty is that we have been only able to record the types of cholinergic responses evoked from cells in the main cellular tectal layer. These responses may not be reflective of responses evoked in other tectal cell populations: specifically, the interneurons that are scattered throughout the superficial tectal layers or cells in the other, smaller cellular layers (layers 2 and 4; Székely and Lázár, 1976). Indeed, a previous observation that atropine suppressed visual responses in tectal layer 8, suggests that some cells in that layer may be excited by muscarinic receptor activation (Fite and Wang, 1986). Since interneurons in layer 8 are thought to be inhibitory in nature, their activation would be expected to decrease visual responses. Muscarinic receptor activation does in fact reduce light-evoked responses in the superficial layers of the rat superior colliculus (Kasser and Cheney, 1987).
Also necessary to help resolve results of cholinergic receptor activation at the cellular and system levels is a knowledge of whether different cholinergic pathways can selectively elicit different cholinergic responses. Although the nucleus isthmi supplies the tectum with over 90% of its ACh (Ricciuti and Gruberg, 1985), that ACh is provided by cholinergic projections from both the ipsilateral and contralateral isthmi. These projections have differing distributions in the superficial tectal lamina making it possible that different responses arise from the activation of each of these pathways (Desan et al., 1987; Udin et al., 1990; Gruberg et al., 1994). The non-repeatability of the nicotinic receptor-evoked response versus the relative stability of the muscarinic one, also raises the possibility that sustained cholinergic input might lead to an increasingly muscarinic receptor-dominated response as nicotinic receptors desensitize. The nucleus isthmi projections may also produce different effects from the cholinergic input to deep tectal layers, possibly provided by two tegmental nuclei that contribute a small cholinergic projection to the tectum (Marín and González, 1999).
Conclusions
Our work suggests that nicotinic and muscarinic receptors have both presynaptic and postsynaptic locations in the optic tectum. Nicotinic receptor activation functions to increase both glutamate and GABA release and depolarize tectal cells. On the other hand, the predominant effect of muscarinic receptor activation is tectal cell hyperpolarization although these receptors apparently also have a limited ability to affect neurotransmitter release. Since not all cells are directly affected by cholinergic receptor activation, the effects of the activation of nicotinic and muscarinic receptors on tectal activity at the system level remain to be elucidated.
Acknowledgments
This work was supported by a grant from the National Eye Institute (EY11913).
Abbreviations
- ACh
acetylcholine
- CNQX
6-cyano-7-nitroquinoxaline-2,3-dione
- EGTA
ethylene glycol-bis(2-aminoethylether)-N,N,N′,N-tetraacetic acid
- HEPES
HCO(3-)-free N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid
- sPSCs
spontaneous postsynaptic currents
- TTX
tetrodotoxin
References
- Aubert I, Cecyre D, Gauthier S, Quirion R. Characterization and autoradiographic distribution of [3H]AF-DX 384 binding to putative muscarinic M2 receptors in the rat brain. Eur J Pharmacol. 1992;217:173–184. doi: 10.1016/0014-2999(92)90843-s. [DOI] [PubMed] [Google Scholar]
