

Abstract
Folding and self-assembly of the 42-residue amyloid β-protein (Aβ) are linked to Alzheimer's disease (AD). The 21–30 region of Aβ, Aβ(21–30), is resistant to proteolysis and is believed to nucleate the folding of full-length Aβ. The conformational space accessible to the Aβ(21–30) peptide is investigated by using replica exchange molecular dynamics simulations in explicit solvent. Conformations belonging to the global free energy minimum (the “native” state) from simulation are in good agreement with reported NMR structures. These conformations possess a bend motif spanning the central residues V24–K28. This bend is stabilized by a network of hydrogen bonds involving the side chain of residue D23 and the amide hydrogens of adjacent residues G25, S26, N27, and K28, as well as by a salt bridge formed between side chains of K28 and E22. The non-native states of this peptide are compact and retain a native-like bend topology. The persistence of structure in the denatured state may account for the resistance of this peptide to protease degradation and aggregation, even at elevated temperatures.
Keywords: Alzheimer Aβ peptide, replica exchange molecular dynamics simulations, sampling of conformational space
Alzheimer's disease (AD), the leading cause of late-life dementia, belongs to a class of neurodegenerative disorders involving abnormal protein assembly. Brains of Alzheimer patients contain extracellular deposits, amyloid plaques, that comprise large amounts of fibrillar aggregates of the amyloid β-protein (Aβ). Aβ is a proteolytic product of the amyloid β-protein precursor (Aβ PP) and exists in vivo predominantly as a 40- or 42-residue peptide, Aβ40 or Aβ42, respectively. A variety of Aβ assemblies, including oligomers and fibrils, have been found to be potent neurotoxins (Hardy and Selkoe 2002; Temussi et al. 2003; Klein et al. 2004; Lazo et al. 2005b). The seminal step in the Aβ assembly process is monomer folding. Recently, we (Lazo et al. 2005a) investigated the folding of Aβ and identified, through limited proteolysis, a 10-residue region within both Aβ40 and Aβ42 that was resistant to degradation and was soluble in aqueous solution. This segment, AEDVGSNKGA, comprises residues 21–30 of the full-length peptide. Our solution NMR studies indicate that Aβ(21–30) forms a turn-like structure involving the five central residues of this peptide, V24–K28. Two conformational families were identified in which long-range Coulombic interactions occurred between K28 and either E22 (Family I) or D23 (Family II). In both families, the turn appeared to be stabilized by hydrophobic interactions between the side chains of V24 and K28. We postulated that this V24-to-K28 turn nucleates the folding of full-length Aβ. Formation of aggregates would involve the partial unfolding of this turn in the monomer, with possible population of a helical intermediate structure on route to fibrillization (Kirkitadze et al. 2001).
It is striking that a number of familial forms of AD and cerebral amyloid angiopathy (CAA) involve single amino acid substitutions in the 22–23 segment of Aβ. The mutations causing these diseases are referred to as the Dutch (E22Q), Italian (E22K), Arctic (E22G), and Iowa (D23N). The mutant Aβ peptides display altered biophysical and biological behaviors. For example, the Dutch peptide has significantly higher rates of fibril nucleation and elongation (Teplow et al. 1997), and deposits in cerebral vessels significantly more (Wisniewski and Frangione 1992), than does the wild-type peptide. The Arctic peptide displays an increased propensity to form protofibrils (Harper et al. 1997; Walsh et al. 1997) and may produce the first recognized “protofibril” form of AD (Nilsberth et al. 2001). We speculate that these mutations affect the stability of the turn structure in the 21–30 segment of Aβ and thereby lead to the folding of the Aβ monomer into a conformation with an increased propensity to form toxic assemblies.
Here, we probe the conformational space accessible to Aβ(21–30) in solution using replica exchange molecular dynamics (REMD) simulations. In contrast to ensemble-averaging NMR studies that can only provide information about the highest-populated state, REMD simulations deal with “single molecule” conformations and enable a complete exploration of conformational space. Our simulations show that the most populated conformations adopted by Aβ(21–30) at room temperature possess a stable bend at positions V24–K28. These structures are stabilized by hydrophobic and electrostatic interactions, including a salt bridge between K28 and E22 and a hydrogen bond network involving residues D23 and the central residues G25–K28. Less populated structures show similarity to the most populated structures in terms of overall dimension and the presence of a turn but differ in the details of the interactions stabilizing the turn region. The roles of electrostatic and hydrophobic interactions in stabilizing the Aβ(21–30) bend conformations and the implications for familial forms of AD and CAA are discussed.
Materials and methods
The amino acid sequence of Aβ(21–30) is AEDVGSNKGA. The peptide corresponds to the protease-resistant segment of low-molecular-weight preparations of Aβ40 and Aβ42 (Lazo et al. 2005a). Considering the protonation states of all titratable residues to be appropriate for neutral pH levels, including charged termini, we found that the peptide should carry −1 total electric charge. To render the simulated system electrically neutral, this charge was compensated for by an addition of one sodium ion. By interaction through the OPLS/AA all-atom empirical force-field (Kaminski et al. 2001), a total of 127 peptide atoms were solvated in a cubic box of 1677 TIP3P (Jorgensen et al. 1983) water molecules. The total number of atoms in the simulated system, including the sodium ion, was 5159. The size of the simulation box, 37.4 Å, was determined in short constant pressure simulations at T = 300 K, which were equilibrated at a physiological external pressure of 1 atm.
