

SUMMARY
Proper connections between centromeres and spindle microtubules are of critical importance in ensuring accurate segregation of the genome during cell division. Using an in vitro approach based on the sequence-specific budding yeast centromere, we identified a complex of the chromosomal passenger proteins Bir1 and Sli15 (Survivin and INCENP) that links centromeres to microtubules. This linkage does not require Ipl1/Aurora B kinase, whose targeting and activation are controlled by Bir1 and Sli15. Ipl1 is the tension-dependent regulator of centromere-microtubule interactions that ensures chromosome bi-orientation on the spindle. Elimination of the linkage between centromeres and microtubules mediated by Bir1-Sli15 phenocopies mutations that selectively cripple Ipl1 kinase activation. These findings lead us to propose that the Bir1-Sli15 mediated linkage, which bridges centromeres and microtubules and includes the Aurora kinase-activating domain of INCENP family proteins, is the tension sensor that relays the mechanical state of centromere-microtubule attachments into local control of Ipl1 kinase activity.
Keywords: kinetochore, centromere, microtubule, chromosomal passenger, tension, aurora, survivin
INTRODUCTION
During mitosis, kinetochores assemble on the centromeric regions of each sister chromatid to act as the primary chromosomal attachment sites for spindle microtubules (Cleveland et al., 2003; Kline-Smith et al., 2005; Maiato et al., 2004). Kinetochores exhibit both end-on and lateral interactions with microtubules. End-on connections between the outer kinetochore and microtubule plus ends couple chromosome motility to changes in the polymerization and depolymerization of bound microtubules (Inoue and Salmon, 1995). Lateral interactions between kinetochores and spindle microtubules are associated with poleward as well as equatorial chromosome movements that facilitate the establishment of stable end-on connections (Kapoor et al., 2006; Rieder and Alexander, 1990; Tanaka et al., 2005).
In addition to their mechanical role in segregation, kinetochores serve as signaling hubs that inhibit anaphase onset until every chromosome in the cell is properly connected. The kinetochore-based mitotic checkpoint pathway relays the presence of any unattached kinetochores into inhibition of the ubiquitin protein ligase that triggers sister chromatid separation and mitotic exit (Cleveland et al., 2003; Nasmyth, 2005; Pinsky and Biggins, 2005). Classic micromanipulation studies in insect spermactocytes as well as recent work using chromosome engineering in budding yeast have highlighted the importance of tension in selectively stabilizing correctly bi-oriented chromatid pairs (Dewar et al., 2004; Li and Nicklas, 1995; Nicklas and Koch, 1969; Stern and Murray, 2001). Bi-orientation places kinetochore-microtubule connections under tension whereas incorrect syntelic attachments, where the kinetochores on both sisters are connected to the same spindle pole, do not. The conserved Aurora B kinase is required to eliminate syntelic attachments, facilitating new connection attempts until the correct configuration is achieved (Biggins and Murray, 2001; Dewar et al., 2004; Ditchfield et al., 2003; Hauf et al., 2003; Kallio et al., 2002; Lampson et al., 2004; Pinsky et al., 2003; Tanaka et al., 2002). The action of Aurora B generates unoccupied kinetochores that in turn signal via the mitotic checkpoint pathway to prevent anaphase onset (Pinsky et al., 2006). Thus, the choreography of chromosome segregation is comprised of an intimate feedback between the mechanics of kinetochore-microtubule connections and localized signaling pathways.
Efforts to reconstitute the mechanical and regulatory functions of kinetochores in vitro have been limited by the complexity of the underlying centromeric DNA (Cleveland et al., 2003). An exception to this complexity is budding yeast, where centromeres consist of a well-defined ∼125 base pair region (Clarke, 1998; McAinsh et al., 2003). The biochemical identification of CBF3, the protein complex that directly binds the key cis-acting CDEIII domain (Lechner and Carbon, 1991), provided further impetus for analyzing kinetochore-microtubule interactions in vitro. Previous studies have demonstrated that budding yeast centromeric (CEN) DNA will bind to microtubules following incubation in a cell extract (Hyman et al., 1992; Kingsbury and Koshland, 1991; Severin et al., 1997; Sorger et al., 1994). This interaction requires the CBF3 complex and is subject to regulation by Ipl1, the budding yeast Aurora B kinase, and the counteracting phosphatase PP1/Glc7 (Biggins et al., 1999; Sassoon et al., 1999). However, CBF3 is not sufficient, indicating that other factor(s) are necessary to link CEN DNA-CBF3 to microtubules (Sorger et al., 1994). Here, we extend this in vitro approach to biochemically identify the missing factor(s). Our results reveal that a complex of two chromosomal passenger proteins, Bir1/Survivin and Sli15/INCENP, connects CEN DNA-CBF3 to microtubules in vitro. This connection is independent of Ipl1, whose activation and targeting are controlled by Bir1 and Sli15. In vivo analysis of Sli15 mutants that eliminate the in vitro activity leads us to propose that the Bir1-Sli15 mediated linkage between CBF3-CEN DNA and microtubules acts as a tension sensor that activates Ipl1 in the vicinity of incorrect syntelic attachments.
RESULTS
A Quantitative In Vitro Assay for the Interaction of CBF3-bound CEN DNA with Microtubules
In the presence of a yeast cell extract, fluorescent beads coupled to CEN DNA will bind to immobilized microtubules adsorbed to a coverslip surface. Although required, CBF3 is not sufficient for binding, indicating the presence of additional factors(s) in the extract that connect CBF3-bound CEN DNA to microtubules (Sorger et al., 1994). To identify these factor(s), we adapted the bead assay to enable rapid, reproducible testing of column fractions (Fig. 1A). As expected, beads coated with CEN DNA exhibited robust microtubule binding in the presence of wild-type extract, whereas beads coated with a mutant CEN DNA that renders it non-functional in vivo failed to bind (Fig. 1B).
Fig. 1.
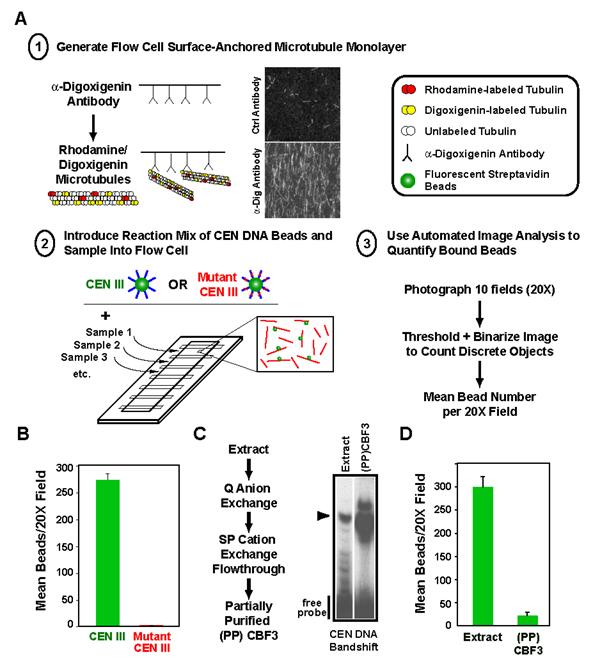
A quantitative in vitro assay for the binding of budding yeast CEN DNA to microtubules. (A) Schematic of the in vitro assay. Adaptations to the original scheme important for quantitative biochemical analysis are emphasized here and include: 1) stable adsorption of microtubules using tubulin covalently modified with digoxigenin; 2) multiplexing of flow cells on a single slide; and 3) automated image analysis to measure number of bound beads. For quantitation, 10 fields at 20X magnification are photographed per sample and averaged. (B) Linkage of beads to microtubules is observed with wild-type but not mutant CEN DNA. The mutant harbors a deletion of the central CCG in CDEIII that prevents binding of the CBF3 complex and abolishes centromere activity in vivo. Error bars=SD. (C) Partial purification of CBF3 using a CEN DNA gel-shift assay. The flowchart describes the chromatography steps and the gel panel shows enrichment of the CEN DNA bandshift relative to starting extract in the partially purified (PP) fraction. The arrowhead marks the CEN DNA-CBF3 complex. (D) Partially purified CBF3 does not link CEN DNA beads to microtubules. Note that the volume of starting extract used to prepare the CBF3 added to the (PP)CBF3 reaction is ∼25-fold greater than that assayed in the extract reaction. If equivalent extract volumes are assayed, no binding is observed with (PP)CBF3. Error bars=SD.