- Bear MF, Singer W. Modulation of visual cortical plasticity by acetylcholine and noradrenaline. Nature. 1986;320:172–176. doi: 10.1038/320172a0. [DOI] [PubMed] [Google Scholar]
- Bickford ME, Ramcharan E, Godwin DW, Erisir A, Gnadt J, Sherman SM. Neurotransmitters contained in the subcortical extraretinal inputs to the monkey lateral geniculate nucleus. J Comp Neurol. 2000;424:701–717. doi: 10.1002/1096-9861(20000904)424:4<701::aid-cne11>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- Binns KE. The synaptic pharmacology underlying sensory processing in the superior colliculus. Prog Neurobiol. 1999;59:129–159. doi: 10.1016/s0301-0082(98)00099-9. [DOI] [PubMed] [Google Scholar]
- Binns KE, Salt TE. The functional influence of nicotinic cholinergic receptors on the visual responses of neurones in the superficial superior colliculus. Vis Neurosci. 2000;17:283–289. doi: 10.1017/s0952523800172116. [DOI] [PubMed] [Google Scholar]
- Birdsall NJ, Burgen AS, Hulme EC. The binding properties of muscarinic receptors in the brain of the frog (R. temporaria) Brain Res. 1980;184:385–393. doi: 10.1016/0006-8993(80)90807-0. [DOI] [PubMed] [Google Scholar]
- Butt CM, Pauly JR, Debski EA. Distribution and development of nicotinic acetylcholine receptor subtypes in the optic tectum of Rana pipiens. J Comp Neurol. 2000;423:603–618. doi: 10.1002/1096-9861(20000807)423:4<603::aid-cne6>3.0.co;2-f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butt CM, Pauly JR, Wilkins LH, Dwoskin LP, Debski EA. Pharmacology, distribution and development of muscarinic acetylcholine receptor subtypes in the optic tectum of Rana pipiens. Neuroscience. 2001;104:161–179. doi: 10.1016/s0306-4522(01)00048-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caine HS, Gruberg ER. Ablation of nucleus isthmi leads to loss of specific visually elicited behaviors in the frog Rana pipiens. Neurosci Lett. 1985;54:307–312. doi: 10.1016/s0304-3940(85)80096-3. [DOI] [PubMed] [Google Scholar]
- Clarke PB, Schwartz RD, Paul SM, Pert CB, Pert A. Nicotinic binding in rat brain: autoradiographic comparison of [3H]acetylcholine, [3H]nicotine, and [125I]-alpha-bungarotoxin. J Neurosci. 1985;5:1307–1315. doi: 10.1523/JNEUROSCI.05-05-01307.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debski EA, Liu Q, Chapman AM. Non-uniform distribution of cellular phenotypes in the optic tectum of the leopard frog. J Comp Neurol. 1995;360:671–684. doi: 10.1002/cne.903600411. [DOI] [PubMed] [Google Scholar]
- Desan PH, Gruberg ER, Grewell KM, Eckenstein F. Cholinergic innervation of the optic tectum in the frog Rana pipiens. Brain Res. 1987;413:344–349. doi: 10.1016/0006-8993(87)91026-2. [DOI] [PubMed] [Google Scholar]
- Edwards JA, Cline HT. Light-induced calcium influx into retinal axons is regulated by presynaptic nicotinic acetylcholine receptor activity in vivo. J Neurophysiol. 1999;81:895–907. doi: 10.1152/jn.1999.81.2.895. [DOI] [PubMed] [Google Scholar]
- Felder CC. Muscarinic acetylcholine receptors: signal transduction through multiple effectors. FASEB J. 1995;9:619–625. [PubMed] [Google Scholar]
- Fite KV, Wang SR. Microiontophoresis and single-unit analysis of cholinergic drugs in the optic tectum of the frog. Brain Behav Evol. 1986;28:198–206. doi: 10.1159/000118703. [DOI] [PubMed] [Google Scholar]
