

Abstract
The receptor-type protein tyrosine phosphatase (RPTP) CD148 is thought to have an inhibitory function in signaling and proliferation in non-hematopoietic cells. However, its role in the immune system has not been thoroughly studied. Our analysis of CD148 loss of function mice suggests that CD148 has a positive regulatory function in B-cells and macrophages, similar to the role of CD45 as a positive regulator of Src-family kinases (SFKs). Analysis of CD148/CD45 doubly-deficient B cells and macrophages revealed hyperphosphorylation of the C-terminal inhibitory tyrosine of SFKs accompanied by substantial alterations in B and myeloid lineage development and defective immunoreceptor signaling. These findings suggest the C-terminal tyrosine of SFKs is a common substrate for both CD148 and CD45 phosphatases and that a reassessment of the function of CD45 in the B and myeloid lineages is warranted.
Introduction
The state of tyrosine phosphorylation is a tightly regulated equilibrium, coordinated by the actions of protein tyrosine kinases (PTKs) and protein tyrosine phosphatases (PTPs). Src family kinases (SFKs) play essential roles in the initiation of immunoreceptor signaling (Iwashima et al., 1994). SFKs have two regulatory tyrosine phosphorylation sites. Autophosphorylation of the tyrosine in the kinase activation loop leads to an increase in catalytic activity. Dephosphorylation of this tyrosine has an inhibitory effect and can be accomplished by several PTPs, the best characterized being PEP (Cloutier and Veillette, 1999). When phosphorylated by Csk, the second regulatory site, in the C-terminal tail of SFKs binds its own SH2 domain, leading to an autoinhibited “closed” conformation (Sicheri and Kuriyan, 1997). The CD45 RPTP, highly expressed on hematopoietic cells, is known to dephosphorylate this tyrosine in SFKs (Hermiston et al., 2003).
CD45 is critical for the immune system since both CD45 deficient mice (Byth et al., 1996; Kishihara et al., 1993; Mee et al., 1999) and humans develop severe combined immunodeficiency (SCID) (Kung et al., 2000; Tchilian et al., 2001). CD45 deficiency affects T lymphocytes most profoundly. Thymocyte development is severely blocked at the positive selection stage (Byth et al., 1996; Kishihara et al., 1993; Mee et al., 1999). In surprising contrast, CD45 deficient B cells develop relatively normally in the bone marrow, although splenic B cell development is partially blocked at transitional stages (Byth et al., 1996; Kishihara et al., 1993). Moreover, while in CD45 deficient T cells TCR-mediated increase in free intracellular Ca2+ concentration is almost completely abolished (Koretzky et al., 1991; Mee et al., 1999), in CD45 deficient B cells it is largely preserved (Benatar et al., 1996; Hermiston et al., 2005). Nevertheless, CD45 deficient B cells do not proliferate well in vitro after stimulation with anti-IgM antibodies (Benatar et al., 1996; Byth et al., 1996; Kishihara et al., 1993). However, in vivo responses to T-dependent and T-independent antigens are almost normal, with the exception that germinal centers are less persistent, when CD45+/+ T cells are supplied (Huntington et al., 2006; Kong et al., 1995). Collectively, these observations suggest that CD45 deficiency substantially increases the threshold for TCR signaling but has diminished or more complex effects on BCR signaling. Even though SFKs are substrates of CD45, in contrast to the partial defect in B cell development in CD45 deficient mice, loss of B cell specific SFKs blocks B cell development at the pro-B /pre-B cell transition (Saijo et al., 2003). The different outcomes from CD45 and SFK deficiencies on B cell development suggest that there may be another phosphatase involved. In the myeloid lineage, no major developmental defects were observed, although myelopoiesis was slightly increased, in CD45-/- mice (Kishihara et al., 1993). Macrophages from these mice displayed alterations in integrin-mediated adhesion (Roach et al., 1997). The variable effects of CD45 deficiency in different lineages have led to varied interpretations of its functions. One possible explanation for these inconsistencies is that other mechanisms in B cells and in myeloid cells are able to compensate for the loss of CD45 in these lineages.
As suggested above, one possibility is another PTP. A candidate RPTP is CD148. CD148 is expressed in most leukocyte lineages including B cells and myeloid cells, but in T cells its expression is induced after activation (Lin et al., 2004). It differs substantially from CD45, having 8-9 fibronectin domains in its extracellular domain and a single (rather than tandem) PTP homology domain intracellularly (Supplementary Fig. 1). Very limited data address the function of CD148 in the immune system. In the Jurkat T cell line, induced expression of CD148 led to selective dephosphorylation of phospholipase Cγ1 (PLCγ1) and linker for activation of T cells (LAT) resulting in downregulation of TCR-dependent signaling (Baker et al., 2001). Its function in the B and myeloid lineages is largely uncharacterized. CD148 is also expressed in the cells outside the immune system, including epithelial and endothelial cells as well as fibroblasts, where it was shown to inhibit cell growth and promote differentiation, potentially acting as a tumor suppressor (Balavenkatraman et al., 2006; Keane et al., 1996; Ruivenkamp et al., 2002; Trapasso et al., 2000; Trapasso et al., 2004). It has also been reported to dephosphorylate the inhibitory C-terminal tyrosine of c-Src in a rat thyroid carcinoma, leading to increased c-Src activity and cell-substratum adhesion (Pera et al., 2005).
The replacement of the CD148 PTP domain with GFP in mice led to early embryonic lethality due to vascular defects, thus preventing any study of the immune system (Takahashi et al., 2003). Here, we attempted to overcome this difficulty by generating mice in which the transmembrane (TM) exon of CD148 is conditionally or constitutively deleted. Surprisingly, our constitutively deficient mice (TM-KO) were viable without any gross abnormalities. Recently, an independent CD148 deficient mouse showed similar viability (Trapasso et al., 2006). Our detailed analysis of the immune system in CD148 TM-KO mice revealed a partial block in peripheral B cell development, resembling the B cell phenotype in CD45 deficient mice. This raised the possibility that these structurally distinct RPTPs share overlapping functions. The CD148 TM-KO/CD45 doubly-deficient (DKO) mice had substantial defects in B and myeloid cell lineage development and immunoreceptor signaling. Strong hyperphosphorylation of the negative regulatory C-terminal tyrosine of SFKs in DKO B cells and macrophages suggests that failure to dephosphorylate the inhibitory tyrosine of SFKs leads to the decreased activity of these kinases in vivo and results in early defects in the signaling pathways mediated by BCR and Fc receptors.
Results
Generation of CD148 deficient mice
Because of the reported embryonic-lethality of CD148 deficiency in mice, we designed a lineage-specific inactivation of CD148. We inserted loxP recombination sites flanking the transmembrane (TM) exon, along with a floxed neo cassette, and isolated mouse embryonic stem cells with the properly targeted locus (Supplementary Fig. 2). This deletion, if expressed, would result in a truncated secreted protein. From breeding mice with the targeted CD148 gene with CMV-Cre mice, we obtained mice not only with floxed alleles, but also mice with a constitutively deleted transmembrane domain allele (TM-KO). Only mice with the constitutively deleted allele will be characterized here. Surprisingly, the CD148 TM-KO mice were born at a normal Mendelian frequency and with a grossly normal life span. As expected, only a secreted truncated protein representing the extracellular portion of CD148 protein was observed in the serum of CD148 TM-KO mice (Supplementary Fig. 2). Importantly however, CD148 protein was not detected on any cells of hematopoietic origin in these mice by either western blotting or flow cytometry (Supplementary Fig. 2 and data not shown). Moreover, we have not noted any phenotype in mice heterozygous for the TM-KO allele. Thus, the secreted protein does not appear to bind to hematopoietic cells, or cause any physiologic consequences.
B cell development in CD148 deficient mice is partially impaired
The thymus, spleen and lymph nodes from the CD148 TM-KO mice did not exhibit any phenotypic changes throughout T cell development (Fig. 1A, supplementary table 1 and data not shown). There was no obvious defect in B cell development in the bone marrow (BM; data not shown). Even though the total T and B cell number in the spleen and lymph node was unchanged, a partial peripheral B cell developmental block was observed. Immature B cells that migrate from the BM to the spleen continue to develop through transitional stages (T1 and T2), becoming either follicular mature B cells (CD19+ IgMlo IgDhi) or marginal zone B cells (CD19+ CD1dhi) (Martin and Kearney, 2000). Spleens from TM-KO mice contained more T1 B cells (IgMhi IgDlo) and fewer follicular mature (FM) B cells (IgMlo IgDhi), implying a partial block at the T1 stage (Fig. 1B and supplementary table 1). Moreover, CD148 TM-KO mice also had increased splenic marginal zone B cells (CD19+ CD1dhi) (Fig. 1C and supplementary table 1). Skewing towards the B2 lineage at the expense of the B1 lineage was noted in peritoneal lavage B cells (Fig. 1D and supplementary table 1). This phenotype in the CD148 TM-KO B cell lineage was remarkably reminiscent of that reported in CD45 KO mice (Byth et al., 1996; Hermiston et al., 2005).