The simulations were carried out by using GROMACS software (Berendsen et al. 1995; Lindahl et al. 2001). During the simulations, covalent bonds of the water molecules were held constant by the SETTLE algorithm (Miyamoto and Kollman 1992). The bonds involving hydrogens were constrained according to the LINCS protocol (Hess et al. 1997). These approximations allowed a relatively long simulation time step of 2 fsec to be employed. Nonbonded Lennard-Jones interactions were tapered starting at 11 Å and extending to 12 Å cut-off. Neighbor lists for the nonbonded interactions were updated every 10 simulation steps. Electrostatic interactions were included by using the PME (Essmann et al. 1995) approach. In this approach, an 8 Å cut-off for the real space force contributions and 1.2 Å grid spacing for the Fourier transform in the reciprocal space were used. Fourth-order cubic interpolations were used for off-grid positions. The temperature was controlled by the Nose-Hoover algorithm (Nosé 1991) with a 0.05-psec time constant.
The replica-exchange algorithm (REMD) (Sugita and Okamoto 1999; Garcia and Onuchic 2003) was employed to improve equilibration. In total, 36 replicas of the original system were considered, at temperatures exponentially spaced between 300 K and 600 K. Exchanges of replicas at adjacent temperatures were attempted every 500 simulation steps. The same time interval was used to periodically save atomic coordinates. Acceptance ratios for replica swaps observed in our simulations ranged between 24% at low temperatures and 50% at higher temperatures. The simulations were started from a random extended conformation, the same for all replicas, and were run for a total of 24 nsec. The first 4 nsec of the simulation were treated as an equilibration phase. All the statistical analyses reported in this article were performed on the last 20 nsec of the trajectory. In our analysis of hydrogen bond formation, we assumed that a hydrogen bond exists for a triplet of donor, hydrogen and acceptor (D-H-A) atoms if the distance between hydrogen and acceptor is <2.5 Å and the angle formed by the triplet is <60°.
Results
Clustering of structures
Our replica exchange sampling procedure generated a very large number of conformations of the peptide Aβ(21–30). To sort through and analyze these structures, we grouped the entire pool of conformational states into clusters using the root mean square deviations (RMSDs) among conformations as a measure of structural similarity (Daura et al. 1999). Conformations with Cα RMSD > 1 Å over residues 22–28 were considered to be structurally distinct. The most representative structure (or “centroid”) of the most populated cluster (cluster C1) was used as the reference structure for subsequent principal component (PC) analyses (Jolliffe 1986; van der Spoel et al. 2004). The free energy surface at T = 300 K, calculated as a function of the first two PCs, is given in Figure 1A, while Figure 1B shows the location of the 10 most frequently visited conformational clusters (C1 to C10) in the PC space in decreasing order of population.
Figure 1.

Conformational ensembles from replica exchange molecular dynamics simulations of Aβ(21–30). (A) Free energy (in units of kT) as a function of the first two principal components PC1 and PC2 at T = 300 K. (B) Projections of the 10 most populated conformational clusters C1–C10 onto PC1–PC2 space. The most frequently visited cluster C1 is ∼30% populated.
Our results indicate that under ambient conditions, cluster C1 is ∼30% populated, cluster C2 is 10% populated, and the rest of the clusters C3–C10 are visited <5% of the time each. Comparison of the panels of Figure 1, A and B, reveals that the most populated conformational ensemble from the clustering analysis, (at approximate coordinates −0.1,−0.1), corresponds to the lowest free energy region in the PC1–PC2 map and that other significantly populated clusters correspond to local free energy minima. These results indicate that the chosen PCs are indeed useful order parameters as they correctly partition the conformational space into its most thermodynamically stable substates.
Lowest free energy minimum: Most populated cluster
We now turn to an analysis of the most frequently visited cluster, C1. The most representative structure (“centroid”) of this cluster is shown in Figure 2A, with Figure 2B illustrating the extent of conformational fluctuations present in this cluster.
Figure 2.

The most populated structure observed in replica exchange simulations of Aβ(21–30). (A) Centroid conformation of the cluster (cluster C1) and (B) a number of cluster members superimposed onto each other to demonstrate the extent of structural variability.