To generate a source of CBF3 for in vitro complementation assays, we optimized a partial purification using CEN DNA-bandshift to monitor CBF3 activity (Fig. 1C). This procedure yielded ∼50-fold partially purified (PP)CBF3 that exhibited a robust bandshift (Fig. 1C), but did not support binding of CEN DNA beads to microtubules on its own (Fig. 1D). Extracts prepared from strains harboring mutations in CBF3 lacked CEN DNA bandshift but complemented (PP)CBF3 in the bead-microtubule binding assay (data not shown, (Sorger et al., 1994)). Trypsin treatment indicated that the complementing activity is protease-sensitive (Suppl. Fig. 1). Taken together, these results confirmed the existence of an unknown protein(s) that connects CBF3-bound CEN DNA to microtubules in vitro and established a robust assay that could be used for its identification.
A Conventional Purification Strategy Identifies Bir1 as a Candidate for the Activity that Links CBF3-bound CEN DNA to microtubules
To identify the protein(s) that connect CBF3-bound CEN DNA to microtubules we utilized two strategies. First, we tested CBF3 complementing activity in extracts either prepared from mutant strains or immunodepleted of candidate kinetochore and microtubule-binding proteins. At the budding yeast kinetochore, the Dam1 ring complex plays an important role in bi-oriented microtubule attachments and the Mis12, Ctf19 and Ndc80 complexes are suggested to direct assembly of the microtubule-binding interface (reviewed in (De Wulf et al., 2003; McAinsh et al., 2003; Tanaka et al., 2005)). However, neither mutations in nor immunodepletions of these complexes perturbed the in vitro linkage between CEN DNA and microtubules (Suppl. Table 1). Other candidates, including motor and non-motor microtubule-binding proteins, were similarly excluded. These results indicated the presence of a CBF3-dependent linkage between CEN DNA and microtubules that did not involve any of the obvious candidates suggested by prior studies.
Parallel to the candidate analysis, we pursued an unbiased conventional purification, using the quantitative in vitro assay (Fig. 2A). Complementing activity was measured relative to a standard curve generated by serially diluting the starting material for the purification step into a constant amount of (PP)CBF3 (Fig. 2B, C). Negative controls in which mutant CEN DNA beads were used, or (PP)CBF3 was omitted, verified the specificity of binding. By combining 3 purification steps in series, we enriched the complementing activity ∼400 fold (Fig. 2D). However, attempts at additional purification resulted in a significant loss of specific activity that did not appear to be due to separation of different components.
Figure 2.
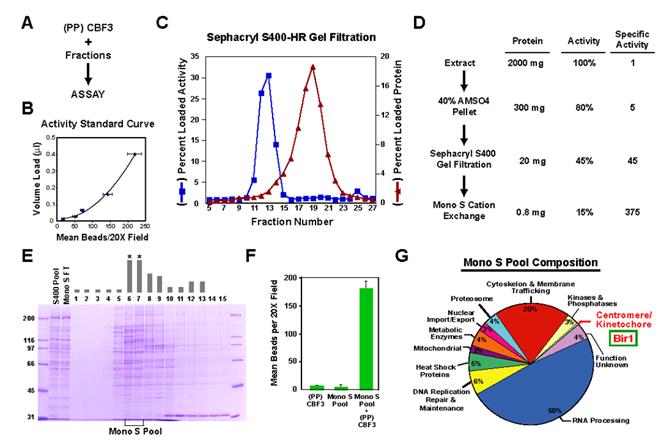
Conventional purification of an activity that complements CBF3 in the in vitro assay. (A) Schematic of the in vitro complementation approach. (B) Example of a standard curve used to quantitatively monitor fractionation of the complementing activity. The starting material, in this case the gel filtration load, is serially diluted into a constant amount of (PP)CBF3 and the points fitted to a polynomial curve. Complementing activity in each fraction measured after adding the same amount of (PP)CBF3 is converted to a percentage of total loaded activity. Error bars=SD. (C) Column profile of Sephacryl S400HR gel filtration. The percentage of loaded activity calculated from the standard curve and the percentage of total loaded protein is plotted for each fraction. (D) Summary of the complementing activity purification. The activity column lists the percentage yield, relative to the starting extract, after each step. (E) Fractions from the MonoS gradient elution stained with Coomassie Blue. The complementing activity is indicated with the gray bars above each fraction. No activity is detected in the column flowthrough. Asterisks denote the two fractions that constitute the MonoS Pool. (F) The MonoS Pool complements (PP)CBF3. Error bars=SD. (G) Annotation-based classification of proteins identified by mass spectrometry of the MonoS pool. The 247 proteins that showed >10% sequence coverage are represented in the pie chart (see also Suppl. Table 2).
Since the purification did not achieve sufficient enrichment to directly correlate complementing activity with co-purifying proteins, we identified candidates that could be functionally tested by performing mass spectrometry (Washburn et al., 2001) on the highest specific activity material—the Mono S cation exchange pool (Fig. 2E, F). 247 polypeptides with greater than 10% sequence coverage were detected in the Mono S pool (Suppl. Table 2). Database functional annotations identified a single protein within this large set previously implicated in centromere function (Fig. 2G; Suppl. Table 2). This protein was Bir1, the budding yeast homolog of the Survivin subunit of the chromosomal passenger complex. This finding led us to focus on Bir1 as a candidate for the CBF3 complementing activity.
Bir1 is an Essential Component of the CBF3 Complementing Activity
To test whether Bir1 is required to link CEN DNA to microtubules in vitro, we generated a strain in which bir1 was deleted. Sporulation of heterozygous diploids yielded two wild-type and two apparently inviable spores (n=105 tetrads), as expected for an essential gene. However, consistent with previous conflicting studies describing bir1 as both essential (Li et al., 2000; Widlund et al., 2006) and non-essential (Uren et al., 1999; Yoon and Carbon, 1999), ∼10% of bir1∆ spores formed tiny colonies after extended incubation. The bir1∆ cells derived from these colonies grew, but had aberrant, polyploid DNA content (data not shown). Similar aberrant cells are obtained if the deletion is covered with a centromeric plasmid expressing Bir1 and the plasmid is removed from the deleted haploid using negative selection after tetrad dissection, indicating that neither passage through meiosis nor haploinsufficiency in the diploid state are required for the observed phenotype. Immunoblotting using an affinity-purified anti-Bir1 antibody confirmed that a series of bands present in wild-type extracts, likely reflecting differentially phosphorylated forms of Bir1, are absent in bir1∆ extracts (Fig. 3A).
Figure 3.
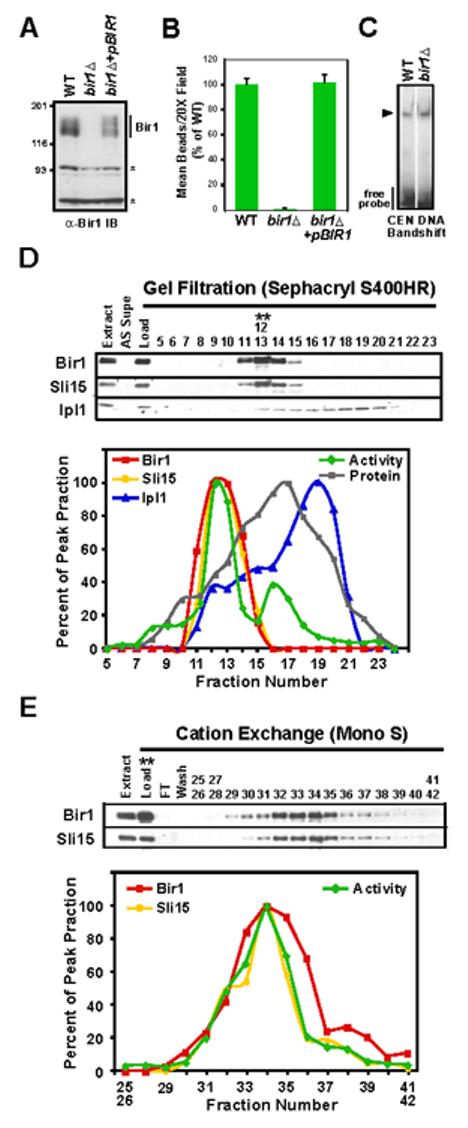
Bir1 is required for linking CEN DNA to microtubules in vitro. (A) bir1∆ cells lack Bir1 protein. Western blot of extracts prepared from WT (ODY49), bir1∆(ODY65), and bir1∆+pCEN-BIR1 (ODY114) strains probed with an anti-Bir1 antibody. Asterisks indicate background bands that serve as loading controls. (B) Bir1 is required for linking CEN DNA to microtubules. Extracts indicated in (A) were analyzed using the bead assay. Activity was normalized relative to the wild-type extract. Error bars=SD. (C) Bir1 does not affect the ability of CBF3 to bind to CEN-DNA. Arrowhead indicates position of the CBF3-CEN DNA complex. (D) Bir1 and Sli15, but not Ipl1, co-fractionate with the complementing activity. Gel filtration fractions of extracts prepared from BIR1:6HA; SLI15:13Myc (ODY97) were analyzed by western blotting using anti-HA, anti-Myc and anti-Ipl1 antibodies. The blot signal intensity for all 3 proteins, as well as activity in the bead assay, is plotted as a percentage of the respective peak fractions (12/13 for Sli15, Bir1, and activity; 18 for Ipl1). (E) Bir1 and Sli15 continue to co-fractionate with the complementing activity during the cation exchange step. The activity peak from gel filtration (fractions 12/13 in (D)) was further fractionated using a MonoS cation exchange column and analyzed as in (D).
