

Abstract
The mechanisms by which G-protein-coupled receptors (GPCRs) activate G-proteins are not well understood due to the lack of atomic structures of GPCRs in an active form or in GPCR/G-protein complexes. For study of GPCR/G-protein interactions, we have generated a series of chimeras by replacing the third cytoplasmic loop of a scaffold protein bacteriorhodopsin (bR) with various lengths of cytoplasmic loop 3 of bovine rhodopsin (Rh), and one such chimera containing loop 3 of the human β2-adrenergic receptor. The chimeras expressed in the archaeon Halobacterium salinarum formed purple membrane lattices thus facilitating robust protein purification. Retinal was correctly incorporated into the chimeras, as determined by spectrophotometry. A 2D crystal (lattice) was evidenced by circular dichroism analysis, and proper organization of homotrimers formed by the bR/Rh loop 3 chimera Rh3C was clearly illustrated by atomic force microscopy. Most interestingly, Rh3C (and Rh3G to a lesser extent) was functional in activation of GTPγ35S/GDP exchange of the transducin α subunit (Gαt) at a level 3.5-fold higher than the basal exchange. This activation was inhibited by GDP and by a high-affinity peptide analog of the Gαt C terminus, indicating specificity in the exchange reaction. Furthermore, a specific physical interaction between the chimera Rh3C loop 3 and the Gαt C terminus was demonstrated by cocentrifugation of transducin with Rh3C. This Gαt-activating bR/Rh chimera is highly likely to be a useful tool for studying GPCR/G-protein interactions.
Keywords: G-protein-coupled receptors, chimera, bacteriorhodopsin, rhodopsin, transducin, GTP exchange
G-protein-coupled receptors (GPCRs) constitute a superfamily of >800 membrane proteins that directly interact with intracellular proteins including guanine nucleotide-binding proteins, or G-proteins. GPCRs mediate numerous cellular signal transductions triggered by a variety of stimulants (Lundstrom 2005), and therefore serve as the targets of nearly half of all known drugs. GPCRs are heptahelical proteins that transverse the cell membrane seven times. With an extracellular N terminus and an intracellular C terminus, GPCRs contain three extracellular loops and three or four cytoplasmic loops (depending on whether or not the C-terminal tail is post-translationally modified). It is through the cytoplasmic loops that GPCRs interact with G-proteins. A critical region appears to be the cytoplasmic loop 3 including the sequences located in the helix 6 juxtaposed on the membrane (O'Dowd et al. 1988; Luttrell et al. 1993; Kudo et al. 1996; Cai et al. 2001; Natochin et al. 2003; Janz and Farrens 2004).
Enormous efforts have been made to study the physical characteristics of atomic interactions between GPCRs and G-proteins to fully understand the nature of the protein–protein interactions that lead to activation of G-proteins following ligand binding and activation of GPCRs. These studies include high-resolution crystallography of bovine rhodopsin (Palczewski et al. 2000; Teller et al. 2001; Okada et al. 2002; Li et al. 2004; Okada et al. 2004), mutational studies of both G-proteins and GPCRs in conjunction with functional studies to determine the role of specific residues (Dixon et al. 1988; Franke et al. 1988, 1990; Fraser et al. 1988; Cai et al. 2001; Itoh et al. 2001; Natochin et al. 2003; Herrmann et al. 2004; Janz and Farrens 2004; Pereira and Cerione 2005), peptide studies in which the interaction domains are mimicked (Hamm et al. 1988; Martin et al. 1996; Terakita et al. 2002), structural determinations by NMR (Yeagle et al. 1997; Choi et al. 2002; Koenig et al. 2002; Kisselev et al. 2004; Klein-Seetharaman et al. 2004; Ridge et al. 2006), electron spin resonance studies identifying residue interactions and distance geometries in both the active and the inactive states of GPCRs (Altenbach et al. 1996; Farrens et al. 1996; Kim et al. 2004), and the construction of GPCR/GPCR chimeras for the purpose of establishing the domain properties important for G-protein specificity (Frielle et al. 1988; Kobilka et al. 1988; Cotecchia et al. 1990; Wong et al. 1990; Ahumada et al. 2002; Kim et al. 2005).
While all these studies have provided a macroscopic view of how GPCRs interact with G-proteins, they do not provide resolution at the atomic level. Several efforts are currently under way to obtain high-resolution crystal structures to elucidate this interaction, but none as yet has been successful due to either the inability to generate sufficient quantities of protein for crystal generation or the lack of availability of proteins with the necessary physical properties needed for forming a resolvable crystal (Ridge et al. 2003; Lundstrom 2005). The only crystal structure of a GPCR, bovine rhodopsin in an inactive form, has provided a great deal of information for understanding GPCR structures (Palczewski et al. 2000). However, since a crystal structure of the active form rhodopsin has not been available due to the instability of the rhodopsin crystal, characterization of the rhodopsin–transducin interactions remains a challenging task.
One potential solution to the problem of obtaining sufficient quantities of a protein that has the necessary physical properties for forming high-resolution crystals is to create a chimera by inserting a GPCR domain of interest into a well-characterized protein with the properties suited for structural study. For this reason, bacteriorhodopsin (bR) can be used as a scaffold upon which various GPCR cytoplasmic domains could be integrated. bR is a seven transmembrane helices protein expressed in high levels in the archaeon Halobacterium salinarum (Oesterhelt and Stoeckenius 1974; Lanyi 2004). The bR molecules are incorporated into a highly structured lattice of trimers in the membrane of H. salinarum. Since bR can be overexpressed and forms highly resolvable crystals (Luecke et al. 1998, 1999), bR/GPCR chimeras would provide for the possibilities of obtaining atomic-level structural information of GPCR domains and developing constitutively active signaling to a heterotrimeric G-protein.
While attempts have been previously made for overexpressing heterologous proteins in H. salinarum using bR as a scaffold, for the purpose of structural determinations, none of these studies showed function of the chimeric proteins. A fusion protein of bR-aspartyl transcarbamylase is successfully expressed in H. salinarum in large quantity, but the function of this fusion protein is not reported (Turner et al. 1999). The bR/GPCR chimeras containing the cytoplasmic loop 3 of bovine rhodopsin (Heymann et al. 2000 and the loop 3 of the aminergic α2b-adrenergic receptor (Jaakola et al. 2005), respectively, have been reported. However, no G-protein activation function has been demonstrated with these bR/GPCR chimeras. Encouraging progress has been made, in that a chimera of bR/CCR5 extracellular segments confers HIV-1 coreceptor activity (Abdulaev et al. 2002), although the chimera is expressed in COS-1 cells, which may not be able to provide sufficient protein for structural studies.
Therefore, the objective of this study is to generate bR/GPCR chimeras in large quantity that have functional attributes similar to native GPCRs. The highly structured membrane lattice, in which bR is expressed, was used as a scaffold into which the third cytoplasmic loop of rhodopsin (Fig. 1) or the β2-adrenergic receptor (β2AR) was integrated to substitute for the corresponding loop of bR. The work presented here focuses primarily on the biochemical properties of a functionally active bR/Rh chimera (Rh3C) that could lead to high-resolution structural characterization of GPCR/G-protein coupling.
Figure 1.