The structures of cluster C1 are seen to form a stable core in the middle of the sequence, stretching from E22–K28, with the three terminal residues subject to significant fluctuations. The core of Aβ(21–30) forms a bend at positions 24–28 (VGSNK), according to the DSSP (Definition of Secondary Structure of Proteins) classification (Kabsch and Sander 1983), while the rest of the sequence populates random coils. The centroid of cluster C1 is stabilized by a network of hydrogen bonds involving residue D23 and a number of the neighboring residues along the sequence. Specifically, strong hydrogen bonds are formed between the carboxylate oxygens of D23 (Oδ) and amide hydrogens of G25, S26, N27, and K28 with probabilities of 55%, 80%, 58%, and 48%, respectively. Slightly less populated bonds are observed between D23:Oδ and V24 (25%) and G29 (30%). The side chains of D23 and S26 are also seen to engage in a hydrogen bond that is populated ∼50% of the time. The role of salt bridges in stabilizing the structure is investigated by monitoring the distances separating the charged groups in the peptide. Distance distributions between atoms K28:Nζ and E22:Cδ (carbon atom of carboxylate group), shown in Figure 3, reveal two maxima at positions 3.4 Å and 6.3 Å. These maxima indicate two possible ways of forming a K28–E22 bridge, either through close interatomic contact or through a water-mediated contact. The close contact salt bridge is seen to be formed ∼56% of the time, and the solvent-mediated one ∼24% of the time, while the remaining 20% of the time the salt bridge is not present. In Figure 3 we also examine two other possible salt bridges formed in this peptide, involving the ion pairs K28–D23 and K28–C terminus. The distance distribution for K28–D23 is sharply peaked at ∼6.5 Å. However, a direct contact salt bridge is not formed between these two residues as the side chain of D23 is involved in an extensive hydrogen bond network and buried in the core of the peptide. Inspection of the C1 conformations did not reveal any bridging waters for K28–D23 residues. Hence the observed strong correlation between K28 and D23 must be due to an indirect interaction (possibly electrostatic in nature), mediated by other atoms of the peptide and the solvent. The second possible salt bridge shown in Figure 3, K28–C terminus, is mostly absent, with a formation probability of ∼10%. Half of the time that K28 is not forming a salt bridge with E22, it is forming one with the C terminus. Other possible choices of salt-bridging, N terminus–E22, N terminus–D23, and N terminus–C terminus, were not observed for the cluster C1.
Figure 3.

Formation of salt bridges in the most populated cluster C1 between the positively-charged side chain of K28 and a number of negatively-charged residues and the C terminus. Histograms are shown for distances between K28:Nζ and E22:Cδ, K28:Nζ and D23:Cγ, and K28:Nζ and A10:C. Data for the K28–E22 pair reveal both close contact and solvent-mediated salt bridges.
Free energy local minima
Conformations of the next most populated cluster, cluster C2, are shown in Figure 4. As was the case for cluster C1, the most structured region of C2 is located in the middle of the sequence, while the termini remain rather flexible. C2 shows great similarity to C1 over residues 2–7, with a bend present at V24 and G25. However, the hydrogen bond between D23:Oδ and K28:H is absent in C2, leading to a partial destabilization of the bend.
Figure 4.

The second most-populated structure observed in replica exchange simulations of Aβ(21–30). (A) Centroid conformation of the cluster (cluster C2) and (B) a number of cluster members superimposed onto each other to demonstrate the extent of structural variability.
This destabilization is offset by the presence of an additional hydrogen bond between G25:O and K28:H, involving two backbone atoms. The S26–K28 segment immediately following the bend in C2 adopts a 310-helix according to the DSSP classification. Despite sharing the bend motif, conformations of clusters C1 and C2 are quite dissimilar. The centroids of these clusters have a substantial Cα RMSD of ∼4.2 Å. Large mutual RMSDs on the same order of magnitude are found for the other, less significantly populated clusters (C3-C10). The K28–E22 salt bridge found in cluster C1 is absent in cluster C2. The only other populated salt bridge we observe for C2 is K28–C terminus, with a population of ∼14%. The disruption of salt bridges and a hydrogen bond energetically disfavor C2 conformations over their C1 counterparts, raising the free energy of these conformations (as can be seen in Fig. 1).
We note that if we consider the structures from all clusters combined, we find that the K28–E22 salt bridge is populated ∼24% of the time and the K28–D23 salt bridge ∼11% of the time.
Hydrophobic interactions
Our analysis in the previous sections revealed that the structured conformations of Aβ(21–30) are stabilized by a network of hydrogen bonds and formation of salt bridges. Here, we examine the contribution of hydrophobic interactions in stabilizing and shaping the conformational ensembles of this peptide.
Hydrophobic interactions are solvent mediated, and the magnitude and functional dependence of these interactions depend on the shape and the size of the solvated peptide. Hydrophobic interactions reduce the size of solvated objects and are proportional to the solvent-accessible surface area (SASA) for solutes of ∼20 Å in diameter or larger and to the molecular volume (MV) for solutes of smaller dimensions (Lum et al. 1999; Rein ten Wolde et al. 2001). Because our Aβ(21–30) peptide can fall into both size categories depending on whether it is folded or unfolded, we use both definitions to investigate the role of the hydrophobic effect. To probe the relative contributions of hydrophobic interactions to the free energy, we plot in Figure 5 projections of mean MV and SASA onto the first two PCs, PC1 and PC2. From Figure 5A, we see that the areas of low MV in the (PC1,PC2) space coincide with the location of the free energy minima observed in Figure 1A. In particular, both the MV map and the free energy map have their global minimum at (−0.1,−0.1), which corresponds to cluster C1. It is interesting to note that the hydrophobic and other interactions discussed in the previous sections have minima at conformations corresponding to cluster C1. As a consequence, cluster C1 becomes the global free energy minimum for the peptide—the “native state” in protein folding terminology. Our simulations provide direct support for the principle of minimal frustration (Bryngelson and Wolynes 1987) that states that conflicts among many different types of interactions operating in proteins are minimized for the native state.
Figure 5.
