

Abstract
High-level expression of G-protein-coupled receptors (GPCRs) in functional form is required for structure–function studies. The main goal of the present work was to improve expression levels of β2-adrenergic receptor (β2-AR) so that biophysical studies involving EPR, NMR, and crystallography can be pursued. Toward this objective, the total synthesis of a codon-optimized hamster β2-AR gene suitable for high-level expression in mammalian systems has been accomplished. Transient expression of the gene in COS-1 cells resulted in 18 ± 3 pmol β2-AR/mg of membrane protein, as measured by saturation binding assay using the β2-AR antagonist [3H] dihydroalprenolol. Previously, we reported the development of an HEK293S tetracycline-inducible system for high-level expression of rhodopsin. Here, we describe construction of β2-AR stable cell lines using the HEK293S-TetR-inducible system, which, after induction, express wild-type β2-AR at levels of 220 ± 40 pmol/mg of membrane protein corresponding to 50 ± 8 μg/15-cm plate. This level of expression is the highest reported so far for any wild-type GPCR, other than rhodopsin. The yield of functional receptor using the single-step affinity purification is 12 ± 3 μg/15-cm plate. This level of expression now makes it feasible to pursue structure–function studies using EPR. Furthermore, scale-up of β2-AR expression using suspension cultures in a bioreactor should now allow production of enough β2-AR for the application of biophysical techniques such as NMR spectroscopy and crystallography.
Keywords: GPCR, synthetic gene, β-adrenergic receptor, expression, mammalian cell lines
G-protein-coupled receptors (GPCRs) sense biological signals and represent the largest known family of cell surface receptors (Takeda et al. 2002; Schoneberg et al. 2004). β2-adrenergic receptor (β2-AR) is a prototypic and a well-characterized member of the GPCR family. However, there is little information available on the structure of this receptor. This is in part due to limitation in the amounts of the protein available for structural studies. GPCRs, with the exception of the light-sensitive opsins found in bacteria and in the retinae of higher organisms, occur at very low levels (Khorana 1992). Further, there are very few expression systems, except for rhodopsin, that yield reproducible levels of GPCRs. In addition, following their expression in bacterial, yeast, or insect cells, GPCRs often undergo protein misfolding and aggregation (Tate and Grisshammer 1996; Stanasila et al. 1998).
Codon optimization provides a means to increase the expression of genes. However, reports on the use of this approach in the GPCR field are limited (Mirzabekov et al. 1999; Babcock et al. 2001; Farrens et al. 2002). Recently, Joseph Sodroski's group (Mirzabekov et al. 1999; Babcock et al. 2001) showed that optimization of codon usage for two GPCRs, the HIV-1 coreceptors CCR5 and CXCR4, increased their expression in mammalian cells. Similarly, the expression of a 7TM receptor encoded by the U51 open reading frame from human herpesviruses HHV-6 and HHV-7 increased 10- to 100-fold when the codon-optimized gene was expressed in mammalian cells (Bradel-Tretheway et al. 2003).
Like most GPCRs, β2-AR is expressed at very low levels in mammalian cells. This is in part due to the presence, in all mammalian β2-AR mRNAs, of a small open reading frame in the 5′-untranslated region (5′-UTR) (Kobilka et al. 1987; Parola and Kobilka 1994), and a 3′-UTR sequence (Subramaniam et al. 2004), both of which are known to repress translation of the mRNA. Additionally, translation of β2-AR mRNA could be reduced by the presence of suboptimal codons. Therefore, removal of sequences in the 5′- and 3′-UTR and codon optimization would be necessary for optimal expression of β2-AR in mammalian cells.
High-level expression of membrane proteins is required for studies of their structure and function, as demonstrated in previous studies of rhodopsin and bacteriorhodopsin (Khorana 1992; Farrens et al. 1996; Klein-Seetharaman et al. 2002; Patel et al. 2005). Previously, using the yeast and baculovirus systems, expression levels of wild-type β2-AR in the range of 20–120 pmol/mg of membrane protein have been reported by different groups (Parker et al. 1991; Sizmann et al. 1996; Weiss et al. 1998; for a recent review, see Sarramegna et al. 2003). The main goal of the present work is to improve the expression level of β2-AR, so that NMR spectroscopic studies and crystallization attempts can be pursued. Toward this objective, the total synthesis of a codon-optimized hamster β2-AR gene suitable for expression in mammalian systems has been accomplished. Transient expression of the gene in COS-1 cells yields 18 ± 3 pmol β2-AR/mg of membrane protein, as measured by saturation binding assay using the β2-AR antagonist, [3H] dihydroalprenolol. For biophysical studies, tetracycline-inducible HEK293S stable cell lines, which express the gene to give 220 ± 40 pmol β2-AR/mg of membrane protein, have been constructed. This level of expression is the highest reported so far for any wild-type GPCR, other than rhodopsin.