Extracts prepared from bir1∆ cells exhibit no activity in the in vitro CEN DNA-microtubule interaction assay (Fig. 3B), whereas the ability of the CBF3 to bind CEN DNA was unaffected (Fig. 3C). Addition of (PP)CBF3 to bir∆ extracts failed to restore CEN-DNA microtubule binding activity (data not shown). Transformation with a plasmid encoding Bir1 under control of its endogenous promoter fully restored activity in the assay (Fig. 3B) as well as the bands detected in the Bir1 molecular weight range in immunoblots (Fig. 3A). We conclude that Bir1, a candidate identified by mass spectrometry of a fraction enriched for the complementing activity by conventional purification, is an essential component of the link between CBF3-bound CEN DNA and microtubules in vitro.
Sli15 and Bir1, but not Ipl1, are Required for the CBF3 Complementing Activity
Bir1, like its homologue Survivin in higher eukaryotes, is a subunit of the chromosomal passenger complex. In budding yeast, this complex includes Ipl1, the single Aurora family kinase in budding yeast, and Sli15, the homologue of the Aurora B activator and targeting subunit INCENP (Adams et al., 2000; Cheeseman et al., 2002; Kim et al., 1999). Since the complexity of the Mono S fraction could have prevented identification of the other two subunits by mass spectrometry, we repeated the purification and monitored the fate of each subunit. Bir1 and Sli15 showed a fractionation profile that was similar to the activity throughout the purification (Fig. 3D, E). In contrast, the majority of Ipl1 fractionated away from Bir1-Sli15 into a distinct pool of smaller hydrodynamic radius during the gel filtration step (Fig 3D).
The fractionation analysis suggested that a complex containing Bir1 and Sli15 is responsible for the in vitro complementing activity, and that Ipl1 might be dispensable. Since Sli15 and Ipl1 are essential genes, and the available conditional alleles do not affect protein levels (see below), we tested this by immunodepleting >90% of Sli15 or Ipl1, each tagged with 6 copies of the hemagglutinin (HA) epitope (Fig. 4A). Consistent with their fate during purification, depletion of Sli15 dramatically reduced activity whereas depletion of Ipl1 had no effect (Fig. 4A). The assays were performed without ATP addition, and normal binding was observed at all tested dilutions, arguing against the catalytic activity of residual Ipl1 kinase accounting for the difference between extracts depleted of Sil15 and Ipl1.
Figure 4.
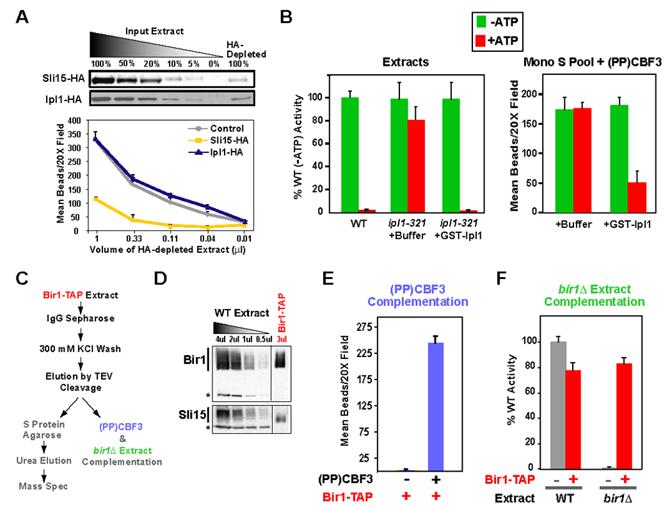
Sli15, but not Ipl1, is required for the complementing activity. (A) Extracts prepared from SLI15:6HA (ODY54) and IPL1:6HA (ODY55) cells were immunodepleted using an anti-HA affinity resin. The depleted supernatants were serially diluted and complementing activity measured with constant (PP)CBF3. Comparing a standard curve of the input extract to the depleted extract using anti-HA immunoblotting assessed depletion efficiency. Both Sli15 and Ipl1 were successfully depleted by >90% but only the Sli15 depletion resulted in a severe activity reduction. (B) Ipl1 kinase activity regulates the binding observed in the in vitro assay. ipl1-321 mutant extracts are insensitive to ATP addition, which is in contrast to wild-type extracts. Addition of purified GST-Ipl1 protein restores ATP sensitivity to the mutant extracts. The MonoS pool combined with (PP)CBF3 is ATP-insensitive in the bead assay but becomes sensitive following addition of GST-Ipl1. (C) Flowchart describing the purification and use of the Bir1-TAP complex. (D) Immunoblot of Bir1-TAP after elution by TEV cleavage. Both Sli15 and Bir1 are present in the elution. (E) Bir1-TAP complements (PP)CBF3. (F) Bir1-TAP complements loss of activity in bir1∆ extracts. In both (E) and (F), the amount of Bir1-TAP added is similar to that present in 2 μl of extract, which is the standard amount analyzed in the bead assay (D).
Although Ipl1 does not appear to be required to connect CEN DNA to microtubules in vitro, its kinase activity regulates this linkage (Biggins et al., 1999). Addition of ATP to wild-type extracts severely inhibits binding of CEN DNA beads to microtubules (Fig. 4B; (Biggins et al., 1999)). Sensitivity to ATP addition is lost in extracts prepared from ipl1 mutants with compromised kinase activity, but can be restored following addition of purified Ipl1 protein (Fig. 4B). This result predicts that if Ipl1 is absent from fractions enriched for the complementing activity, they should be insensitive to ATP. We tested this prediction using the Mono S pool (Fig. 2E, F). The binding observed in mixtures of the Mono S pool and (PP)CBF3 is insensitive to ATP, but becomes sensitive following addition of purified Ipl1 protein (Fig. 4B). These results suggest that the CBF3 complementing activity is comprised of a complex containing Bir1-Sli15 that is independent of Ipl1 but subject to regulation by its kinase activity.
A Purified Endogenous Bir1-Sli15 Complex Complements CBF3 and bir1∆ Extracts
To determine if a purified Bir1-Sli15 complex complements (PP)CBF3, we generated extracts from a strain expressing TAP (tandem affinity purification)-tagged Bir1. Following incubation with IgG resin, the TAP-Bir1 was eluted by cleavage with TEV protease. A fraction of the elution was subjected to the second affinity purification step and analyzed by mass spectrometry; the rest of the elution was used for in vitro experiments (Fig. 4C). Both immunoblotting and mass spectrometry indicated that the TAP prep of Bir1 contains Sli15 (Fig. 4D), but not Ipl1, as expected from a previous study (Widlund et al., 2006). Neither protein was detectable in the (PP)CBF3 fraction (Suppl. Table 3). The purified Bir1-Sli15 complex had no activity on its own, but exhibited robust activity when mixed with (PP)CBF3 (Fig. 4E). Mass spectrometry of (PP)CBF3 indicated that no other known kinetochore proteins were present in this fraction (Suppl. Table 3). Addition of Bir1-Sli15 also complemented the activity loss in bir1∆ extracts (Fig. 4F). These findings strongly suggest that a complex containing Bir1 and Sli15 directly connects CBF3-bound CEN-DNA to microtubules in vitro. This conclusion is supported by previous work documenting an interaction between Bir1 and the Ndc10 subunit of CBF3 (Bouck and Bloom, 2005; Gillis et al., 2005; Yoon and Carbon, 1999) and direct binding of Sli15 to microtubules (Kang et al., 2001).