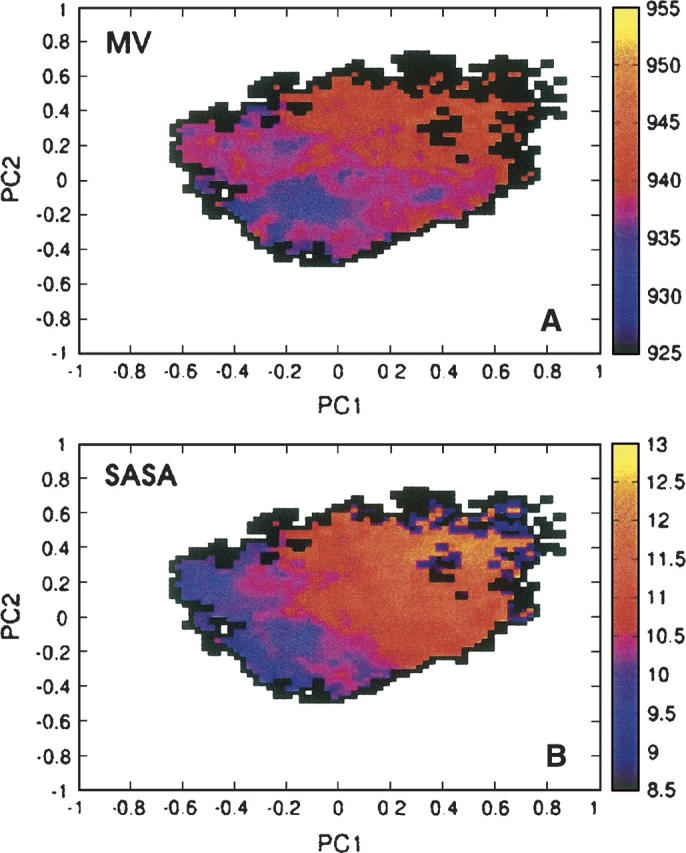
Mean molecular volume (in units of Å3) (A) and solvent-accessible surface area (in units of nm2) (B), as a function of the two first principal components PC1,PC2 generated for conformations sampled in the replica exchange simulations.
A similar conclusion regarding the importance of the hydrophobic interactions for stabilizing the conformations belonging to C1 can be reached from the SASA plot shown in Figure 5B. The SASA and MV results are similar for the less populated clusters, although the MV approach attributes a stronger stabilizing effect from the hydrophobic forces for clusters C4 and C7 (see Fig. 1B) than does the SASA approach. It is unclear which method is better suited for our peptide, but we note that the MV-based method provides a better correlation with the free energy map.
Comparison with experimentally derived NMR structures
In earlier work, we (Lazo et al. 2005a) reported a solution NMR study of the Aβ(21–30) peptide. Ratios of ROE intensities for αH − NH(i,i + 1)/αH − NH(i,I) resonances showed distinct patterns characteristic of turn structures for residues G25–N27. Structural refinement using ROE constraints obtained in the experiment were consistent with the existence of a turn at V24–N27. The observation of a turn-like structure at this location is supported by the simulations presented in this article. We limit our comparison to the highest populated cluster C1 because it corresponds to the lowest free energy minimum and because the other clusters are not sufficiently populated to give rise to a discernible NMR signal. It is important to note that a direct comparison between experiment and simulation is difficult. NMR provides an ensemble average measurement of a specific subset of structures that show relatively time-independent conformations. These structures may not have a one-to-one mapping with the native conformation, as there may be a significant fraction of non-native structures in this subset that maintain a certain local structure in the turn region. With this caveat in mind, we compared the interproton distances obtained from simulation for the conformations belonging to cluster C1 with those obtained experimentally (Lazo et al. 2005a) in Table 1.
Table 1.
Comparison of the interproton distances obtained for the highest-populated cluster through the <1/r6>–1/6 averaging with the weak, medium-, and long-range ROE constraints observed for the Aβ(21–30) peptide (Lazo et al. 2005a)

In total, four medium range (i,i + 2) constraints and one long range (i,i + 8) constraint were found in the NMR study. Of these, the 21α–23N and 28α–30N constraints are reproduced well by conformations of cluster C1. The constraint 24α–26N is insignificantly violated, by <0.5 Å, while the remaining medium-range 22α–24N and long-range 22α–30N constraints show a more significant violation by ∼1.5 Å. The NMR experiments revealed two families of conformations stabilized by hydrophobic interactions between the isopropyl and butyl side chains of V24 and K28, respectively. The families differed in the packing of the K28 side chain, with one family showing long-range Coulombic interactions between K28 and E22, the other between K28 and D23. Cα RMSD between the centroid of C1 and the first NMR family is 2.5 Å. Although this RMSD may appear large, the deviations are mostly due to the flexibility of the termini. This deviation drops to 0.7 Å when only the most structured residues belonging to the turn region (V24–N27) are considered. The flexibility of the termini found in our simulations is consistent with recent molecular dynamics simulations initiated from the NMR structure (Borreguero et al. 2005; Cruz et al. 2005). The deviation of C1 from the second NMR-derived structure is slightly higher, ∼3.1 Å. As in the NMR structures, we see Coulombic interactions between K28–E22 and K28–D23 in our simulated structures. Our simulations show both close contact and water-mediated geometries for the K28–E22 salt bridge and suggest an indirect interaction for K28–D23 mediated by atoms of the peptide or solvent.