Results and Discussion
Synthesis and expression of hamster β2-AR
The synthetic DNA corresponding to the β2-AR gene consisted of 1310 bp, of which 1278 bp represents the open reading frame (Fig. 1). To facilitate future subcloning and mutagenesis, by design, the synthetic gene has evenly spaced unique restriction sites. The other salient features of the codon-optimized and epitope-tagged β2-AR gene include: (1) a Kozak consensus sequence (GCCACCATGG) 5′ to the ATG start codon; (2) a bovine rhodopsin octapeptide tag (ETSQVAPA) immediately 5′ to the natural stop codon of β2-AR; (3) an increase in the GC content of the gene from 51% to 57%, and restriction sites, for EcoRI at the 5′ end and NotI site at the 3′ end. The use of repetitive codons for any particular amino acid was avoided wherever possible to ensure that the total tRNA pool of the cell was not adversely affected (Young and Dong 2004). Transient expression of this synthetic gene in COS-1 cells yielded 18 ± 3 pmol β2-AR/mg of membrane protein. While this level of expression is adequate for structure–function studies using membrane preparations, milligram quantities of the receptor will be required for biophysical studies. Therefore, construction of β2-AR stable cell lines using the HEK293S-TetR inducible system was explored. Using these cell lines, after induction, β2-AR was expressed at levels of 220 ± 40 pmol/mg of membrane protein, which is 10–15-fold higher than the level of protein observed following transient expression of β2-AR in COS-1 cells (Table 1).
Figure 1.

Nucleotide sequence of the codon-optimized hamster β2-AR gene, and the corresponding amino acid sequence. The locations of the restriction sites in the gene are shown above the DNA sequence. The synthetic gene contains at its C terminus the rhodopsin C-terminal octapeptide sequence (underlined) to facilitate detection and purification of the protein using the monoclonal antibody rho-1D4.
Table 1.
Ligand binding properties of synthetic β2-AR expressed in COS-1 cells, HEK293S cells, and HEK293S-TetR stable cell line

Immunoblot analysis of β2-AR expressed in the HEK293S (GnTI−) cell line
Immunoblot analysis showed that β2-AR expressed in HEK293S cells consists of two major protein bands with an apparent molecular mass in the range of 47–60 kDa (Fig. 2, lane A). Presumably, the two protein bands result from heterogeneous N-glycosylation of the two asparagine residues in the N terminus. Previously, it has been shown that removal of these N-glycosylation sites at the N terminus of hamster β2-AR did not affect ligand binding (Dixon et al. 1987; Rands et al. 1990). While heterogeneous glycosylation would not interfere with NMR experiments, it could cause potential problems in crystallization. Therefore, we transiently expressed β2-AR in the HEK293S (GnT−) cell line, previously generated in this laboratory, that is resistant to ricin as a consequence of loss of N-acetylglucosamine transferase 1 (GnT−) activity (Reeves et al. 2002a). β2-AR, as expressed in this cell line, shows homogenous glycosylation and migrates predominantly as a single band with an apparent molecular mass of ∼50 kDa, as judged by immunoblotting (Fig. 2, lane B). Construction of a tetracycline-inducible GnT− cell line containing the codon-optimized gene is under progress.
Figure 2.
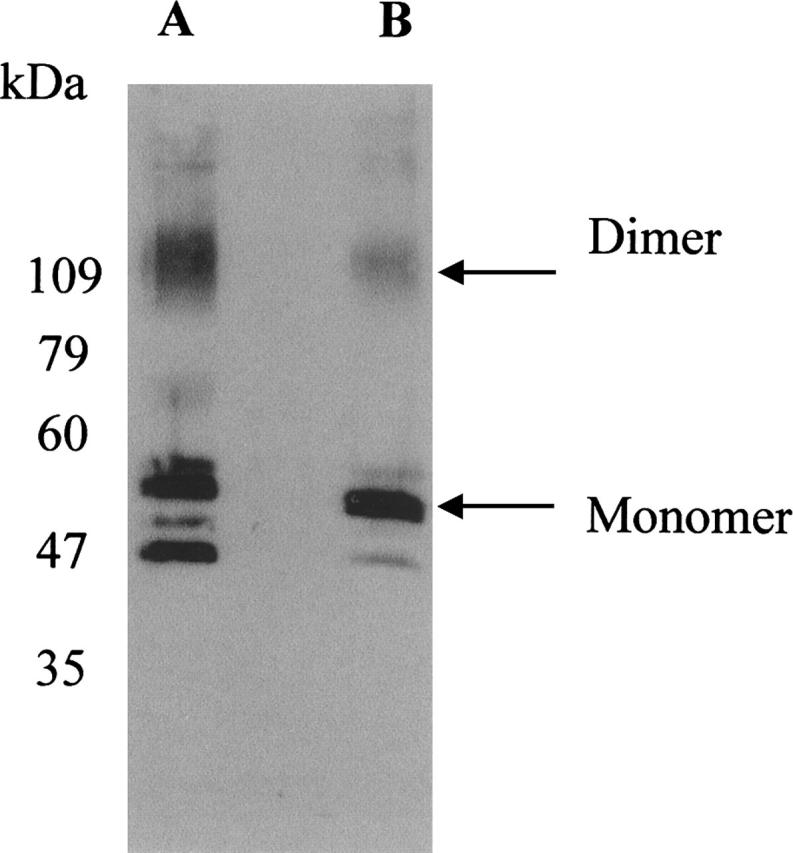
Immunoblot analysis of β2-AR using the monoclonal antibody rho-1D4 (the epitope tag for this antibody was added to the C-terminal tail of β2-AR gene). β2-AR transiently expressed in HEK293S (lane A). By using a HEK293S (GnT−) cell line defective in N-acetylglucosamine transferase I, β2-AR was expressed with restricted and homogeneous N-glycosylation (lane B). Mobility of molecular weight standards is indicated on the left of the gel.
