

Abstract
The transcription factors encoded by the E2A gene have been shown to play essential roles in the initiation and progression of lymphocyte development. However, there is still a lack of comprehensive understanding of E2A downstream genes in B-cell development. We previously developed a gene tagging-based chromatin immunoprecipitation (ChIP) system to directly evaluate E2A target genes in B-cell development. Here, we have improved this ChIP strategy and used it in conjunction with microarray analysis on E2A-deficient pre-B-cell lines to determine E2A target genes in lymphocyte development. Both microarray data and ChIP studies confirmed that E2A directly controls IgH gene expression. The microarray assay also revealed genes that were significantly up-regulated after E2A disruption. ChIP analysis showed that E2A was most likely to be directly involved in repression of some of these target genes such as Nfil3 and FGFR2. An inducible E2A reconstitution system further demonstrated that E2A-mediated repression of Nfil3 and FGFR2 was reversible. Collectively, these findings indicate that E2A is a positive regulator for one set of genes and a negative regulator for another set of genes in developing B lymphocytes.
Lymphocytes develop from multipotent hematopoietic stem cells to provide cell-mediated and humoral immunological protection against foreign antigens. Hematopoietic stem cells give rise to mature B- and T-lymphocytes through a series of well defined and highly ordered differentiation stages in the bone marrow and thymus, respectively (1). These stages are functionally defined by the progressive assembly and expression of the lymphocyte antigen receptors. These antigen receptors are generated through tightly regulated genomic recombination events at the B- and T-lymphocyte antigen receptor loci to establish a diverse pool of receptor specificities (2).
Antigen receptor gene recombination and thus lymphocyte differentiation requires the activity of numerous broadly expressed and lineage-specific transcription factors. The basic helix-loop-helix transcription factors encoded by the E2A gene have been identified as essential regulators of gene expression during lymphocyte development (3). The E2A proteins were initially characterized as immunoglobulin heavy (IgH)1 and light chain enhancer-binding factors and have since been shown to bind consensus E-box motifs (CANNTG) in the promoters and enhancers of numerous lymphoid lineage-specific genes (4, 5). E2A proteins are highly expressed in lymphoid progenitor populations (6) and are required for the initiation of B-lymphocyte development in the bone marrow. Mice deficient for E2A show a complete and persistent block at the earliest stage of B-cell development prior to the initiation of immunoglobulin heavy chain rearrangements (7, 8).
E2A has been implicated in regulating the expression of multiple BCR complex components, including the surrogate light chain λ5, the BCR signaling molecule mb-1, and the immunoglobulin heavy chain (IgH) and k light chain genes (9-15). Whereas early ectopic expression and gene targeting studies indicated that these genes were downstream of E2A function, it was not known whether E2A directly regulated their transcription. A gene tagging-based chromatin immunoprecipitation system was recently used to confirm that E2A is physically associated with many of these B-cell-specific genes (16). The ChIP-based approach was also applied to identification of novel E2A targets in B-cell development. However, the total number of E2A target genes recovered in this study is still limited due to low cloning efficiency and binding specificity.
Previous investigations of E2A-deficient mice clearly demonstrated the importance of E2A in lymphocyte development (7, 8). However, the strong block in B-lymphocyte development, complex thymocyte phenotype, and poor postnatal survival seen in these mice precluded more detailed studies on functions for E2A in lymphocyte differentiation and lymphoid gene regulation. A conditional E2A deletion model was developed in our laboratory to evaluate roles for E2A specifically in developing lymphocytes (17). Mice carrying the loxp-flanked E2A allele (E2Aloxp) enabled lineage-restricted deletion of the E2A gene based upon ectopic expression of the Cre recombinase. Here we describe a conditional E2A deletion system based upon the E2Aloxp mouse model for studying E2A-mediated gene regulation in B-lymphocyte development. We have utilized this conditional E2A knockout system in conjunction with our gene tagging-based ChIP strategy to investigate novel functions for E2A proteins in gene regulation during early B-lymphocyte development. Microarray analysis on E2A-deficient pre-B-cell lines has revealed numerous genes with E2A-dependent expression patterns. We have further evaluated a subset of these genes that appear to be directly suppressed by E2A in pre-B-cells. Chromatin immunoprecipitation analysis on the regulatory elements of these genes provides the first physiological evidence for direct gene repression by E2A proteins. These studies demonstrate novel functions for E2A proteins in both positive and negative gene regulation during lymphocyte development.