“Denatured” state ensemble
An interesting experimental observation regarding the Aβ(21–30) peptide is that it does not aggregate under the experimental conditions employed (Lazo et al. 2005a). It is clear that the conformations belonging to the structured clusters at room temperature do not have the correct geometry (e.g., as would be the case for a β-hairpin or a β-strand) to self-assemble into fibrillar aggregates rich in β-structure (Serpell 2000). Aggregation would have to involve the partial unfolding of these conformations for fibrillization to occur. We now turn to an examination of the denatured states to determine whether any of these conformations could serve as possible starting points for aggregation. Considering the C1 conformations to constitute the “native” state, we construct a denatured state by selecting all conformations sampled in the simulations that differ by >1 Å in Cα RMSD from the centroid of C1 over residues E22–K28. We find that, on average, the denatured conformations, while structurally dissimilar from the native state, are of comparable dimension. The radius of gyration over all non-hydrogen atoms is 4.5 Å for the native state and 5.1 Å for the unfolded state, the MV of the denatured state is 937.9 Å3 compared with 933.5 Å3 in the native state, and the SASA of the unfolded states is 10.6 nm2, while that of the folded state is 10.0 nm2.
In Figure 6 we analyze the secondary structure of the two ensembles at T = 300 K. Figure 6A confirms that the folded state forms a stable bend at residues V24–K28 while the rest of the sequence is random coil. No other secondary structural elements are significantly populated. As can be seen in Figure 6B, the structured central segment resists denaturation. In the denatured state, the central residues V24–K28 have a significant bend structural content (as high as 80% for G25). Smaller amounts of random coil (up to 20%) and β-turn (up to 20%) are observed for these residues, while the amount of other secondary structure types is negligible. The terminal residues A21, E22, G29, and A30 mostly remain as random coils, as was the case for the native state ensemble. A picture of the denatured state of Aβ(21–30) emerges from Figure 6 in which the main secondary structural characteristics of the native state are maintained. The non-native states of this peptide are not random coils but rather preserve both the topology and, to a large extent, the dimensions of the native state. The resistance of Aβ(21–30) peptide to aggregation can hence be explained not only by its ability to form a tightly folded native state but also by its failure to significantly populate unfolded conformations that can potentially give rise to polymerization, such as β-hairpins or fully extended states.
Figure 6.

Secondary structure assignments for the individual amino acid residues of Aβ(21–30), determined by using the DSSP algorithm (Kabsch and Sander 1983). The assignments—(A-Helix) α–helix; (3-Helix) 310-helix; (Beta-S) β-sheet; (Beta-B) β–bridge; (Bend) bends; (Turns) β–turns; (Coil) all other conformations—are shown separately for the most populated cluster C1 (“native” state) (A), and all other conformations sampled in the simulations (“denatured” state) (B).
Discussion and Conclusions
Our REMD simulations of Aβ(21–30) reveal that this peptide adopts a structured turn in solution, in agreement with recently reported NMR studies (Lazo et al. 2005a). The most populated conformational cluster found in the simulations, which corresponds to the free energy minimum (the “native” state), possesses a stable bend over the central five residues of its amino acid sequence, VGSNK. The negatively charged side chain of D23 is involved in a network of hydrogen bonds with the backbone hydrogens of residues G25, S26, N27, and K28. As in the NMR structure, we find that the folded conformation is stabilized by a significantly populated salt bridge formed between oppositely charged side chains of residues E22 and K28. Hydrophobic interactions are seen to provide additional energetic stabilization to the folded state. Our simulations do not show a direct salt bridge between residues D23 and K28 for cluster 1, but they do indicate that these two residues are strongly correlated in space. We attribute this spatial correlation to indirect interactions (possibly electrostatic) between K28 and D23, which are mediated through other atoms of the peptide and the solvent. If we consider all structures from all clusters, then a salt bridge is formed 24% of the time between E22 and K28 and 11% of the time between D23 and K28. The centroid of the most populated conformational ensemble identified from simulation has a low (0.7 Å) RMSD over the turn region Cα atoms from the representative conformations derived from NMR, indicating structural similarity between computationally and experimentally obtained conformations.
The denatured state ensemble for this peptide possesses similar dimensions to the native state. In addition, the denatured state retained native-like elements of secondary structure, with the central residues of the unfolded state populating bend configurations similar to those seen for the native state. As a result, these less-structured Aβ(21–30) conformations had an overall native-like topology and did not significantly populate conformations that could serve as initiation sites for polymerization, (such as, e.g., extended coils or β-hairpins). This may explain the enhanced solubility of Aβ(21–30) in water (Lazo et al. 2005a). In addition, we note that because denatured conformations are mostly determined by local interactions, conclusions drawn about the denatured state for this fragment may also be applicable to full-length Aβ peptides.
The presence of structured turn or bend motifs in the central part of the sequence is not unique to Aβ(21–30) and has been observed for a number of other monomeric Aβ peptides in aqueous solutions (Zhang et al. 2000; Riek et al. 2001; Hou et al. 2004). NMR-derived structures of Aβ(10–35) by Zhang et al. (2000) contain a turn over the central EDVG region, which appears to be stabilized by a K28–D23 salt bridge. NMR experiments (Riek et al. 2001) found that residues 20–24 of the Met-oxidized form of the 40- and 42-residue-long amyloid β peptides, [Met35(O)] Aβ40 and [Met35(O)] Aβ42, form a stable FAEDV α-helical turn. In addition, another NMR study on Aβ40/42 and [Met35(O)] Aβ40/42 peptides (Hou et al. 2004) has found a structured turn/bend motif stretching over residues 20–26. These experiments indicate that the formation of a turn/bend structure in a region spanning residues 20–30 is a feature common to many monomeric Aβ peptides. Local interactions present in the 21–30 fragment of Aβ appear to be sufficient to induce a substantial propensity for forming structured turns or bends, irrespective of whether this segment belongs to a fragment or to the full-length Aβ peptide.