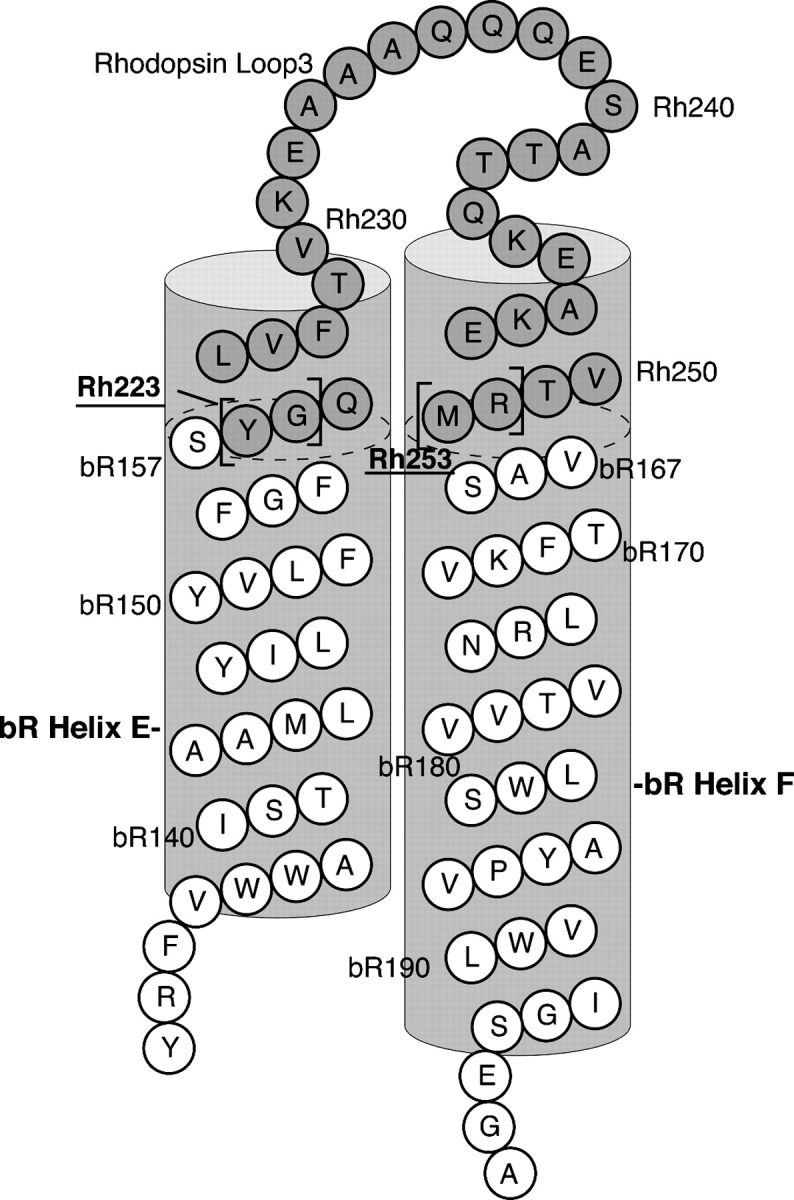
Schematic of bR/Rh chimera construction. Shown is the bR/Rh chimera, Rh3C. Amino acid residue numbers are noted for bR (Luecke et al. 1999) and Rh (Palczewski et al. 2000). Shaded amino acids are the Rh residues added into the bR sequence. The Rh residues were added onto the ends of the corresponding bR helices (shown as dotted lines in the helices). The Rh residues shown in brackets are the amino acids that were systematically removed from each end of the insert, which resulted in the other bR/Rh chimeric constructs with shorter inserts (Table 1) and thus different “twisting” of the helical region of rhodopsin. To distinguish the nomenclatures of the bR and Rh transmembrane helices, we use A–G for bR helices and 1–7 for Rh helices.
Results
PM-forming constructs were selected by colony phenotyping of H. salinarum expressing bR/GPCR chimeras
It has been previously shown that the bop gene deletion strain of H. salinarum, MPK40, when transformed with the bop gene on the carrier plasmid pMPK85, which has a mevinolin-resistance gene, will successfully integrate the bop gene into the H. salinarum genome (Peck et al. 2000). Successive rounds of plating H. salinarum colonies from mevinolin and CM-containing agar plates, to only CM-containing plates, results in the stable integration of the bop gene into the H. salinarum genome. For this strain, it has been shown that using the CM plates from the final rounds of colony re-plating, under conditions of constant exposure to white light, will lead to overexpression of the bop gene product, bR, in the colonies. When observed against a white background, the overexpression of bR on the cellular surfaces of these colonies makes them appear purple (Fig. 2A).
Figure 2.

Purification of bR/GPCR chimeras expressed in H. salinarum PM. (A) Purple phenotype of a bR/Rh chimera (only Rh3C data are presented here). A bR-deficient strain of H. salinarum, MPK40, was transformed with the chimeric gene through a homologous recombination protocol (Peck et al. 2000). The transformed cells were plated on a mevinolin selection media, and the plate was incubated at 37°C under illumination for 3 d. A purple colony, indicating proper formation of the bR lattice structure, and a wild-type colony with no transformation (not expressing any bR), are indicated by arrows. (B) Wild-type bR, bR/GPCR chimeras Rh3C and β2L3 were collected from H. salinarum cells and purified by equilibrium centrifugation over a sucrose density gradient. The PM migrated in the middle of the tube as a purple band. (C) Purified bR and chimeras were incubated with Laemmli buffer containing SDS and DTT at room temperature overnight, and then subjected to 12% SDS-PAGE. Rh3G ran at the same position as Rh3C (data not shown).
We generated chimeric bR constructs in which bR was used as a scaffold upon which to place portions of GPCRs, such as bovine rhodopsin, human β2-adrenergic receptor, human adenosine A1 receptor, and human adrenocorticotropic hormone receptor. bR/Rh chimeras generated included loop 2, loop 3, loop 2 plus 3, the C terminus, and loop 3 plus the C terminus of bovine rhodopsin inserted into the bR sequence at the appropriate position, while the other bR/GPCR chimeras contained only GPCR loop 3. All the constructs generated were confirmed by DNA sequencing and characterized by several different assays. The data are summarized in Supplemental Table SI. However, most of our interest was in generating and characterizing bR/Rh loop 3 chimeras, which are summarized in Table 1. Since the position of the junction is an α-helix as demonstrated by the crystal structure of both bR (Luecke et al. 1999) and rhodopsin (Palczewski et al. 2000), we reasoned that changing the starting rhodopsin position by up to three amino acids would introduce almost a complete rotation of the helix, and likewise with the C-terminal junction. With three amino acids in the N terminus and three amino acids in the C terminus, there are nine possible permutations (RhA–I). A similar approach was used for the generation of rhodopsin loop 2 constructs as well as the C terminus constructs.
Table 1.
Characterization of PM-forming bR/Rh and bR/β2AR chimeras

All the bR/Rh loop 3 chimeras (except Rh2E), as well as the bR/β2AR loop 3 chimera showed a purple phenotype (Table 1). However, the chimeras of bR/Rh loop 2, bR/Rh loop 2 + 3, bR/Rh C-term, bR/Rh C-term + loop 3, and the bR/human adenosine A1 receptor loop 3 chimera (AA1AI3) failed to show purple colonies (Supplemental Table SI), even though DNA sequencing of the transformed H. salinarum genome confirmed proper insertion in all cases. Interestingly, the bR/human adenocorticotropic hormone receptor loop 3 chimera (ACTRI3) also formed purple colonies but was not further characterized. The chimeras that did not form purple colonies were not subjected to further investigation for the obvious reason of selection and protein purification.
bR/GPCR chimeric proteins were readily purified from PMs
It has been previously described that bR forms a two-dimensional crystal lattice in the membrane of H. salinarum (Oesterhelt and Stoeckenius 1974; Heymann et al. 2000). This lattice confers the ability to purify bR from other cellular constituents by the use of equilibrium centrifugation with a continuous sucrose density gradient. The importance of this observation for the present study was twofold: First, it provided a convenient purification strategy for the chimeras, and second, it is an early indicator of the potential for the chimera to form two-dimensional crystals that may be useful later in structure determination experiments. The migration of the chimeric proteins with respect to bR on the sucrose density gradients is shown in Figure 2B. The chimeric proteins migrate identically compared to bR, indicating proper lattice formation.
Analysis of the sucrose-gradient-purified bR/GPCR chimeras Rh3C and β2L3 by SDS-PAGE showed the migration of highly purified protein bands of the appropriate molecular weights (Fig. 2C). Chimera Rh3C adds an additional 21 amino acids; Rh3G, an additional 17 amino acids; and the β2AR chimera, an additional 58 amino acids, to bR.
The bR/GPCR chimeras form a lattice that is similar to bR
UV/Vis spectrophotometry of the bR/GPCR chimeras is a useful measure of the correct incorporation of the retinal into the transmembrane domains. Analysis of sucrose-density-gradient-purified chimeras Rh3C, Rh3G, and β2L3 revealed that the generated pigment was identical to that of bR (Fig. 3A). The maximal absorbance peak at 570 nm is consistent with the correct incorporation of retinal into the protein.
Figure 3.

Spectroscopic characterization of bR/GPCR chimeras. The spectroscopic properties of the purified bR and bR/GPCR chimeras were compared, by scanning their UV/Vis absorbance (A) and CD spectroscopy (B). The experiments were performed as described in Materials and Methods. As can be seen in this figure, bR and the chimeras exhibit similar spectra.