MATERIALS AND METHODS
Pre-B-cell Lines
The Abelson murine leukemia virus-transformed pre-B-cell lines E2AFH1B and E2AGFP were previously derived from bone marrow of mice carrying the dual affinity-tagged E2A allele (E2AFH/FH) (16) or from E2AGFP/GFP mice (18). For gene array studies, pre-B-cell lines were derived from bone marrow of a mouse carrying the loxp-flanked E2A allele (E2Aloxp/loxp) (17). Transformed cells were then transduced with a retrovirus encoding Cre recombinase as well as a puromycin resistance cassette. A control cell line was established in parallel by infecting the E2Aloxp/loxp pre-B-cells with a retrovirus encoding Cre recombinase in the antisense orientation. Transduced cells were selected by puromycin treatment and three different clonal E2A-deficient populations were established by limiting dilution plating. All cell lines were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum, 100 units/ml penicillin, 100 μg/ml streptomycin, and 55 μm 2-mercaptoethanol.
RT-PCR Assay
Total RNA was purified using Trizol reagent (Invitrogen) from cultured Abelson transformed pre-B-cell lines, and cDNA was synthesized with random hexamers. The following oligonucleotide primers were used in PCR analysis of serial 3-fold cDNA dilutions: E2A-E47, E8 (5′-AGG TCC CAC GCA CGC GCA CC-3′) and E20 (5′-CGC CTG CTG CAG GAT GAG CA-3′); EBF, EBF for (5′-TCC AGG AAA GCA TCC AAC GG-3′) and EBF rev (5′-TCG TGT GTG AGC AAT ACT CGG C-3′); Igα, mb-1 for (5′-CCT CTC CTC CTC TTC TTG TCA TAC G-3′) and mb-1 rev (5′-CCC CTG TGT TCT TGT TTA CTT CGG-33); Igβ, B29 for (5′-TGT TCC TGC TGC TGC TCT TCT C-3′) and B29 rev (5′-TCG GTG ACA TTA TGG TTG GCG-3′); Pax-5, Pax-5 for (5′-CCG CCA AAG GAT AGT GGA AAC TTG-3′) and Pax-5 rev (5′-CAC AGT GTC ATT GTC ACA GAC TCG C-3′); EF1α, EF1α-for (5′-AGT TTG AGA AGG AGG CTG CT-3′) and EF1α-rev (5′-CAA CAA TCA GGA CAG CAC AGT C-3′); FGFR2, FGFR2A (5′-GCG CTT CAT CTG CCT GGT CTT GG-3′) and FGFR2B (5′-TCC AAC TGA TCA CGG CGG CAT CT-3′); NFIL3, NFIL3A (5′-AGC GCC GCC TCA ATG ACC TGG TT-3′) and NFIL3B (5′-GCT CAC GGC AGC CTT GGA TGT CT-3′).
Flow Cytometry Assay
FACS analysis of the parental E2A wild type cells and the E2A-deficient lines was performed on a FACSCalibur (BD Biosciences) using the following fluorescent antibody conjugates: CD19-PE and CD43-PE (Pharmingen).