It is interesting to note that a turn involving residues 21–30 may also be present in early prefibrillar intermediates of Aβ40 (Chimon and Ishii, 2005) and in fibrils of Aβ40 (Petkova et al. 2002), Aβ42, and Aβ(10–35) (Antzutkin et al. 2002). A recent structural model of Aβ40 fibrils based on solid-state NMR studies (Petkova et al. 2002) suggests that residues 25–29 contain a bend that brings the two adjacent β-sheets in contact through side-chain–side-chain interactions. This structural motif also appears to be present in fibrils of Aβ42 and Aβ(10–35) (Antzutkin et al. 2002) peptides. A recent solid-state NMR study by Riek and coworkers (Lührs et al. 2005) suggests that [Met35(O)] Aβ42 protofibrils present a β-strand-loop β-strand motif, with the loop spanning residues 27–30. Another model of amyloid fibrils derived from proline scanning methods for Aβ40 (Guo et al. 2004; Williams et al. 2004) and for Aβ42 (Morimoto et al. 2004) predicts that residues E22 and D23 occupy turn positions between two structured β-strand motifs. A turn formed within the 21–30 segment of the full-length Aβ peptide, after cleavage from the amyloid β-protein precursor (Aβ PP), may be required for fibrillation to proceed. We emphasize that the presence of a turn alone is not sufficient to induce aggregation. Indeed, the 21–30 fragment cannot aggregate because this motif, present in both the native and non-native states, does not have the correct geometrical properties to self-assemble. Additional interactions present in the full-length Aβ40/42 peptide (e.g., those involving the central hydrophobic core or the C terminus), along with possible rearrangements of the turn region, are required in order for fibrillization to proceed.
It is also possible that the presence or absence of the bend may affect the kinetics of fibril assembly. The degree of similarity between the bend observed for the Aβ(21–30) monomer and the one formed in Aβ40/42 fibrils is not known at present as the structure of the fibrils has not been solved. Complicating matters is the fact that different preparations of amyloid fibrils seem to lead to different structural characteristics of the 25–29 segment (Petkova et al. 2005). We have suggested that the monomer nucleus (the turn region) rearranges during the fibril formation process (Lazo et al. 2005a). If so, then Aβ fibril formation should be manipulable through rational amino acid substitutions in the 21–30 region within both the fragment and the full-length peptide. Such mutations could affect the formation of the turn region. As noted in our earlier work (Lazo et al. 2005a), a particularly interesting issue that arises in this regard is whether the familial forms of AD resulting from mutations at E22 or D23 can be explained in terms of conformational preferences of the monomeric Aβ peptides. Our recent experimental data obtained by limited proteolysis and solution-state NMR (M.A. Grant, N.D. Lazo, M.C. Condron, A.C. Rigby, and D.B. Teplow, in prep.) indicate that substitutions in the turn region identified in this work, including those mutations associated with early-onset AD and CAA (such as the Arctic [E22G], Dutch [E22Q], and Iowa [D23N] mutations), affect the stability of the Val24–Lys28 turn. Our simulations suggest that mutations at D23 may lead to a change in stability of the bend region because the D23 side chain is involved in a strong network of intramolecular hydrogen bonds involving this bend region. This hypothesis can be tested through investigations of the conformations populated by the Iowa form of the Aβ peptide (Grabowski et al. 2001). In this mutant, N is substituted for D at position 23, leading to enhanced fibrillation and increased toxicity in mutants of both Aβ40 (Van Nostrand et al. 2001) and Aβ42 (Murakami et al. 2003). The study of familial mutants of residues 22 and 23 is the subject of our ongoing theoretical and experimental work.
Acknowledgments
We thank M. Griffin for helpful discussions. J.E.S. acknowledges support from the NSF Career Award no. 0133504, the A.P. Sloan Foundation, and the David and Lucile Packard foundation. Computational resources of the California NanoSystems Institute (CNSI) obtained through a NSF grant (CHE-0321368) are also acknowledged. Part of the simulations were performed by using the NSF TERAGRID facilities, through the allocation grant no. MCA055027. M.T.B. acknowledges support from NSF grants CHE-0140215 and CHE-0503728. D.B.T. acknowledges support from the NIH (grants NS38328, NS44147, and AG18921) and from the Foundation for Neurologic Diseases.
Footnotes
Reprint requests to: Joan-Emma Shea, Department of Chemistry and Biochemistry, University of California at Santa Barbara, Santa Barbara, CA 93106; e-mail: shea@chem.ucsb.edu; fax: (805) 893-4120.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062076806.