Bir1-Sli15 and Ipl1 are chromosomal passenger proteins that localize prominently to the anaphase spindle. Dephosphorylation of Sli15 by the phosphatase Cdc14 is required for this localization (Pereira and Schiebel, 2003). Recent work has shown that the CBF3 complex also localizes to the anaphase spindle (Bouck and Bloom, 2005; Gillis et al., 2005). These observations raised the possibility that the in vitro CEN DNA-microtubule linkage assay is detecting microtubule-binding activity of the pool of Bir1-Sli15 present in anaphase. To test this, we analyzed cdc14-1 and cdc15-2, two mutants in which Sli15 anaphase spindle localization is not observed (Pereira and Schiebel, 2003; Stoepel et al., 2005). Extracts prepared from these mutants exhibited wild-type CEN DNA-microtubule linkage (Suppl. Fig 2A). Linkage activity is also low in G1, when Sli15 is dephosphorylated, but high in mitosis, when Sli15 is hyperphoshorylated (Suppl. Fig. 2B). Together with the in vivo analysis described below, these results support the conclusion that the CEN DNA-microtubule linkage mediated by Bir1-Sli15 is of critical importance during chromosome segregation.
Shutting off Sli15 or Deleting its Microtubule-Binding Domain Eliminates the Linkage Between CEN DNA and Microtubules in Vitro
To investigate the physiological role of the Bir1-Sli15 mediated linkage between CBF3-CEN DNA and microtubules, we focused our efforts on Sli15 because of the abnormal polyploidy observed in bir1 mutants. Extracts prepared from strains harboring the only previously characterized mutant allele of SLI15, sli15-3 (Kim et al., 1999), exhibited normal CEN DNA-microtubule binding, but were insensitive to the addition of ATP (Fig. 5A). sli15-3 extracts are therefore similar to extracts prepared from ipl1 mutants, but distinct from extracts immunodepleted of Sli15 (see Fig. 4A). This difference was explained by sequencing sli15-3, which revealed that the critical mutation is within the IN box domain that binds to and activates Ipl1 kinase (Suppl Fig. 3A, B; (Sessa et al., 2005). Thus, the mutant protein encoded by the sli15-3 allele, while defective in Ipl1 activation, is present at normal levels (Suppl Fig. 3C) and capable of linking CEN DNA to microtubules in vitro.
Figure 5.
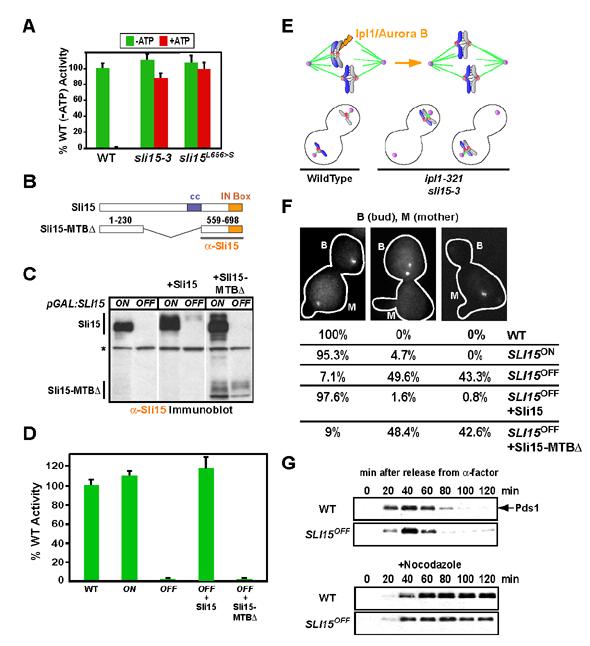
Sli15 mutants that eliminate CEN DNA-microtubule interactions in vitro phenocopy Ipl1 kinase activation mutants in vivo. (A) sli15-3 extracts behave similarly to Ipl1 kinase mutant extracts. sli15L656>S is an engineered mutant with only the IN box amino acid change in sli15-3 (see Suppl. Fig. 2). (B) Schematic of Sli15 and the Sli15-MTB-binding domain. The C-terminal region used to generate the anti-Sli15 antibody is underlined. (C) Immunoblotting of ODY155 (pGAL-SLI15), ODY192 (pGAL-SLI15+Sli15) and ODY193 (pGAL-SLI15+Sli15-MTB∆) in galactose (ON) or glucose (OFF) medium. All SLI15OFF samples were prepared from cells arrested with nocodazole to maintain viability. Asterisk denotes the background band that serves as a loading control. See also Suppl. Fig. 4 and Suppl. Fig. 5. (D) Depletion of Sli15 or deletion of its microtubule-binding domain eliminate the CEN DNA-microtubule linkage in vitro. (E) Ipl1/Aurora B is required for the correction of attachment errors where both sister chromatids are connected to the same spindle pole (syntelic attachment; upper panels). Mutants that perturb Ipl1 kinase activity (ipl1-321, ipl1-2, sli15-3), fail to correct syntelic attachments, resulting in sister chromatid missegregation (lower panels). (F) SLI15OFF and SLI15OFF+Sli15-MTB∆ both phenocopy Ipl1 kinase activity mutants in vivo. Segregation of a marked Chr IV was monitored 120 minutes after release from alpha factor arrest. Between 100-200 cells for each strain and growth condition were analyzed. The mother cell (M) was identified by residual alpha factor-induced shmoos (projections in cell outline). (G) SLI15OFF cells do not arrest in the cell cycle despite having a functional checkpoint. WT (SBY818) and pGAL:SLI15 cells expressing Pds1-Myc18 (ODY181) were grown as described above. Nocodazole was added to one set of cultures at 10 μg/ml upon release from alpha factor. Lysates were immunoblotted with an anti-Myc antibody.
Since sli15-3 encodes a protein that retains the ability to link CEN DNA to microtubules, a new mutant was needed to investigate the in vivo role of the Bir1-Sli15 mediated linkage. To obtain such a mutant, we generated a strain where the galactose promoter is integrated upstream of the SLI15 coding region (pGAL-SLI15). Sli15 is overexpressed when this mutant is grown in galactose (SLI15ON; Fig. 5C, Suppl. Fig. 4A) but this does not result in a significant phenotype. Switching this strain to glucose medium (SLI15OFF) causes a dramatic loss in Sli15 protein levels (Fig. 5C, Suppl. Fig. 4A). Importantly, in agreement with results obtained from immunodepleting Sli15, no CEN DNA-microtubule linkage activity was detected in SLI15OFF extracts (Fig. 5D).
The Sli15 shutoff mutant offered the opportunity to directly test the role of the Sli15 microtubule-binding domain in the in vitro linkage. Sli15 is a multi-domain protein with a centrally located microtubule-binding domain (Kang et al., 2001). The C-terminus of Sli15, like all INCENP family proteins, contains the Aurora-activating IN box which binds to and activates Aurora B (Kang et al., 2001; Sessa et al., 2005). To test the function of the microtubule-binding domain, we integrated a mutant (Sli15-MTB∆; Fig. 5B) into the URA3 locus of the strain harboring the pGAL-SLI15 shutoff allele. Wild-type Sli15 coding sequence was integrated in parallel as a control. Both the mutant and wild-type coding regions were under control of the endogenous SLI15 promoter.
Immunoblots confirmed expression of Sli15-MTB pGAL-SLI15 (Fig. 5C) and the mutant protein retained the ability to interact with Bir1 (Suppl. Fig. 5B), indicating that it is not misfolded. When expression of wild-type Sli15 was turned off, cells expressing the Sli15-MTB contrast to controls expressing the wild-type protein, indicating that the microtubule-binding domain of Sli15 is essential for viability (Suppl. Fig. 5A). Analysis of nocodazole-arrested extracts prepared after shutting off pGAL-SLI15 showed that the Sli15-MTB -microtubule linkage in contrast to full-length Sli15, which supported normal binding (Fig. 5D). Thus, the linkage detected in vitro requires the microtubule-binding domain of Sli15.