- Fucile S, Lax P, Eusebi F. Nicotine modulates the spontaneous synaptic activity in cultured embryonic rat spinal cord interneurons. J Neurosci Res. 2002;67:329–336. doi: 10.1002/jnr.10124. [DOI] [PubMed] [Google Scholar]
- Fujino K, Oertel D. Cholinergic modulation of stellate cells in the mammalian ventral cochlear nucleus. J Neurosci. 2001;21:7372–7383. doi: 10.1523/JNEUROSCI.21-18-07372.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel R, Volgyi B, Pollak E. Calretinin-immunoreactive elements in the retina and optic tectum of the frog, Rana esculenta. Brain Res. 1998;782:53–62. doi: 10.1016/s0006-8993(97)01261-4. [DOI] [PubMed] [Google Scholar]
- Gordon B, Mitchell B, Mohtadi K, Roth E, Tseng Y, Turk F. Lesions of nonvisual inputs affect plasticity, norepinephrine content, and acetylcholine content of visual cortex. J Neurophysiol. 1990;64:1851–1860. doi: 10.1152/jn.1990.64.6.1851. [DOI] [PubMed] [Google Scholar]
- Greuel JM, Luhmann HJ, Singer W. Pharmacological induction of use-dependent receptive field modifications in the visual cortex. Science. 1988;242:74–77. doi: 10.1126/science.2902687. [DOI] [PubMed] [Google Scholar]
- Groos G, Mason R, Meijer J. Electrical and pharmacological properties of the suprachiasmatic nuclei. Fed Proc. 1983;42:2790–2795. [PubMed] [Google Scholar]
- Gruberg ER, Hughes TE, Karten HJ. Synaptic interrelationships between the optic tectum and the ipsilateral nucleus isthmi in Rana pipiens. J Comp Neurol. 1994;15:353–364. doi: 10.1002/cne.903390305. [DOI] [PubMed] [Google Scholar]
- Gruberg ER, Wallace MT, Caine HS, Mote MI. Behavioral and physiological consequences of unilateral ablation of the nucleus isthmi in the leopard frog. Brain Behav Evol. 1991;37:92–103. doi: 10.1159/000114350. [DOI] [PubMed] [Google Scholar]
- Hohmann CF, Berger-Sweeney J. Cholinergic regulation of cortical development and plasticity: new twists to an old story. Perspect Dev Neurobiol. 1998;5:401–425. [PubMed] [Google Scholar]
- Hickmott PW, Constantine-Paton M. The contributions of NMDA, non-NMDA, and GABA receptors to postsynaptic responses in neurons of the optic tectum. J Neurosci. 1993;13:4339–4353. doi: 10.1523/JNEUROSCI.13-10-04339.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes TE. A light- and electron-microscopic investigation of the optic tectum of the frog, Rana pipiens. II: The neurons that give rise to the crossed tecto-bulbar pathway. Vis Neurosci. 1990;4:519–531. doi: 10.1017/s0952523800005733. [DOI] [PubMed] [Google Scholar]
- Kasser RJ, Cheney PD. DFP action on rat superior colliculus: localization and role of cholinergic receptors. Neurotoxicology. 1987;8:607–622. [PubMed] [Google Scholar]
- King WM, Schmidt JT. The long latency component of retinotectal transmission: enhancement by stimulation of nucleus isthmi or tectobulbar tract and block by nicotinic cholinergic antagonists. Neuroscience. 1991;40:701–712. doi: 10.1016/0306-4522(91)90006-a. [DOI] [PubMed] [Google Scholar]
- Kuras A, Gutmaniene N. N-cholinergic facilitation of glutamate release from an individual retinotectal fiber in frog. Vis Neurosci. 2001;18:549–558. doi: 10.1017/s0952523801184051. [DOI] [PubMed] [Google Scholar]
- Lauder JM, Schambra UB. Morphogenetic roles of acetylcholine. Environ Health Perspect. 1999 107:165–69. doi: 10.1289/ehp.99107s165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lázár G. Peptides in frog brain areas processing visual information. Microsc Res Tech. 2001;54:201–219. doi: 10.1002/jemt.1133. [DOI] [PubMed] [Google Scholar]