Ligand binding characteristics of β2-AR expressed in HEK293S-TetR cells
Figure 3 shows binding of the β-adrenergic antagonist [3H] DHA to β2-AR expressed in COS-1 and HEK293S-TetR cells. The affinity of [3H] DHA for the receptor expressed in these two cell lines is comparable, with Kd values in the range of 2–5 nM (Table 1). In general, the Kd values are similar to those previously reported for β2-AR (Benovic et al. 1984; Parker et al. 1991). In addition, β2-AR, as expressed in the HEK293S-TetR cell line and in COS-1 cells, showed similar affinities toward various agonists such as salbutamol, formoterol, and isoproteronol (data not shown).
Figure 3.
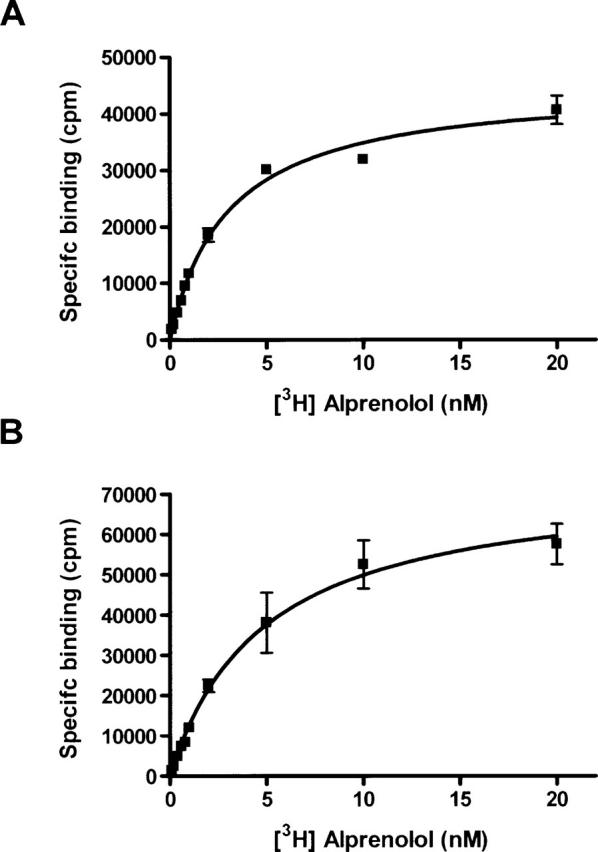
Saturation binding assays for membrane bound β2-AR from COS-1 (A), and HEK293S-TetR stable cell line (B) were performed using [3H] dihydroalprenolol as the radioligand (TRK 649, Amersham). Saturation assays were performed using 0.1–20 nM [3H] DHA, and 2–20-μg membrane protein/well. Binding of [3H] DHA in the presence of 10 μM alprenolol was used as a measure of nonspecific binding. Specific binding was derived by subtracting nonspecific binding from the total binding. Saturation binding data were obtained from a minimum of two independent determinations done in triplicate, and are shown as mean ± SE. Nonspecific binding was always <10% of total binding. Data were analyzed by nonlinear regression using PRISM software (Graph Pad Software).
Solubilization and purification of β2-AR expressed in HEK293S-TetR-inducible stable cell lines
HEK293S-TetR cells expressing the codon-optimized β2-AR gene were grown as monolayers in 15-cm dishes. When the cell density reached 3 × 106 cells per dish (95% confluence), the medium was replaced with fresh DMEM F-12 medium containing 1 μg/mL tetracycline and 7.5 mM sodium butyrate. After induction for 44–48 h, the cells were collected by centrifugation and the cell pellet was snap frozen and stored at −80°C. For receptor purification, membranes were prepared from the cell pellet and solubilized using 1% DM. As shown in Table 2, following solubilization of the receptor from the HEK293S membrane preparations, the yield as determined by ligand binding assay was found to be ∼70%. In the next step, which involves rho-1D4-Sepharose affinity purification, the concentration of DM was reduced to 0.05% in order to minimize the detrimental effects of high concentrations of DM in the purified samples. The receptor purified by rho-1D4-Sepharose affinity was found to be >90% pure, as determined by 10% SDS-PAGE (Fig. 4). The receptor produced in HEK293S-TetR was glycosylated and migrated as two bands on the SDS-PAGE, with the minor band around 47 kDa and the major band showing an apparent molecular mass of around 60 kDa.
Table 2.
Purification of β2-adrenergic receptor

Figure 4.

SDS-PAGE (10%) of the solubilized (lane A) and rho-1D4-affinity purified β2-AR (lane B). Proteins were detected by Coomassie staining. (Lane A) 5 μL of solubilized membrane protein, (lane B) 5 μL of rho-1D4-purified protein (25 pmol of receptor), (lane M) molecular weight standards. The receptor produced in HEK293S-TetR was glycosylated and migrated as two bands on the SDS-PAGE with the minor band around 47 kDa and the major band showing an apparent molecular mass of around 60 kDa.
The measured specific activity of the rho-1D4-Sepharose affinity purified β2-AR was found to be 10–12 nmol/mg (Table 2). While this value is less than the theoretical specific activity, 21 nmol/mg, assuming a single binding site per receptor of molecular weight of 47,000 Da, it compares favorably with the previously published values (Benovic et al. 1984; Kobilka 1995; Warne et al. 2003). The specific activity of hamster β2-AR purified by alprenolol-agarose chromatography and HPLC was found to be 12–16 nmol/mg (Benovic et al. 1984), with a yield of 7 μg β2-AR from 40–60 g of tissue. Similarly, the specific activity of β2-AR purified from baculovirus-infected cells using a combination of heparin agarose and alprenolol affinity chromatography was found to be 2 nmol/mg, corresponding to 10% of theoretical (Parker et al. 1991).