Figure 1. T and B cell phenotypes in CD148 TM-KO mice.

(A) Representative FACS analyses of thymocytes from 8-week-old B6 WT and CD148 TM-KO mice. (B) Splenocytes, pre-gated on CD19+ CD1dlo to exclude MZ cells, were further separated into T1 (IgMhi IgDlo) and T2 + FM (follicular mature) B cells (IgMhi IgDhi for T2 and IgMlo IgDhi for FM). (C) Splenocytes from the indicated mice were stained with mAb to CD19 and CD1d. Percentages of MZ (CD1dhi) cells in the CD19+ gate are shown. (D) FACS analysis of peritoneal lavage from 8-week-old B6 WT and CD148 TM-KO mice, stained with mAb to CD19, CD5 and CD23. Percentages of B1b (CD5+, CD23-), B1a (CD5-, CD23-) and B2 (CD5-, CD23+) cells in the CD19+ gate are shown. (E,F,G) Quantitation of percentages and cell numbers of T1 and T2+FM (E), MZ (F), B1a and B2 (G) are indicated. Complete quantification of all subsets is provided in supplementary table 1.
Generation of CD148/CD45 double deficient mice
The phenotypic similarity of the CD148 TM-KO and CD45 KO mouse lines lent support to the idea that CD148 and CD45 have overlapping functions in B cells. We generated CD148 TM-KO and CD45 doubly deficient mice (DKO), backcrossed for 6 generations onto the C57BL/6 background. Due to the infertility of CD148 TM-KO mice, we have used CD148 TM+/- CD45+/- or CD148 TM+/- as breeders and wild type or single KO littermates as controls. CD148 TM+/+ and CD148 TM+/-, or CD45+/+ and CD45+/- were used interchangeably, since they shared phenotypes. DKO mice were significantly under-weight (60% of WT mice, Fig. 2A) and died prematurely, between 25 to 65 days (Fig. 2B). DKO mice also developed B cell lymphopenia. Total cell numbers in spleens and lymph nodes were significantly lower than in the WT, CD148-TM KO or CD45 KO mice (Fig. 2C,D, and data not shown). Before death, all DKO mice developed a myeloproliferative disorder with extramedullary hematopoiesis and distortion of splenic architecture (Supplementary Fig. 3), as well as extensive myeloid infiltration of the liver (Fig. 2E). Scattered myeloid infiltrates in the lung were also detected (Fig. 2F and data not shown). Varying degrees of anemia, which could contribute to the accelerated mortality, were observed in pre-terminal DKO mice.
Figure 2. CD45/CD148 DKO mice on the B6 background die prematurely.
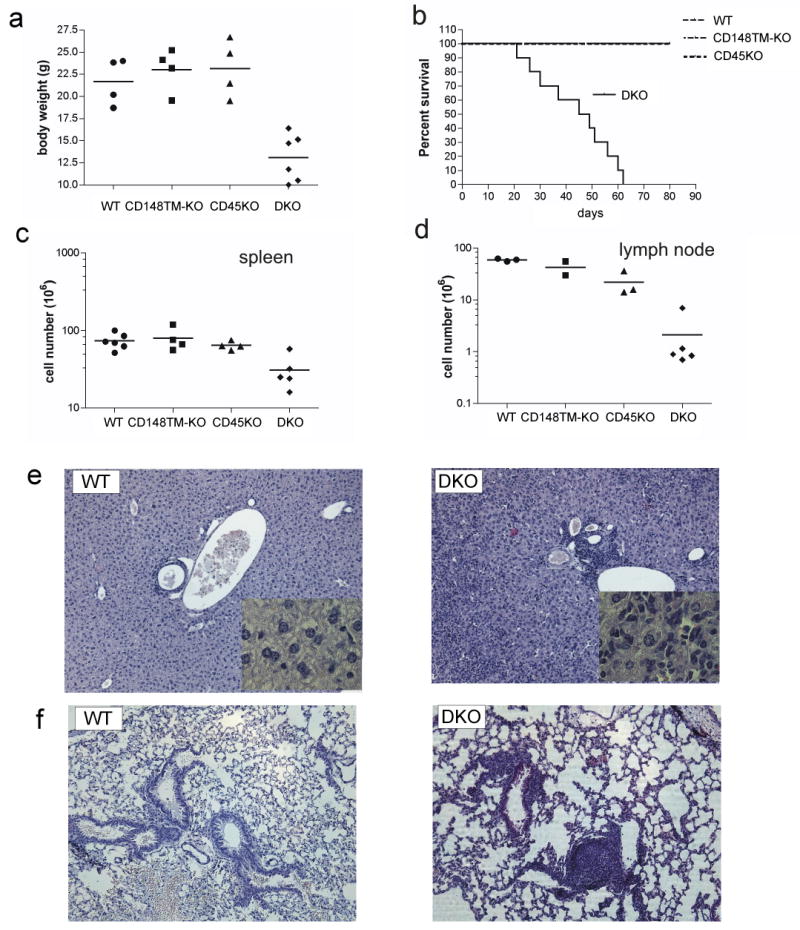
(A) Body weight of 4-6 week old mice of the indicated genotypes. (B) Percent survival of mice of the indicated genotypes. (C,D) Total cell numbers of spleens (C) and lymph nodes (D) from 4-6 week old mice of the indicated genotypes. (E) Liver sections (10X) from 5 week old mice of the indicated genotypes stained with H&E. Inserts are 63X. (F) Lung sections (10X) from 5 week old mice of the indicated genotypes stained with H&E.
Severe B cell developmental block in DKO mice
To better understand the B cell developmental defect, we analyzed the B-cell compartment of C57BL/6 DKO mice. As previously reported (Byth et al., 1996; Kishihara et al., 1993) and confirmed here, spleens from CD45 KO mice contain a higher percentage of B cells due to the T cell developmental block (Fig. 3A). In contrast, spleens from the DKO mice showed a marked decrease in the percentage, as well as the number of B cells (Fig. 3A,B,C). Similar to CD45 KO mice, DKO mice also have higher proportion of transitional B cells (IgMhi IgDlo and IgMhi IgDhi), but the overall B cell number is much lower (Fig. 3A,B,C). In the spleens of DKO mice, we also observed an unusual cell population that expresses CD19, but not IgM nor IgD. They are slightly bigger than normal B cells (as detected by flow cytometer forward scatter), and also express CD43, CD11b and Gr1 antigens (data not shown). These cells may represent an unusual myeloid cell population. We excluded this population from the B cell gate based on forward and side scatter intensities during FACS analysis. Further analysis of these cells is in progress.
Figure 3. B cell developmental block in CD45/CD148 DKO mice on the B6 background.

(A) Percentages and cell numbers of CD19+ splenocytes from 4-6 week-old mice of the indicated genotypes. (B) Representative FACS analyses of CD19+ splenocytes stained with mAb to CD19, IgM and IgD. Percentages of CD19+ B cells, T1 (IgMhi IgDlo) cells and T2 (IgMhi IgDhi) plus follicular mature (FM) (IgMlo IgDhi) B cells are shown. (C) Percentages and absolute numbers of each B cell subset are shown. (D) Percentages and cell numbers of CD19+ lymphocytes in the BM from 4-6 week old mice of the indicated genotypes. (E) Representative FACS analyses of BM cells stained with mAb to CD19 and CD43. Percentages of CD19+ B cells and CD43- or CD43+ in CD19+ gate are shown. (F) Based on the FACS analysis, CD19+ BM B cells were identified as fraction B (CD43+ CD24+, BP1-), fraction C (CD43+ CD24+, BP1+), fraction D (CD43-IgM- IgD-), fraction E (CD43- IgM+ IgD-), fraction F (CD43- IgM+ IgD+) (see also Supplementary Fig. 2). Fraction A (B220+, CD19-) could not be analyzed due to the lack of the B220 marker in DKO mice (ND)(Hardy et al., 2000).
Decreased B cell numbers in the peripheral organs from DKO mice indicates that there might be a severe block in early B cell development in the BM. Indeed, marked decreases in both B cell percentages as well as absolute numbers were observed in the DKO BM (Fig.3D and data not shown). CD19+ CD43+ pre-B cell numbers (fraction B and C) were similar in DKO and WT mice. However, very small numbers of B cells developed further to CD19+CD43- in DKO mice (fraction D) (Fig. 3D,E,F and Supplementary Fig. 4). Additionally, there was a substantial decrease in immature (IgM+IgD-) and almost no recirculating mature IgM+ IgD+ B cells were detected in the DKO BM (Figure 3E,F and data not shown).