Circular dichroism (CD) analysis of the purified bovine rhodopsin loop 3 chimeras Rh3C and Rh3G showed a bilobed pattern of spectra, similar to that found in bR (Fig. 3B). The bilobed spectrum is indicative of exciton coupling, which for bR is caused by the proper packing of bR monomers into a homotrimer arrangement in the membrane of H. salinarum. If bR were in the monomeric form (i.e., non-lattice-forming), it would produce a monolobed spectrum.
An atomic force microscopy topograph further confirmed the proper homotrimer arrangement of Rh3C. The subnanometer high-resolution image of the Rh3C extracellular surface is shown in Figure 4. The trimers and the loops of the single monomers are clearly discernible, indicating a highly ordered 2D crystal. These data demonstrate that the crystallinity of the PM was not disturbed by the rhodopsin loop 3 insertion.
Figure 4.

AFM imaging of Rh3C purple membrane. The AFM topograph of Rh3C extracellular surface shows that the homotrimers of chimera Rh3C are well organized in the 2D crystal. A trimer of Rh3C is magnified as shown in the circle. AFM imaging was conducted as described in Materials and Methods.
bR/Rh chimeras activate transducin
Activation of transducin by the bR/Rh chimeras was measured by the ability of the chimera to stimulate the exchange of GTPγ35S on Gαt. As can be seen in Figure 5A, the Rh3C chimera stimulated GTPγ35S exchange on transducin by 3.5-fold. The chimera Rh3G, which is four amino acids shorter than Rh3C (Table 1), stimulated GTPγ35S exchange by 2.3-fold. Since in addition to the cytoplasmic loop 3, the other regions of rhodopsin, such as the cytoplasmic loop 2 and the fourth cytoplasmic loop (or helix 8), also contribute to transducin activation (Franke et al. 1990; Marin et al. 2000; Terakita et al. 2002; Natochin et al. 2003), a 3.5-fold activation above the basal activity of transducin by the Rh3C chimera containing only loop 3 of rhodopsin is significant compared to native rhodopsin (12.75-fold activation). Furthermore, this stimulation is deemed specific, since it is inhibited both by GDP (but much less by ADP) (Fig. 5B) and a high-affinity analog of a Gαt C-terminal peptide (Fig. 5C; Martin et al. 1996). The C terminus of Gαt has been suggested to interact with rhodopsin loop 3 (Cai et al. 2001). In contrast, a nonspecific peptide in which the high-affinity sequence has been scrambled inhibited the exchange to a much less extent. The ability of the β2AR loop 3 chimera (β2L3) to stimulate GTPγ35S exchange of Gαs was also measured (data not shown), but no activation of Gαs was observed.
Figure 5.
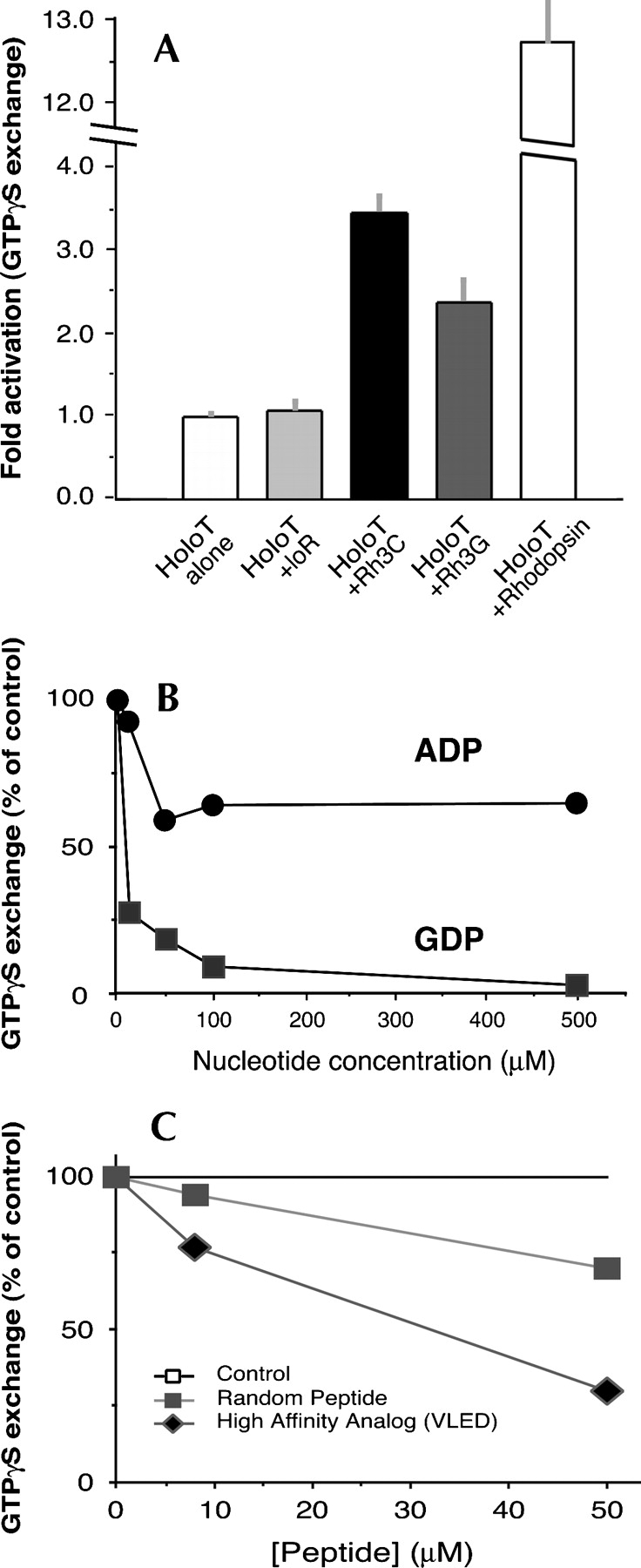
GTPγ35S exchange assay showing activation of transducin by the bR/Rh chimeras. (A) The activation of transducin by the purified bR or chimeras Rh3C and Rh3G was assayed by incubating the chimeras (0.6 μM each) with 0.2 μM transducin and 5 μM GTPγ35S for 10 min at 30°C. The samples were rapidly vacuum-filtered through Millipore cellulose acetate filters (0.22-μm pore size). The 35S content on the filters was determined by liquid scintillation counting. The basal activation is represented by the condition of holoT alone (one fold; first bar). Native rhodopsin was used for the assay as a positive control (last bar), at a molar ratio of 2.3:1 vs. transducin. All the assays were performed under room light. Error bars represent standard deviations (SDs). (B) Chimera Rh3C-catalyzed GTPγ35S exchange on transducin was completely inhibited by the addition of GDP in a dose-dependent manner, but to a much less extent by ADP. Data are presented as the percentage of the GTPγ35S exchange control without adding any nucleotide. (C) Chimera Rh3C-catalyzed GTPγ35S exchange on transducin was inhibited by the addition of a high affinity transducin C-terminal peptide analog (termed VLED) identified by Martin et al. (1996), but only minimally by a randomized version of the peptide. Data are presented as the percentage of the GTPγ35S exchange in the absence of any peptide (the top line).
Transducin activation by the bR/Rh chimeras was also measured by using the photoaffinity label [32P]-(4-azidoanilido)-GTP (Rasenick et al. 1994). This compound is used to “trap” the activated Gαt in an active state. Only activated Gαt will bind [32P]-(4-azidoanilido)-GTP. The covalently derivatized Gαt was separated from the βγ subunits using SDS-PAGE, and the [32P] incorporated was determined using a PhosphorImager (Fig. 6). The level of activation of transducin by chimeras Rh3C and Rh3G was measured to be 2.25-fold and 1.75-fold, respectively.
Figure 6.

Transducin activation by bR/Rh chimeras assayed by photolabeling using [32P]-(4-azidoanilido)-GTP. Holotransducin was incubated with the photoactivatable GTP analog, [32P]-(4-azidoanilido)-GTP, either alone or in the presence of purified chimeras (Rh3C or Rh3G) at an approximate molar ratio of 1:1, as can be seen from the Coomassie-stained bands (left panel). The samples were photoactivated using a 1-kW mercury vapor lamp from a distance of 10 cm. Samples were electrophoresed on a 12% SDS-polyacrylamide gel, and radioactivity was measured using a Molecular Dynamics PhosphorImager (pseudocolor is depicted; right panel). The photolabeling of Gαt was quantitated using ImageQuant and plotted as fold labeling with each bar (±SD) on top of its corresponding triplet lanes.