Preparation of membranes, solubilization, and purification of the receptor were done on the same day to minimize proteolysis, which was observed when preparations were carried out over prolonged periods of time (data not shown). The overall recovery of β2-AR obtained after purification by rho-1D4-Sepharose affinity chromatography was ∼20%. The yield of functional receptor using the single-step affinity purification is 12 ± 3 μg/15-cm plate. This level of expression using the 15-cm plates now makes it feasible to pursue structure–function studies on β2-AR using EPR spectroscopy.
Effect of optimization of codon sequences in β2-AR on its expression in a tetracycline-inducible mammalian stable cell line
Previously, using the HEK293S-TetR-inducible system, opsin was expressed at levels of up to 10 mg/L (Reeves et al. 2002b). The high-level expression resulted, at least in part, because the codons in naturally occurring opsin are biased toward those shown to be optimal for efficient translation in mammalian cells. However, a significant proportion of codons in genes encoding other GPCRs, including β2-AR, are suboptimal for high-level expression in mammalian cells. Our initial attempts at expression of the nonoptimized synthetic hamster β2-AR gene using the HEK293S-TetR-inducible system resulted in expression levels of 130 pmol β2-AR/mg of membrane protein (Chelikani et al. 2004). Following gene optimization and construction of stable cell lines, we observed an increase in expression to 220 ± 40 pmol β2-AR/mg of membrane protein. While the differences in expression levels using stable cell lines could also be due to positional effects of the stably integrated optimized β2-AR gene, the level of expression obtained is the highest reported so far for any wild-type GPCR other than rhodopsin.
In conclusion, we have reported on the synthesis of a β2-AR gene codon optimized for expression in mammalian cells, its expression in HEK293S stable cell lines, and purification of β2-AR. Purification of the receptor in milligram amounts is now feasible. Scale-up of β2-AR expression using suspension cultures in a bioreactor using established techniques should allow production of β2-AR at levels suitable for the application of biophysical techniques, such as NMR spectroscopy, and attempts at crystallization. The one-step purification method also enables convenient labeling of the purified β2-AR with either an agonist or an antagonist. In addition, β2-AR can be further purified by alprenolol-affinity chromatography (Caron et al. 1979), to yield the antagonist-bound form of the receptor. Current efforts are directed toward enhancing the stability of the purified receptor by exploring the use of lipids and various detergents.
Materials and methods
Materials
Synthetic oligonucleotides were purchased from Invitrogen. Sepharose 4B was purchased from Sigma. The detergent n-dodecyl-β-D-maltoside (DM) was purchased from Anatrace. The monoclonal antibody, rho-1D4, was prepared by the Cell Culture Center (Minneapolis) from a cell line provided by R.S. Molday (University of British Columbia. FBS, tetracycline, and dextran sulfate (average Mr 5000) were purchased from Sigma, and sodium butyrate was from J.T.Baker (Mallinckrodt Baker). Primatone RL-UF was a gift from Quest International, calcium-free DMEM was from Atlanta Biologicals, and Ham's F-12/DME High Glucose was from Irvine scientific. Geneticin (G418), Blasticidin S-HCl was from Invitrogen. The nonapeptide corresponding to the C-terminal sequence of rhodopsin, which was used to elute β2-AR samples from the antibody 1D-4 sepharose matrix, was prepared at the MIT Biopolymers Laboratory.
The β2-AR antagonist [3H] dihydroalprenolol was purchased from Amersham (TRK 649). Alprenolol, propranolol, ICI 118,551, formoterol, procaterol, isoproterenol, and salbutamol were purchased from Sigma. Protease inhibitors and common chemicals were purchased either from Sigma or Invitrogen. Restriction enzymes were purchased from NEB and the buffers NEB2 and NEB3 were used.
Buffers used were as follows: PBS buffer, 137 mM NaCl, 2.7 mM KCl, 1.8 mM KH2PO4, 10 mM Na2HPO4 (pH 7.4); Buffer A (lysis buffer), 10 mM Tris-HCl (pH 7.4), containing protease inhibitors (1 mM EDTA, 10 μg/mL benzamidine, 10 μg/mL leupeptin, 20 μg/mL soybean trypsin inhibitor, 5 μg/mL aprotinin, and 0.2 mM phenylmethylsulfonyl fluoride); Buffer B (storage buffer), 50 mM Tris-HCl (pH 7.4), 12.5 mM MgCl2, containing protease inhibitors as in Buffer A; Buffer C (binding buffer), 75 mM Tris-HCl (pH 7.4), 12.5 mM MgCl2, containing protease inhibitors as in Buffer A; Buffer D, 20 mM Tris-HCl (pH 7.4), containing 100 mM NaCl and 1 mM EDTA; Buffer E (solubilization buffer), 20 mM Tris-HCl (pH 7.4), containing 500 mM NaCl, 10% glycerol, 1% DM and the protease inhibitors as in Buffer A; Buffer F (no-salt buffer), 20 mM Tris-HCl (pH 7.4); Buffer G (high-salt buffer), 20 mM Tris-HCl (pH 7.4) containing 500 mM NaCl.