References
- Antzutkin O.N., Leapman R.D., Balbach J.J., Tycko R. 2002. Supramolecular structural constraints on Alzheimer's β-amyloid fibrils from electron microscopy and solid-state nuclear magnetic resonance Biochemistry 41 15436–15450. [DOI] [PubMed] [Google Scholar]
- Berendsen H.J.C., van der Spoel D., van Drunen R. 1995. GROMACS: A message-passing parallel molecular dynamics implementation Comput. Phys. Commun. 91 43–56. [Google Scholar]
- Borreguero J.M., Urbanc B., Lazo N.D., Buldyrev S.V., Teplow D.B., Stanley H.E. 2005. Folding events in the 21-30 region of amyloid β-protein (Aβ) studied in silico Prot. Natl. Acad. Sci. 102 6015–6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryngelson J.D. and Wolynes P.G. 1987. Spin glasses and the statistical mechanics of protein folding Proc. Natl. Acad. Sci. 84 7524–7528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chimon S. and Ishii Y. 2005. Capturing intermediate structures of Alzheimer's β-amyloid, Aβ (1-40), by solid-state NMR spectroscopy J. Am. Chem. Soc. 127 13472–13473. [DOI] [PubMed] [Google Scholar]
- Cruz L., Urbanc B., Borreguero J.M., Lazo N.D., Teplow D.B., Stanley H.E. 2005. Solvent and mutation effects on the nucleation of amyloid β-protein folding Proc. Natl. Acad. Sci. 102 18801–18806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daura X., Gademann K., Jaun B., Seebach D., van Gunsteren W.F., Mark A.E. 1999. Peptide folding: When simulation meets experiment Angew. Chem. Int. Ed. Engl. 38 236–240. [Google Scholar]
- Essmann U., Perera L., Berkowitz M.L., Darden T., Lee H., Pedersen L.G. 1995. A smooth particle mesh Ewald method J. Chem. Phys. 103 8577–8593. [Google Scholar]
- Garcia A.E. and Onuchic J.N. 2003. Folding a protein in a computer: An atomic description of the folding/unfolding of protein A Proc. Natl. Acad. Sci. 100 13898–13903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabowski T.J., Cho H.S., Vonsattel J.P.G., Rebeck G.W., Greenberg S.M. 2001. Novel amyloid precursor protein mutation in an Iowa family with dementia and severe cerebral amyloid angiopathy Ann. Neurol. 49 697–705. [DOI] [PubMed] [Google Scholar]
- Guo J.T., Wetzel R., Ying X. 2004. Molecular modeling of the core of Aβ amyloid fibrils Proteins 57 357–364. [DOI] [PubMed] [Google Scholar]
- Hardy J. and Selkoe D.J. 2002. The amyloid hypothesis of Alzheimer's disease: Progress and problems on the road to therapeutics Science 297 353–357. [DOI] [PubMed] [Google Scholar]
- Harper J.D., Wong S.S., Lieber C.M., Lansbury P.T. Jr. 1997. Observation of metastable Aβ amyloid protofibrils by atomic force microscopy Chem. Biol. 4 119–125. [DOI] [PubMed] [Google Scholar]
- Hess B., Bekker H., Berendsen H.J.C., Fraaije J.G.E.M. 1997. LINCS: A linear constraint solver for molecular simulations J. Comput. Chem. 18 1463–1472. [Google Scholar]
- Hou L., Shao H., Zhang Y., Li H., Menon N.K., Neuhaus E.B., Brewer J.M., ByeonaLxRay D.G., Vitek M.P.et al. 2004. Solution NMR studies of the Aβ (1-40) and Aβ (1-42) peptides establish that the MET35 oxidation state affects the mechanism of amyloid formation J. Am. Chem. Soc. 126 1992–2005. [DOI] [PubMed] [Google Scholar]
- Jolliffe I.T. In Principal component analysis . 1986. Springer-Verlag, New York.
- Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L. 1983. Comparison of simple potential functions for simulating liquid water J. Chem. Phys. 79 926–935. [Google Scholar]
- Kabsch W. and Sander C. 1983. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features Biopolymers 22 2577–2637. [DOI] [PubMed] [Google Scholar]
- Kaminski G.A., Friesner R.A., Tirado-Rives J., Jorgensen W.L. 2001. Evaluation and reparametrization of the opls-aa force field for proteins via comparison with accurate quantum chemical calculations on peptides J. Phys. Chem. B 105 6474–6487. [Google Scholar]
- Kirkitadze M., Condron M.M., Teplow D.B. 2001. Identification and characterization of key kinetic intermediates in amyloid β-protein fibrillogenesis J. Mol. Biol. 312 1103–1119. [DOI] [PubMed] [Google Scholar]
- Klein W.L., Stine Jr. W.B., Teplow D.B. 2004. Small assemblies of unmodified amyloid β-protein are the proximate neurotoxin in Alzheimer's disease Neurobiol. Aging 25 569–580. [DOI] [PubMed] [Google Scholar]
- Lazo N.D., Grant M.A., Condron M.C., Rigby A.C., Teplow D.B. 2005a. On the nucleation of amyloid β-protein monomer folding Protein Sci. 14 1581–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazo N.D., Maji S.K., Fradinger E.A., Bitan G., Teplow D.B. 2005b. The amyloid β-protein In Amyloid proteins: The β-sheet conformation and disease pp. 385–491. Wiley-VCH Verlag GmbH, Weinheim, Germany Vol. 2. [Google Scholar]