A physical interaction between chimera Rh3C and transducin evidenced by cocentrifugation experiments
To further confirm that the activation of transducin by chimeras Rh3C and Rh3G was specific, physical interaction between transducin and bR/Rh chimera was investigated. As detected by Western blotting analysis using anti-Gαt raised against the N terminus of Gαt (Fig. 7A, C), transducin was cocentrifuged with Rh3C at a significantly higher level than the background detected for wild-type bR (Fig. 7B). The background for bR was likely due to a nonspecific interaction between transducin and the purple membrane lipid. The high-affinity Gαt C-terminal analog peptide (VLED) prevented the interaction between Rh3C and transducin, as indicated by the data from cocentrifugation of transducin with Rh3C in the presence of the peptide VLED (Fig. 7A, lane 4). Furthermore, the peptide did not reduce the background signal from bR (Fig. 7A, lane 2), indicating a specific competition between the C terminus of Gαt and Rh3C loop 3. Interestingly, when loop 3 in chimera Rh3C was cleaved by V8 protease, Gαt cocentrifuged with Rh3C only at a background level similar to that detected with the bR control (Fig. 7C, cf. lanes 3 and 1), indicating that loop 3 in Rh3C is the domain that specifically interacted with Gαt. V8 protease treatment removes eight to 10 amino acids from loop 3 in the bR/Rh loop 3 chimera, while the two fragments generated by V8 cleavage (Fig. 7D, lane 3) are still held integrated by the membrane (Heymann et al. 2000).
Figure 7.

Cocentrifugation of transducin and chimera Rh3C. (A) Cocentrifugation of transducin with bR (lane 1) and Rh3C (lane 3) was detected by Western blotting. The Gαt C terminus analog VLED (in 10-fold molar excess over Gαt) protected pull-down of transducin by Rh3C (lane 4) but not the background by bR (lane 2). (B) Quantitation of transducin pull-down. The intensity of Western signal from Gαt is normalized by the amount of protein in the bR or Rh3C band as detected by Coomassie staining. Each bar represents an average (±SD) of three lanes, and all the lanes are from the same blot. (*) p < 0.05. (C) Cocentrifugation of transducin with V8 protease-cleaved Rh3C (lane 3) is compared to that with uncleaved Rh3C (lane 2) and bR (lane 1). Western blotting was performed as in A. (D) A Coomassie-stained SDS gel (16%) shows cleavage of Rh3C loop 3 by V8 protease. Lane 3 indicates that Rh3C was cleaved into two bands, a higher band (∼18 kDa) and a lower one (∼6 kDa). bR (lane 1) and Rh3C (lane 2) were also run on the same gel to indicate the position of uncleaved Rh3C.
Discussion
The ultimate goal of this study was to select a bR/Rh chimera that can facilitate robust production of chimeric protein sufficient both in quantity and quality for possible structural studies. More importantly, the chimera should be functional in activating transducin and thus be useful for investigation of rhodopsin–transducin interactions.
GPCRs initiate a wide variety of cellular events by activating G-proteins; how these proteins interact is therefore the focus of intense interest. Rhodopsin, the prototypical member in the GPCR superfamily, is the only member with a resolved atomic structure (Palczewski et al. 2000). However, since this structure was obtained from the inactive form, the GPCR/G-protein activation process still remains largely unknown. Using chimeric GPCRs provides alternate approaches for obtaining structural information for the interactions of GPCRs/G-proteins (Frielle et al. 1988; Kobilka et al. 1988; Cotecchia et al. 1990; Wong et al. 1990; Ahumada et al. 2002; Kim et al. 2005). Since bR has been proven to be highly stable, simple in purification, and well characterized structurally (Luecke et al. 1999; Lanyi 2004), it is an ideal scaffold for constructing chimeras that contain the elements of rhodopsin (Heymann et al. 2000) or other GPCRs (Jaakola et al. 2005). Multiple lines of evidence suggest that the rhodopsin cytoplasmic loop 3 is an important domain for transducin activation (Franke et al. 1990; Cai et al. 2001; Natochin et al. 2003; Janz and Farrens 2004).
In this study, by selecting from a family of bR/Rh loop 3 chimeras, we obtained two rhodopsin loop 3 chimeras (Rh3C and Rh3G) that could be readily purified in large amounts that formed a correctly organized 2D crystal and, most interestingly, demonstrated a substantial capacity to activate transducin.
The successful selection of the Rh3C (and Rh3G) chimera was facilitated by an efficient screening method using the purple colony phenotype assay (Fig. 2A), for the following reasons: First, it was an indicator that the chimeric gene was successfully being integrated into the genome of H. salinarum. Second, it was an indicator that our chimeric protein is being folded in the membrane of H. salinarum in a manner analogous to that of bR; that is, it was forming a seven transmembrane helices structure, capable of binding retinal. Third, it was an indicator that expression levels of the chimeric protein were significantly abundant. And fourth, it provided an early decision point to determine whether or not the expression of a particular chimera or group of chimeras could be successfully accomplished with H. salinarum. We were therefore able to select from several constructs and focused on characterization of some chimeras that formed PMs (Supplemental Table SI).
One of the major advantages of bR/GPCR chimeras is the robust purification, benefiting from the unique property of bR in forming the highly organized lattice, as has also been proven by other studies (Luecke et al. 1999; Heymann et al. 2000). Since the highly organized bR homotrimers are enriched in the PM, they can be easily separated from the contaminating proteins by sucrose gradient centrifugation, and the purple color is a discernible indicator for the purified chimeric protein (Oesterhelt and Stoeckenius 1974). We can therefore readily obtain many milligrams of >95% pure Rh3C by sucrose gradient centrifugation without using a secondary purification method (Fig. 2).
Since the purified chimeras are able to maintain the physical properties similar to bR, as determined by UV/Vis and CD spectroscopy (Fig. 3), and a similar migration through sucrose density gradients (Fig. 2B), we suggest that (as also previously suggested by Jaakola et al. 2005) it is possible to overexpress and purify sufficient bR/GPCR chimeras suitable for structural determination. Homotrimers of Rh3C formed in the PM lattice were clearly revealed by the AFM imaging (Fig. 4), indicating that the crystallinity of the lattice was not disrupted by an insertion of the bovine rhodopsin loop 3. The combined data strongly suggest that our bR/Rh chimeras are forming a high-order 2D crystal similar to that of wild-type bR, which may allow for structural determination of the bovine rhodopsin cytoplasmic loop 3 domain.
Most importantly, we have also demonstrated that the bR/Rh chimera Rh3C (and Rh3G) expressed in the PM lattice is functionally active with regard to its ability to catalyze GTP exchange for GDP in transducin, and that this exchange is specific as demonstrated by the ability of GDP and a Gαt C terminus analog to inhibit this exchange (Fig. 5B, C). The data from Gαt photolabeling using the GTP analog [32P]-(4-azidoanilido)-GTP are further consistent with this conclusion (Fig. 6). Furthermore, the specific activation of transducin by chimera Rh3C is supported by the physical interaction observed between transducin and Rh3C (Fig. 7). On one hand, the high-affinity peptide analog of the Gαt C terminus prevented this interaction, strongly arguing that this interaction involved the Gαt C terminus. On the other hand, the chimera Rh3C with loop 3 cleaved by V8 protease failed to “pull down” transducin, indicating that it was loop 3 that specifically interacted with Gαt.
Therefore, these data (Fig. 7) together with the evidence from the GTPγ35S binding assay (Fig. 5) support a point-to-point interaction between Gαt and Rh3C; that is, the rhodopsin loop 3 interacts with the C terminus of Gαt. This is consistent with a recent study showing that a direct interaction occurs between the C terminus of Gαt and a hydrophobic patch involving loop 3 in the metarhodopsin II (Meta II) cytoplasmic face that is exposed during receptor activation (Janz and Farrens 2004). Although the other regions of rhodopsin have also been suggested to contribute to the interactions between these two signaling proteins (Franke et al. 1990; Marin et al. 2000; Cai et al. 2001; Hamm 2001; Terakita et al. 2002; Natochin et al. 2003), our data suggest that the loop 3/Gαt C terminus interaction is prominent.