Electrophoretic Mobility Shift Assay
Abelson transformed pre-B-cells were cultured and treated with 1 μm tamoxifen dissolved in Me2SO or Me2SO alone as indicated. Nuclei were isolated, and nuclear extracts were prepared as previously described (19). Extracts were incubated with 32P-labeled μE5 oligonucleotide probe with or without anti-E2A monoclonal antibody YAE (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) and resolved on a 5% polyacrylamide gel containing Walsh TBE buffer. Gels were dried and exposed to a phosphor screen (Amersham Biosciences) for analysis.
Microarray Analysis
RNA was isolated from three independent E2A-deficient pre-B-cell lines and the antisense Cre-infected control cell line using Trizol solution followed by isopropyl alcohol precipitation. RNA was then resuspended in diethyl pyrocarbonate-treated distilled H2O for global differential gene expression analysis using the Affymetrix murine microarray (U74Av2). All samples were reverse-transcribed, analyzed, and processed by the Duke University Microarray Core Facility. Subsequent data analysis was conducted with the R statistical package using the Bioconductor library (20). Briefly, gene expression levels were quantified by the gcRMA model (n = 3). The p values from the t test were adjusted by false discovery rate (21) for multiple comparisons.
Chromatin Extracts and Immunoprecipitations
Chromatin extracts were prepared using a modified version of the protocol described by Fernandez et al. (22). Approximately 5 × 107 pre-B-cells (E2AFH1B and E2AGFP) were harvested and fixed by adding one-tenth culture volume of fixing buffer (11% formaldehyde, 100 mm NaCl, 50 mm Tris-HCl, pH 7.9). After fixing for 15 min rocking gently at room temperature, one-twentieth volume of 2.5 m glycine quenching buffer was added, and the cells were incubated for 5 min rocking at room temperature (all subsequent steps were performed at 4 °C). Fixed cells were harvested by centrifugation, washed with PBS, resuspended in 15 ml of Triton buffer (10 mm Tris-HCl, pH 8.0, 0.25% Triton X-100), and incubated on ice for 15 min. Cells were then harvested, resuspended in 15 ml of NaCl buffer (10 mm Tris-HCl, pH 8.0, 200 mm NaCl) and incubated for an additional 15 min on ice. Washed cells were harvested, resuspended in 1 ml of sonication buffer (10 mm Tris-HCl, pH 8.0, 1% SDS; supplemented with phenylmethylsulfonyl fluoride, aprotinin, leupeptin, and pepstatin), and sonicated (Fisher Scientific 550 tapered microtip probe, setting 4.5) for 14 cycles of 25 s on a cold block with 15 s of cooling between each cycle. Sonicates were centrifuged at 14,000 × g for 10 min, and bulk chromatin extracts were harvested as supernatants and stored at −80 °C.
E2A-bound DNA fragments were isolated from chromatin extracts using either the previously described anti-FLAG one-step purification protocol (16) or the new dual epitope isolation protocol. For dual epitope purifications, bulk chromatin extracts (2 mg) from the E2AFH1B pre-B-cell line and the E2AGFP control cell line were thawed on ice, and one-tenth volume of 5× extraction/wash buffer (250 mm NaH2PO4, pH 7.0, 1.5 m NaCl) was added to each. Samples were then incubated with 100 μl of washed Talon cobalt affinity resin for 30 min rotating slowly at room temperature. Bound resin was then harvested by centrifugation, washed twice for 10 min each with 1× extraction/wash buffer (50 mm NaH2PO4, pH 7.0, 300 mm NaCl), and transferred to gravity flow chromatography columns (Bio-Rad polyprep 0.8 × 4.0 cm). After the resins settled, the column end caps were removed, and the resins were washed with 3 ml of 1× extraction/wash buffer. Bound protein-DNA complexes were then eluted three times with 500 μl of 100 mm EDTA, pH 8.0, and the eluted materials were collected into 15-ml conical tubes.
