

Abstract
Objective
To investigate whether variable antidepressant response may be influenced by an interaction between the serotonin transporter promoter polymorphism (5-HTTLPR) and antidepressant concentration.
Methods
Elderly subjects with depression treated with paroxetine (n = 110) were genotyped and assessed with the Hamilton Rating Scale for Depression (HAMD). A mixed-effect analysis of repeated measures was used.
Results
There was an interaction between early paroxetine concentration and 5-HTTLPR genotype on symptomatic improvement over 12 weeks (F18,59.5 = 1.8, p < 0.05), as well as main effects of both paroxetine concentration (F68,55.3 = 2.4, p < 0.005) and genotype (F2,74.2 = 5.7, p < 0.005). Paroxetine concentrations were correlated with change in HAMD scores after 2 weeks of treatment in subjects with the short (s) allele (r = 0.31, p < 0.05) but not in subjects homozygous for the long (l) allele.
Conclusion
The results demonstrate a concentration–response relation for paroxetine in late-life depression and support the hypothesis for both a direct main effect and a moderating influence of 5-HTTLPR alleles on this concentration–response relation.
Medical subject headings: serotonin, paroxetine, depression, aged
Abstract
Objectif
Savoir si la réponse variable aux antidépresseurs peut être influencée par une interaction entre le polymorphisme du promoteur du transporteur de la sérotonine (5 HTTLPR) et la concentration d'antidépresseur.
Méthodes
On a déterminé le génotype de sujets âgés atteints de dépression traitée à la paroxétine (n = 110) et on les a évalués au moyen de l'échelle de dépression de Hamilton (HAMD). On a utilisé une analyse à effets mixtes de mesures répétées.
Résultats
On a constaté qu'une interaction entre la concentration de paroxétine au début et le génotype de la 5 HTTLPR était associée à une amélioration des symptômes en 12 semaines (F18,59,5 = 1,8, p < 0,05), et que la concentration de paroxétine (F68,55,3 = 2,4, p < 0,005) et le génotype (F2,74,2 = 5,7, p < 0,005) étaient associés à des effets principaux. On a établi une corrélation entre les concentrations de paroxétine et l'évolution des résultats du test HAMD après deux semaines de traitement chez les sujets qui avaient l'allèle (s) court (r = 0,31, p < 0,05), mais non chez les sujets homozygotes pour l'allèle long (l).
Conclusion
Les résultats démontrent l'existence d'un lien concentration-réponse pour la paroxétine dans le cas de la dépression chez les personnes âgées et appuient l'hypothèse selon laquelle les allèles de la 5 HTTLPR ont à la fois un effet principal direct et une influence modératrice sur cette relation concentration-réponse.
Introduction
Many patients with depression do not achieve full remission with selective serotonin reuptake inhibitors (SSRIs) or else improve very slowly.1–3 Hypotheses for this response variability invoke differences in SSRI levels, because there can be a wide range of SSRI concentrations2,4 (hypothesis A), or genetic differences in pharmacodynamic sensitivity5,6 (hypothesis B), or both. In addition to having a main effect on response, genetic variability could also influence the concentration–response relation (hypothesis C), that is, affect SSRI potency and thus shift the concentration–response curve. In combination, all 3 hypotheses may potentially be clinically useful in guiding treatment approaches to SSRI nonresponders.
The following findings are consistent with hypothesis A (a role for differences in SSRI levels):
• Dropout rates for SSRIs can have dose–response relations.7,8
• There is a concentration–response relation for SSRIs on acute neuroendocrine response.9
• Obese patients may have a poorer antidepressant response to fluoxetine.10
• There is evidence for a dose–response relation for the antidepressant effect of fluoxetine.11
• In the treatment of neuropathic pain, a concentration–response relation has been described for paroxetine.12
• Brain paroxetine concentrations were found to be correlated with brain activity as assessed with the use of fluorine magnetic resonance spectroscopy.13
• In mice, there is an association between paroxetine concentration, brain serotonin transporter binding and changes in behaviour.14
• Finally, consistent with these various findings, a recent meta-analysis supported a dose–response relation for SSRIs.15
Nonetheless, most clinical trials have generally failed to observe a concentration dependence for SSRIs.16–19 One explanation is that there may be genetic heterogeneity that obscures evidence for it.7 It is plausible that different SSRI concentration–response curves can result from genetic differences in serotonin transporter (SERT) function (hypothesis C).
In addition to a possible main effect of concentration, a main effect of genetic differences (hypothesis B) has been observed. Genetic variation in the SERT promoter (5-HTTLPR) can influence response to SSRIs, as replicated in several studies20 although certainly not in all.21,22 Specifically, in late-life depression, patients of European ethnicity with the short (s) allele in 5-HTTLPR responded less quickly to both paroxetine23 and sertraline.24
Consistent with this, the acute functional effect of SSRIs on various brain regions as determined by positron emission tomography is associated with genetic differences in 5-HTTLPR.25 At a cellular level, cells with the short (s) allele can have 50% less SERT expression than cells that are homozygous for the long (l) allele,26 and they also have lower maximal transport of serotonin.27
A potential clinical implication of these findings is that, if the SSRI concentration is not at a maximally inhibiting level for a particular genotype, then an increased concentration could have a beneficial effect for individuals with that genotype. Some elderly subjects prescribed low dosages or who are poorly adherent to treatment may be in this category. Conversely, if the SSRI concentration is already achieving maximal inhibition of SERT for a given genotype, then increasing the concentration should have no additional benefit.