Both the Sli15 Shutoff and the Sli15 Microtubule-Binding Domain Deletion Mutant Phenocopy Ipl1 Kinase Activation Mutants In Vivo
The Sli15 shutoff and the Sli15-MTB CEN DNA and microtubules in vitro. Analyzing the phenotype of these mutants would therefore make it feasible to distinguish between two potential models for the physiological role of this linkage. First, as anticipated during the initial development of the bead assay, the Bir1-Sli15 linkage could play an Ipl1-independent role in the force-transducing connection of centromeres to spindle microtubules, which we will refer to as the “core” attachment. Alternatively, the role of Sli15 as an activator of Ipl1 raised the attractive possibility that Bir1-Sli15 mediate a “tension-sensing” attachment that ensures chromosome bi-orientation by locally controlling Ipl1 kinase activation.
To distinguish between these models, we used a GFP-marked chromosome to follow the segregation of sister chromatids. The failure to correct syntelic attachments in ipl1 mutants results in sister chromatids co-segregating to the mother or the bud with roughly equal frequency (Fig. 5E; (Biggins and Murray, 2001; Biggins et al., 1999; Tanaka et al., 2002)). In both the SLI15OFF and SLI15OFF+Sli15-MTB∆cells, the marked chromosome exhibited a missegregation pattern similar to ipl1 kinase activity mutants (Fig. 5F). This missegregation pattern is distinct from different classes of kinetochore mutants required for the core attachment (reviewed in (Biggins and Walczak, 2003)). By contrast, the majority of sisters segregated properly in SLI15ON and SLI15OFF+Sli15 cells (Fig. 5F).
In ipl1 mutants, despite a functional checkpoint and nearly 100% missegregation, the cell cycle is not delayed because core attachments, although incorrect, are present. This is in contrast to mutations that compromise the core attachment, which either delay in the cell cycle due to checkpoint activation, or lack a functional checkpoint due to severe defects in kinetochore structure (Biggins and Walczak, 2003). To determine if cells lacking Sli15 have a functional checkpoint and/or experience a cell cycle delay, we monitored the levels of securin (Pds1) after releasing SLI15OFF cells from a G1 arrest in the presence or absence of nocodazole. SLI15OFF mutants arrested normally in nocodazole, indicating the presence of a functional checkpoint (Fig. 5G). However, in the absence of nocodazole, SLI15OFF cells do not exhibit a significant cell cycle delay (Fig. 5G) despite extensive missegregation of sister chromatids (Fig. 5F), and exhibit a rapid loss of cell viability (Suppl. Fig. 4C). These results indicate that cells lacking Sli15 behave similarly to ipl1 kinase activity mutants (ipl1-321, ipl1-2, sli15-3), but distinctly from mutants in kinetochore proteins important for the core attachment. We conclude that the Bir1-Sli15 mediated linkage between CEN DNA and microtubules is not part of the core attachment. Instead our results indicate that the Bir1-Sli15 mediated linkage is essential for the tension-dependent regulation of microtubule attachments by Ipl1 that ensures bi-orientation
DISCUSSION
A Complex of Bir1-Sli15 Links CBF3-CEN DNA to Microtubules in Vitro
The ability of budding yeast CEN DNA to interact with microtubules following incubation in a cell extract was first documented more than a decade ago. The importance of CBF3, the sequence-specific scaffold of the budding yeast kinetochore, was immediately apparent. However, the identity of an additional necessary component remained elusive. Here, we used a biochemical complementation assay to show that a complex of Bir1-Sli15 links CBF3-CEN DNA to microtubules in vitro. Bir1 directly interacts with the Ndc10 subunit of CBF3 (Bouck and Bloom, 2005; Gillis et al., 2005; Yoon and Carbon, 1999) and the central region of Sli15, required for the in vitro CEN DNA-microtubule linkage (this study), directly binds microtubules (Kang et al., 2001). Although a full reconstitution using recombinant proteins remains to be achieved, the evidence strongly suggests that the linkage observed in the in vitro assay can be accounted for by Bir1-Sli15 directly connecting CBF3-CEN DNA to microtubules.
It is interesting to note that the majority of proteins required for kinetochore assembly and function in vivo, do not play a role in the in vitro linkage. This result was anticipated by the earlier finding that the CDEIII element of CEN DNA is sufficient in the in vitro assay but is not sufficient for centromere function in cells (Sorger et al., 1994). We suspect that the specialized chromatin domain on which kinetochores assemble, which includes nucleosomes containing the centromere-specific histone CENP-A, will be important for achieving in vitro reconstitution of the core attachment.
Ipl1 Kinase Regulates but is Not Required for the CEN DNA-Microtubule Linkage In Vitro
Ipl1 kinase is dispensable for connecting CBF3-bound CEN-DNA to microtubules but regulates this linkage (this study; (Biggins et al., 1999)). This observation raises the question as to what specific components are phosphorylated by Ipl1. In the original study describing Ipl1 regulation, the Ndc10 subunit of CBF3 was suggested to be the critical target (Biggins et al., 1999). Identification of Bir1-Sli15 as the linker between CBF3-CEN DNA and microtubules raises the possibility that phosphorylation of these components may also be important. In particular, Sli15 is a well-established Ipl1 substrate in addition to being its activator (Cheeseman et al., 2002; Kang et al., 2001). The binding of CBF3 to CEN DNA is unaffected in Ipl1 kinase mutant or bir1∆ extracts (this study; (Biggins et al., 1999)). An important future direction will be to determine if Ipl1 phosphorylation regulates the interaction between CBF3 and Bir1-Sli15, between Bir1 and Sli15, or between Bir1-Sli15 and microtubules.
In Vivo Function of the Bir1-Sli15 Mediated Linkage Between CBF3-bound CEN DNA and Microtubules
In considering the in vivo role of the Bir1-Sli15 mediated linkage in chromosome segregation, we focused on the connection to Ipl1, which is known to play a central role in bi-orientation. In the bi-oriented state, sister chromatids are connected to opposite spindle poles, placing centromere-spindle attachments under tension. Improper syntelic attachments are detected and eliminated in an Ipl1-dependent manner, facilitating new connection attempts until all chromosomes are bi-oriented. This paradigm for the mechanism of bi-orientation raises the question as to how changes in tension at centromere-microtubule connections are relayed to control Ipl1 kinase activity. The centromeric cohesion protector Sgo1 has been suggested to act as a tension sensor (Indjeian et al., 2005), but this function appears to be distinct from the Ipl1-dependent mechanism (Pinsky et al., 2006).
Our in vivo analysis indicates that the Bir1-Sli15 mediated linkage between CEN DNA and microtubules is required for Ipl1-dependent correction of core attachments lacking tension. One possibility to explain these findings is that the Bir1-Sli15 linkage facilitates recognition of Ipl1 substrates at syntelic attachments. Alternatively, this linkage, which includes the Aurora-activating IN box domain, may constitute the tension-sensitive activator of Ipl1. We favor the latter possibility and discuss it further below although direct evidence for a tension-induced change in the Bir1-Sli15 linkage is necessary to distinguish between these alternatives.
A Speculative Model for Tension-Regulated Ipl1 Activation by Bir1-Sli15
The independent bi-orientation of multiple chromosomes on the same spindle poses a major challenge to understanding tension-regulated Ipl1 activation. We propose that Bir1-Sli15 links centromeres to microtubules in a manner that allows the IN box of Sli15 to locally activate Ipl1 when core attachments are not under tension (Fig. 6). The finding that deletion of the microtubule-binding domain of Sli15 phenocopies ipl1 kinase mutants suggests that formation of the ternary complex between Bir1-Sli15, CBF3-CEN DNA, and microtubules is necessary for robust Ipl1 activation. Such a mechanism could contribute to restricting Ipl1 kinase activity to the vicinity of attachments lacking tension. A potentially intriguing mechanistic parallel exists with Aurora A, the other major Aurora kinase present in metazoans but absent in fungi, that is activated by the TPX2 protein (Kufer et al., 2002; Tsai et al., 2003). Like INCENPs, TPX2 directly binds microtubules and the activation of Aurora A by TPX2 is strongly enhanced by microtubule-binding (Tsai et al., 2003).
Figure 6.