Secondary anti-FLAG immunoprecipitations were then performed on the eluted fractions from the Cobalt affinity resin (or on bulk chromatin extracts for single-epitope purifications). Eluted solutions were diluted 1:10 with ChIP buffer (140 mm NaCl, 100 μg/ml bovine serum albumin, 100 μg/ml yeast tRNA, 1% Triton X-100, 1 mm phenylmethylsulfonyl fluoride) and incubated with 50 μl of washed anti-FLAG-conjugated M2-agarose (Sigma) for 2 h at 4 °C rotating slowly. Bound agarose was harvested by centrifugation, washed once with 10 ml of ChIP buffer, and then transferred to microcentrifuge tubes. The bound agarose was washed twice with 1 ml of ChIP buffer, twice with 1 ml of ChIP buffer containing 500 mm NaCl, twice with 1 ml of wash buffer (10 mm Tris-HCl, pH 8.0, 250 mm LiCl, 1 mm EDTA), and twice with 1 ml of TE buffer (10 mm Tris-HCl, pH 8.0, 1 mm EDTA). Bound DNA was eluted for 15 min at 65 °C in 250 μl of elution buffer (50 mm Tris-HCl, pH 8.0, 10 mm EDTA, 1% SDS), and both bound and input DNA samples were adjusted to 0.5% SDS in 500 μl. Samples were incubated at 65 °C overnight to reverse cross-links and then RNase-treated, deproteinized, and precipitated as described by Fernandez et al. (22). Processed DNA fragments were resuspended in 50 μl of distilled water.
PCR Analysis of Immunoprecipitated DNA
PCR screening for enrichment of target regulatory regions was performed using a slightly modified version of the original protocol (16). Serial 3-fold dilutions of input chromatin and immunoprecipitated DNA were PCR-amplified for 32–34 cycles (94 °C for 45 s, 57 °C for 45 s, 72 °C for 45 s, with a 2-min final extension at 72 °C) in 20 μl of PCR buffer containing 1.5 mm MgCl2 and Platinum Taq polymerase (Invitrogen). PCR samples were then resolved on 1% agarose gels and visualized by ethidium bromide staining. Previously published oligonucleotide sequences were used in PCR screening of IgH enhancers and λ5 and mb-1 promoters (16). The following primer sequences were used to screen for E2A binding to novel target loci in ChIP-PCR analysis: FGFR2-A (5′-TGG TGG CTT GCG GCT GTC CAC TT-3′) and FGFR2-B (5′-CAG AGA GGC TGT GCC TCC AGA GC-3′); pNfil3-A (5′-GCC ACT GAA GTA GAA GGG TTT GT-3′) and pNfil3-B (5′-GTC CCT TGG GCA GCT GTG TGC TA-3′).
Inducible E2A Reconstitution System
293T cells were cotransfected with the GFP-expressing E47-estrogen receptor fusion construct MigR1-E47R (23) along with the vesicular stomatitis virus glycoprotein coat protein and gag/pol. Retroviral supernatant was harvested, concentrated by ultracentrifugation, and used to transduce an E2A-deficient clonal pre-B-cell line. Transduced cells were selected by FACS based upon GFP expression to establish a stable bulk population of E47R-reconstituted cells. For induction of E47R activity, cells were cultured in the presence of 1 μm tamoxifen (resuspended in Me2SO) for 5 h.