Supporting an interaction between SERT genotype and SSRI concentration (hypothesis C), we initially noted that patients with low paroxetine plasma levels may have a slower antidepressant response if they have the 5-HTTLPR s allele.28 Therefore, to explicitly test whether there is a concentration by genotype interaction, we combined 2 cohorts involving paroxetine treatment of older adults: a Maintenance Treatment of Late-Life Depression-2 (MTLD-2)29 cohort and a cohort from a double-blind study of paroxetine and nortiptyline.30 The specific hypothesis was that lower paroxetine plasma concentrations would more adversely affect subjects with the s allele, whereas subjects with the l/l genotype would be less affected by low concentrations. This would result in a concentration by genotype interaction. We also tested for main effects of both concentration and genotype. These results may have relevance for guiding the early (within the first few weeks) individualized treatment of major depression in those who are poor responders to SSRIs.
Methods
Subjects
Participants provided written informed consent, as approved by the University of Pittsburgh Medical Center Institutional Review Board, as well as similar additional consent for genotyping. The first cohort was limited to genotyped subjects from a clinical trial comparing paroxetine and nortriptyline30 who had 2-week paroxetine levels available (n = 47). They were aged 60 years or older and started on 20 mg paroxetine. Paroxetine dosages were increased to 30 mg after 5 weeks in subjects who still had a score of 15 or above on the 17-item Hamilton Rating Scale for Depression (HAMD) or who had experienced a decrease in HAMD score of less than 50%. The patients in the second cohort, from the MTLD-2,29 were started on open-label paroxetine treatment. The subset included in this study were those for whom genotyping was obtained, along with acute plasma paroxetine levels at either week 2 (n = 49) or week 3 (n = 14). Subjects without blood samples available between days 12 and 24 were not included in these analyses. These subjects, aged 70 years or older, were initially treated with paroxetine in dosages that were openly adjusted and augmented by weekly interpersonal psychotherapy.
All subjects met DSM-IV31 criteria for a major depressive episode and were without psychotic features or any unstable medical condition. Many had 4–6 chronic medical problems typical of this age group and scored 15 or higher on the 17-item HAMD as well as 18 or higher on the Folstein Mini-Mental State Examination. Benzodiazepines could be provided as needed.29,30
Paroxetine concentration and genotyping
In both cohorts, plasma samples for paroxetine levels were planned and were obtained regardless of treatment response. Neither paroxetine levels nor genotypes were available to the clinicians involved in treating the patients and therefore did not influence treatment decisions. We assessed paroxetine concentrations from plasma, using high performance liquid chromatography with ultraviolet detection according to methods previously described.32 Paroxetine levels were obtained weekly for all subjects in cohort 1, all of whom therefore had paroxetine levels available for week 2. In cohort 2, blood samples were obtained intermittently and variably during treatment. Of 63 subjects, 49 had week 2 (day 12–18 after the start of treatment) paroxetine levels available; for the other 14 subjects, week 3 levels (day 19–24 after the start of treatment) were used. Other than plasma levels, objective adherence assessments, such as electronic monitoring, were not routinely included in these studies.
For genotyping, high-molecular-weight DNA was isolated from blood lymphocytes with the PureGene kit (Gentra Systems, Minneapolis, MN) and amplified (polymerase chain reaction) with the flanking primers 5′-CTTGTTGGGGATTCTCCCGCCTGGCGT T-3′ (forward) and 5′-TCGAGGCTGAGCGTCTAGAG-GGACTGAGCTGG-3′ (reverse). Amplification products were resolved by electrophoresis on 2% agarose gels and visualized with ethidium bromide staining, as previously described.23 Alleles were designated s (484 bp) and l (528 bp) by direct comparison with control samples run on the same gel. Both concentration measures and genotyping were performed blind to treatment and treatment outcome.
Statistics
Analyses used SPSS 13.0. The primary outcome was the change in baseline HAMD score over the course of 12 weeks of treatment for major depression. Sixty-three percent (29/47) of cohort 1 and 92% (58/63) of cohort 2 completed at least 10 weeks of treatment for major depression. Mixed-effect analyses of repeated measures were performed, with genotype and early exposure as fixed effects. Early exposure was defined as the plasma paroxetine concentration at week 2 (or week 3 when week 2 plasma levels were unavailable). Because of the repeated-measures design, a first-order antedependence covariance structure was used33; according to Akaike's information criteria, it provided a better fit than either autoregressive or heterogeneous Toeplitz structures. The primary hypothesis was that there would be a genotype × exposure interaction, although both main effects were also examined. We also included time as a factor, but to maximize statistical power, we did not assess interactions with time. The analysis controlled for multiple covariates: the cohort, baseline HAMD score, cumulative history of past antidepressant treatment obtained from the Antidepressant Treatment History Form (ATHF),34 strength of prior antidepressant treatment obtained from the ATHF, use of benzodiazepines during the trial (yes or no), any use of estrogens and any use of thyroid hormone. The analysis was also performed without these covariates and then performed including them individually. Mixed-effect analyses of repeated measures were also used to examine whether any of the covariates individually influenced symptomatic improvement and also to compare drug levels and dosages between groups over time.