The success in generation of functionally active bR/Rh chimeras in this study may be attributed to our appropriate strategy for chimera construction. The choices of amino acids and the construction of the bR/Rh chimeras were systematically approached based on the data of Farrens et al. (1996) and Luecke et al. (1999). Additional data from Choi et al. (2002) indicate a structural change in loop 3 upon activation of rhodopsin to the Meta II conformation, which may be induced by a “twist” of rhodopsin helix 6 upon activation. To mimic this twisting action, residues of rhodopsin were added systematically onto either the N or the C terminus of the loop 3 sequence introduced into bR, in such a way as to replace the bR helix E and F residues (Fig. 1). Since the junction positions between bR and rhodopsin (at the N and C termini) are within rhodopsin helices 5 and 6, respectively (Palczewski et al. 2000), the addition of one amino acid would induce a twisting of the helical portion of rhodopsin. The addition of three amino acids would produce almost a complete rotation of the helix. With the possibility of three amino acids added to the N terminus and three amino acids to the C terminus, there are nine possible permutations of the combinations (chimeras A–I) (see Table 1; Supplemental Table SI). Importantly, the rhodopsin loop 3 chimeras included the residues Leu226–Val230 at the N-terminal side and Val250 at the C-terminal side of the loop 3 insertion. The residues Leu226, Thr229, and Val230 are reported by Janz and Farrens (2004) to play a key role for transducin–MetaII interaction by imparting high affinity binding for the Gαt C-terminal tail with each of these hydrophobic residues contributing up to 3 kcal/mol to the binding energy. In addition, a movement of the V250 region in helix 6 toward the helix 5 residues Leu226–Val230 due to light activation is suggested to form a hydrophobic patch in the rhodopsin cytoplasmic face, which directly interacts with the Gαt C terminus and increases the affinity of transducin binding. It is also consistent with the observations by Natochin et al. (2003); that is, the reverse substitutions of the loop 3 residues Thr229/Val230 and Ser240/Thr242/Thr243/Gln244 on a poly-Ala background produced a rhodopsin mutant with a full capacity for transducin activation. We therefore reason that constitutively active rhodopsin loop 3 chimeras could have been generated in this study. Among the several bR/Rh chimeras that have similar lengths of rhodopsin loop 3 inserts, Rh3C showed the highest activity in stimulating GTP/GDP exchange of Gαt. This likely reflects that the proper orientation and rigidity of rhodopsin loop 3 are required for interacting with and activating Gαt (Farrens et al. 1996; Choi et al. 2002; Janz and Farrens 2004).
In summary, we have demonstrated the ability for producing large quantities of bR chimeric proteins suitable for the structural study of transducin–rhodopsin interactions in the activated state. Our strategy should also be applicable for generating other bR/GPCR chimeras functional for G-protein activation. It is likely that the structural determination (i.e., crystallography) of the bR/GPCR chimeras alone or in complex with G-proteins will provide important new knowledge regarding the mechanisms involved in G-protein activation.
Materials and methods
Construction of bR/GPCR chimeras
Using the crystal structures of bacteriorhodopsin (Luecke et al. 1999) and bovine rhodopsin (Palczewski et al. 2000), a chimera was modeled in which the third cytoplasmic loop of bovine rhodopsin was exchanged for the third intracellular loop of bR (see Fig. 1 for a schematic representation). In an effort to select functional bR/GPCR chimeras, various bR/GPCR chimeras were constructed using the same method, as listed in Table 1 and Supplemental Table SI. The construction of chimera Rh3C is described here as an example for all the constructs. Rh3C was constructed by replacing amino acid residues S158 to E166 in bR with residues Y223 to M253 from bovine rhodopsin. The DNA template for the bovine rhodopsin loop 3 insert was designed using synthetic oligonucleotides that incorporated optimum codon usage for H. salinarum as previously described (Robb 1995). The following complementary oligonucleotides were used to generate the bovine rhodopsin loop 3 template:
5′-GGGTTCACCGGCCAGCTCGTCTTCACGGTCAAGGAGGCGGCGGCGCAGCAGCAGGAGTCGGCGACGACGCAGAAGGCGGAGAAG-3′ and
5′-GGACCACAACACAACGGTAACGTTACGCAGTACTTTGAACGTGGATGCGACGCGCGTGACCTCCTTCTCCGCCTTCTGCGTCGTCGCCGA-3′.
These oligonucleotides were annealed and extended using Taq Plus Precision DNA Polymerase (Stratagene). The product was gel-purified on a 2% agarose gel and used as a template for a second primer extension using the following oligonucleotides:
5′-GCTGCCGATCAGCCACACGACTGGATACGCGGACCACAACACAACGGTAACGTTACGCAGTACTTTGAACGTGGATGCGACCATGCGCGTGACCTCCTTC-3′ and
5′-TTTGTACATGTACATCCTGTACGTGCTGTTCTTCGGGTTCACCTACGGCCAGCTCGTCTTCACGGTCAAGGAGG-3′.
The product from the second extension was double digested with BsrGI and BstXI to generate the rhodopsin loop 3 insert. To generate a vector with the bop (gene for bR) sequence containing both BsrGI and BstXI restriction sites (pAHGA), the bop gene from pMPK85 was subcloned into pGEM11ZF+ (Promega), and a BstXI restriction site was introduced into the transmembrane domain 6 of bR by a silent mutation following the protocol of the GENE EDITOR kit (Promega) using primer M1 (5′-CGCGTATCCAGTCGTGTGGC-3′). With a BsrGI site present in the transmembrane domain 5 of bR, the construct pAHGA conferred double restriction sites suitable for replacing the third cytoplasmic loop of bR. The bR/Rh chimeric construct (pAHGA-Rh3C) was thus obtained by a ligation of the rhodopsin loop 3 insert into BsrGI/BstXI double-digested pAHGA.
For transforming H. salinarum, a transformation construct (pMPK-Rh3C) was generated by ligating the bR/Rh chimeric bop insert (obtained by BamHI digestion) into the BamHI-digested pMPK85 vector. pMPK-Rh3C was amplified in Escherichia coli and then purified using a Midiprep Spin Plasmid Extraction kit (Qiagen). The plasmid was used without further manipulation for transforming H. salinarum as described below.
All the constructs generated in this study were confirmed by DNA sequencing, which was conducted at the Biotechnology Center, University of Wisconsin–Madison.
Transformation of H. salinarum
Transformation of the bR/GPCR chimera constructs into the bop deletion strain of H. salinarum MPK40 was performed as previously described (Peck et al. 2000). Each transformation was first selected on an agar plate containing CM (4.2 M NaCl, 8 mM MgSO4, 10 mM trisodium citrate, 27 mM KCl, 0.3% yeast extract, 0.5% tryptone) and 4 μg/mL mevinolin (Lovastatin, Merck). Small colonies were observed after 7 d of incubation at 37°C. These colonies were then re-plated onto the CM plates with no mevinolin. Colonies were observed after 7 d at 37°C. The plates were then exposed to continuous white light at room temperature for an additional 3–4 d. Colonies successfully integrating and expressing a chimera on the cell membrane and binding retinal in an appropriate chromophore environment exhibited a purple phenotype. These purple colonies were then picked and plated for the second round. After 7 d, colonies were observed, and the plates were then exposed to continuous white light at room temperature for 3–4 d. All the colonies on these second-round CM plates exhibited a purple phenotype. Integration of the chimeric DNA into H. salinarum’s genome was characterized by DNA sequencing.
Expression and purification of bR/GPCR chimeras
Expression and purification of bR/GPCR chimeras were performed as previously described for bR (Oesterhelt and Stoeckenius 1974). Briefly, a single purple colony from a CM plate was used to inoculate 5 mL of CM media, which was grown for 4 d at 40°C with 270 rpm shaking, and then used to inoculate 1.8 L of RM (CM containing 1% OXOID bacteriological peptone) media in a 2-L flask. This culture was grown for 4 d in an illuminated shaker at 40°C with shaking at 250 rpm, and then harvested by centrifugation at 4000g for 35 min at 5°C. The cell pellet was resuspended in 18 mL of water containing PMSF and DNase I, and dialyzed against 2 L of water containing an EDTA-free protease inhibitor cocktail (EDTA-Free COMPLETE, Boeringer Mannheim) in a dialysis tubing (Visking) for 18 h at 4°C. The dialysate was suspended in 5 mM NaCl containing a complete protease inhibitor cocktail (COMPLETE, Boeringer Mannheim) and the purple membranes (PMs) then collected by ultracentrifugation in a Ti 45 rotor at 27,000 rpm for 40 min at 4°C. The purple pellet was resuspended in a 5 mM NaCl solution, carefully avoiding disturbance of the grayish cell debris. The resuspended PMs were then recentrifuged as just described. The pellet from the second spin was resuspended in 2 mL of NaCl solution and loaded onto 15 mL of a 30%–58% continuous sucrose density gradient containing the complete protease inhibitor cocktail. The sample was then centrifuged in an SW28 swinging bucket rotor at 27,000 rpm for 17 h. PM-containing bands were carefully syringed out by puncturing the polyallomer centrifuge tubes (25 × 89 mm [Beckman]) with an 18-gauge needle. Samples not for immediate use were stored at −80°C in the sucrose mixture. Samples that were to be used immediately had the sucrose removed by performing two rounds of NaCl solution centrifugation in the Ti 45 rotor under the conditions described above.