Accelerated phenotypes in DKO mice backcrossed onto the C57BL/6 background
While backcrossing DKO mice, we noted that the B cell phenotype, described above, appeared much earlier in DKO mice that were backcrossed for 6 generations to C57BL/6 background when compared to mice of mixed genetic background. DKO mice that were backcrossed to C57BL/6 for only 2-3 generations lived up to 6 months and were grossly healthy until 2 months of age. Young mixed lineage background mice had a mild reduction in B cell number, similar to that seen in CD45 KO mice (Supplementary Fig. 5). Moreover, follicular lymph node B cells were quite homogeneous across all genotypes, except that both CD45 KO and DKO B cells expressed higher levels of IgM compared with WT and CD148 TM-KO B cells (Supplementary Fig. 5). However, upon aging these mice also developed B cell lymphopenia with a profound developmental block at the pre-B cell stage in the BM, accompanied by the myeloproliferative disorder (data not shown). This suggested that the severe B cell phenotype and lethality was only delayed in mixed lineage mice.
Impaired BCR-mediated increases in intracellular free Ca2+ in DKO mice
The attenuated B cell phenotype in less fully backcrossed DKO mice allowed us to perform biochemical assays using purified peripheral B cells from these young mixed lineage mice (6-8 weeks of age), where B cell development is relatively normal (Supplementary Fig. 5). We analyzed B cell receptor mediated elevation of intracellular free calcium, using purified B cells from lymph nodes. Strikingly, DKO B cells showed a markedly delayed Ca2+ flux following BCR crosslinking when compared to WT and singly-deficient mice (Fig. 4A). Since there were no significant phenotypic differences in the lymph node B cell populations across all genotypes in the mixed lineage background, the observed delay in Ca2+ flux is likely to be the result of a defect in BCR-mediated signaling rather than a consequence of differences in the B cell populations.
Figure 4. Impaired BCR-mediated signaling in CD45/CD148 DKO mice.
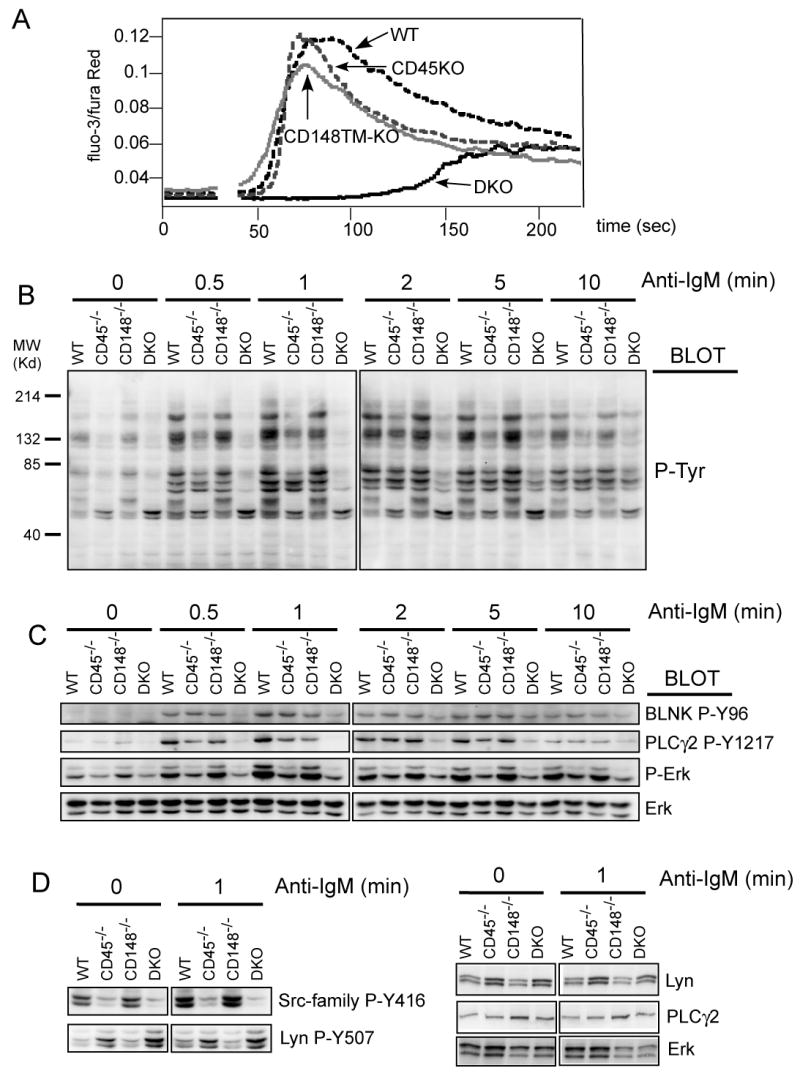
(A) Purified lymph node B cells of the indicated mice were loaded with Fluo3-AM and Fura Red and intracellular free Ca2+ concentrations were monitored before and after addition of IgM F(ab')2 (5μg/ml). (B) Purified lymph node B cells of all four genotypes were stimulated with F(ab')2 fragments of anti-IgM antibody (5 μg/ml). At the indicated time points, the cells were lysed in SDS-PAGE sample buffer and the lysates were analyzed by immunoblotting with phosphotyrosine antibody. (C) Samples from panel (B) were subjected to immunoblotting with phospho-specific antibodies as indicated. Staining of total Erk served as a loading control. (D) Samples from panel (B) (the 0 min and 1 min timepoints) were stained with site-specific antibody against phosphorylated activation loop of SFKs (Src-family P-Y416, this antibody crossreacts with multiple SFKs) and against C-terminal inhibitory phosphotyrosine of Lyn (Lyn P-Y507). The right hand panels are protein controls in the various genotypes. See quantification for westerns shown in (C) and (D) in supplementary Fig. 6.
Impaired SRC family kinase-dependent signaling pathway in DKO B cells
To understand the signaling defect, we investigated the BCR induced protein tyrosine phosphorylation involved in the initiation of the calcium response. Mixed background mice were used since it was not possible to obtain sufficient numbers of B cells from backcrossed mice. Consistent with the calcium data, general tyrosine phosphorylation was severely impaired in DKO B cells (Fig. 4B). We noted diminished phosphorylation and activation of BLNK and PLCγ2, proteins upstream of the calcium response. In addition, activation of the MAP kinase pathway was also impaired as documented by decreased ERK phosphorylation in DKO B cells (Fig. 4C and supplementary Fig. 6), although in this case lack of CD45 alone was already responsible for a significant part of the defect.
The general decrease in tyrosine phosphorylation in DKO B cells suggested a proximal signaling defect, most likely at the level of SFKs. The negative regulatory phosphotyrosine of the SFKs is a primary substrate of CD45. Indeed, we noted increased phosphorylation of the C-terminal inhibitory tyrosine of the major B cell SFK Lyn (Y507) in CD45-/- mice. This was further increased in the DKO B cells (Fig. 4D and supplementary Fig. 6). In contrast, phosphorylation of the SFK activation loop tyrosine was not induced in DKO B cells. Collectively, these data suggest that CD45 and CD148 both dephosphorylate negative regulatory tyrosines of the SFKs in B cells and, thus, decrease the threshold for SFK activation.
Defects in BCR-mediated signaling are B cell intrinsic
Because of the different phenotypes observed on mixed versus C57BL/6 genetic backgrounds, we tested whether the defect in BCR mediated signaling is B cell intrinsic. We performed mixed BM transplantation by transferring WT together with fully backcrossed DKO BM cells into lethally irradiated C57BL/6 Ly5.1 (CD45.1+) recipients at a 1:1 ratio. Six to eight weeks after BM transplantation, we purified lymph node B cells and analyzed BCR-mediated responses. Although the ratio of DKO to wild type B cells in lymph node of the recipient was 30% versus 70%, sufficient numbers of B cells were available for analysis. Following BCR crosslinking, we observed normal Ca2+ increases in B cells from the WT donor (identified as CD45.2+). In sharp contrast, Ca2+ flux in B cells from DKO donor (CD45.2-) was substantially delayed, similar to that observed in mixed background B cells (Fig. 5A), despite the fact that B cells from the DKO donor had slightly higher BCR levels on their surface than their WT counterparts. This result confirms that the calcium signaling defect in DKO B cells is B cell intrinsic.
Figure 5. BCR mediated signaling and B cell development in competitively reconstituted chimeras.