To help us interpret the results from these primary analyses, we then examined improvement in HAMD scores at week 2 for correlations with paroxetine levels during treatment for major depression (defined as above). In this exploratory analysis, we elected to combine the s/s and s/l groups for consistency with prior findings in geriatric treatment response,23,24 and on the basis of power considerations (there are relatively few s/s subjects). We then creaed a receiver operating curve (ROC) for subjects with the s allele, using early paroxetine levels, and defining a HAMD score < 7 by week 12 as categorical remission.
We used a χ2 test to assess for Hardy–Weinberg equilibrium, and to compare demographic characteristics between groups. We also used t tests and analyses of variance to compare demographic characteristics between groups. Unless indicated otherwise, results are reported as mean and standard error of the mean (SEM).
Results
Demographics
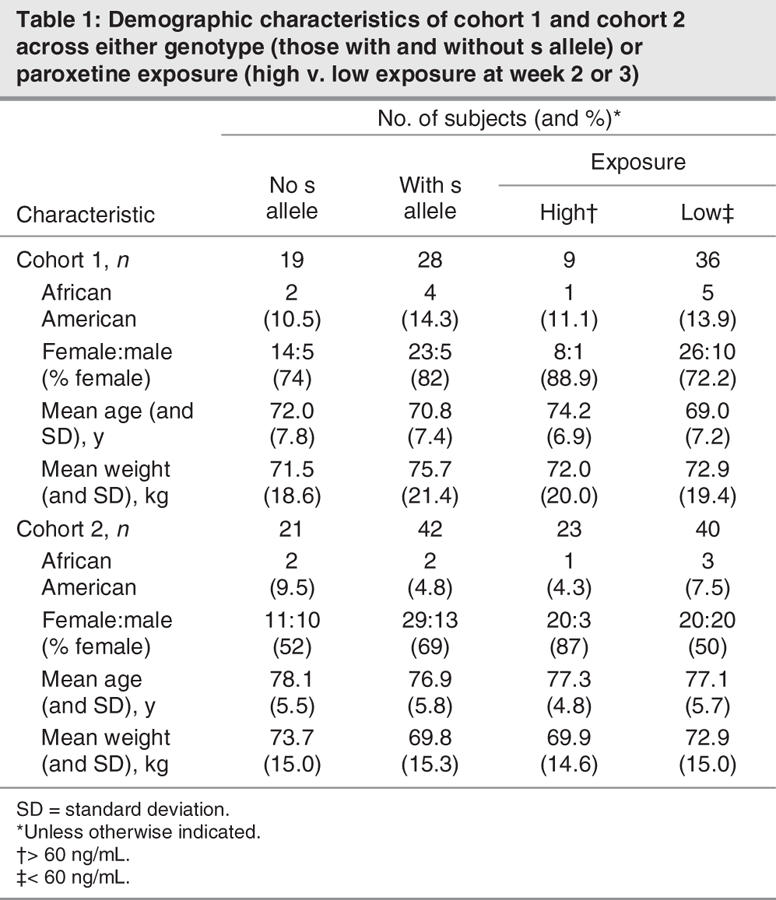
The 5-HTTLPR was in Hardy–Weinberg equilibrium in both cohorts (19 l/l, 22 s/l, 6 s/s; and 21 l/l, 35 s/l, 7 s/s, respectively), and the frequencies of the s and l alleles were similar to those reported for populations of European ethnicity.35 In both cohorts, the 2 genetic groups (those with an s allele and those without s alleles) did not significantly differ in age, weight, percentage of women or percentage of African Americans (Table 1), nor were there any demographic differences between groups with low paroxetine exposure (< 60 ng/mL at week 2 or 3) and high paroxetine exposure (> 60 ng/mL at week 2 or 3). Finally, there were no significant demographic differences between subjects in our study compared with subjects from the 2 studies29,30 who had not been genotyped.
Table 1
Paroxetine exposure
The average paroxetine levels in cohort 2 subjects did not significantly differ between the 2 genetic groups over time, although levels did rise over time (Table 2), similar to prior published observations in cohort 1.23 This is consistent with paroxetine inhibiting its own metabolism. Notably, low-exposure subjects continued to have low paroxetine levels throughout the entire treatment period, differing from highexposure subjects (F1,60.4 = 159, p < 0.001) (Table 2). Lower plasma levels were associated with lower dosages. At week 2, plasma levels also trended to being inversely associated with weight (r = 0.2, p = 0.05). The role of adherence (measured by electronic cap monitoring) or variability related to time of last dosage could not be explicitly assessed.
Table 2

Improvement in depression symptoms
The mixed-effect analysis of repeated measures controlling for all the covariates (Table 3) supported the primary hypothesis of an interaction between early paroxetine exposure and genotype on symptomatic improvement (F18,59.5 = 1.8, p < 0.05). There was also a main effect of concentration (F68,55.3 = 2.4, p < 0.005) and of genotype (F2,74.2 = 5.7, p < 0.005). The results were not affected by excluding African Americans. Notably, the results were also not significantly affected by excluding the covariates or including them singly. Exploratory post hoc assessments of individual genotypes using the mixed-effect analyses indicated that early paroxetine exposure influenced symptomatic improvement in all 3 genotypes (l/l, s/l and s/s: F33,6.37 = 4.3, p < 0.05; F43,18.1 = 4.6, p < 0.0005; and F10,31.1 = 6.3, p < 0.0005, respectively), although the significant interaction was because of a larger effect size in those with the s allele, as hypothesized.