UV/Vis spectroscopy
Retinal binding and chromophore generation in the bR/GPCR chimeric proteins were characterized by UV/Vis spectrophotometry as described previously (Ahl et al. 1990; Chen and Gouaux 1996), with a Perkin-Elmer Lambda 2 spectrophotometer at room temperature. Prior to measurement, purified PMs were diluted 500-fold into water from a stock solution, and light-adapted for 5 min under illumination >520 nm. A scan speed of 480 nm/min was used with 0 nm smoothing and a reading slot width of 2 nm.
Circular dichroism (CD) spectroscopy
For characterization of the homotrimer formation and exciton coupling in the chimeric proteins, CD spectroscopy was performed as previously described (Brith-Linder and Rosenheck 1977) on an AVIV 202 DS circular dichroism spectrometer. The sample suspended in a 25 mM sodium acetate solution (pH 5.0) at a concentration of ∼18 μM was contained in a 1-cm quartz microcell. Scans were recorded from 700 nm to 450 nm at 25°C using a 4-sec averaging time, a 1-nm bandwidth, a 2-nm sampling interval, and a 1-sec settling time. All data values plotted were corrected by subtracting the baseline absorbance of a 25 mM sodium acetate solution.
Atomic force microscopy (AFM)
The AFM imaging of the purified Rh3C PM was performed according to Heymann et al. (2000). All images were recorded in the imaging buffer (10 mM Tris-HCl at pH 7.8, 100–150 mM KCl) at room temperature under ambient pressure. The diluted Rh3C PM (∼10 μg/mL) was adsorbed to freshly cleaved mica for 20 min. The sample was washed with the imaging buffer and then mounted on the piezoelectric scanner of the atomic force microscope (Nanoscope III, Digital Instruments), which was equipped with a liquid cell. The cantilevers used had nominal force constants of k = 0.09 N/m or k = 0.02 N/m and oxide sharpened Si3N4 tips (Olympus Ltd.). Prior to scanning the sample, the operating point of the microscope was set to forces <0.5 nN.
GTPγS-binding assay for bR/chimeras
GTPγS binding was assayed according to a previously described method for bovine rhodopsin activation of transducin (Wessling-Resnick and Johnson 1987). Holotransducin (HoloT) and stripped membranes containing rhodopsin were prepared from rod outer segments (ROS) isolated from frozen, dark-adapted bovine retinas (W.L. Lawson Co.) following the established protocols (Fung 1983; Grant et al. 2006). For optimization of the assay conditions, holoT was reconstituted at 0.1–1.0 μM with varying concentrations of rhodopsin, bR, or chimeras (0.5–10.0 μM) in a 200-μL assay buffer (20 mM HEPES at pH 7.5, 5.0 mM MgCl2, 20 mM NaCl, and 1.0 mM DTT). GTPγ35S (50.0 nM to 5.0 μM) was added, and the mixture was incubated at 30°C for 1–60 min. Aliquots of the reaction mixture (50–150 μL) were vacuum-filtered through nitrocellulose filters (HAWP 45/Millipore) on a Millipore 1225 vacuum manifold and washed with 15 mL of ice-cold kill buffer (10 mM Tris at pH 7.5, 100.0 mM NaCl, and 5.0 mM MgCl2). Filters were then placed in scintillation fluid overnight, and the radioactivity was quantitated by liquid scintillation counting. For the GDP and ADP inhibition of transducin activation by the rhodopsin loop 3 chimera, GDP and ADP were added to the GTPγS assay at concentrations ranging from 10 μM to 500 μM.
For the peptide inhibition studies, peptides were added to the GTPγS-binding assay at concentrations ranging from 0.125 μM to 50 μM. The peptide sequences are as follows: high-affinity analog of the C terminus of transducin α subunit (Gαt), VLEDLKSCGLFG (termed VLED in this study) (Martin et al. 1996); a random peptide, SSVFLVVDRSR.
Photoaffinity labeling of bR/Rh–stimulated transducin with [32P]-(4-azidoanilido)-GTP
Chimera activation of transducin was additionally characterized using [32P]-(4-azidoanilido) guanosine 5′-triphosphate (GTP) as previously described (Rasenick et al. 1994). Briefly, holotransducin (0.625 μM) was added to 0.625 μM bR/bovine rhodopsin loop 3 chimera Rh3C or Rh3G, in 200-μL assay buffer (20 mM HEPES at pH 7.5, 5 mM MgCl2, 20 mM NaCl, and 1 mM DTT). Varying concentrations of [32P]-(4-azidoanilido)-GTP (45 nM to 360 nM) were added to the mixture, and the mixture was incubated for 10 min at 30°C. The entire reaction mixture was then transferred to thick-wall Pyrex centrifuge tubes and photolyzed at 4°C for 10 sec at 10 cm from a 1-kW mercury vapor lamp. Laemmli (1976) buffer was added to final 1% SDS and 50 mM DTT, and the samples were run on SDS-PAGE (12%). After Coomassie blue staining, the gels were dried and subjected to PhosphorImager analysis. The radioactive content of the protein band corresponding to the α-subunit of transducin (∼40 kDa) was quantitated using ImageQuant software.
Cocentrifugation of transducin with chimera Rh3C
Holotransducin of 2.5 μM was incubated with bR and Rh3C, respectively, in the HEPES buffer (20 mM HEPES, 120 mM NaCl, 5 mM MgCl2) at pH 7.5. In order to minimize possible nonspecific interactions caused by the PM lipid, 2% glycerol and additional 500 mM NaCl were added into the reactions, and a low molar ratio of transducin versus bR and Rh3C (1:3) was used. After 10 min of incubation at room temperature, the reactions were centrifuged at 16,000g and 5°C for 25 min. The supernatant was carefully removed, and 50 mL of HEPES buffer was added to resuspend the PM pellet by gentle vortex, which was then centrifuged again for 25 min. The supernatant was discarded; the PM pellet was incubated with Laemmli buffer and then subjected to 10% SDS-PAGE. The PMs of bR and Rh3C were recovered at a similar ratio (∼85%) by centrifugation at 16,000g for 25 min, as estimated by Coomassie staining of the SDS-PAGE gel.
Other methods
To cleave the rhodopsin loop 3 on the Rh3C chimera (Heymann et al. 2000), 50 μg of Rh3C was treated with 5 μg of V8 protease (Sigma) at room temperature for 16 h in the pH 7.5 HEPES buffer. The PM was spun down at 16,000g and 5°C for 25 min, washed twice by repeating the centrifugation process, and then resuspended in the HEPES buffer.
For Western blotting detection of transducin that cocentrifuged with Rh3C, following SDS-PAGE the proteins were electrotransferred to a PVDF membrane (0.45 μm [Millipore]), and then immunodetected using polyclonal anti-Gαt antibody raised to the N terminus of Gαt (Affinity Bioreagents) and the chemiluminescence reagents from Pierce (Guo et al. 2005). Anti-Gαt was diluted 2000-fold; the secondary antibody, horse radish peroxidase, was diluted 200,000-fold.
Protein concentrations of the PMs were determined based on the absorption at 570 nm. Gαt concentration was measured by the Bradford method using BSA as a standard. The Western signal intensity and Coomassie-stained protein bands on the PVDF membrane were quantitated using NIH 1.62.
Acknowledgments
This work was supported by NIH grant GM33138 (to A.E.R.). We thank Ron F. Peck for his help in transformation of H. salinarum and helpful information. CD data were obtained at the University of Wisconsin–Madison Biophysics Instrumentation Facility, which is supported by the University of Wisconsin–Madison and grants BIR-9512577 (NSF) and S10 RR13790 (NIH).
Footnotes
Supplemental material: see www.proteinscience.org
Reprint requests to: Lian-Wang Guo, Department of Pharmacology, University of Wisconsin–Madison, 1300 University Avenue, Madison, WI 53706, USA; e-mail: lianwangguo@wisc.edu; fax: (608) 262-1257.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062192306.