Synthesis of codon optimized hamster β2-AR gene
Gene design
The synthetic β2-AR gene consisted of 1310 bp, of which the 1278 bp encodes β2-AR. The salient features of the codon-optimized β2-AR gene include a Kozak consensus (GCCACCATGG) 5′ to the ATG start codon (Kozak 1991), a bovine rhodopsin C8 peptide tag (ETSQVAPA) immediately 5′ to the natural stop codon of β2-AR, an increase in the GC content of the gene from 51% to 57%, and restriction sites for EcoRI at the 5′ end and NotI at the 3′ end to facilitate cloning. The use of repetitive codons for any particular amino acid was also avoided wherever possible, to ensure that the total tRNA pool of the cell was not adversely affected. The sequence encoding the hamster β2-AR gene was optimized for mammalian cell codon usage (Fig. 5 of Supplemental Material) by utilizing the codons predicted to occur frequently in mammals (Nakamura et al. 2000). PCR primers were designed using the Upgene algorithm, and some of them were later refined manually (Gao et al. 2004). A total of 70 oligonucleotides were synthesized at 10-nmol scale; of these, 68 oligonucleotides were 38 nucleotides (nt) in length while two oligonucleotides were 18 nt in length (Table 3 of Supplemental Material). The sense strand (referred to as SS) and the antisense strand (referred to as AS) consists of 35 oligonucleotides, each configured in such a way that upon assembly, oligonucleotides overlap by 10 nt at each end and with 18 bp in the middle, except for the two outer primers, which overlap by only 10 nt with one of their neighboring oligonucleotides.
Gene synthesis
Construction of the hamster β2-AR gene was by PCR (Stemmer et al. 1995). The overall procedure involves gene assembly, gene amplification, and cloning. For the gene assembly, the reaction mixture (50 μL) contained, 20 nM of each oligonucleotide, 20 mM Tris-HCl (pH 8.4), 50 mM KCl, 2.2 mM MgCl2, 0.2 mM each dNTP (Stratagene), 1 U of Taq polymerase (Invitrogen), and 0.02 U of PfuTurbo polymerase (Stratagene). The PCR program consisted of 55 cycles, each cycle involved heating at 94°C for 30 sec, annealing at 52°C for 30 sec, and extension at 72°C for 30 sec (MJ Research PTC-100). For PCR amplification of the synthetic gene, 2.5 μL of the gene assembly reaction mixture was diluted 40-fold in 100 μL of PCR mix containing 20 mM Tris-HCl (pH 8.4), 50 mM KCl, 2.2 mM MgCl2, 0.2 mM each dNTP (Stratagene), 0.1% Triton X-100, 5 U of Taq polymerase (Invitrogen), 0.1 U of PfuTurbo polymerase (Stratagene), and two flanking primers at a concentration of 1 μM. The two flanking primers can be the same as the two oligonucleotides representing the 5′ ends of the sense and antisense strands. The PCR program consisted of 25 cycles; each cycle involved heating at 94°C for 30 sec, annealing at 50°C for 30 sec, and extension at 72°C for 60 sec.
Cloning and sequencing
For cloning, the PCR product was separated on 1% agarose gel, and the ∼1.3-kb band was excised and gel-purified (QIAquick gel extraction kit). The purified DNA was subjected to dA tailing and cloned into pGEM-T vector (Promega), following the manufacturer's protocol. The resulting white colonies were screened for the presence of the β2-AR gene by restriction enzyme digestion. Isolates that showed the correct banding pattern were sequenced (MIT biopolymers facility), and the isolate with the correct sequence was double digested with EcoRI and NotI. The β2-AR gene thus released was cloned into the mammalian expression vector pMT4 (Oprian et al. 1987).
Cell culture
The wild-type β2-AR gene was expressed in COS-1 cells using a DEAE–dextran-based transient transfection method (Oprian et al. 1987). For transient transfection of HEK293S cells, the plasmid pMT4 was cotransfected with pRSVTAg plasmid (a gift from Prof. Elliot Ross [The University of Texas Southwestern Medical Center, Dallas]) and lipofectamine 2000 (Invitrogen) mediated transfection was used as described by the manufacturer. The transfection was allowed to proceed for 44–48 h, the transfected cells were harvested, and membranes were prepared immediately or the cell pellets were snap frozen in liquid nitrogen and stored at −80°C.
Construction of a tetracycline-inducible HEK293S stable cell line expressing the codon-optimized β2-AR gene
The plasmid, pMT4, containing the codon-optimized β2-AR gene was digested with EcoRI for 1 h in 20 μL of buffer NEB2 at standard concentration. The enzyme was then removed by using the Qiagen gel extraction kit, and the linearized plasmid was eluted in 20 μL of water. The EcoRI sticky end in the DNA was repaired by incubation with the Klenow fragment of Escherichia coli DNA Polymerase I at 22°C for 30 min in a 30-μL reaction volume containing each dNTP at 0.5 mM concentration and the buffer NEB2. After removal of the enzyme using the Qiagen gel purification kit, the DNA was eluted in 20 μL of water. The DNA was next digested with the enzyme NotI, in the buffer NEB3 containing BSA for 1 h in a total volume of 30 μL. The EcoRI (blunt)–NotI fragment was purified by gel electrophoresis using 0.7% agarose, and DNA was extracted from the gel using the Qiagen gel purification kit in a total volume of 20 μL of water. This fragment was then ligated into the plasmid pACMVtetO (Reeves et al. 2002b), which had been previously digested with KpnI (repaired to blunt) and then digested with NotI. The ligation mixtures were used directly for transformation of competent E. coli DH5α. The transformants were screened for the presence of the 1.3-kb β2-AR gene following digestion with EcoRI and NotI, and their identity was confirmed by DNA sequencing. The plasmid with the correct sequence was then transfected into HEK293S (WT)-TetR cells (10 μg of plasmid/10-cm plate). Transfection was carried out by the calcium phosphate precipitation method using DMEM medium (Reeves et al. 2002b). An untransfected 10-cm plate containing HEK293S (WT) cells served as the control. After incubation of the cells for 20 h at 36°C (1.5% CO2) the medium was changed to DMEM F-12, and the cells were further incubated for another 24 h under 5% CO2. After a total of 44 h following transfection, the cells were trypsinized and split using DMEM F-12 medium supplemented with 20% conditioned medium. Two days later the medium was replaced with media (DMEM F-12/20% conditioned) containing the antibiotic, G418 (1 mg/mL). The cells were re-fed every 3 d with fresh medium containing G418 (1 mg/mL) for 2 wk or until large single colonies resulted (the cells in the untransfected control plate died after 1 wk). About 18 large, single, and visible colonies were selected by using cloning rings. The cells were trypsinized and the suspension transferred into 24-well dishes for growth. When they reached confluence, the cells in each well were further split into three wells and the latter incubated until the cells reached confluence. Then, one well of cells was frozen for storage, while the cells in a second well were fed with DMEM containing both tetracycline (1 μg/mL) and sodium butyrate (7.5 mM). The cells in the third well were not induced, and used as the control. After induction for 44–48 h, the cells were harvested in 1 mL of PBS, and the pellet was either frozen for storage at −80°C or solubilized in 200 μL of PBS containing protease inhibitors and 1% DM by end-over-end mixing for 1 h at 4°C. The cell extract thus obtained was centrifuged for 10 min at 13,000 rpm (4°C) and the supernatant fraction was serially diluted three times, 1 to 5 each time, and transferred to a nitrocellulose membrane by dot blot. β2-AR was detected by the antibody rho-1D4 and visualized by chemiluminescence (ECL, Amersham).