Table 3
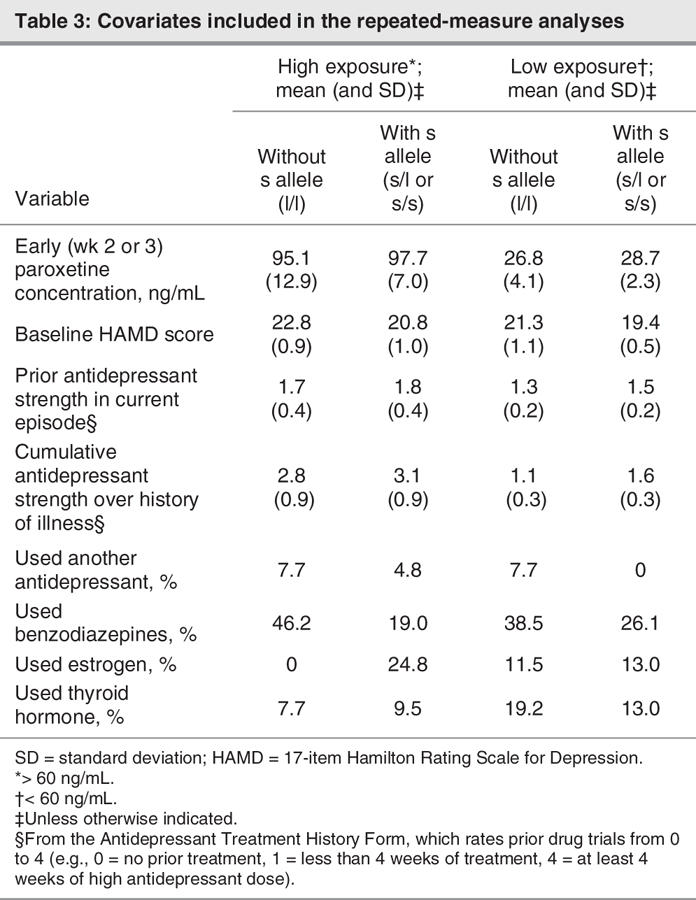
There were no significant differences between genotypes in any of the covariates examined. Examination of individual covariates as fixed factors in separate mixed-effect analyses indicated that neither cumulative antidepressant treatment history nor the strength of prior treatment influenced improvement in symptoms in these studies (F12,126.98 = 0.8 and F4,129.28 = 1.2, respectively). However, baseline HAMD score was associated with symptomatic improvement over 12 weeks (F21,307.8 = 6.6, p < 0.005). The use of benzodiazepines was also favourably associated with treatment outcome (F1,145.8 = 6.5, p < 0.05).
Examination of early symptomatic improvement at week 2
Paroxetine concentration was significantly correlated with improvement in HAMD scores at week 2 in subjects with the s allele (intercept 4.2, slope 0.04, r = 0.31, p < 0.05) but not in subjects with l/l alleles (intercept = 7.9; slope = 0.01; r = 0.12; p = 0.45). The s/l and s/s subjects had fairly similar intercepts and slopes (for s/l subjects, 4.3 and 0.04, respectively; for s/s subjects, 3.6 and 0.04, respectively), supporting their inclusion together. Despite this post hoc evidence, because there were only 39 l/l subjects, there was only 52% power to detect a slope of 0.036 (given α = 0.05 and the measured variability in l/l subjects). For the combined group of s allele subjects (given α = 0.05 and the measured variability in s subjects), the power to detect a slope of 0.036 was estimated to be 72%. Moreover, with this type of regression analysis, to detect statistically different concentration–response slopes (0.036 v. 0.012) between the 2 genetic groups would require 202 l/l subjects for 80% power. The differences in concentration–response at 2 weeks are illustrated in Figure 1.

Fig. 1: Improvement in HAMD scores (mean and SEM) at week 2 of paroxetine treatment increases with acute paroxetine concentrations in s allele subjects (circles, linear r = 0.31 p < 0.05) but not l/l subjects (squares, r = 0.12 p = 0.45). HAMD = Hamilton Rating Scale for Depression; SEM = standard error of the mean.
ROC
Defining categorical remission as HAMD scores less than or equal to 7 by week 12, we generated an ROC to explore the subsequent predictive ability of early 2-week paroxetine levels. ROCs are plots of true positives versus false positives and are designed to examine the predictive ability of different values (e.g., early paroxetine concentrations) to determine a categorical outcome (e.g., remission by week 12). A relation with zero predictive ability (where the percentages of true and false positives are always equal) will have an area under the curve (AUC) of 0.5. The ROC for subjects with the s allele had an AUC of 0.63 (p < 0.05, compared with 0.5). When the acute paroxetine level was < 60 ng/mL, there was about 88% sensitivity for identifying categorical nonremitters, albeit with a low specificity of 64% (Fig. 2). The AUC for l/l subjects did not differ significantly from 0.5. As illustrated in Figure 3, subjects with the s allele improved more slowly than l/l subjects when acute levels were < 60 ng/mL, consistent with the findings of the mixed-effect analysis. At higher concentrations, all genotypes responded to SSRI treatment similarly.