RESULTS
A conditional E2A deletion mouse model was previously established in our laboratory by introducing the loxp-flanked E2A allele (E2Aloxp) into the genomic E2A locus (17). This E2Aloxp model was adapted to facilitate studies on E2A-mediated gene regulation in B-lymphocyte development. Pre-B-cell lines were first derived by transforming E2Aloxp/loxp bone marrow with the Abelson murine leukemia virus. Abelson-transformed pre-B-cells were then transduced with a retrovirus encoding a puromycin resistance cassette along with the Cre recombinase in either the sense (E2A-deleted cell lines) or antisense (control line) orientation. Transduced cells were selected by puromycin treatment, and three independent clonal E2A-deficient populations were subsequently established. Complete deletion of E2A in each clone was confirmed by genomic PCR specific for either the deleted or undeleted alleles (Fig. 1A). The absence of E2A expression in each E2A-deficient cell line was confirmed by RT-PCR analysis (Fig. 1B). FACS analysis demonstrates that E2A-deficient B-cells maintain expression of the surface antigens CD43 and CD19, indicating that these cells retain a phenotype indicative of pro- or early pre-B-cells (Fig. 1C). In order to confirm that E2A−/− Abelson B-cells maintain their global B-cell gene expression profile, we evaluated the relative transcript levels of important B-cell-specific genes. Transcript levels of the genes mb-1 and B29, which encode the B-cell receptor-associated signaling molecules Igα and Igβ, respectively, were not significantly altered by E2A deletion (Fig. 1D). Expression levels of the Pax-5 gene, which encodes the B-cell-specific transcription factor BSAP and EBF, another important B-cell transcription factor, were also unaffected by E2A deletion. These data strongly indicate that E2A deficiency in pre-B-cells does not cause an overall shift in cell identity away from the B-cell lineage.
FIG. 1. Analysis of E2A-deficient Abelson pre-B-cells.
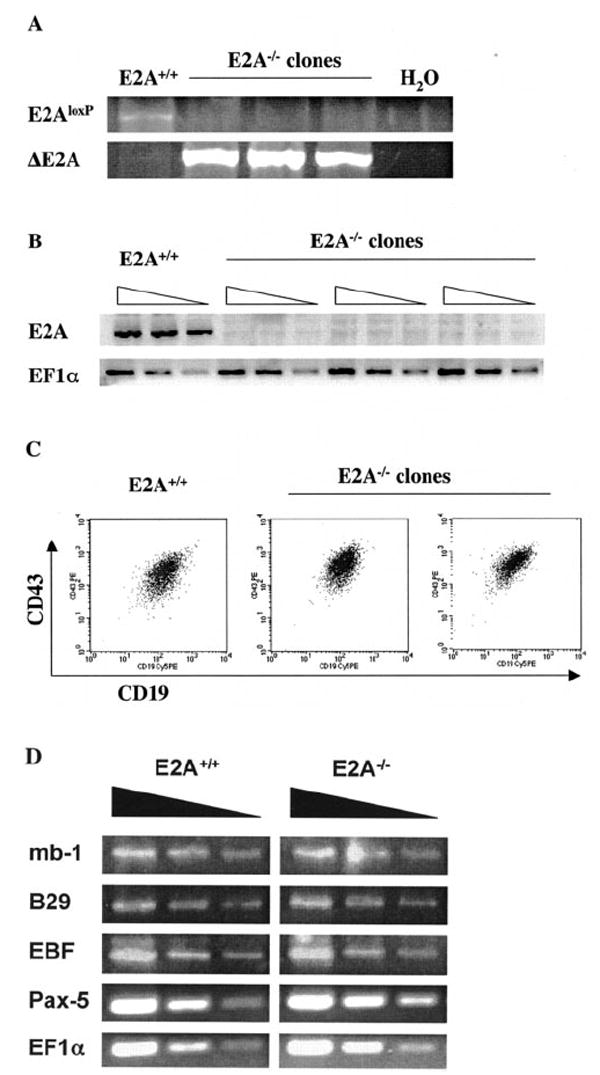
A, genomic PCR of control and Cre-transduced Abelson B-cells to detect the unrearranged (E2Aloxp) and deleted (ΔE2A) conditional E2A allele (17). B, RT-PCR analysis of total RNA from control Abelson pre-B-cells and three E2A−/− pre-B-cell lines. PCR for E2A (E47) and EF1α expression was performed on serial 3-fold dilutions of cDNA from each cell line. C, FACS analysis of E2A+/+ and E2A−/− pre-B-cell lines. Cells were analyzed for surface expression of the common pro-/pre-B-cell markers CD19 and CD43. D, expression of B-cell-specific genes in E2A−/− cell lines. Semiquantitative PCR was used to assess the relative expression levels of mb-1/Igα, B29/Igβ, EBF, and Pax-5/BSAP in control and E2A−/− cell lines. EF1α expression was used to verify equivalent sample loading.