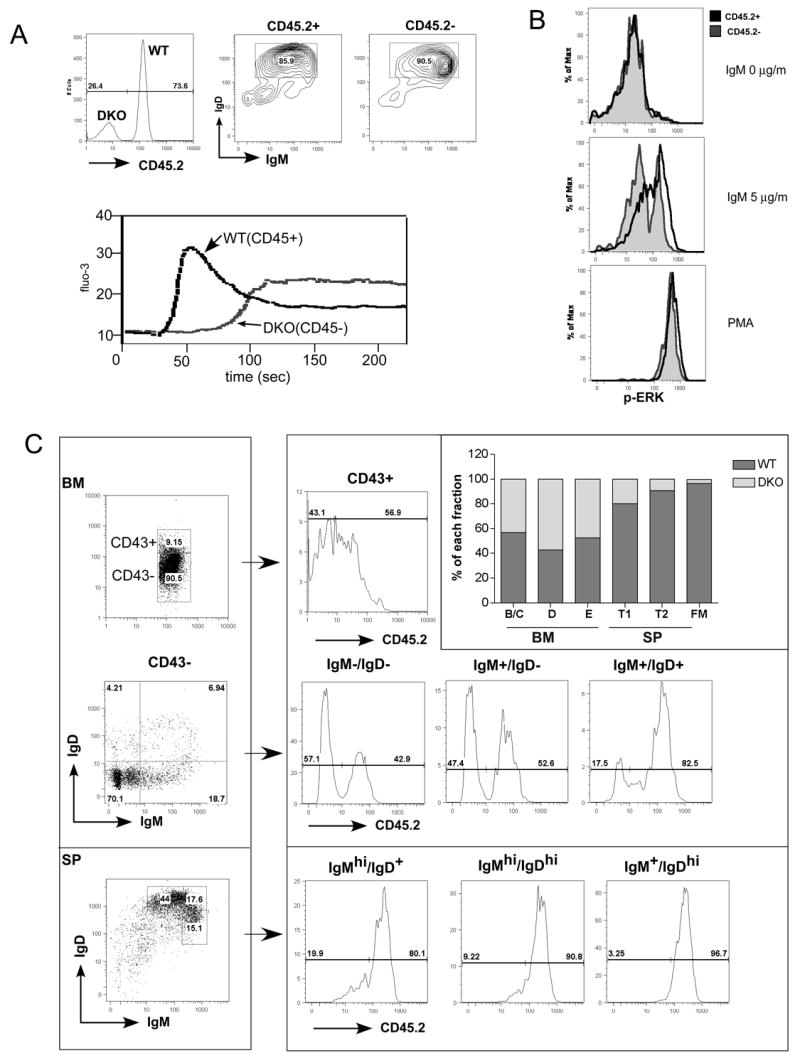
(A) Cell autonomous defect in intracellular free Ca2+ increase. Lethally irradiated recipient mice were reconstituted with BM from CD45/CD148 DKO and WT control mice. The donor origins of reconstituted B cells were identified using CD45.2 as a congenic marker: WT B cells were CD45.2+ and DKO B cells were CD45.2-. Lymph node B cells were stained with CD45.2 (upper left) and IgM /IgD (upper right) antibodies. The intracellular free Ca2+ levels were monitored before and after addition of IgM F(ab')2 (5μg/ml). (B) Cell autonomous defect in activation of ERK. Purified B cells as in (A) were stimulated with IgM F(ab')2 (5μg/ml) or PMA for 2.5 minutes, fixed, permeabilized and then stained with phospho-ERK along with other surface makers. Follicular mature B cells (IgM+IgD+CD23+) were separated based on the origin of the donors (same as in (A)). Phospho-ERK levels of CD45.2+ (WT origin) and CD45.2- (DKO origin) upon indicated stimulation were overlaid in histograms. (C) B cell development in competitively reconstituted chimera. Cells from BM and spleen were isolated and stained with antibodies as indicated. For BM, CD19+ B cells were gated to separate CD43+, and CD43-. The CD43- cells were further subdivided based on expression of IgM and IgD. For the spleen, CD19+ B cells were separated based on IgM and IgD expression. The donor origin of each subset was then identified by CD45.2 staining. Percentages of WT and DKO B cells of each sunset were presented in the overlaid bar graph. Results in (C) are representive of 4 independent experiments.
We used such chimeras to test further downstream signaling by measuring the phosphorylation of ERK kinase using a flow cytometry based assay. Following BCR crosslinking, most follicular mature B cells from the WT donor (CD45.2+) responded to IgM stimulation, but only 32% of B cells from DKO donor upregulated their phospho-ERK level. The majority of the DKO B cells retained basal levels of ERK phosphorylation. Thus, similar to calcium signaling, the defect in BCR-mediated ERK activation is also B cell autonomous. Moreover, since fully backcrossed mice were used, this study also demonstrates that the observed signaling defects are independent of the genetic background of the mouse strains.
The B cell developmental defect is caused by both intrinsic and extrinsic factors
To further test whether the observed developmental block is B cell autonomous, we used the same chimera as described before and analyzed the chimeras 6 weeks post transplantation. Surprisingly, in the chimeric BM, the ratio of WT and DKO B cells remained close to 1:1 throughout development in the BM (Fig 5C). In contrast, as B cells progressed through further maturation in the spleen, the ratio became 80% to 20% (wt to DKO) at the T1 stage, 90% to 10% at T2 and was even more skewed in the mature B cell compartment (Fig 5C). These results reveal that the B cell intrinsic developmental block occurs much later at the level of the immature to mature B cell transition while the defects observed in DKO BM described above are caused, at least in part, by extrinsic factors, possibly as a consequence of the dysregulated myeloid compartment.
CD148 and CD45 regulate Fc receptor mediated phagocytosis and cytokine production in BM-derived macrophages
Since both CD45 and CD148 are also expressed in myeloid cells, we examined whether they play similar roles in this lineage by studying Fc receptor-mediated pathways ex vivo in BM-derived macrophages (BMDM). Fc receptor-mediated signaling is a well-defined ITAM/SFK-dependent pathway. Activation of this pathway results in diverse responses including phagocytosis, cytokine production, and MAP kinase activation. We first investigated Fc-receptor mediated phagocytosis using opsonized human erythrocytes. Fc receptor mediated phagocytosis was severely impaired in DKO BMDM (40% of WT) (Fig. 6A), whereas only mild defects were observed in singly-deficient cells. Moreover, Fc receptor-induced TNFα production was also severely impaired in DKO BMDMs (Fig. 6B). In contrast, after LPS treatment, TNFα production in DKO BMDM was equivalent to WT cells (Fig. 6C). We measured the Fc receptor levels by flow cytometry and found them to be similar across all genotypes (Fig. 6D). The selective defect in the Fc receptor-mediated signaling in DKO BMDM suggests a requisite role of for both CD148 and CD45 in this pathway, but not in Toll-like receptor 4 pathways.
Figure 6. CD148 and CD45 regulate Fc receptor mediated phagocytosis and cytokine production in BMDMs.

(A) BMDMs were incubated with human red blood cells coated with mouse IgG2a mAb. BMDMs with ingested erythrocytes were fixed, labeled with Hoechst and quantified under fluorescent microscope. Phagocytic index was determined as [number of erythrocytes] / [number of nuclei]. Phagocytic index of WT macrophages was then set as 100%. * p< 0.001 (B,C) BMDMs were stimulated with plate-bound mAb to FcR II/III (2.4G2) (B) or LPS (C) in the presence of brefeldin A for 4 hours. TNFα production was detected by intracellular staining with TNFα-specific antibody. (D) BMDMs were stained with mAb to FcγR II/III (2.4G2) or mouse IgG. The expression levels on the BMDMs of the indicated genotypes over the negative control (shaded histogram) are shown.
We determined whether Fc receptor signaling by BMDMs had signaling defects similar to B cells. Indeed, overall tyrosine phosphorylation was substantially reduced in DKO BMDMs following Fc receptor stimulation, whereas no reproducible differences were observed in singly-deficient cells (Fig. 7A). Similar results were obtained with ERK and Akt phosphorylation, where the maximum response was diminished and substantially delayed in DKO BMDMs (Fig. 7B). Strong hyperphosphorylation of the Lyn inhibitory tyrosine (Y507) in the DKO cells and mild hyperphosphorylation of this site in the singly-deficient cells (Fig. 7C) was observed in the BMDM. This lent strong support to the idea that defective SFK activation, caused by hyperphosphorylation of the C-terminal inhibitory tyrosine, is responsible for the diminished Fc-receptor mediated signaling. The functional and signaling defects in DKO BMDMs were independent of the genetic backgrounds tested (data not shown).
Figure 7. Impaired FcR mediated signaling in DKO BMDMs.