Fig. 2: Receiver operating curve for categorical remission (HAMD < 7), as predicted by early paroxetine levels for treatment of major depression (n = 69; area under the curve = 0.63; p < 0.05). HAMD = Hamilton Rating Scale for Depression.

Fig. 3: The interaction between genotype and nondichotomized paroxetine levels was significant. To illustrate this finding, mean (and SEM) improvement in HAMD scores for s allele carriers (circles) and l/l homozygotes (squares) was dichotomized into 2 paroxetine exposure groups: (A) describes subjects with paroxetine concentrations < 60 ng/mL, and (B) describes subjects with concentrations > 60 ng/mL. HAMD = Hamilton Rating Scale for Depression.
Discussion
The primary hypothesis of an interaction between genotype and concentration was supported: the 5-HTTLPR genotype influenced the SSRI concentration–response relation in elderly patients with depression. Conversely, the SSRI concentration can influence whether 5-HTTLPR is associated with antidepressant outcome. Of clinical importance, these finding suggest that it may be feasible to rationally decide whether to increase dosage or change medication in poorly responding patients, particularly because early improvement is often predictive of later response.36 At week 2, if the poorly responding patient has both the s allele and levels well below 60 ng/mL, the dosage may need to be increased or adherence problems addressed, or both. If the concentration is above 60 ng/mL or the patient has the l/l genotype, then increasing the concentration may be less effective. Additional studies are needed to confirm the utility of this hypothesis and to determine the optimal concentration threshold for clinical decision making. A value of about 60 ng/mL is very preliminary at this point. Moreover, before clinical implementation, these initial results will need to be replicated, in particular with other SSRIs and in other age groups.
We also confirmed that there is a main effect of early paroxetine concentration, an observation not always made in clinical trials.16–19 Notably, we explicitly focused on early (week 2) paroxetine concentrations rather than on later time points. The decision for this was multifold. First, these were not fixed-dose studies. Because dosages could be adjusted in reaction to adverse side effects or poor response, the clinical status of the subjects likely influenced concentrations at later time points rather than vice versa. Second, paroxetine is capable of inhibiting its own metabolism.37 At later time points, most 5-HT transporters are likely to be occupied regardless of genotype,38 and paroxetine may even start to inhibit norepinephrine reuptake.39 The results of this study are therefore limited to concentrations in early treatment for major depression. We also did not fully explain the sources of variable paroxetine exposure. Electronic cap monitoring could have provided information regarding adherence but was not used. The dosage partially accounted for plasma levels, but poor adherence, higher weight and genetic differences in metabolism37 could be additional reasons for low plasma levels.
We also replicated prior findings of a main effect of genotype on response.23,24,40–43 5-HTTLPR is associated with functional differences in response to SSRIs used to treat major depression25 despite the fact that these functional differences may not be the result of global changes in SERT expression.44 Although the association with response has not always been observed,21 SSRI concentrations were not obtained in most studies. It is thus not clear whether past negative findings are the result of confounds associated with concentration differences. This is particularly salient in primary care settings, where adherence can be problematic.45
However, there are some caveats that require comment. First, possible population stratification was not accounted for.46 Although patients were from the same cohort (this is not a case–control study), which may mitigate against this prospect, population stratification is still a possibility that may confound the results. Because there were only 10 African-American subjects in this study, excluding them from the analyses did not significantly affect the results. However, we cannot make any definitive statements about concentrations or genotype in African Americans. We must limit our conclusions to subjects of European ethnicity. In a cohort of Japanese subjects, the influence of 5-HTTLPR differed depending on whether paroxetine or fluvoxamine was used.47 It is possible that P450 alleles can have variable effects on paroxetine concentrations in either Japanese48 subjects or those of European ethnicity.49 It is therefore conceivable that the 5-HTTLPR effects may vary among different ethnic groups.
Second, 5-HTTLPR may be in linkage disequilibrium with another polymorphism that may be causally responsible for the influence on concentration–response. In addition to other plausible linked and unlinked SERT polymorphisms, several other factors may influence SERT expression and function,27 including kinases and cytokines. Moreover, there is evidence for a role of other genes in SSRI response.50 It is likely that these other influences could explain some of the additional variability in response that was not explained by 5-HTTLPR, concentration, baseline HAMD and benzodiazepine use.