Preparation of membranes containing β2-AR
Cells (COS-1 or HEK293S) expressing β2-AR were grown in 10-cm dishes. The dishes were rinsed, using ice-cold PBS buffer, and the cells were then harvested in Buffer A. All further steps were carried out at 0°–4°C. The cell suspension was next poured into a 15-mL dounce tissue homogenizer, and the cells were homogenized using 30 strokes. The suspension was centrifuged in a tabletop centrifuge at 1500 rpm (300g) for 10 min, and the pellet was discarded. The supernatant fraction was centrifuged at 48,000g for 20 min. The resulting pellet was resuspended in 10 mL of buffer B, and the suspension again centrifuged at 48,000g for 20 min. The resulting pellet was resuspended in 1 mL of the above buffer, and aliquots of the suspension were snap-frozen and stored at −80°C. The protein concentration in the resuspended membrane pellet was determined using a modified DC protein assay kit from Bio-Rad Laboratories.
Immunoblot analysis
One to five micrograms of the protein sample were resolved by 10% SDS-PAGE gel. The protein was then transferred from the gels onto a nitrocellulose membrane by electroblotting, and β2-AR was visualized by immunodetection with the monoclonal antibody, rho-1D4, the epitope tag for this antibody had been incorporated into the C terminus of the β2-AR gene.
Purification of β2-AR
Cell pellets from 10 to 40 dishes (15 cm) were resuspended using 100 mL of buffer A, and the suspension was homogenized using a dounce homogenizer (20 strokes). The suspension was centrifuged at 48,000g for 30 min, the pellets were resuspended in 100 mL of buffer A, and the suspension poured into preweighed centrifuge tubes, which were centrifuged at 48,000g for 30 min. Following centrifugation, the supernatant solution was discarded, while the membrane pellet, after weighing, was resuspended in buffer E using a dounce homogenizer (20 strokes) and the suspension mixed by nutation at 4°C for 1 h. Ten milliliters of buffer E were used for each gram of the crude membrane pellets. The solution was then centrifuged at 48,000g for 30 min to remove any insoluble particulate material.
Subsequent purification of β2-AR by rho-1D4-affinity chromatography was carried out as follows: The solubilized β2-AR receptor was adsorbed to rho-1D4-Sepharose beads in batch mode (binding capacity, 1.5 mg β2-AR/mL of beads) with slow nutation for 2 h at 4°C. The rho-1D4-beads were then collected by centrifugation at 1500g and washed with buffer G until the absorbance of the wash at 280 nm was <0.01. Elution was carried out with buffer G containing 100 μM nonapeptide. The fractions were then assayed for receptor binding using [3H] DHA while their protein concentration was determined by Biorad DC protein assay. The receptor appeared in the effluent, following elution with two bed volumes of the eluant.
Radioligand binding assays
Radioligand binding studies were carried out in Buffer C at 37°C for 60 min, using 2–20 μg of membrane protein. Saturation binding assays were performed using 0.1–20 nM [3H] DHA. Binding of [3H] DHA in the presence of 10 μM alprenolol was used as a measure of nonspecific binding. Competition binding assays were performed using 5 nM [3H] DHA and different concentrations of unlabeled agonists (10−2–10−9 M) and antagonists (10−5–10−11 M). Binding was terminated by filtering under vacuum on GF/A filters (Millipore). Filter-bound radioactivity was determined using a liquid scintillation counter. All data shown are mean values of ±SE for n determinations. Equilibrium dissociation constants (Kd) were determined from saturation isotherms.
Binding assays on solubilized receptors were performed using a 20-nM concentration of [3H] DHA in a total volume of 100 μL of buffer D containing 0.05% DM, which was then incubated for 1 h. Bound and free radioligand were separated by gel filtration on 2-mL Sephadex G-50 columns using ice-cold buffer D. Binding of [3H] DHA in the presence of 10 μM alprenolol was used as a measure of nonspecific binding. Radioligand binding data obtained from competition curves were analyzed by nonlinear regression analysis to determine the EC50 values and Ki values using PRISM software version 4.03 (GraphPad Software Inc.).