Abbreviations: GPCR, G-protein-coupled receptors; bR, bacteriorhodopsin; Rh, rhodopsin; PM, purple membrane; bop, gene for bR; β2AR, β2-adrenergic receptor; Rh3C, the bR chimera with Rh cytoplasmic loop 3 (Y223–M253) replacing the corresponding third loop of bR (S158–E166); Rh3G, the bR chimera differing with Rh3C only in the Rh cytoplasmic loop 3 Q225–T251; β2L3, the bR chimera with β2AR cytoplasmic loop 3 R221–L275 replacing the corresponding third loop of bR (S158–E165); HoloT, transducin heterotrimer containing the α, β, and γ subunits; Gαt, transducin α subunit; VLED, the high-affinity analog peptide of the Gαt C terminus; CD, circular dichroism; AFM, atomic force microscopy.
References
- Abdulaev N.G., Strassmaier T.T., Ngo T., Chen R., Luecke H., Oprian D.D., Ridge K.D. 2002. Grafting segments from the extracellular surface of CCR5 onto a bacteriorhodopsin transmembrane scaffold confers HIV-1 coreceptor activity. Structure 10 515–525. [DOI] [PubMed] [Google Scholar]
- Ahl P.L., Price R., Smuda J., Gaber B.P., Singh A. 1990. Insertion of bacteriorhodopsin into polymerized diacetylenic phosphatidylcholine bilayers. Biochim. Biophys. Acta 1028 141–153. [DOI] [PubMed] [Google Scholar]
- Ahumada A., Slusarski D.C., Liu X., Moon R.T., Malbon C.C., Wang H.Y. 2002. Signaling of rat Frizzled-2 through phosphodiesterase and cyclic GMP. Science 298 2006–2010. [DOI] [PubMed] [Google Scholar]
- Altenbach C., Yang K., Farrens D.L., Farahbakhsh Z.T., Khorana H.G., Hubbell W.L. 1996. Structural features and light-dependent changes in the cytoplasmic interhelical E-F loop region of rhodopsin: A site-directed spin-labeling study. Biochemistry 35 12470–12478. [DOI] [PubMed] [Google Scholar]
- Brith-Linder M. and Rosenheck K. 1977. The circular dichroism of bacteriorhodopsin: Asymmetry and light-scattering distortions. FEBS Lett. 76 41–44. [DOI] [PubMed] [Google Scholar]
- Cai K., Itoh Y., Khorana H.G. 2001. Mapping of contact sites in complex formation between transducin and light-activated rhodopsin by covalent crosslinking: Use of a photoactivatable reagent. Proc. Natl. Acad. Sci. 98 4877–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G.Q. and Gouaux J.E. 1996. Overexpression of bacterio-opsin in Escherichia coli as a water-soluble fusion to maltose binding protein: Efficient regeneration of the fusion protein and selective cleavage with trypsin. Protein Sci. 5 456–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi G., Landin J., Galan J.F., Birge R.R., Albert A.D., Yeagle P.L. 2002. Structural studies of metarhodopsin II, the activated form of the G-protein coupled receptor, rhodopsin. Biochemistry 41 7318–7324. [DOI] [PubMed] [Google Scholar]
- Cotecchia S., Exum S., Caron M.G., Lefkowitz R.J. 1990. Regions of the α 1-adrenergic receptor involved in coupling to phosphatidylinositol hydrolysis and enhanced sensitivity of biological function. Proc. Natl. Acad. Sci. 87 2896–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon R.A., Sigal I.S., Strader C.D. 1988. Structure–function analysis of the β-adrenergic receptor. Cold Spring Harb. Symp. Quant. Biol. 53 487–497. [DOI] [PubMed] [Google Scholar]
- Farrens D.L., Altenbach C., Yang K., Hubbell W.L., Khorana H.G. 1996. Requirement of rigid-body motion of transmembrane helices for light activation of rhodopsin. Science 274 768–770. [DOI] [PubMed] [Google Scholar]
- Franke R.R., Sakmar T.P., Oprian D.D., Khorana H.G. 1988. A single amino acid substitution in rhodopsin (lysine 248-leucine) prevents activation of transducin. J. Biol. Chem. 263 2119–2122. [PubMed] [Google Scholar]
- Franke R.R., Konig B., Sakmar T.P., Khorana H.G., Hofmann K.P. 1990. Rhodopsin mutants that bind but fail to activate transducin. Science 250 123–125. [DOI] [PubMed] [Google Scholar]
- Fraser C.M., Chung F.Z., Wang C.D., Venter J.C. 1988. Site-directed mutagenesis of human β-adrenergic receptors: Substitution of aspartic acid-130 by asparagine produces a receptor with high-affinity agonist binding that is uncoupled from adenylate cyclase. Proc. Natl. Acad. Sci. 85 5478–5482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frielle T., Daniel K.W., Caron M.G., Lefkowitz R.J. 1988. Structural basis of β-adrenergic receptor subtype specificity studied with chimeric β 1/β 2-adrenergic receptors. Proc. Natl. Acad. Sci. 85 9494–9498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung B.K. 1983. Characterization of transducin from bovine retinal rod outer segments. I. Separation and reconstitution of the subunits. J. Biol. Chem. 258 10495–10502. [PubMed] [Google Scholar]
- Grant J.E., Guo L.-W., Vestling M.M., Martemyanov K.A., Arshavsky V.Y., Ruoho A.E. 2006. The N-terminus of GTPγ S-activated transducin α-subunit interacts with the C-terminus of the cGMP phosphodiesterase γ-subunit. J. Biol. Chem. 281 6194–6202. [DOI] [PubMed] [Google Scholar]
- Guo L.-W., Hajipour A.R., Gavala M.L., Arbabian M., Martemyanov K.A., Arshavsky V.Y., Ruoho A.E. 2005. Sulfhydryl-reactive, cleavable, and radioiodinatable benzophenone photoprobes for study of protein–protein interaction. Bioconjug. Chem. 16 685–693. [DOI] [PubMed] [Google Scholar]
- Hamm H.E. 2001. How activated receptors couple to G proteins. Proc. Natl. Acad. Sci. 98 4819–4821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamm H.E., Deretic D., Arendt A., Hargrave P.A., Koenig B., Hofmann K.P. 1988. Site of G protein binding to rhodopsin mapped with synthetic peptides from the α subunit. Science 241 832–835. [DOI] [PubMed] [Google Scholar]
- Herrmann R., Heck M., Henklein P., Henklein P., Kleuss C., Hofmann K.P., Ernst O.P. 2004. Sequence of interactions in receptor-G protein coupling. J. Biol. Chem. 279 24283–24290. [DOI] [PubMed] [Google Scholar]
- Heymann J.B., Pfeiffer M., Hildebrandt V., Kaback H.R., Fotiadis D., Groot B., Engel A., Oesterhelt D., Muller D.J. 2000. Conformations of the rhodopsin third cytoplasmic loop grafted onto bacteriorhodopsin. Structure 8 643–653. [DOI] [PubMed] [Google Scholar]
- Itoh Y., Cai K., Khorana H.G. 2001. Mapping of contact sites in complex formation between light-activated rhodopsin and transducin by covalent crosslinking: Use of a chemically preactivated reagent. Proc. Natl. Acad. Sci. 98 4883–4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaakola V.P., Rehn M., Moeller M., Alexiev U., Goldman A., Turner G.J. 2005. G-protein-coupled receptor domain overexpression in Halobacterium salinarum: Long-range transmembrane interactions in heptahelical membrane proteins. Proteins 60 412–423. [DOI] [PubMed] [Google Scholar]
- Janz J.M. and Farrens D.L. 2004. Rhodopsin activation exposes a key hydrophobic binding site for the transducin α-subunit C terminus. J. Biol. Chem. 279 29767–29773. [DOI] [PubMed] [Google Scholar]
- Kim J.M., Altenbach C., Kono M., Oprian D.D., Hubbell W.L., Khorana H.G. 2004. Structural origins of constitutive activation in rhodopsin: Role of the K296/E113 salt bridge. Proc. Natl. Acad. Sci. 101 12508–12513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.M., Hwa J., Garriga P., Reeves P.J., Rajbhandary U.L., Khorana H.G. 2005. Light-driven activation of β 2-adrenergic receptor signaling by a chimeric rhodopsin containing the β 2-adrenergic receptor cytoplasmic loops. Biochemistry 44 2284–2292. [DOI] [PubMed] [Google Scholar]