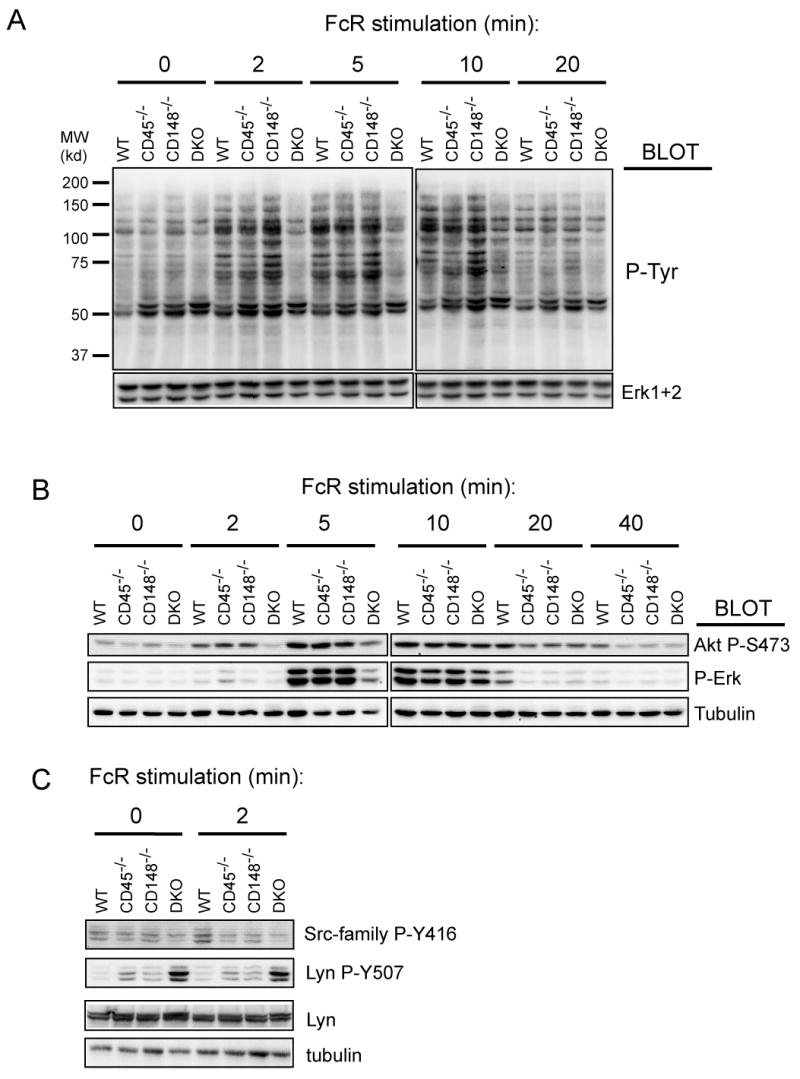
(A) BMDMs of all four genotypes were sensitized with purified mouse IgG (20 μg/ml) and then stimulated with anti-mouse secondary antibody (10 μg/ml). Cells were lysed in SDS-PAGE sample buffer and the lysates were analyzed by immunoblotting with phosphotyrosine antibody (4G10). Staining of total Erk served as a loading control. (B) Samples from BMDMs stimulated the same way as in (a) were subjected to immunoblotting with phospho-specific antibodies as indicated. Staining of tubulin represents a loading control. (C) Samples from panel (B) 0 min and 2 min timepoints were stained with site-specific antibody against phosphorylated activation loop of SFKs (Src-family P-Y416) and against the C-terminal inhibitory phosphotyrosine of Lyn (Lyn P-Y507). See quantification for westerns shown in (B) and (C) in supplementary Fig. 6.
Discussion
We describe new roles for the RPTP CD148 in the regulation of SFK activity in the immune system. Our studies of CD148 TM-KO mice showed a phenotype similar to mice deficient in CD45 suggesting that CD148 is a positive regulator of ITAM-dependent signaling in the B cell and myeloid lineages. Importantly, analyses of the signaling pathways in B cells and macrophages from CD45/CD148 DKO mice revealed substantial defects not seen in the singly deficient mice. At the molecular level, DKO B cells displayed impaired calcium flux and MAP kinase phosphorylation. Importantly, CD148 and CD45 are both required to optimally dephosphorylate the C-terminal negative regulatory tyrosine of SFKs, such as Lyn, in B cells. Very similar defects were also observed in Fc receptor-mediated activation and phagocytosis in BMDMs. These data suggest a high level of redundancy between the two structurally distinct RPTPs CD148 and CD45 in regulating SFKs. Moreover, these findings suggest a reinterpretation is needed of the function of CD45 in non-T cell lineages where CD148 is constitutively expressed and may compensate at least partially for some of the described phenotypes seen in the absence of CD45.
One of the striking features of the DKO mice is their early mortality. We speculate the development of the myeloproliferative syndrome with liver and lung infiltration likely contributed to their premature death. Interestingly, the myeloproliferative syndrome is transplantable with BM and can be observed even in aged chimeric mice (data not shown). The animals also develop anemia, which could contribute to their death. However, the precise cause of death could be complex since multiple lineages and pathways may be dysregulated. Further analysis will be aided by the lineage specific inactivation of CD148.
It is tempting to speculate that the observed defects in SFK signaling are responsible for the phenotype. Indeed, many features of the DKO mice resemble those of mice deficient in different combinations of SFKs. A milder myeloproliferative syndrome was also observed in Lyn deficient mice (Harder et al., 2001), suggesting that the reduced activity of SFKs in CD45/CD148 DKO mice could explain this phenotype. The myeloproliferative disease in DKO mice was accompanied by a severe block in early B cell development in the C57BL/6 genetic background. However, a different phenotype was observed in the Lyn singly-deficient mice, which instead display B-cell hyperactivity and B-cell mediated autoimmunity leading to severe glomerulonephritis (Hibbs et al., 1995; Nishizumi et al., 1995). CD45/CD148 deficiency likely affects many SFKs and so the balance between the activities of the individual SFKs governs the outcome. Deficiencies in different combinations of SFKs can result in variable phenotypes, ranging from immunodeficiency (Saijo et al., 2003) to autoimmunity (Hibbs et al., 1995; Nishizumi et al., 1995) and leukemogenesis (Marth et al., 1988; Marth et al., 1986). Due to the very limited number of B cells we obtained from DKO mice, we could not address the phosphorylation status of the individual SFKs. However, it is possible that different SFK members are affected to a varying degree by deficiency in each RPTP. In addition to SFKs, there may be other substrates of CD148 and CD45 that contribute to the phenotype we observed in DKO mice.
Our studies of BMDM support a role for CD45 and CD148 in ITAM-receptor mediated signaling in the myeloid lineage. The defective Fc receptor-mediated phagocytosis and TNFα production in macrophages derived from the CD45/CD148 DKO is reminiscent of SFK and Syk deficiency in macrophages. Previous studies of lyn/hck/fgr triple KO mice demonstrated a defect in FcR-mediated phagocytosis (Fitzer-Attas et al., 2000). An even more profound defect in FcR-mediated phagocytosis was observed in Syk deficient mice (Crowley et al., 1997). Thus, it seems likely that the functions of CD45 and CD148 overlap and converge on SFKs in ITAM-dependent BCR- and FcR-mediated functions in B cells and macrophages.
It is intriguing that while DKO mice have a severe myeloproliferative disease they also develope a B cell developmental block and impaired BCR and FcR signaling. The myeloproliferative disease in DKO may involve cytokine- and growth factor receptor-induced pathways, in which CD45 and CD148 may play different roles. While it is possible that these RPTPs directly act on cytokine receptor associated-JAK kinases and growth factor receptor kinases (Irie-Sasaki et al., 2001), our preliminary data suggest an effect of SFK on inhibitory control of growth factor receptors.
Strikingly, in mice of a mixed genetic background (B6, Balb/c and 129) the B cell developmental block in the BM and the myeloproliferative syndrome were much delayed. Genetic modifiers may contribute to the severity of the disease. The discordant phenotypes also suggest that the early B cell developmental block in the DKO mice is not B-cell autonomous. A number of cytokines produced by myeloid cells such as type I interferons can suppress B cell development (Lin et al., 1998). Relatively normal B-cell development in the BM of the young mixed lineage mice suggests that lack of CD148 and CD45 per se is not sufficient to block very early B cell development and that other factors contribute to the phenotype as the disease progresses. In addition, data from our competitively reconstituted chimeras more clearly demonstrated that the very early B cell developmental block seen in B6 mice is secondary to some other not fully understood effects, possibly coming from a dysregulated myeloid compartment. In fact, the only B cell developmental block that is most likely intrinsic was observed during immature to mature B cell transition, given the discordant results obtained between wild-type and DKO B cells in healthy chimeric mice at week 6 post transplantation. It is not clear how much of this developmental defect is due to the CD45 deficiency alone. Further clarification of this issue will require B cell lineage-specific conditional DKO mice.