Electronic supplemental material
The electronic supplemental material contains a figure showing the nucleotide sequence alignment of the optimized gene with the other two nonoptimized hamster β2-AR genes, and a table listing the PCR primers used for the synthesis of the hamster β2-AR gene.
Acknowledgments
The research reported here was supported by grants RO1-EY11716 (NIH) and EIA-0225609 (NSF). Ms. Judy Carlin's assistance during the preparation of the manuscript is gratefully acknowledged.
Footnotes
Supplemental material: see www.proteinscience.org
Reprint requests to: H. Gobind Khorana, Room 68-680, Departments of Biology and Chemistry, Massachusetts Institute of Technology, Cambridge, MA 02139, USA; e-mail: khorana@mit.edu; fax: (617) 253-0533.
Abbreviations: β2-AR, β2-adrenergic receptor; TM, transmembrane; [3H] DHA, tritium-labeled dihydroalprenolol; Bmax, binding maximum of the ligand for the receptor; UTR, untranslated-region; HEK293S, human embryonic kidney cells–suspension adapted; COS-1, monkey kidney cells expressing the SV40 T antigen.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062080006.
References
- Babcock G.J., Mirzabekov T., Wojtowicz W., Sodroski J. 2001. Ligand binding characteristics of CXCR4 incorporated into paramagnetic proteoliposomes J. Biol. Chem. 276 38433–38440. [DOI] [PubMed] [Google Scholar]
- Benovic J.L., Shorr R.G., Caron M.G., Lefkowitz R.J. 1984. The mammalian β2-adrenergic receptor: Purification and characterization Biochemistry 23 4510–4518. [DOI] [PubMed] [Google Scholar]
- Bradel-Tretheway B.G., Zhen Z., Dewhurst S. 2003. Effects of codon-optimization on protein expression by the human herpesvirus 6 and 7 U51 open reading frame J. Virol. Methods 111 145–156. [DOI] [PubMed] [Google Scholar]
- Caron M.G., Srinivasan Y., Pitha J., Kociolek K., Lefkowitz R.J. 1979. Affinity chromatography of the β-adrenergic receptor J. Biol. Chem. 254 2923–2927. [PubMed] [Google Scholar]
- Chelikani P., Kota P., Cao Z., Huang Y., Kim J., Reeves P.J., Khorana H.G. 2004. Expression, purification and crystallization trials on β2–adrenergic receptor FASEB J. 18–C281.
- Dixon R.A., Kobilka B.K., Strader D.J., Benovic J.L., Dohlman H.G., Frielle T., Bolanowski M.A., Bennett C.D., Rands E., Diehl R.E.et al. 1986. Cloning of the gene and cDNA for mammalian β-adrenergic receptor and homology with rhodopsin Nature 321 75–79. [DOI] [PubMed] [Google Scholar]
- Dixon R.A., Sigal I.S., Candelore M.R., Register R.B., Scattergood W., Rands E., Strader C.D. 1987. Structural features required for ligand binding to the β-adrenergic receptor EMBO J. 6 3269–3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrens D.L., Altenbach C., Yang K., Hubbell W.L., Khorana H.G. 1996. Requirement of rigid-body motion of transmembrane helices for light activation of rhodopsin Science 274 768–770. [DOI] [PubMed] [Google Scholar]
- Farrens D.L., Dunham T.D., Fay J.F., Dews I.C., Caldwell J., Nauert B. 2002. Design, expression, and characterization of a synthetic human cannabinoid receptor and cannabinoid receptor/G-protein fusion protein J. Pept. Res. 60 336–347. [DOI] [PubMed] [Google Scholar]
- Gao W., Rzewski A., Sun H., Robbins P.D., Gambotto A. 2004. UpGene: Application of a web-based DNA codon optimization algorithm Biotechnol. Prog. 20 443–448. [DOI] [PubMed] [Google Scholar]
- Khorana H.G. 1992. Rhodopsin, photoreceptor of the rod cell. An emerging pattern for structure and function J. Biol. Chem. 267 1–4. [PubMed] [Google Scholar]
- Klein-Seetharaman J., Reeves P.J., Loewen M.C., Getmanova E.V., Chung J., Schwalbe H., Wright P.E., Khorana H.G. 2002. Solution NMR spectroscopy of [α-15N]lysine-labeled rhodopsin: The single peak observed in both conventional and TROSY-type HSQC spectra is ascribed to Lys-339 in the carboxyl-terminal peptide sequence Proc. Natl. Acad. Sci. 99 3452–3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobilka B.K. 1995. Amino and carboxyl terminal modifications to facilitate the production and purification of a G protein-coupled receptor Anal. Biochem. 231 269–271. [DOI] [PubMed] [Google Scholar]
- Kobilka B.K., Frielle T., Dohlman H.G., Bolanowski M.A., Dixon R.A., Keller P., Caron M.G., Lefkowitz R.J. 1987. Delineation of the intronless nature of the genes for the human and hamster β2-adrenergic receptor and their putative promoter regions J. Biol. Chem. 262 7321–7327. [PubMed] [Google Scholar]
- Kozak M. 1991. Structural features in eukaryotic mRNAs that modulate the initiation of translation J. Biol. Chem. 266 19867–19870. [PubMed] [Google Scholar]
- Mirzabekov T., Bannert N., Farzan M., Hofmann W., Kolchinsky P., Wu L., Wyatt R., Sodroski J. 1999. Enhanced expression, native purification, and characterization of CCR5, a principal HIV-1 coreceptor J. Biol. Chem. 274 28745–28750. [DOI] [PubMed] [Google Scholar]