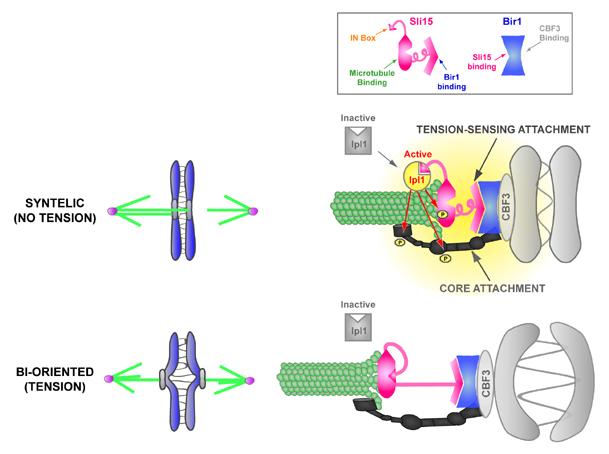
A model for tension-regulated Ipl1 activation by the Bir1-Sli15 mediated linkage between centromeres and microtubules. The central tenet of the model is based on the finding that the Bir1-Sli15 mediated linkage between CBF3-CEN DNA and microtubules is required for correction of syntely but not for the core attachment between centromeres and microtubules in vivo. We propose that this linkage constitutes a tension-sensing attachment that is modulated by the primary force-generating (or force-transducing) core attachment such that syntely (no tension) promotes Ipl1 activation. Active Ipl1 phosphorylates multiple targets (red arrows) to dissociate the centromere from the microtubule. Bi-orientation (tension) silences Ipl1 activation, stabilizing the correct configuration.
Ipl1 activated in the vicinity of syntelic attachments is predicted to phosphorylate components of both the core attachment and the tension-sensing attachment to release the centromere from the microtubule (Fig. 6; (Cheeseman et al., 2002; Kang et al., 2001)). Phosphorylation-triggered detachment of the tension-sensing attachment, presumably recapitulated in the in vitro assay when ATP is present, may reduce Ipl1 activation and promote dephosphorylation to allow new attachments to be formed.
Once a chromosome is bi-oriented and the core attachment is under tension, this in turn could modulate the tension-sensing attachment such that Sli15 is no longer able to activate Ipl1 (Fig. 6). The modulation could occur by a tension-induced conformational change that buries the IN box, as depicted in Fig. 6. Alternatively, tension may detach Bir1-Sli15 from the microtubule or from CBF3, preventing formation of the ternary complex that promotes activation (not depicted). Exploring potential tension regulation will be facilitated by the in vitro CEN DNA bead-microtubule interaction assay, which is amenable to direct biophysical analysis.
Significance to metazoan chromosome segregation
The role of Aurora B in correction of syntelic attachments and the activation of Aurora B by INCENP family proteins are widely conserved (reviewed in (Andrews et al., 2003; Meraldi et al., 2004; Vagnarelli and Earnshaw, 2004)). Drosophila and mammalian INCENPs directly bind to microtubules, indicating that this biochemical activity is also conserved (Adams et al., 2001; Wheatley et al., 2001). Although the CBF3-Bir1/Survivin interaction is restricted to budding yeast, Survivin and another accessory subunit Borealin/Dasra target Aurora B to centromeres in metazoans (reviewed in (Vagnarelli and Earnshaw, 2004). Thus, we suspect that the general principle of utilizing a direct connection between centromeres and spindle microtubules that includes the activator of Aurora B will be widely relevant for understanding how tension helps ensure the correct distribution of chromosomes.
In summary, our work highlights the existence of a direct linkage between centromeres and microtubules that includes the activating scaffold protein for Ipl1/Aurora B. Such a linkage provides the potential for mechanically-sensitive control of kinase activation. The concept of kinase-activating scaffolds acting as local mechanical sensors controlling phosphorylation cascades is likely to also be significant in other cellular contexts.
EXPERIMENTAL PROCEDURES
Strains & Extract Preparation
Strains are summarized in Supplemental Table 4. For Pds1 blots, 1.5ml culture was processed at each time point as described (Pinsky et al., 2006). To test candidate mutants in the bead assay, 200-500 ml cultures were grown to mid-log phase. Cells were washed in water, resuspended in a volume equal to the cell pellet of 2X Breakage Buffer (100mM BisTrisPropane, 200mM β-glycerophosphate, 400mM KCl, 10mM EDTA, 10mM EGTA, 20% glycerol, 1mM PMSF; pH 7.6), frozen as drops in liquid nitrogen, and ground in a liquid nitrogen cooled mortar and pestle. The crude lysate was centrifuged at 15,000g for 2 x 15 min and the supernatant used for assays. Alpha factor was used at 1 μg/ml; nocodazole at 10 μg/ml.
Partial Purification of CBF3
CEN DNA bandshift assays were performed as described (Severin et al., 1999; Severin et al., 1997; Sorger et al., 1994). For the partial CBF3 purification, 50 ml extract was precipitated with 60% ammonium sulfate, resuspended in 40 ml BB150 (50 mM BisTrisPropane, 100 mM β-glycerophosphate, 150 mM KCl, 2 mM EDTA, 2 mM EGTA, 1 mM DTT, 10% glycerol, and protease inhibitors; pH 7.0), dialyzed into BB150, loaded onto a 20 ml Poros HQ/20 column and eluted with a KCl gradient. Fractions with peak activity were pooled, flowed through a 5ml HiTrap SP cation exchange column and concentrated on a 1.6ml Poros HQ/2O column by elution with a KCl step gradient.
In Vitro CEN DNA-Microtubule Interaction Assay
The in vitro assay was performed as described (Severin et al., 1999) with the following modifications. Double-stick tape flow cells were constructed such that 6 samples could be analyzed on a single slide using a multichannel pipette. Taxol microtubules, polymerized using unlabeled, digoxigenin-labeled and rhodamine-labeled tubulin (30:9:1), were adsorbed to anti-digoxigenin (10 μg/ml) antibody-coated flow cell surfaces for 5 min. Unbound microtubules were washed out and the chamber blocked for 5 min in BRB80 (80 mM PIPES, 1 mM MgCl2, 1 mM EGTA, pH 6.8) plus10 μM taxol and 1.5 mg/ml casein. In parallel, 2 μl of ∼20 mg/ml yeast extracts were incubated in 15 μl reactions in Binding Buffer (10 mM HEPES pH 8.0, 6 mM MgCl2, 10% glycerol, ∼120-140 mM KCl); ∼3 μg sheared salmon sperm DNA was added as a non-specific competitor. For complementation assays, (PP)CBF3 was added to ∼3X the bandshift activity present in a typical extract assay, casein was added to 0.3 mg/ml, and the final reaction included 15 mM BisTrisPropane, 30 mM β-glycerophosphate, 0.6 mM EGTA, 0.6 mM EDTA, pH 7.0, in addition to the Binding Buffer components. After 45 min, an equal volume of reaction buffer plus 10 μM taxol was added and reactions were introduced into the flow cell with adsorbed microtubules. 15 min later unbound beads were washed out and 10 fields of the coverslip surface per sample photographed using a 20X, 0.7 NA objective. Bead binding was stable and did not change appreciably for 1-2 hours after the final wash. A custom macro was written to threshold and binarize each image and quantify bead number.
Enrichment of the CBF3 Complementing Activity and Mass Spectrometry
Conventional purification of the CBF3 complementing activity was performed using strain PS886. A fermentor was used to grow 100L cultures to mid-log phase. Cells were harvested using an ultrafiltration cassette followed by centrifugation. After a water wash, cell paste was directly extruded as fine vermicelli-like threads into liquid nitrogen using a compressor driven caulk gun. Extracts were prepared by grinding the frozen cell paste threads in a liquid nitrogen chilled stainless steel Waring blender. The fine cell powder was weighed and thawed by addition of an equivalent volume of 2X Breakage Buffer. Crude extract was clarified in 2 sequential 45,000 rpm spins (2 hrs each, 4°C) and frozen in 50 ml aliquots. For a standard prep, 125-150 ml extract was precipitated with 40% ammonium sulfate, the pellet resuspended in 0.5X extract volume of BB300, loaded onto a 1L S400HR gel filtration column volume (in BB300), peak activity fractions dialyzed into SB150 (100 mM BisTrisPropane, 150 mM KCl, 2 mM EDTA, 2 mM EGTA, 0.5 mM DTT, 10% glycerol, and protease inhibitors; pH 7.0), loaded onto a 1 ml Mono S column and eluted with a 20 column volume SB150 to SB1000 gradient. TCA was added to a final concentration of 20% to 50 micrograms of the Mono S pool and the sample left overnight on ice. The precipitate was washed in cold acetone and analyzed by MudPIT mass spectrometry (Washburn et al., 2001).