Total RNA was isolated from three independent E2A-deficient pre-B-cell lines, and gene expression levels were compared with that of the antisense Cre-transduced control cell line using Affymetrix microarrays. These microarray experiments revealed numerous genes with E2A-dependent expression patterns in the deficient pre-B-cell lines. For the initial analysis, we chose to use these relatively high stringent criteria: 1) average -fold change larger than 3; 2) average difference larger then 48 intensity units; 3) adjusted p value smaller than 0.01; and 4) detectable hybridization signal in at least one of the four hybridizations. This analysis yielded 32 independent probe sets that are down-regulated and 35 that are up-regulated due to E2A disruption (Table I). The previously characterized E2A target gene immunoglobulin heavy chain and a number of novel punitive E2A target genes were significantly down-regulated in the absence of E2A (Table I, upper section).
TABLE I. Summary of microarray analysis.
Shown is a summary of microarray analysis on E2A-deficient pre-B cell lines. Global gene expression profiles from three independent E2A-deficient Abelson pre-B cell clones were analyzed using Affymetrix microarrays (U74Av2) and compared with the gene expression profile from E2A+/+ (antisense Cre-transduced) pre-B cells. The table indicates transcripts whose expression was significantly altered (average 3-fold or greater) across three E2A-deficient pre-B cell clones compared with the E2A+/+ pre-B cell line. Transcripts down-regulated 3-fold or greater in the E2A-deficient cell lines are shown in the upper section of the table. The lower section of the table includes transcripts up-regulated 3-fold or greater in the E2A-deficient cell lines.
| Accession number | Gene name | Change | p value |
|---|---|---|---|
| -fold | |||
| AF099973 | Schlafen 2 | −261.7 | 0.000123 |
| X04653 | Lymphocyte antigen 6 complex, locus A | −222.1 | 5.58E-05 |
| AA204265 | B lymphoid kinase | −112.4 | 3.04E-05 |
| AA795946 | Immune associated nucleotide 1 | −101.6 | 0.000143 |
| X98471 | Epithelial membrane protein 1 | −81.1 | 8.09E-05 |
| X02466 | Unknown mRNA | −68.7 | 0.001694 |
| X03453 | Unknown mRNA | −65.5 | 0.000141 |
| AI844939 | CREBBP/EP300-inhibitory protein 1 | −53.9 | 6.21E-05 |
| X02463 | Unknown mRNA | −48.9 | 0.002875 |
| AI844839 | RIKEN cDNA 6330442E10 gene | −48.2 | 0.000444 |
| X00496 | Ia-associated invariant chain | −26.5 | 0.002311 |
| M30903 | B lymphoid kinase | −24.7 | 0.000245 |
| AF099973 | Schlafen 2 | −22.6 | 0.000535 |
| U81453 | Myosin VIIa | −22.4 | 0.000337 |
| X67210 | Unknown (protein for MGC:68300) | −22 | 0.002151 |
| AI835274 | Cytoplasmic FMR1-interacting protein 2 | −19.3 | 0.000711 |
| AF065324 | Immunoglobulin heavy chain (J558 family) | −18.1 | 0.000273 |
| AJ007970 | Guanylate nucleotide-binding protein 2 | −12.6 | 0.012706 |
| L38444 | T-cell-specific GTPase | −12 | 0.011238 |
| U09816 | GM2 ganglioside activator protein | −11.6 | 0.002324 |
| AI553536 | RIKEN cDNA E030024M05 gene | −6 | 0.007887 |
| M38700 | Thyroid autoantigen | −5.6 | 0.001055 |
| L29441 | Tumor differentially expressed 1 | −5.3 | 0.004501 |
| AW125333 | Ceramide kinase | −5 | 0.003614 |
| AI852553 | Thymosin, β 10 | −3.9 | 0.016597 |