At the molecular level, we observed clear BCR signaling defects in DKO peripheral B cells. In the absence of CD45 and CD148, the cells maintained a high phosphorylation level of the C-terminal tyrosine of Lyn, thus keeping the kinase in the inactive conformation. As a likely consequence, the DKO B cells had impaired calcium flux following BCR ligation and failed to activate PLCγ2 and ERK. Importantly, the same defects in calcium flux and ERK activation in the DKO B cells occurred in competitively reconstituted chimeras, suggesting that the BCR signaling defects are in fact B cell intrinsic. Moreover, the signaling defects observed in the in vitro expanded BMDMs were very similar to the defects observed in B cells. Finally, our results from DKO B cells are also consistent with mutants of the DT40 chicken B-cell line, which does not express endogenous CD148 (J.L. unpublished observation). CD45 deficient and Lyn deficient DT40 lines share very similar BCR-induced calcium signaling defects to our DKO B cells, exhibiting delayed and decreased peak responses (Takata et al., 1994; Yanagi et al., 1996). In contrast, calcium flux in Syk deficient DT40 cells is completely abolished (Takata et al., 1994). These data suggest that via the regulation of SFKs, CD148 and CD45 contribute to the early peak calcium flux following BCR stimulation, whereas Syk may be responsible for sustaining the calcium flux after receptor clustering. A similar model may hold for the partial defects seen in the FcR-mediated signaling in macrophages. It is also important to stress that most of the signaling defects we observed in both B cells and macrophages were independent of the genetic background.
Although our study demonstrates that in B cells and macrophages CD148 and CD45 have similar overlapping functions, it is not clear whether there is any unique role for CD148 or CD45 in these lineages. Their very distinct structures suggest that they may be subject to different regulatory mechanisms. Previously, CD148 was considered to have an inhibitory role. In non-hematopoietic cells, CD148 has been reported to act as a tumor suppressor and inhibit proliferation of many different cell types predominantly of epithelial origin, most likely via dephosphorylation of different growth factor receptors (Balavenkatraman et al., 2006; Jandt et al., 2003; Keane et al., 1996; Kovalenko et al., 2000; Lesueur et al., 2005; Palka et al., 2003; Ruivenkamp et al., 2003; Ruivenkamp et al., 2002; Trapasso et al., 2000; Trapasso et al., 2004). It is possible that CD148 plays different roles depending on cell type and redundancy with other proteins. The overall phenotype reflects a net effect on the entire spectrum of available substrates that may be different in distinct cell types. It is possible that in B cells and myeloid cells the positive regulatory role of CD148 on Src-kinases prevails. Indeed, a study on rat thyroid carcinoma showed that overexpression of CD148 led to specific dephosphorylation of the C-terminal regulatory phosphotyrosine of c-Src that led to increased substratum adhesion (Pera et al., 2005).
Our study identifies CD148 as an important regulator of SFKs and suggests that it does so in concert with CD45 in at least some hematopoietic lineages. In B cells and macrophages, CD148 dephosphorylates the negative regulatory phosphotyrosine of SFKs and therefore together with CD45 helps to set the threshold for SFK activation and, hence, the threshold for the immunoreceptor signaling in these cells. Since SFKs are important components of many other signaling pathways in hematopoietic cells, it is reasonable to expect that CD148 may have complex effects at multiple levels of immune cell regulation. Future studies of CD148 may provide not only deeper insights into the function of this RPTP itself but also of SFKs and immune cell regulation, in general.
Methods
Generation of CD148 TM-KO Mice
Detailed method for generation of CD148 TM-KO mice is described in Supplementary Fig. 2. CD148 TM-KO mice were backcrossed to C57BL/6 mice for at least six generations. To generate CD148/CD45 doubly deficient mice, CD148TM-KO mice were crossed to CD45 KO mice (exon 6 disruption, in C57BL/6 strain, obtained from E. Brown, UCSF). All animals used in the experiments were at 6-10 weeks of age. All animals were housed in a specific pathogen-free facility at UCSF according to University and NIH guidelines.
Antibodies and Reagents
Antibodies to murine CD1d, CD3, CD4, CD5, CD8, CD19, CD21, CD23, CD24, CD43, CD45.1, CD45.2, BP-1, Gr1, IgM, IgD, Mac1, TNFα conjugated to FITC, PE, PerCP, APC, APC-Alexa750 or Pacific Blue were from BD Biosciences. Anti-phosphotyrosine antibody 4G10 was from Upstate Biotechnology. Antibodies to phospho-BLNK (Tyr96), phospho-PLCγ2 (Tyr1217), phospho-Erk (Thr202/Tyr204), phospho-Src Family (Tyr416), phospho-Lyn (Tyr507), and phospho-Akt (Ser473) were purchased from Cell Signaling Technology. Goat anti-mouse IgG, Goat anti-mouse IgM (F(ab')2 fragments), and purified mouse IgG and were from Jackson Immunoresearch. Mouse IgG2a antibody to human CD59 (MEM-43) was kindly provided by V. Horejsi (Institute of Molecular Genetics, Prague, Czech Republic). Erk1 and Erk2 antibodies were from Santa Cruz Biotechnology. Tubulin antibody was obtained from Sigma.
Isolation and activation of primary B Cells
Mouse lymph node B cells were purified by negative selection with CD11b- and CD43-conjugated magnetic beads (Miltenyi Biotec). Purified B cells were resuspended in serum-free RPMI (108 per ml) and activated by 5 μg/ml Goat anti-mouse IgM (F(ab')2) (Jackson Immunoresearch). Aliquots of 5 × 106 cells were transferred directly to SDS-PAGE sample buffer.
Flow cytometry
Single-cell suspensions were prepared from peritoneal lavage, lymph nodes, spleen, and BM. Fc receptors were blocked with rat anti-CD16/CD32 (2.4G2). 106 cells were stained with indicated antibodies and analyzed on a FACSCalibur (BD Biosciences) or CyAn ADP (Dako) flow cytometers.
Intracellular phospho-ERK staining
Spleen cells were stained with FITC conjugated mouse IgM (F(ab)') monomer, then stimulated with 5 μg/ml Goat anti-mouse IgM (F(ab')2) (Jackson Immunoresearch) or PMA for 2.5 minutes, and fixed with 2% paraformaldehyde. Cells were permeabilized with 100% ice cold methanol, stained with anti-phospho-ERK (Cell Signaling Technology), IgD, and CD23 antibody and then analyzed on the CyAn ADP (Dako).
Calcium flux
Mouse lymph node B cells were loaded with a mixture of Fluo-3 (2 μg/ml) and Fura Red (4 μg/ml), 30 min at 37°C in complete culture media. Cells were analyzed by flow cytometry (FACS Calibur) at 37°C and activated by 5 μg/ml F(ab')2 goat anti-mouse IgM. Calcium flux was monitored as a ratio between Fluo-3 and Fura Red fluorescence. Purified lymph node B cells derived from adoptively transferred BMs were loaded with Fluo-3 only and labeled with APC-conjugated CD45 antibody to distinguish wild-type and DKO cells and the concentration of intracellular free calcium was measured as Fluo-3 fluorescence.
Differentiation and activation of BMDM
BMDMs were prepared by culturing mouse BM cells in BMDM media (RPMI-1640 with 10% FCS and 10% culture supernatant from L929 CMG cells producing M-CSF). Macrophages were used for experiments between days 7 and 9 of culture. For immunoblot analysis, BMDMs were plated in 6-well tissue culture plates (Costar) in BMDM media, 106 per well. The next day, the cells were washed with serum free RPMI and incubated with 20 μg/ml purified mouse IgG (Jackson Immunoresearch), 30 min, 4°C. Cells were washed, incubated for 15 min at 37°C and then activated with Goat anti-mouse antibody (Jackson Immunoresearch), 10 μg/ml. The stimulation was stopped by adding SDS-PAGE sample buffer. Samples were analyzed by SDS-polyacrylamide gel electrophoresis (PAGE) and immunoblotting. To assess TNFα production, 24-well non-tissue culture plates were coated with DOTAP (1mg/ml) (Sigma D-6182) for 10 min followed by incubation with anti-FcγR antibody (2.4G2), 10 μg/ml, overnight at 4°C. 2×105 BMDMs were added to each well together with brefeldin A 10 μg/ml and incubated for 4 hours at 37°C. For LPS stimulation, LPS (List Biological Laboratories Inc. #304) was directly added to the cells together with brefeldin A, 10 μg/ml. Cells were then harvested and stained intracellularly with TNFα Ab and analyzed by flow cytometry.
Phagocytosis assay
Human erythrocytes were prepared from human blood from healthy donors over Histopaque 1119 (Sigma) and labeled with FITC (0.1 mg/ml in PBS, overnight at 4°C) followed by opsonization with 10 μg/ml mouse IgG2a antibody against human CD59 (MEM-43, V.Horejsi). BMDMs were plated on sterile glass coverslips in a 6-well plate, 5 × 105 per well, and cultured overnight at 37°C. Then BMDMs were incubated with 7 × 106 opsonized human erythrocytes, 30 min, 37°C, washed 2 × with PBS and non-internalized erythrocytes were lysed in a hypotonic ACK buffer. The macrophages were fixed and labeled with Hoechst (1 μg/ml). Slides were analyzed on a fluorescent microscope (Marianas Turn-Key system using Slidebook software, Intelligent Imaging Innovations). The phagocytic index represents the number of erythrocytes per number of nuclei and normalized as a percentage of the phagocytic index of wild-type BMDMs.