- Levey AI, Edmunds SM, Heilman CJ, Desmond TJ, Frey KA. Localization of muscarinic m3 receptor protein and M3 receptor binding in rat brain. Neuroscience. 1994;63:207–221. doi: 10.1016/0306-4522(94)90017-5. [DOI] [PubMed] [Google Scholar]
- Li Z, Fite KV. Distribution of GABA-like immunoreactive neurons and fibers in the central visual nuclei and retina of frog, Rana pipiens. Vis Neurosci. 1998;15:995–1006. doi: 10.1017/s0952523898155207. [DOI] [PubMed] [Google Scholar]
- Li Z, Fite KV. GABAergic visual pathways in the frog Rana pipiens. Vis Neurosci. 2001;18:457–464. doi: 10.1017/s0952523801183124. [DOI] [PubMed] [Google Scholar]
- Liu Q, Debski EA. Origins of serotonin-like immunoreactivity in the optic tectum of Rana pipiens. J Comp Neurol. 1995;352:280–296. doi: 10.1002/cne.903520210. [DOI] [PubMed] [Google Scholar]
- Malayev A, Debski EA. Serotonin modulates induced synaptic activity in the optic tectum of the frog. Brain Res. 1998;781:167–181. doi: 10.1016/s0006-8993(97)01230-4. [DOI] [PubMed] [Google Scholar]
- Marín O, González A. Origin of tectal cholinergic projections in amphibians: a combined study of choline acetyltransferase immunohistochemistry and retrograde transport of dextran amines. Vis Neurosci. 1999;16:271–283. doi: 10.1017/s0952523899162084. [DOI] [PubMed] [Google Scholar]
- Marks MJ, Smith KW, Collins AC. Differential agonist inhibition identifies multiple epibatidine binding sites in mouse brain. J Pharmacol Exp Ther. 1998;285:377–386. [PubMed] [Google Scholar]
- Nakagawa H, Kikkawa S, Matsumoto N. Synaptic connection patterns between frog retinal ganglion cells and tectal neurons revealed by whole-cell recordings in vivo. Brain Res. 1994;665:319–322. doi: 10.1016/0006-8993(94)91355-2. [DOI] [PubMed] [Google Scholar]
- Nistri A, Sivilotti L, Welsh DM. An electrophysiological study of the action of N-methyl-D-aspartate on excitatory synaptic transmission in the optic tectum of the frog in vitro. Neuropharmacology. 1990;29:681–687. doi: 10.1016/0028-3908(90)90030-u. [DOI] [PubMed] [Google Scholar]
- Nobili L, Sannita WG. Cholinergic modulation, visual function and Alzheimer's dementia. Vision Res. 1997;37:3559–3571. doi: 10.1016/S0042-6989(97)00076-X. [DOI] [PubMed] [Google Scholar]
- Perry DC, Kellar KJ. [3H]epibatidine labels nicotinic receptors in rat brain: an autoradiographic study. J Pharmacol Exp Ther. 1995;275:1030–1034. [PubMed] [Google Scholar]
- Prusky GT, Cynader MS. [3H]nicotine binding sites are associated with mammalian optic nerve terminals. Vis Neurosci. 1988;1:245–248. doi: 10.1017/s0952523800001504. [DOI] [PubMed] [Google Scholar]
- Ricciuti AJ, Gruberg ER. Nucleus isthmi provides most tectal choline acetyltransferase in the frog Rana pipiens. Brain Res. 1985;341:399–402. doi: 10.1016/0006-8993(85)91083-2. [DOI] [PubMed] [Google Scholar]
- Sargent PB, Pike SH, Nadel DB, Lindstrom JM. Nicotinic acetylcholine receptor-like molecules in the retina, retinotectal pathway, and optic tectum of the frog. J Neurosci. 1989;9:565–573. doi: 10.1523/JNEUROSCI.09-02-00565.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastry BV. Cholinergic systems and multiple cholinergic receptors in ocular tissues. J Ocul Pharmacol. 1985;1:201–226. doi: 10.1089/jop.1985.1.201. [DOI] [PubMed] [Google Scholar]