Third, our findings may specifically pertain to older patients treated in a closely monitored research setting with careful attention to identifying and treating potential side effects. In some clinical situations, higher SSRI levels may be counterproductive because of their side effects. Elevated SSRI levels may confound the HAMD score (e.g., by adversely influencing sleep and agitation) or result in poor adherence in some patients,51,52 or both; the s allele has been associated with more side effects in some studies.22,53 It is therefore plausible that, in some clinical settings, elevated paroxetine levels could “paradoxically” adversely affect response.54 In 1 study, ideal paroxetine concentrations at 2 weeks were 43 ng/mL, with poorer outcomes at either higher or lower concentrations.54 Other analyses have similarly suggested that paroxetine levels in treatment for chronic symptoms should be between 20 and 70 ng/mL.55 Both of our cohorts were permitted to use benzodiazepines, the use of which was favourably associated with outcome. It is plausible that the role of 5-HTTLPR can be influenced by the clinical attention given to SSRI-induced side effects. Similarly, given potentially increased sensitivity to side effects in the elderly,2 it plausible that the role of 5-HTTLPR may differ depending on the patient's age. Whether our results can be extended to other treatment situations (e.g., where benzodiazepines are not as permitted or in younger patients) will require further examination.
In summary, with the above considerations in mind, the results suggest that it may be possible to identify at week 2 those poorly responding patients who would simply benefit from increased paroxetine levels (achieved by targeting either increased dosage or improved adherence) and those for whom another intervention would be more beneficial. Replicating these results and then extending these findings to younger ages, other SSRIs, other patient populations and other clinical protocols will be an important next step. To date, few clinical studies have included regular and early antidepressant concentration measurements in their protocols. Therefore, an important implication of our findings is that the role of antidepressant concentrations should be included in future antidepressant clinical trials, particularly those studies examining the role of pharmacogenetics and individualized treatment.
Acknowledgments
This research was supported by NIMH grants K23MH074012, K24MH065416, R01MH043832, P30MH071944, the John A. Hartford Foundation Center of Excellence in Geriatric Psychiatry, the University of Pittsburgh Medical Center Endowed Chair in Geriatric Psychiatry and the Sandra A. Rotman Chair in Neuropsychiatry. The results were preliminarily reported in abstract form at Collegium Internationale Neuropsychopharmacologicum 2006.
Footnotes
Contributors: Dr. Lotrich designed the study. Drs. Pollock, Ferrell and Reynolds, and Ms. Kirshner acquired the data, which Drs. Lotrich, Pollock and Reynolds analyzed. Dr. Lotrich wrote the article, and Drs. Pollock, Ferrell and Reynolds, and Ms. Kirshner revised it. All authors gave final approval for the article to be published.
Competing interests: Bruce Pollock has received research support from Janssen Pharmaceuticals and is a consultant to Lundbeck and Forest Pharmaceuticals and until August 2004 was a consultant to SmithKline Beecham. Charles Reynolds has received research support from GSK, Forest, Pfizer, BMS and Lilly. SmithKline Beecham donated the paroxetine used in the trials.
Correspondence to: Dr. F.E. Lotrich, Western Psychiatric Institute and Clinics, 3811 O'Hara St., Pittsburgh PA 15213; fax 412 246-6260; lotrichfe@upmc.edu
References
- 1.Driscoll HC, Basinski J, Mulsant BH, et al. Late-onset major depression: clinical and treatment-response variability. Int J Geriatr Psychiatry 2005;20:661-7. [DOI] [PubMed]
- 2.Lotrich FE, Pollock BG. Aging and clinical pharmacology: implications for antidepressants. J Clin Pharmacol 2005;45:1106-22. [DOI] [PubMed]
- 3.Whyte EM, Dew MA, Gildengers AG, et al. Time course of response to antidepressants in late-life major depression: therapeutic implications. 2004;21:531-554. Drugs Aging 2004;21:531-54. [DOI] [PubMed]
- 4.Bies RR, Feng Y, Lotrich FE, et al. Utility of sparse concentration samploing for citalopram in elderly cinical trial subjects. J Clin Pharmacol 2004;44:1352-9. [DOI] [PubMed]
- 5.Serretti A, Artioli P, Quartesan R. Pharmacogenetics in the treatment of depression: pharmacodynamic studies. Pharmacogenet Genomics 2005;15:61-7. [DOI] [PubMed]
- 6.Binder EB, Holsboer F. Pharmacogenomics and antidepressant drugs. Ann Med 2006;38:82-94. [DOI] [PubMed]
- 7.Preskorn SH. Recent dose-effect studies regarding antidepressants. In: Balant LP, editor. Clinical pharmacology in psychiatry: finding the right dose of psychotropic drugs conference held in Badajoz, Spain, October 23-25, 1997. Luxembourg: European Commission, Directorate-General Science, Research and Development; 1998. p. 43-62.