| J02980 | Alkaline phosphatase 2, liver | −3.7 | 0.011843 |
| M35244 | Histocompatibility 2, T region locus 10 | −3.3 | 0.000239 |
| U72519 | Ena-vasodilator-stimulated phosphoprotein | −3.2 | 0.005328 |
| AI046826 | Growth factor receptor-bound protein 2-associated protein 1 | −3.1 | 0.006082 |
| AW125526 | RIKEN cDNA 0610038D11 gene | −3.1 | 0.004102 |
| U10410 | Recombinant antineuraminidase single chain Ig VH and VL domains | −3 | 0.002839 |
| AI850401 | DNA segment, Chr 15, Wayne State University 75, expressed | −3 | 0.009169 |
|
| |||
| Accession number | Gene name | Change | p value |
|
| |||
| -fold | |||
| M34141 | Prostaglandin-endoperoxide synthase 1 | 15.1 | 0.010314 |
| U04354 | Scinderin | 11.8 | 0.004649 |
| AF019385 | Heparan sulfate (glucosamine) 3-O-sulfotransferase 1 | 8.2 | 0.000513 |
| M13352 | Thymidylate synthase | 8.1 | 0.005353 |
| U92454 | WW domain binding protein 5 | 7.4 | 0.00198 |
| D12646 | Kinesin family member 4 | 7.2 | 0.000119 |
| AA590345 | RIKEN cDNA C330027C09 gene | 7.2 | 0.008416 |
| AW123907 | Kelch domain containing 2 | 5.6 | 0.00714 |
| U80932 | Serine/threonine kinase 6 | 5.4 | 0.01059 |
| AF002823 | Budding uninhibited by benzimidazoles 1 homolog (S. cerevisiae) | 5.1 | 0.006987 |
| U19596 | Cyclin-dependent kinase inhibitor 2C (p18, inhibits CDK4) | 5.1 | 0.001504 |
| AW123269 | RIKEN cDNA 2900037I21 gene | 4.6 | 8.83E-06 |
| AA184423 | Unknown mRNA | 4.5 | 0.00087 |
| U83902 | MAD2 (mitotic arrest-deficient, homolog)-like 1 (yeast) | 4.5 | 0.004558 |
| X64713 | Cyclin B1 | 4.4 | 0.008331 |
| L29480 | Serine/threonine kinase 18 | 4.3 | 0.003255 |
| X75483 | Cyclin A2 | 4.3 | 0.006751 |
| AJ223293 | Kinesin family member 11 | 4.1 | 0.001694 |
| AA007891 | Kinesin family member 2C | 4 | 0.01095 |
| AI853802 | Phosphofructokinase, platelet | 3.9 | 0.005044 |
| U13174 | Solute carrier family 12, member 2 | 3.9 | 0.004716 |
| M86377 | Ttk protein kinase | 3.8 | 0.008107 |
| M23362 | Fibroblast growth factor receptor 2 | 3.8 | 0.001661 |
| AW122347 | Rac GTPase-activating protein 1 | 3.8 | 0.002574 |
| L11316 | ect2 oncogene | 3.8 | 0.015746 |
| M15668 | Phosphoglycerate kinase 1 | 3.7 | 0.016656 |
| X82786 | Antigen identified by monoclonal antibody Ki 67 | 3.7 | 0.004989 |
| U01915 | Topoisomerase (DNA) II alpha | 3.5 | 0.003634 |
| Z35401 | H2A histone family, member X | 3.4 | 0.003817 |
| AA275196 | RIKEN cDNA 2610201A12 gene | 3.3 | 0.015762 |
| X66032 | Cyclin B2 | 3.3 | 0.011732 |
| M25944 | Carbonic anhydrase 2 | 3.2 | 0.008015 |
| U28656 | Eukaryotic translation initiation factor 4E-binding protein 1 | 3.1 | 0.007679 |
| AB017189 | Solute carrier family 7+B100, member 5 | 3 | 0.009212 |
| X54327 | Glutamyl-prolyl-tRNA synthetase | 3 | 0.013704 |
The expression of another subset of genes was found to be dramatically up-regulated in the absence of E2A (Table I, lower section). These up-regulated genes represented nearly one-half of the genes with significant and consistent changes in E2A-dependent expression in the pre-B-cell lines. Although E2A is generally considered a positive regulator of transcription, these findings suggested that E2A proteins might be involved in the lineage-specific repression of certain genes as well.