- Sherman SM, Koch C. The control of retinogeniculate transmission in the mammalian lateral geniculate nucleus. Exp Brain Res. 1986;63:1–20. doi: 10.1007/BF00235642. [DOI] [PubMed] [Google Scholar]
- Stuesse SL, Adli DS, Cruce WL. Immunohistochemical distribution of enkephalin, substance P, and somatostatin in the brainstem of the leopard frog, Rana pipiens. Microsc Res Tech. 2001;54:229–245. doi: 10.1002/jemt.1135. [DOI] [PubMed] [Google Scholar]
- Székely G, Lázár G. Cellular and synaptic architecture of optic tectum. In: Llinás R, Precht W, editors. Frog neurobiology. New York: Springer; 1976. pp. 407–434. [Google Scholar]
- Titmus MJ, Tsai HJ, Lima R, Udin SB. Effects of choline and other nicotinic agonists on the tectum of juvenile and adult Xenopus frogs: a patch-clamp study. Neuroscience. 1999;91:753–769. doi: 10.1016/s0306-4522(98)00625-3. [DOI] [PubMed] [Google Scholar]
- Tostivint H, Lihrmann I, Bucharles C, Vieau D, Coulouarn Y, Fournier A, Conlon JM, Vaudry H. Occurrence of two somatostatin variants in the frog brain: characterization of the cDNAs, distribution of the mRNAs, and receptor-binding affinities of the peptides. Proc Natl Acad Sci USA. 1996;93:12605–12610. doi: 10.1073/pnas.93.22.12605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu S, Butt CM, Pauly JR, Debski EA. Activity-dependent regulation of substance P expression and topographic map maintenance by a cholinergic pathway. J Neurosci. 2000;20:5346–5357. doi: 10.1523/JNEUROSCI.20-14-05346.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udin SB, Fisher MD, Norden JJ. Ultrastructure of the crossed isthmotectal projection in Xenopus frogs. J Comp Neurol. 1990;292:246–254. doi: 10.1002/cne.902920207. [DOI] [PubMed] [Google Scholar]
- Vogt KE, Regehr WG. Cholinergic modulation of excitatory synaptic transmission in the CA3 area of the hippocampus. J Neurosci. 2001;21:75–83. doi: 10.1523/JNEUROSCI.21-01-00075.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SR, Matsumoto N. Postsynaptic potentials and morphology of tectal cells responding to electrical stimulation of the bullfrog nucleus isthmi. Vis Neurosci. 1990;5:479–488. doi: 10.1017/s0952523800000602. [DOI] [PubMed] [Google Scholar]
- Wenzel B, Elsner N, Heinrich R. mAChRs in the grasshopper brain mediate excitation by activation of the AC/PKA and the PLC second-messenger pathways. J Neurophysiol. 2002;87:876–888. doi: 10.1152/jn.00312.2001. [DOI] [PubMed] [Google Scholar]
- Whiteaker P, McIntosh JM, Luo S, Collins AC, Marks MJ. 125I-alpha-conotoxin MII identifies a novel nicotinic acetylcholine receptor population in mouse brain. Mol Pharmacol. 2000;57:913–925. [PubMed] [Google Scholar]
- Xiao J, Wang Y, Wang SR. Effects of glutamatergic, cholinergic and GABAergic antagonists on tectal cells in toads. Neuroscience. 1999;90:1061–1067. doi: 10.1016/s0306-4522(98)00474-6. [DOI] [PubMed] [Google Scholar]
- Yeh JJ, Yasuda RP, Davila-Garcia MI, Xiao Y, Ebert S, Gupta T, Kellar KJ, Wolfe BB. Neuronal nicotinic acetylcholine receptor alpha3 subunit protein in rat brain and sympathetic ganglion measured using a subunit-specific antibody: regional and ontogenic expression. J Neurochem. 2001;77:336–346. doi: 10.1046/j.1471-4159.2001.t01-1-00259.x. [DOI] [PubMed] [Google Scholar]
- Zubieta JK, Frey KA. Autoradiographic mapping of M3 muscarinic receptors in the rat brain. J Pharmacol Exp Ther. 1993;264:415–422. [PubMed] [Google Scholar]