- 8.Bollini P, Pampallona S, Tibaldi G, et al. Effectiveness of antidepressants. Meta-analysis of dose-effect relationships in randomised clinical trials. Br J Psychiatry 1999;174:297-303. [DOI] [PubMed]
- 9.Lotrich FE, Bies R, Muldoon M, et al. Neuroendocrine response to intravenous citalopram in healthy control subjects: pharmacokinetic influences. Psychopharmacology (Berl) 2005;178:268-75. [DOI] [PubMed]
- 10.Papakostas GI, Petersen T, Iosifescu DV, et al. Obesity among outpatients with major depressive disorder. Int J Neuropsychopharmacol 2005;8:59-63. [DOI] [PubMed]
- 11.Rausch JL, Johnson ME, Fei Y-J, et al. Initial conditions of serotonin transporter kinetics and genotype: influence on SSRI treatment trial outcome. Biol Psychiatry 2002;51:723-32. [DOI] [PubMed]
- 12.Sindrup SH, Grodum E, Gram LF, et al. Concentration-response relationship in paroxetine treatment of diabetic neuropathy symptoms: a patient-blinded dose-escalation study. Ther Drug Monit 1991;13:408-14. [DOI] [PubMed]
- 13.Henry ME, Moore CM, Kaufman MJ, et al. Brain kinetics of paroxetine and fluoxetine on the third day of placebo substitution: a fluorine MRS study. Am J Psychiatry 2000;157:1506-8. [DOI] [PubMed]
- 14.Hirano K, Kimura R, Sugimoto Y, et al. Relationship between brain serotonin transporter binding, plasma concentration and behavioural effect of selective serotonin reuptake inhibitors. Br J Pharmacol 2005;144:695-702. [DOI] [PMC free article] [PubMed]
- 15.Baker CB, Tweedie R, Duval S, et al. Evidence that the SSRI dose response in treating major depression should be reassessed: a meta-analysis. Depress Anxiety 2003;17:1-9. [DOI] [PubMed]
- 16.Tasker TC, Kaye CM, Zussman BD, et al. Paroxetine plasma levels: lack of correlation with efficacy or adverse events. Acta Psychiatr Scand Suppl 1989; 350:152-5. [DOI] [PubMed]
- 17.Amsterdam JD, Fawcett J, Quitkin FM, et al. Fluoxetine and norfluoxetine plasma concentrations in major depression: a multicenter study. Am J Psychiatry 1997;154:963-9. [DOI] [PubMed]
- 18.Normann C, Horn M, Hummel B, et al. Paroxetine in major depression: correlating plasma concentrations and clinical response. Pharmacopsychiatry 2004;37:123-6. [DOI] [PubMed]
- 19.Reis M, Aberg-Wistedt A, Agren H, et al. Serum disposition of sertraline, N-desmethylsertraline and paroxetine: a pharmacokinetic evaluation of repeated drug concentration measurements during 6 months of treatment for major depression. Hum Psychopharmacol 2004;19:283-91. [DOI] [PubMed]
- 20.Lotrich FE, Pollock BG, Ferrell RE. Polymorphism of the serotonin transporter: implications for the use of selective serotonin reuptake inhibitors. Am J Pharmacogenomics 2001;1:153-64. [DOI] [PubMed]
- 21.Kraft JB, Peters EJ, Slager SL, et al. Analysis of association between the serotonin transporter and antidepressant response in a large clinical sample. Biol Psychiatry 2007;61:734-42. [DOI] [PubMed]
- 22.Murphy GMJ, Hollander SB, Rodrigues HE, et al. Effects of the serotonin transporter gene promoter polymorphism on mirtazapine and paroxetine efficacy and adverse events in geriatric major depression. Arch Gen Psychiatry 2004;61:1163-9. [DOI] [PubMed]
- 23.Pollock BG, Ferrell RE, Mulsant BH, et al. Allelic variation in the serotonin transporter promoter affects onset of paroxetine treatment response in late-life depression. Neuropsychopharmacology 2000;23:587-90. [DOI] [PubMed]
- 24.Durham LK, Webb SM, Milos PM, et al. The serotonin transporter polymorphism, 5HTTLPR, is associated with a faster response time to sertraline in an elderly population with major depressive disorder. Psychopharmacology (Berl) 2004;174:525-9. [DOI] [PubMed]
- 25.Smith GS, Lotrich FE, Malhotra AK, et al. The effects of serotonin transporter promoter polymorphisms on serotonin function. Neuropsychopharmacology 2004;29:2226-31. [DOI] [PubMed]
- 26.Heils A, Mossner R, Lesch KP. The human serotonin transporter gene polymorphism — basic research and clinical implication. J Neural Transm Gen Sect 1997;104:1005-14. [DOI] [PubMed]
- 27.Rausch JL. Initial conditions of psychotropic drug response: Studies of serotonin transporter long promoter region (5-HTTLPR), serotonin transporter efficiency, cytokine and kinase gene expression relevant to depression and antidepressant outcome. Prog Neuropsychopharmacol Biol Psychiatry 2005;29:1046-61. [DOI] [PubMed]
- 28.Lotrich FE, Bies RB, Smith GS, et al. Relevance of assessing drug concentration exposure in pharmacogenetic and imaging studies. J Psychopharmacol 2006;20:33-40. [DOI] [PubMed]
- 29.Reynolds CF, Dew MA, Pollock BG, et al. Maintenance treatment of major depression in old age. N Engl J Med 2006;354:1130-8. [DOI] [PubMed]
- 30.Mulsant BH, Pollock BG, Nebes R, et al. A twelve-week, double-blind, randomized comparison of nortriptyline and paroxetine in older depressed in patients and outpatients. Am J Geriatr Psychiatry 2001;9:406-14. [PubMed]
- 31.American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 4th ed. Washington: The Association; 1994.
- 32.Foglia JP, Sorisio DA, Kirshner M, et al. Quantitative determination of paroxetine in plasma by high-performance liquid chromatography and ultraviolet detection. J Chromatogr B Biomed Sci Appl 1997;693:147-51. [DOI] [PubMed]
- 33.Kenward M. A method for comparing profiles of repeated measurements. Appl Stat 1987;36:296-308.