Adoptive transfer
BM chimeras were generated by transfer of wild type and DKO BM into lethally irradiated Ly5.1 recipients at a 1:1 ratio. Recipients were sacrificed 6-8 weeks later.
Statistical methods
Statistical analyses were performed with Prism4 (GraphPad Software). Student T test analysis was used to calculate the P value. Error bars represent standard errors of the mean.
Supplementary Material
Acknowledgments
We thank A. Roque for excellent assistance with animal husbandry and J. Cyster, A. DeFranco, C. Lowell and Weiss lab members for advice and helpful discussions. We thank E. Brown and V. Horejsi for mice and reagents. The authors declare no financial conflict of interest. J.Z. and T.K are supported by NIH T32 training grant. This work is supported by NIH grant A1066120.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baker JE, Majeti R, Tangye SG, Weiss A. Protein tyrosine phosphatase CD148-mediated inhibition of T-cell receptor signal transduction is associated with reduced LAT and phospholipase Cgamma1 phosphorylation. Mol Cell Biol. 2001;21:2393–2403. doi: 10.1128/MCB.21.7.2393-2403.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balavenkatraman KK, Jandt E, Friedrich K, Kautenburger T, Pool-Zobel BL, Ostman A, Bohmer FD. DEP-1 protein tyrosine phosphatase inhibits proliferation and migration of colon carcinoma cells and is upregulated by protective nutrients. Oncogene. 2006;25:6319–6324. doi: 10.1038/sj.onc.1209647. [DOI] [PubMed] [Google Scholar]
- Benatar T, Carsetti R, Furlonger C, Kamalia N, Mak T, Paige CJ. Immunoglobulin-mediated signal transduction in B cells from CD45-deficient mice. J Exp Med. 1996;183:329–334. doi: 10.1084/jem.183.1.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byth KF, Conroy LA, Howlett S, Smith AJ, May J, Alexander DR, Holmes N. CD45-null transgenic mice reveal a positive regulatory role for CD45 in early thymocyte development, in the selection of CD4+CD8+ thymocytes, and B cell maturation. J Exp Med. 1996;183:1707–1718. doi: 10.1084/jem.183.4.1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloutier JF, Veillette A. Cooperative inhibition of T-cell antigen receptor signaling by a complex between a kinase and a phosphatase. J Exp Med. 1999;189:111–121. doi: 10.1084/jem.189.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley MT, Costello PS, Fitzer-Attas CJ, Turner M, Meng F, Lowell C, Tybulewicz VL, DeFranco AL. A critical role for Syk in signal transduction and phagocytosis mediated by Fcgamma receptors on macrophages. J Exp Med. 1997;186:1027–1039. doi: 10.1084/jem.186.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Facchetti F, Chan JK, Zhang W, Tironi A, Chilosi M, Parolini S, Notarangelo LD, Samelson LE. Linker for activation of T cells (LAT), a novel immunohistochemical marker for T cells, NK cells, mast cells, and megakaryocytes: evaluation in normal and pathological conditions. Am J Pathol. 1999;154:1037–1046. doi: 10.1016/S0002-9440(10)65356-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzer-Attas CJ, Lowry M, Crowley MT, Finn AJ, Meng F, DeFranco AL, Lowell CA. Fcgamma receptor-mediated phagocytosis in macrophages lacking the Src family tyrosine kinases Hck, Fgr, and Lyn. J Exp Med. 2000;191:669–682. doi: 10.1084/jem.191.4.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder KW, Parsons LM, Armes J, Evans N, Kountouri N, Clark R, Quilici C, Grail D, Hodgson GS, Dunn AR, Hibbs ML. Gain- and loss-of-function Lyn mutant mice define a critical inhibitory role for Lyn in the myeloid lineage. Immunity. 2001;15:603–615. doi: 10.1016/s1074-7613(01)00208-4. [DOI] [PubMed] [Google Scholar]
- Hardy RR, Li YS, Allman D, Asano M, Gui M, Hayakawa K. B-cell commitment, development and selection. Immunol Rev. 2000;175:23–32. [PubMed] [Google Scholar]
- Hermiston ML, Tan AL, Gupta VA, Majeti R, Weiss A. The juxtamembrane wedge negatively regulates CD45 function in B cells. Immunity. 2005;23:635–647. doi: 10.1016/j.immuni.2005.11.001. [DOI] [PubMed] [Google Scholar]
- Hermiston ML, Xu Z, Weiss A. CD45: a critical regulator of signaling thresholds in immune cells. Annu Rev Immunol. 2003;21:107–137. doi: 10.1146/annurev.immunol.21.120601.140946. [DOI] [PubMed] [Google Scholar]
- Hibbs ML, Tarlinton DM, Armes J, Grail D, Hodgson G, Maglitto R, Stacker SA, Dunn AR. Multiple defects in the immune system of Lyn-deficient mice, culminating in autoimmune disease. Cell. 1995;83:301–311. doi: 10.1016/0092-8674(95)90171-x. [DOI] [PubMed] [Google Scholar]
- Huntington ND, Xu Y, Puthalakath H, Light A, Willis SN, Strasser A, Tarlinton DM. CD45 links the B cell receptor with cell survival and is required for the persistence of germinal centers. Nat Immunol. 2006;7:190–198. doi: 10.1038/ni1292. [DOI] [PubMed] [Google Scholar]
- Irie-Sasaki J, Sasaki T, Matsumoto W, Opavsky A, Cheng M, Welstead G, Griffiths E, Krawczyk C, Richardson CD, Aitken K, et al. CD45 is a JAK phosphatase and negatively regulates cytokine receptor signalling. Nature. 2001;409:349–354. doi: 10.1038/35053086. [DOI] [PubMed] [Google Scholar]
- Iwashima M, Irving BA, van Oers NS, Chan AC, Weiss A. Sequential interactions of the TCR with two distinct cytoplasmic tyrosine kinases. Science. 1994;263:1136–1139. doi: 10.1126/science.7509083. [DOI] [PubMed] [Google Scholar]
- Jandt E, Denner K, Kovalenko M, Ostman A, Bohmer FD. The protein-tyrosine phosphatase DEP-1 modulates growth factor-stimulated cell migration and cell-matrix adhesion. Oncogene. 2003;22:4175–4185. doi: 10.1038/sj.onc.1206652. [DOI] [PubMed] [Google Scholar]
- Keane MM, Lowrey GA, Ettenberg SA, Dayton MA, Lipkowitz S. The protein tyrosine phosphatase DEP-1 is induced during differentiation and inhibits growth of breast cancer cells. Cancer Res. 1996;56:4236–4243. [PubMed] [Google Scholar]
- Kishihara K, Penninger J, Wallace VA, Kundig TM, Kawai K, Wakeham A, Timms E, Pfeffer K, Ohashi PS, Thomas ML, et al. Normal B lymphocyte development but impaired T cell maturation in CD45-exon6 protein tyrosine phosphatase-deficient mice. Cell. 1993;74:143–156. doi: 10.1016/0092-8674(93)90302-7. [DOI] [PubMed] [Google Scholar]
- Kong YY, Kishihara K, Sumichika H, Nakamura T, Kaneko M, Nomoto K. Differential requirements of CD45 for lymphocyte development and function. Eur J Immunol. 1995;25:3431–3436. doi: 10.1002/eji.1830251234. [DOI] [PubMed] [Google Scholar]
- Koretzky GA, Picus J, Schultz T, Weiss A. Tyrosine phosphatase CD45 is required for T-cell antigen receptor and CD2-mediated activation of a protein tyrosine kinase and interleukin 2 production. Proc Natl Acad Sci U S A. 1991;88:2037–2041. doi: 10.1073/pnas.88.6.2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalenko M, Denner K, Sandstrom J, Persson C, Gross S, Jandt E, Vilella R, Bohmer F, Ostman A. Site-selective dephosphorylation of the platelet-derived growth factor beta-receptor by the receptor-like protein-tyrosine phosphatase DEP-1. J Biol Chem. 2000;275:16219–16226. doi: 10.1074/jbc.275.21.16219. [DOI] [PubMed] [Google Scholar]
- Kung C, Pingel JT, Heikinheimo M, Klemola T, Varkila K, Yoo LI, Vuopala K, Poyhonen M, Uhari M, Rogers M, et al. Mutations in the tyrosine phosphatase CD45 gene in a child with severe combined immunodeficiency disease. Nat Med. 2000;6:343–345. doi: 10.1038/73208. [DOI] [PubMed] [Google Scholar]