- Kisselev O.G., Downs M.A., McDowell J.H., Hargrave P.A. 2004. Conformational changes in the phosphorylated C-terminal domain of rhodopsin during rhodopsin arrestin interactions. J. Biol. Chem. 279 51203–51207. [DOI] [PubMed] [Google Scholar]
- Klein-Seetharaman J., Yanamala N.V., Javeed F., Reeves P.J., Getmanova E.V., Loewen M.C., Schwalbe H., Khorana H.G. 2004. Differential dynamics in the G protein-coupled receptor rhodopsin revealed by solution NMR. Proc. Natl. Acad. Sci. 101 3409–3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobilka B.K., Kobilka T.S., Daniel K., Regan J.W., Caron M.G., Lefkowitz R.J. 1988. Chimeric α2-, β2-adrenergic receptors: Delineation of domains involved in effector coupling and ligand binding specificity. Science 240 1310–1316. [DOI] [PubMed] [Google Scholar]
- Koenig B.W., Kontaxis G., Mitchell D.C., Louis J.M., Litman B.J., Bax A. 2002. Structure and orientation of a G protein fragment in the receptor bound state from residual dipolar couplings. J. Mol. Biol. 322 441–461. [DOI] [PubMed] [Google Scholar]
- Kudo M., Osuga Y., Kobilka B.K., Hsueh A.J. 1996. Transmembrane regions V and VI of the human luteinizing hormone receptor are required for constitutive activation by a mutation in the third intracellular loop. J. Biol. Chem. 271 22470–22478. [DOI] [PubMed] [Google Scholar]
- Laemmli U.K. 1976. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227 680–685. [DOI] [PubMed] [Google Scholar]
- Lanyi J.K. 2004. Bacteriorhodopsin. Annu. Rev. Physiol. 66 665–688. [DOI] [PubMed] [Google Scholar]
- Li J., Edwards P.C., Burghammer M., Villa C., Schertler G.F. 2004. Structure of bovine rhodopsin in a trigonal crystal form. J. Mol. Biol. 343 1409–1438. [DOI] [PubMed] [Google Scholar]
- Luecke H., Richter H.T., Lanyi J.K. 1998. Proton transfer pathways in bacteriorhodopsin at 2.3 Å resolution. Science 280 1934–1937. [DOI] [PubMed] [Google Scholar]
- Luecke H., Schobert B., Richter H.T., Cartailler J.P., Lanyi J.K. 1999. Structure of bacteriorhodopsin at 1.55 Å resolution. J. Mol. Biol. 291 899–911. [DOI] [PubMed] [Google Scholar]
- Lundstrom K. 2005. Structural genomics of GPCRs. Trends Biotechnol. 23 103–108. [DOI] [PubMed] [Google Scholar]
- Luttrell L.M., Ostrowski J., Cotecchia S., Kendall H., Lefkowitz R.J. 1993. Antagonism of catecholamine receptor signaling by expression of cytoplasmic domains of the receptors. Science 259 1453–1457. [DOI] [PubMed] [Google Scholar]
- Marin E.P., Krishna A.G., Zvyaga T.A., Isele J., Siebert F., Sakmar T.P. 2000. The amino terminus of the fourth cytoplasmic loop of rhodopsin modulates rhodopsin–transducin interaction. J. Biol. Chem. 275 1930–1936. [DOI] [PubMed] [Google Scholar]
- Martin E.L., Rens-Domiano S., Schatz P.J., Hamm H.E. 1996. Potent peptide analogues of a G protein receptor-binding region obtained with a combinatorial library. J. Biol. Chem. 271 361–366. [DOI] [PubMed] [Google Scholar]
- Natochin M., Gasimov K.G., Moussaif M., Artemyev N.O. 2003. Rhodopsin determinants for transducin activation: A gain-of-function approach. J. Biol. Chem. 278 37574–37581. [DOI] [PubMed] [Google Scholar]
- O'Dowd B.F., Hnatowich M., Regan J.W., Leader W.M., Caron M.G., Lefkowitz R.J. 1988. Site-directed mutagenesis of the cytoplasmic domains of the human β 2-adrenergic receptor. Localization of regions involved in G protein-receptor coupling. J. Biol. Chem. 263 15985–15992. [PubMed] [Google Scholar]
- Oesterhelt D. and Stoeckenius W. 1974. Isolation of the cell membrane of Halobacterium halobium and its fractionation into red and purple membrane. Methods Enzymol. 31 667–678. [DOI] [PubMed] [Google Scholar]
- Okada T., Fujiyoshi Y., Silow M., Navarro J., Landau E.M., Shichida Y. 2002. Functional role of internal water molecules in rhodopsin revealed by X-ray crystallography. Proc. Natl. Acad. Sci. 99 5982–5987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada T., Sugihara M., Bondar A.N., Elstner M., Entel P., Buss V. 2004. The retinal conformation and its environment in rhodopsin in light of a new 2.2 Å crystal structure. J. Mol. Biol. 342 571–583. [DOI] [PubMed] [Google Scholar]
- Palczewski K., Kumasaka T., Hori T., Behnke C.A., Motoshima H., Fox B.A., Le Trong I., Teller D.C., Okada T., Stenkamp R.E.et al. 2000. Crystal structure of rhodopsin: A G protein-coupled receptor. Science 289 739–745. [DOI] [PubMed] [Google Scholar]
- Peck R.F., Dassarma S., Krebs M.P. 2000. Homologous gene knockout in the archaeon Halobacterium salinarum with ura3 as a counterselectable marker. Mol. Microbiol. 35 667–676. [DOI] [PubMed] [Google Scholar]
- Pereira R. and Cerione R.A. 2005. A switch 3 point mutation in the α subunit of transducin yields a unique dominant-negative inhibitor. J. Biol. Chem. 280 35696–35703. [DOI] [PubMed] [Google Scholar]
- Rasenick M.M., Talluri M., Dunn W.J. III. 1994. Photoaffinity guanosine 5′-triphosphate analogs as a tool for the study of GTP-binding proteins. Methods Enzymol. 237 100–110. [DOI] [PubMed] [Google Scholar]
- Ridge K.D., Abdulaev N.G., Sousa M., Palczewski K. 2003. Phototransduction: Crystal clear. Trends Biochem. Sci. 28 479–487. [DOI] [PubMed] [Google Scholar]
- Ridge K.D., Abdulaev N.G., Zhang C., Ngo T., Brabazon D.M., Marino J.P. 2006. Conformational changes associated with receptor stimulated guanine nucleotide exchange in a heterotrimeric G-protein α-subunit: NMR analysis of GTPγ S-bound states. J. Biol. Chem. 281 7635–7648. [DOI] [PubMed] [Google Scholar]
- Robb F.T., Sowers K.R., DasSharma S., Schreier H.J., Fleischmann E.M. In Archaea: A laboratory manual . 1995. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Teller D.C., Okada T., Behnke C.A., Palczewski K., Stenkamp R.E. 2001. Advances in determination of a high-resolution three-dimensional structure of rhodopsin, a model of G-protein-coupled receptors (GPCRs). Biochemistry 40 7761–7772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terakita A., Yamashita T., Nimbari N., Kojima D., Shichida Y. 2002. Functional interaction between bovine rhodopsin and G protein transducin. J. Biol. Chem. 277 40–46. [DOI] [PubMed] [Google Scholar]
- Turner G.J., Miercke L.J., Mitra A.K., Stroud R.M., Betlach M.C., Winter-Vann A. 1999. Expression, purification, and structural characterization of the bacteriorhodopsin-aspartyl transcarbamylase fusion protein. Protein Expr. Purif. 17 324–338. [DOI] [PubMed] [Google Scholar]
- Wessling-Resnick M. and Johnson G.L. 1987. Allosteric behavior in transducin activation mediated by rhodopsin. Initial rate analysis of guanine nucleotide exchange. J. Biol. Chem. 262 3697–3705. [PubMed] [Google Scholar]
- Wong S.K., Parker E.M., Ross E.M. 1990. Chimeric muscarinic cholinergic: β-Adrenergic receptors that activate Gs in response to muscarinic agonists. J. Biol. Chem. 265 6219–6224. [PubMed] [Google Scholar]
- Yeagle P.L., Alderfer J.L., Albert A.D. 1997. Three-dimensional structure of the cytoplasmic face of the G protein receptor rhodopsin. Biochemistry 36 9649–9654. [DOI] [PubMed] [Google Scholar]