- Nakamura Y., Gojobori T., Ikemura T. 2000. Codon usage tabulated from international DNA sequence databases: Status for the year 2000 Nucleic Acids Res. 28–292. [DOI] [PMC free article] [PubMed]
- Noda K., Saad Y., Graham R.M., Karnik S.S. 1994. The high affinity state of the β2-adrenergic receptor requires unique interaction between conserved and non-conserved extracellular loop cysteines J. Biol. Chem. 269 6743–6752. [PubMed] [Google Scholar]
- Oprian D.D., Molday R.S., Kaufman R.J., Khorana H.G. 1987. Expression of a synthetic bovine rhodopsin gene in monkey kidney cells Proc. Natl. Acad. Sci. 84 8874–8878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker E.M., Kameyama K., Higashijima T., Ross E.M. 1991. Reconstitutively active G protein-coupled receptors purified from baculovirus-infected insect cells J. Biol. Chem. 266 519–527. [PubMed] [Google Scholar]
- Parola A.L. and Kobilka B.K. 1994. The peptide product of a 5′ leader cistron in the β2 adrenergic receptor mRNA inhibits receptor synthesis J. Biol. Chem. 269 4497–4505. [PubMed] [Google Scholar]
- Patel A.B., Crocker E., Reeves P.J., Getmanova E.V., Eilers M., Khorana H.G., Smith S.O. 2005. Changes in interhelical hydrogen bonding upon rhodopsin activation J. Mol. Biol. 347 803–812. [DOI] [PubMed] [Google Scholar]
- Rands E., Candelore M.R., Cheung A.H., Hill W.S., Strader C.D., Dixon R.A. 1990. Mutational analysis of β-adrenergic receptor glycosylation J. Biol. Chem. 265 10759–10764. [PubMed] [Google Scholar]
- Reeves P.J., Callewaert N., Contreras R., Khorana H.G. 2002a. Structure and function in rhodopsin: High-level expression of rhodopsin with restricted and homogeneous N-glycosylation by a tetracycline-inducible N-acetylglucosaminyltransferase I-negative HEK293S stable mammalian cell line Proc. Natl. Acad. Sci. 99 13419–13424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves P.J., Kim J.M., Khorana H.G. 2002b. Structure and function in rhodopsin: A tetracycline-inducible system in stable mammalian cell lines for high-level expression of opsin mutants Proc. Natl. Acad. Sci. 99 13413–13418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarramegna V., Talmont F., Demange P., Milon A. 2003. Heterologous expression of G-protein-coupled receptors: Comparison of expression systems from the standpoint of large-scale production and purification Cell. Mol. Life Sci. 60 1529–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoneberg T., Schulz A., Biebermann H., Hermsdorf T., Rompler H., Sangkuhl K. 2004. Mutant G-protein-coupled receptors as a cause of human diseases Pharmacol. Ther. 104 173–206. [DOI] [PubMed] [Google Scholar]
- Sizmann D., Kuusinen H., Keranen S., Lomasney J., Caron M.G., Lefkowitz R.J., Keinanen K. 1996. Production of adrenergic receptors in yeast Receptors Channels 4 197–203. [PubMed] [Google Scholar]
- Stanasila L., Pattus F., Massotte D. 1998. Heterologous expression of G-protein-coupled receptors: Human opioid receptors under scrutiny Biochimie 80 563–571. [DOI] [PubMed] [Google Scholar]
- Stemmer W.P., Crameri A., Ha K.D., Brennan T.M., Heyneker H.L. 1995. Single-step assembly of a gene and entire plasmid from large numbers of oligodeoxyribonucleotides Gene 164 49–53. [DOI] [PubMed] [Google Scholar]
- Subramaniam K., Chen K., Joseph K., Raymond J.R., Tholanikunnel B.G. 2004. The 3′-untranslated region of the β2-adrenergic receptor mRNA regulates receptor synthesis J. Biol. Chem. 279 27108–27115. [DOI] [PubMed] [Google Scholar]
- Takeda S., Kadowaki S., Haga T., Takaesu H., Mitaku S. 2002. Identification of G protein-coupled receptor genes from the human genome sequence FEBS Lett. 520 97–101. [DOI] [PubMed] [Google Scholar]
- Tate C.G. and Grisshammer R. 1996. Heterologous expression of G-protein-coupled receptors Trends Biotechnol. 14 426–430. [DOI] [PubMed] [Google Scholar]
- Thompson J.D., Gibson T.J., Plewniak F., Jeanmougin F., Higgins D.G. 1997. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools Nucleic Acids Res. 25 4876–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warne T., Chirnside J., Schertler G.F. 2003. Expression and purification of truncated, non-glycosylated turkey β-adrenergic receptors for crystallization Biochim. Biophys. Acta 1610 133–140. [DOI] [PubMed] [Google Scholar]
- Weiss H.M., Haase W., Michel H., Reilander H. 1998. Comparative biochemical and pharmacological characterization of the mouse 5HT5A5-hydroxytryptamine receptor and the human β2-adrenergic receptor produced in the methylotrophic yeast Pichia pastoris Biochem. J. 330 1137–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young L. and Dong Q. 2004. Two-step total gene synthesis method Nucleic Acids Res. 32–e59. [DOI] [PMC free article] [PubMed]