- Lesueur F, Pharoah PD, Laing S, Ahmed S, Jordan C, Smith PL, Luben R, Wareham NJ, Easton DF, Dunning AM, Ponder BA. Allelic association of the human homologue of the mouse modifier Ptprj with breast cancer. Hum Mol Genet. 2005;14:2349–2356. doi: 10.1093/hmg/ddi237. [DOI] [PubMed] [Google Scholar]
- Lin J, Zhu JW, Baker JE, Weiss A. Regulated expression of the receptor-like tyrosine phosphatase CD148 on hemopoietic cells. J Immunol. 2004;173:2324–2330. doi: 10.4049/jimmunol.173.4.2324. [DOI] [PubMed] [Google Scholar]
- Lin Q, Dong C, Cooper MD. Impairment of T and B cell development by treatment with a type I interferon. J Exp Med. 1998;187:79–87. doi: 10.1084/jem.187.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marth JD, Cooper JA, King CS, Ziegler SF, Tinker DA, Overell RW, Krebs EG, Perlmutter RM. Neoplastic transformation induced by an activated lymphocyte-specific protein tyrosine kinase (pp56lck) Mol Cell Biol. 1988;8:540–550. doi: 10.1128/mcb.8.2.540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marth JD, Disteche C, Pravtcheva D, Ruddle F, Krebs EG, Perlmutter RM. Localization of a lymphocyte-specific protein tyrosine kinase gene (lck) at a site of frequent chromosomal abnormalities in human lymphomas. Proc Natl Acad Sci U S A. 1986;83:7400–7404. doi: 10.1073/pnas.83.19.7400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin F, Kearney JF. B-cell subsets and the mature preimmune repertoire. Marginal zone and B1 B cells as part of a “natural immune memory”. Immunol Rev. 2000;175:70–79. [PubMed] [Google Scholar]
- Mee PJ, Turner M, Basson MA, Costello PS, Zamoyska R, Tybulewicz VL. Greatly reduced efficiency of both positive and negative selection of thymocytes in CD45 tyrosine phosphatase-deficient mice. Eur J Immunol. 1999;29:2923–2933. doi: 10.1002/(SICI)1521-4141(199909)29:09<2923::AID-IMMU2923>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Nishizumi H, Taniuchi I, Yamanashi Y, Kitamura D, Ilic D, Mori S, Watanabe T, Yamamoto T. Impaired proliferation of peripheral B cells and indication of autoimmune disease in lyn-deficient mice. Immunity. 1995;3:549–560. doi: 10.1016/1074-7613(95)90126-4. [DOI] [PubMed] [Google Scholar]
- Palka HL, Park M, Tonks NK. Hepatocyte growth factor receptor tyrosine kinase met is a substrate of the receptor protein-tyrosine phosphatase DEP-1. J Biol Chem. 2003;278:5728–5735. doi: 10.1074/jbc.M210656200. [DOI] [PubMed] [Google Scholar]
- Pera IL, Iuliano R, Florio T, Susini C, Trapasso F, Santoro M, Chiariotti L, Schettini G, Viglietto G, Fusco A. The rat tyrosine phosphatase eta increases cell adhesion by activating c-Src through dephosphorylation of its inhibitory phosphotyrosine residue. Oncogene. 2005;24:3187–3195. doi: 10.1038/sj.onc.1208510. [DOI] [PubMed] [Google Scholar]
- Roach T, Slater S, Koval M, White L, Cahir McFarland ED, Okumura M, Thomas M, Brown E. CD45 regulates Src family member kinase activity associated with macrophage integrin-mediated adhesion. Curr Biol. 1997;7:408–417. doi: 10.1016/s0960-9822(06)00188-6. [DOI] [PubMed] [Google Scholar]
- Ruivenkamp C, Hermsen M, Postma C, Klous A, Baak J, Meijer G, Demant P. LOH of PTPRJ occurs early in colorectal cancer and is associated with chromosomal loss of 18q12-21. Oncogene. 2003;22:3472–3474. doi: 10.1038/sj.onc.1206246. [DOI] [PubMed] [Google Scholar]
- Ruivenkamp CA, van Wezel T, Zanon C, Stassen AP, Vlcek C, Csikos T, Klous AM, Tripodis N, Perrakis A, Boerrigter L, et al. Ptprj is a candidate for the mouse colon-cancer susceptibility locus Scc1 and is frequently deleted in human cancers. Nat Genet. 2002;31:295–300. doi: 10.1038/ng903. [DOI] [PubMed] [Google Scholar]
- Saijo K, Schmedt C, Su IH, Karasuyama H, Lowell CA, Reth M, Adachi T, Patke A, Santana A, Tarakhovsky A. Essential role of Src-family protein tyrosine kinases in NF-kappaB activation during B cell development. Nat Immunol. 2003;4:274–279. doi: 10.1038/ni893. [DOI] [PubMed] [Google Scholar]
- Sicheri F, Kuriyan J. Structures of Src-family tyrosine kinases. Curr Opin Struct Biol. 1997;7:777–785. doi: 10.1016/s0959-440x(97)80146-7. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Takahashi K, St John PL, Fleming PA, Tomemori T, Watanabe T, Abrahamson DR, Drake CJ, Shirasawa T, Daniel TO. A mutant receptor tyrosine phosphatase, CD148, causes defects in vascular development. Mol Cell Biol. 2003;23:1817–1831. doi: 10.1128/MCB.23.5.1817-1831.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata M, Sabe H, Hata A, Inazu T, Homma Y, Nukada T, Yamamura H, Kurosaki T. Tyrosine kinases Lyn and Syk regulate B cell receptor-coupled Ca2+ mobilization through distinct pathways. Embo J. 1994;13:1341–1349. doi: 10.1002/j.1460-2075.1994.tb06387.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchilian EZ, Wallace DL, Wells RS, Flower DR, Morgan G, Beverley PC. A deletion in the gene encoding the CD45 antigen in a patient with SCID. J Immunol. 2001;166:1308–1313. doi: 10.4049/jimmunol.166.2.1308. [DOI] [PubMed] [Google Scholar]
- Trapasso F, Drusco A, Costinean S, Alder H, Aqeilan RI, Iuliano R, Gaudio E, Raso C, Zanesi N, Croce CM, Fusco A. Genetic ablation of Ptprj, a mouse cancer susceptibility gene, results in normal growth and development and does not predispose to spontaneous tumorigenesis. DNA Cell Biol. 2006;25:376–382. doi: 10.1089/dna.2006.25.376. [DOI] [PubMed] [Google Scholar]
- Trapasso F, Iuliano R, Boccia A, Stella A, Visconti R, Bruni P, Baldassarre G, Santoro M, Viglietto G, Fusco A. Rat protein tyrosine phosphatase eta suppresses the neoplastic phenotype of retrovirally transformed thyroid cells through the stabilization of p27(Kip1) Mol Cell Biol. 2000;20:9236–9246. doi: 10.1128/mcb.20.24.9236-9246.2000. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Trapasso F, Yendamuri S, Dumon KR, Iuliano R, Cesari R, Feig B, Seto R, Infante L, Ishii H, Vecchione A, et al. Restoration of receptor-type protein tyrosine phosphatase eta function inhibits human pancreatic carcinoma cell growth in vitro and in vivo. Carcinogenesis. 2004;25:2107–2114. doi: 10.1093/carcin/bgh224. [DOI] [PubMed] [Google Scholar]
- Tridandapani S, Lyden TW, Smith JL, Carter JE, Coggeshall KM, Anderson CL. The adapter protein LAT enhances fc gamma receptor-mediated signal transduction in myeloid cells. J Biol Chem. 2000;275:20480–20487. doi: 10.1074/jbc.M909462199. [DOI] [PubMed] [Google Scholar]
- Tsuda H, Maynard-Currie CE, Reid LH, Yoshida T, Edamura K, Maeda N, Smithies O, Jakobovits A. Inactivation of the mouse HPRT locus by a 203-bp retroposon insertion and a 55-kb gene-targeted deletion: establishment of new HPRT-deficient mouse embryonic stem cell lines. Genomics. 1997;42:413–421. doi: 10.1006/geno.1997.4771. [DOI] [PubMed] [Google Scholar]
- Wilde JI, Watson SP. Regulation of phospholipase C gamma isoforms in haematopoietic cells: why one, not the other? Cell Signal. 2001;13:691–701. doi: 10.1016/s0898-6568(01)00191-7. [DOI] [PubMed] [Google Scholar]
- Yanagi S, Sugawara H, Kurosaki M, Sabe H, Yamamura H, Kurosaki T. CD45 modulates phosphorylation of both autophosphorylation and negative regulatory tyrosines of Lyn in B cells. J Biol Chem. 1996;271:30487–30492. doi: 10.1074/jbc.271.48.30487. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.