- 34.Oquendo MA, Baca-Garcia E, Kartachov A. A computer algorithm for calculating the adequacy of antidepressant treatment in unipolar and bipolar depression. J Clin Psychiatry 2003;64:825-33. [DOI] [PubMed]
- 35.Gelernter J, Cubells JF, Kidd JR, et al. Population studies of polymorphisms of the serotonin transporter protein gene. Am J Med Genet 1999;88:61-6. [PubMed]
- 36.Szegedi A, Muller MJ, Anghelescu I, et al. Early improvement under mirtazapine and paroxetine predicts stable response and remission with high sensitivity in patients with major depression. J Clin Psychiatry 2003;64:413-20. [DOI] [PubMed]
- 37.Solai LKK, Pollock BG, Mulsant BH, et al. Effect of nortriptyline and paroxetine on CYP2D6 activity in depressed elderly patients. J Clin Psychopharmacol 2002;22:481-6. [DOI] [PubMed]
- 38.Meyer JH, Wilson AA, Sagrati S, et al. Serotonin transporter occupancy of five selective serotonin reuptake inhibitors at different doses: An [11C]DASB positron emission tomography study. Am J Psychiatry 2004;161:826-35. [DOI] [PubMed]
- 39.Frazer A. Serotonergic and noradrenergic reuptake inhibitors: prediction of clinical effects from in vitro potencies. J Clin Psychiatry 2001;62:16-23. [PubMed]
- 40.Serretti A, Zanardi R, Rossini D, et al. Influence of tryptophan hydroxylase and serotonin transporter genes on fluvoxamine antidepressant activity. Mol Psychiatry 2001;6:586-92. [DOI] [PubMed]
- 41.Smeraldi E, Zanardi R, Benedetti F, et al. Polymorphism within the promoter of the serotonin transporter gene and antidepressant efficacy of fluvoxamine. Mol Psychiatry 1998;3:508-11. [DOI] [PubMed]
- 42.Arias B, Catalan R, Gasto C, et al. 5-HTTLPR polymorphism of the serotonin transporter gene predicts non-remission in major depression patients treated with citalopram in a 12-weeks follow up study. J Clin Psychopharmacol 2003;23:563-7. [DOI] [PubMed]
- 43.Arias B, Gasto C, Catalan R, et al. Variation in the serotonin transporter gene and clinical response to citalopram in major depression. Am J Med Genet 2000;96:536.
- 44.Parsey RV, Hasting RS, Oquendo MA, et al. Effect of a triallelic functional polymorphism of the serotonin-transporter-linked promoter region on expression of serotonin transporter in the human brain. Am J Psychiatry 2006;163:48-51. [DOI] [PubMed]
- 45.Simon GE, VonKorff M, Wagner EH, et al. Patterns of antidepressant use in community practice. Gen Hosp Psychiatry 1993;15:399-408. [DOI] [PubMed]
- 46.Devlin B, Roeder K, Wasserman L. Genomic control, a new approach to genetic-based association studies. Theor Popul Biol 2001;60:156-66. [DOI] [PubMed]
- 47.Kato M, Ikenaga Y, Wakeno M, et al. Controlled clinical comparison of paroxetine and fluvoxamine considering the serotonin transporter promoter polymorphism. Int Clin Psychopharmacol 2005;20:151-6. [DOI] [PubMed]
- 48.Sawamura K, Suzuki Y, Someya T. Effects of dosage and CYP2D6-mutated allele on plasma concentration of paroxetine. Eur J Clin Pharmacol 2004;60:553-7. [DOI] [PubMed]
- 49.Charlier C, Broly F, Lhermitte M, et al. Polymorphisms in the CYP 2D6 gene: association with plasma concentrations of fluoxetine and paroxetine. Ther Drug Monit 2003;25:738-42. [DOI] [PubMed]
- 50.Lotrich FE, Pollock BG. Candidate genes for antidepressant response to selective serotonin reuptake inhibitors. Neuropsychiatr Dis Treat 2005;1:17-35. [DOI] [PMC free article] [PubMed]
- 51.Grasmader K, Verwohlt PL, Rietschel M, et al. Impact of polymorphisms of cytochrome-P450 isoenzymes 2C9, 2C19 and 2D6 on plasma concentrations and clinical effects of antidepressants in a naturalistic clinical setting. Eur J Clin Pharmacol 2004;60:329-36. [DOI] [PubMed]
- 52.Hartter S, Wetzel H, Hammes E, et al. Serum concentrations of fluvoxamine and clinical effects. Pharmacopsychiatry 1998;31:199-200. [DOI] [PubMed]
- 53.Hu X, Rush AJ, Charney D, et al. A functional serotonin transporter promoter polymorphism with reduced side effects but not symptomatic outcome with citalopram in adult outpatients with major depression. In press 2007. [DOI] [PubMed]
- 54.Gilles M, Deuschle M, Kellner S, et al. Paroxetine serum concentrations in depressed patients and response to treatment. Pharmacopsychiatry 2005;38:118-21. [DOI] [PubMed]
- 55.Mauri MC, Laini V, Bitetto A, et al. Long-term efficacy of paroxetine in major depression: a study with plasma levels. Int J Psychiatry Clin Pract 1999;3:115-9. [DOI] [PubMed]