HA-Immunodepletions, TAP Purifications and Antibody Production
For immunodepletions, extracts prepared from 6HA-tagged strains were diluted with an equal volume of Extract Dilution Buffer (50mM BisTrisPropane, 1mM EDTA, 1mM EGTA, 5% glycerol, plus protease inhibitors) clarified by centrifugation at 80,000 rpm for 10 min at 4°C in a TLA100.2 rotor and added to ∼25 μl anti-HA resin (Roche). The bead/extract mix was rotated at 4°C for 2 hours and the supernatant used for assays and quantifying depletions. TAP tagging and purifications were carried out as described (Cheeseman et al., 2002). GST-Ipl1 (full length), GST-Sli15 (aa558-698), and GST-Bir1 (aa700-945) were used as antigens for injection into rabbits. Affinity purifications were performed using standard methods.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Sue Biggins and Iain Cheeseman for advice and encouragement, Paul Maddox for help with imaging, Scott Emr for use of his tetrad dissection scope, David Drechsel for help with the fermentor, and the yeast chromosome segregation community for its generosity, especially the Biggins, Drubin, Barnes, Kilmartin, Nasmyth, and Murray labs. For comments on the manuscript, we thank Don Cleveland, Ben Black, Iain Cheeseman and Reto Gassmann. This work was supported by a training grant from the National Cancer Institute (S.S.), by a grant from the National Center for Research Resources, NIH to Trisha N. Davis (P41 RR11823), by grants from the Human Frontier Science Program and the NIH to A.D., and by the Max Planck Institute (F.S., A.H.). K.O. is a Pew Scholar in the Biomedical Sciences; A.D. is the Connie and Bob Lurie Scholar of the Damon Runyon Cancer Research Foundation (DRS 38-04). K.O. and A.D. receive salary and additional support from the Ludwig Institute for Cancer Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Adams RR, Maiato H, Earnshaw WC, Carmena M. Essential roles of Drosophila inner centromere protein (INCENP) and aurora B in histone H3 phosphorylation, metaphase chromosome alignment, kinetochore disjunction, and chromosome segregation. J Cell Biol. 2001;153:865–880. doi: 10.1083/jcb.153.4.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams RR, Wheatley SP, Gouldsworthy AM, Kandels-Lewis SE, Carmena M, Smythe C, Gerloff DL, Earnshaw WC. INCENP binds the Aurora-related kinase AIRK2 and is required to target it to chromosomes, the central spindle and cleavage furrow. Curr Biol. 2000;10:1075–1078. doi: 10.1016/s0960-9822(00)00673-4. [DOI] [PubMed] [Google Scholar]
- Andrews PD, Knatko E, Moore WJ, Swedlow JR. Mitotic mechanics: the auroras come into view. Curr Opin Cell Biol. 2003;15:672–683. doi: 10.1016/j.ceb.2003.10.013. [DOI] [PubMed] [Google Scholar]
- Biggins S, Murray AW. The budding yeast protein kinase Ipl1/Aurora allows the absence of tension to activate the spindle checkpoint. Genes Dev. 2001;15:3118–3129. doi: 10.1101/gad.934801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggins S, Severin FF, Bhalla N, Sassoon I, Hyman AA, Murray AW. The conserved protein kinase Ipl1 regulates microtubule binding to kinetochores in budding yeast. Genes Dev. 1999;13:532–544. doi: 10.1101/gad.13.5.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggins S, Walczak CE. Captivating capture: how microtubules attach to kinetochores. Curr Biol. 2003;13:R449–460. doi: 10.1016/s0960-9822(03)00369-5. [DOI] [PubMed] [Google Scholar]
- Bouck DC, Bloom KS. The kinetochore protein Ndc10p is required for spindle stability and cytokinesis in yeast. Proc Natl Acad Sci U S A. 2005;102:5408–5413. doi: 10.1073/pnas.0405925102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheeseman IM, Anderson S, Jwa M, Green EM, Kang J, Yates JR, 3rd, Chan CS, Drubin DG, Barnes G. Phospho-regulation of kinetochore-microtubule attachments by the Aurora kinase Ipl1p. Cell. 2002;111:163–172. doi: 10.1016/s0092-8674(02)00973-x. [DOI] [PubMed] [Google Scholar]
- Clarke L. Centromeres: proteins, protein complexes, and repeated domains at centromeres of simple eukaryotes. Curr Opin Genet Dev. 1998;8:212–218. doi: 10.1016/s0959-437x(98)80143-3. [DOI] [PubMed] [Google Scholar]
- Cleveland DW, Mao Y, Sullivan KF. Centromeres and kinetochores: from epigenetics to mitotic checkpoint signaling. Cell. 2003;112:407–421. doi: 10.1016/s0092-8674(03)00115-6. [DOI] [PubMed] [Google Scholar]
- De Wulf P, McAinsh AD, Sorger PK. Hierarchical assembly of the budding yeast kinetochore from multiple subcomplexes. Genes Dev. 2003;17:2902–2921. doi: 10.1101/gad.1144403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewar H, Tanaka K, Nasmyth K, Tanaka TU. Tension between two kinetochores suffices for their bi-orientation on the mitotic spindle. Nature. 2004;428:93–97. doi: 10.1038/nature02328. [DOI] [PubMed] [Google Scholar]
- Ditchfield C, Johnson VL, Tighe A, Ellston R, Haworth C, Johnson T, Mortlock A, Keen N, Taylor SS. Aurora B couples chromosome alignment with anaphase by targeting BubR1, Mad2, and Cenp-E to kinetochores. J Cell Biol. 2003;161:267–280. doi: 10.1083/jcb.200208091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillis AN, Thomas S, Hansen SD, Kaplan KB. A novel role for the CBF3 kinetochore-scaffold complex in regulating septin dynamics and cytokinesis. J Cell Biol. 2005;171:773–784. doi: 10.1083/jcb.200507017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauf S, Cole RW, LaTerra S, Zimmer C, Schnapp G, Walter R, Heckel A, van Meel J, Rieder CL, Peters JM. The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore-microtubule attachment and in maintaining the spindle assembly checkpoint. J Cell Biol. 2003;161:281–294. doi: 10.1083/jcb.200208092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman AA, Middleton K, Centola M, Mitchison TJ, Carbon J. Microtubule-motor activity of a yeast centromere-binding protein complex. Nature. 1992;359:533–536. doi: 10.1038/359533a0. [DOI] [PubMed] [Google Scholar]
- Indjeian VB, Stern BM, Murray AW. The centromeric protein Sgo1 is required to sense lack of tension on mitotic chromosomes. Science. 2005;307:130–133. doi: 10.1126/science.1101366. [DOI] [PubMed] [Google Scholar]
- Inoue S, Salmon ED. Force generation by microtubule assembly/disassembly in mitosis and related movements. Mol Biol Cell. 1995;6:1619–1640. doi: 10.1091/mbc.6.12.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallio MJ, McCleland ML, Stukenberg PT, Gorbsky GJ. Inhibition of aurora B kinase blocks chromosome segregation, overrides the spindle checkpoint, and perturbs microtubule dynamics in mitosis. Curr Biol. 2002;12:900–905. doi: 10.1016/s0960-9822(02)00887-4. [DOI] [PubMed] [Google Scholar]
- Kang J, Cheeseman IM, Kallstrom G, Velmurugan S, Barnes G, Chan CS. Functional cooperation of Dam1, Ipl1, and the inner centromere protein (INCENP)-related protein Sli15 during chromosome segregation. J Cell Biol. 2001;155:763–774. doi: 10.1083/jcb.200105029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor TM, Lampson MA, Hergert P, Cameron L, Cimini D, Salmon ED, McEwen BF, Khodjakov A. Chromosomes can congress to the metaphase plate before biorientation. Science. 2006;311:388–391. doi: 10.1126/science.1122142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Kang JS, Chan CS. Sli15 associates with the ipl1 protein kinase to promote proper chromosome segregation in Saccharomyces cerevisiae. J Cell Biol. 1999;145:1381–1394. doi: 10.1083/jcb.145.7.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsbury J, Koshland D. Centromere-dependent binding of yeast minichromosomes to microtubules in vitro. Cell. 1991;66:483–495. doi: 10.1016/0092-8674(81)90012-x. [DOI] [PubMed] [Google Scholar]