- Lindahl E., Hess B., van der Spoel D. 2001. GROMACS 3.0: A package for molecular simulation and trajectory analysis J. Mol. Model 7 306–317. [Google Scholar]
- Lührs T., Ritter C., Adrian M., Riek-Loher D., Bohrmann B., Dobeli H., Schubert D., Riek R. 2005. Three-dimensional structure of Alzheimer's amyloid-β (1-42) fibrils Proc. Natl. Acad. Sci. 102 17342–17347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum K., Chandler D., Weeks J.D. 1999. Hydrophobicity at small and large length scales J. Phys. Chem. B 103 4570–4577. [Google Scholar]
- Miyamoto S. and Kollman P.A. 1992. SETTLE: An analytical version of the SHAKE and RATTLE algorithms for rigid water models J. Comput. Chem. 13 952–962. [Google Scholar]
- Morimoto A., Irie K., Murakami K., Masuda Y., Ohigashi H., Nagao M., Fukuda H., Shimizu T., Shirasawa T. 2004. Analysis of the secondary structure of β-amyloid (Aβ 42) fibrils by systematic proline replacement J. Biol. Chem. 279 52781–52788. [DOI] [PubMed] [Google Scholar]
- Murakami K., Irie K., Morimoto A., Ohigashi H., Shindo M., Nagao M., Shimizu T., Shirasawa T. 2003. Neurotoxicity and physicochemical properties of Aβ mutant peptides from cerebral amyloid angiopathy: Implication for the pathogenesis of cerebral amyloid angiopathy and Alzheimer's disease J. Biol. Chem. 278 46179–46187. [DOI] [PubMed] [Google Scholar]
- Nilsberth C., Westlind-Danielsson A., Eckman C.B., Condron M.M., Axelman K., Forsell C., Stenh C., Luthman J., Teplow D.B., Younkin S.G.et al. 2001. The ‘arctic’ APP mutation (E693G) causes Alzheimer's disease by enhanced Aβ protofibril formation Nat. Neurosci. 4 887–893. [DOI] [PubMed] [Google Scholar]
- Nosé S. 1991. Constant temperature molecular dynamics methods Prog. Theor. Phys. 103 1–46. [Google Scholar]
- Petkova A.T., Ishii Y., Balbach J.J., Antzutkin O.N., Leapman R.D., Delaglio F., Tycko R. 2002. A structural model for Alzheimer's β-amyloid fibrils based on experimental constraints from solid state NMR Proc. Natl. Acad. Sci. 99 16742–16747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkova A.T., Leapman R.D., Guo Z.H., Yau W.M., Mattson M.P., Tycko R. 2005. Self-propagating, molecular-level polymorphism in Alzheimer's β-amyloid fibrils Science 307 262–265. [DOI] [PubMed] [Google Scholar]
- Rein ten Wolde P., Sun S.X., Chandler D. 2001. Model of a fluid at small and large length scales and the hydrophobic effect Phys. Rev. E 65 011201-1–011201-9. [DOI] [PubMed] [Google Scholar]
- Riek R., Güntert P., Döbeli H., Wipf B., Wütrich K. 2001. NMR studies in aqueous solution fail to identify significant conformational differences between the monomeric forms of two Alzheimer peptides with widely different plaque-competence, Aβ (1-40)ox and Aβ (1-42)ox Eur. J. Biochem 268 5930–5936. [DOI] [PubMed] [Google Scholar]
- Serpell L.C. 2000. Alzheimer's amyloid fibrils: Structure and assembly Biochim. Biophys. Acta 1502 16–30. [DOI] [PubMed] [Google Scholar]
- Sugita Y. and Okamoto Y. 1999. Replica-exchange molecular dynamics method for protein folding Chem. Phys. Lett. 314 141–151. [Google Scholar]
- Temussi P.A., Masino L., Pastore A. 2003. From Alzheimer to Huntington: Why is a structural understanding so difficult? EMBO J. 22 355–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teplow D.B., Lomakin A., Benedek G.B., Kirschner D.A., Walsh D.M. 1997. Effects of β-protein mutations on amyloid fibril nucleation and elongation In Alzheimer's disease: Biology, diagnosis and therapeutics, pp. 311–319. John Wiley & Sons, Chichester, UK.
- van der Spoel D., Lindahl E., Hess B., van Buuren A.R., Apol E., Meulenhoff P.J., Tieleman D.P., Sijbers A.L.T.M., Feenstra K.A., van Drunen R.et al. 2004. GROMACS user manual version 3.2 www.gromacs.org.
- Van Nostrand W.E., Melchor J.P., Cho H.S., Greenberg S.M., Rebeck G.W. 2001. Pathogenic effects of D23N Iowa mutant amyloid β-protein J. Biol. Chem. 276 32860–32866. [DOI] [PubMed] [Google Scholar]
- Walsh D.M., Lomakin A., Benedek G.B., Condron M.M., Teplow D.B. 1997. Amyloid β-protein fibrillogenesis. detection of a protofibrillar intermediate J. Biol. Chem. 272 22364–22372. [DOI] [PubMed] [Google Scholar]
- Williams A.D., Portelius E., Kheterpal I., Guo J.T., Cook K.D., Xu Y., Wetzel R. 2004. Mapping Aβ amyloid fibril secondary structure using scanning proline mutagenesis J. Mol. Biol. 335 833–842. [DOI] [PubMed] [Google Scholar]
- Wisniewski T. and Frangione B. 1992. Molecular biology of Alzheimer's amyloid Dutch variant Mol. Neurobiol. 6 75–86. [DOI] [PubMed] [Google Scholar]
- Zhang S., Iwata K., Lachenmann M.J., Peng J.W., Li S., Stimson E.R., Lu Y.-A., Felix A.M., Maggio J.E., Lee J.P. 2000. The Alzheimer's peptide: Aβ adopts a collapsed coil structure in water J. Struct. Biol. 130 130–141. [DOI] [PubMed] [Google Scholar]