- Kline-Smith SL, Sandall S, Desai A. Kinetochore-spindle microtubule interactions during mitosis. Curr Opin Cell Biol. 2005;17:35–46. doi: 10.1016/j.ceb.2004.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kufer TA, Sillje HH, Korner R, Gruss OJ, Meraldi P, Nigg EA. Human TPX2 is required for targeting Aurora-A kinase to the spindle. J Cell Biol. 2002;158:617–623. doi: 10.1083/jcb.200204155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampson MA, Renduchitala K, Khodjakov A, Kapoor TM. Correcting improper chromosome-spindle attachments during cell division. Nat Cell Biol. 2004;6:232–237. doi: 10.1038/ncb1102. [DOI] [PubMed] [Google Scholar]
- Lechner J, Carbon J. A 240 kd multisubunit protein complex, CBF3, is a major component of the budding yeast centromere. Cell. 1991;64:717–725. doi: 10.1016/0092-8674(91)90501-o. [DOI] [PubMed] [Google Scholar]
- Li F, Flanary PL, Altieri DC, Dohlman HG. Cell division regulation by BIR1, a member of the inhibitor of apoptosis family in yeast. J Biol Chem. 2000;275:6707–6711. doi: 10.1074/jbc.275.10.6707. [DOI] [PubMed] [Google Scholar]
- Li X, Nicklas RB. Mitotic forces control a cell-cycle checkpoint. Nature. 1995;373:630–632. doi: 10.1038/373630a0. [DOI] [PubMed] [Google Scholar]
- Maiato H, DeLuca J, Salmon ED, Earnshaw WC. The dynamic kinetochore-microtubule interface. J Cell Sci. 2004;117:5461–5477. doi: 10.1242/jcs.01536. [DOI] [PubMed] [Google Scholar]
- McAinsh AD, Tytell JD, Sorger PK. Structure, function, and regulation of budding yeast kinetochores. Annu Rev Cell Dev Biol. 2003;19:519–539. doi: 10.1146/annurev.cellbio.19.111301.155607. [DOI] [PubMed] [Google Scholar]
- Meraldi P, Honda R, Nigg EA. Aurora kinases link chromosome segregation and cell division to cancer susceptibility. Curr Opin Genet Dev. 2004;14:29–36. doi: 10.1016/j.gde.2003.11.006. [DOI] [PubMed] [Google Scholar]
- Nasmyth K. How do so few control so many? Cell. 2005;120:739–746. doi: 10.1016/j.cell.2005.03.006. [DOI] [PubMed] [Google Scholar]
- Nicklas RB, Koch CA. Chromosome micromanipulation. 3. Spindle fiber tension and the reorientation of mal-oriented chromosomes. J Cell Biol. 1969;43:40–50. doi: 10.1083/jcb.43.1.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira G, Schiebel E. Separase regulates INCENP-Aurora B anaphase spindle function through Cdc14. Science. 2003;302:2120–2124. doi: 10.1126/science.1091936. [DOI] [PubMed] [Google Scholar]
- Pinsky BA, Biggins S. The spindle checkpoint: tension versus attachment. Trends Cell Biol. 2005;15:486–493. doi: 10.1016/j.tcb.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Pinsky BA, Kung C, Shokat KM, Biggins S. The Ipl1-Aurora protein kinase activates the spindle checkpoint by creating unattached kinetochores. Nat Cell Biol. 2006;8:78–83. doi: 10.1038/ncb1341. [DOI] [PubMed] [Google Scholar]
- Pinsky BA, Tatsutani SY, Collins KA, Biggins S. An Mtw1 complex promotes kinetochore biorientation that is monitored by the Ipl1/Aurora protein kinase. Dev Cell. 2003;5:735–745. doi: 10.1016/s1534-5807(03)00322-8. [DOI] [PubMed] [Google Scholar]
- Rieder CL, Alexander SP. Kinetochores are transported poleward along a single astral microtubule during chromosome attachment to the spindle in newt lung cells. J Cell Biol. 1990;110:81–95. doi: 10.1083/jcb.110.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassoon I, Severin FF, Andrews PD, Taba MR, Kaplan KB, Ashford AJ, Stark MJ, Sorger PK, Hyman AA. Regulation of Saccharomyces cerevisiae kinetochores by the type 1 phosphatase Glc7p. Genes Dev. 1999;13:545–555. doi: 10.1101/gad.13.5.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sessa F, Mapelli M, Ciferri C, Tarricone C, Areces LB, Schneider TR, Stukenberg PT, Musacchio A. Mechanism of Aurora B activation by INCENP and inhibition by hesperadin. Mol Cell. 2005;18:379–391. doi: 10.1016/j.molcel.2005.03.031. [DOI] [PubMed] [Google Scholar]
- Severin F, Kaplan K, Sorger P, Hyman T. In vitro assays for studying Saccharomyces cerevisiae kinetochore activity. Methods Cell Biol. 1999;61:145–153. doi: 10.1016/s0091-679x(08)61979-2. [DOI] [PubMed] [Google Scholar]
- Severin FF, Sorger PK, Hyman AA. Kinetochores distinguish GTP from GDP forms of the microtubule lattice. Nature. 1997;388:888–891. doi: 10.1038/42270. [DOI] [PubMed] [Google Scholar]
- Sorger PK, Severin FF, Hyman AA. Factors required for the binding of reassembled yeast kinetochores to microtubules in vitro. J Cell Biol. 1994;127:995–1008. doi: 10.1083/jcb.127.4.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern BM, Murray AW. Lack of tension at kinetochores activates the spindle checkpoint in budding yeast. Curr Biol. 2001;11:1462–1467. doi: 10.1016/s0960-9822(01)00451-1. [DOI] [PubMed] [Google Scholar]
- Stoepel J, Ottey MA, Kurischko C, Hieter P, Luca FC. The mitotic exit network Mob1p-Dbf2p kinase complex localizes to the nucleus and regulates passenger protein localization. Mol Biol Cell. 2005;16:5465–5479. doi: 10.1091/mbc.E05-04-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Mukae N, Dewar H, van Breugel M, James EK, Prescott AR, Antony C, Tanaka TU. Molecular mechanisms of kinetochore capture by spindle microtubules. Nature. 2005;434:987–994. doi: 10.1038/nature03483. [DOI] [PubMed] [Google Scholar]
- Tanaka TU, Rachidi N, Janke C, Pereira G, Galova M, Schiebel E, Stark MJ, Nasmyth K. Evidence that the Ipl1-Sli15 (Aurora kinase-INCENP) complex promotes chromosome bi-orientation by altering kinetochore-spindle pole connections. Cell. 2002;108:317–329. doi: 10.1016/s0092-8674(02)00633-5. [DOI] [PubMed] [Google Scholar]
- Tsai MY, Wiese C, Cao K, Martin O, Donovan P, Ruderman J, Prigent C, Zheng Y. A Ran signalling pathway mediated by the mitotic kinase Aurora A in spindle assembly. Nat Cell Biol. 2003;5:242–248. doi: 10.1038/ncb936. [DOI] [PubMed] [Google Scholar]
- Uren AG, Beilharz T, O’Connell MJ, Bugg SJ, van Driel R, Vaux DL, Lithgow T. Role for yeast inhibitor of apoptosis (IAP)-like proteins in cell division. Proc Natl Acad Sci U S A. 1999;96:10170–10175. doi: 10.1073/pnas.96.18.10170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagnarelli P, Earnshaw WC. Chromosomal passengers: the four-dimensional regulation of mitotic events. Chromosoma. 2004;113:211–222. doi: 10.1007/s00412-004-0307-3. [DOI] [PubMed] [Google Scholar]
- Washburn MP, Wolters D, Yates JR., 3rd Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol. 2001;19:242–247. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- Wheatley SP, Kandels-Lewis SE, Adams RR, Ainsztein AM, Earnshaw WC. INCENP binds directly to tubulin and requires dynamic microtubules to target to the cleavage furrow. Exp Cell Res. 2001;262:122–127. doi: 10.1006/excr.2000.5088. [DOI] [PubMed] [Google Scholar]
- Widlund PO, Lyssand JS, Anderson S, Niessen S, Yates JR, 3rd, Davis TN. Phosphorylation of the chromosomal passenger protein Bir1 is required for localization of Ndc10 to the spindle during anaphase and full spindle elongation. Mol Biol Cell. 2006;17:1065–1074. doi: 10.1091/mbc.E05-07-0640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon HJ, Carbon J. Participation of Bir1p, a member of the inhibitor of apoptosis family, in yeast chromosome segregation events. Proc Natl Acad Sci U S A. 1999;96:13208–13213. doi: 10.1073/pnas.96.23.13208. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.