E2A-dependent gene expression profiling was combined with chromatin immunoprecipitation analysis to further characterize E2A target genes in B-lymphocyte development. We previously utilized a gene tagging-based ChIP system (E2AFH) to investigate physiological E2A interaction within numerous genomic regulatory regions in pre-B-cells (16). However, the anti-FLAG purification procedure used in our original ChIP study incorporated only one of the two affinity tags expressed on the E2AFH fusion protein. To further improve the specificity of the E2A ChIP, a dual epitope purification methodology was developed for more selective isolation of E2A-bound DNA fragments from the pre-B-cell line E2AFH1B (this line carries dual affinity-tagged E2A alleles) (16). The His and FLAG tags were sequentially used in the affinity purification of E2A bound chromatin. We first evaluated this revised protocol for E2A interaction with the regulatory regions of the λ5 surrogate light chain and the mb-1 genes (13, 14). Physiological E2A interactions were previously demonstrated at the promoters for both λ5 and mb-1 by ChIP-PCR screening (16). As expected, these regulatory regions immunoprecipitated with E2A with high specificity in the dual epitope ChIP assay (Fig. 2B).
FIG. 2.
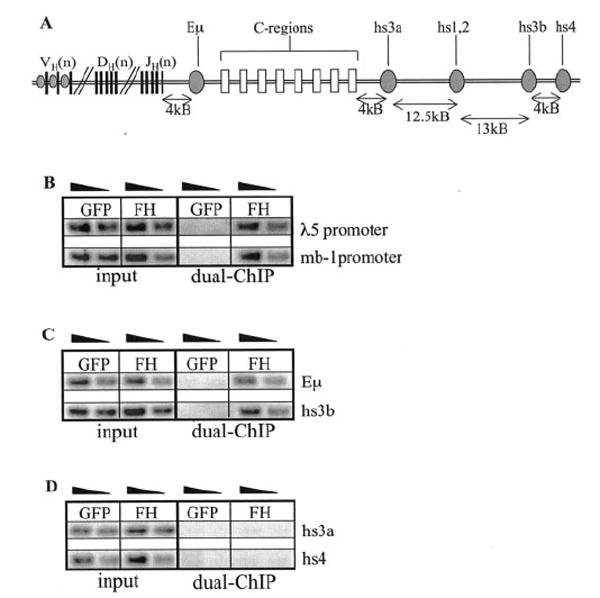
A, schematic representation of the immunoglobulin heavy chain locus. IgH variable (V), diversity (D), and joining (J) exon clusters are shown as vertical boxes. V-region promoters are indicated by small ovals. Constant (C) region exons are shown as filled boxes. IgH intronic (Eμ) and 3′ enhancer regions are represented by large ovals. B–D, dual epitope ChIP-PCR screen for E2A interaction with target regulatory regions in pre-B-cells. Serial 3-fold dilutions of input chromatin and dual ChIP DNA were PCR-amplified using primers covering potential E2A binding sites within the indicated regulatory regions. B, E2A association with the lambda-5 and mb-1 promoters. C, E2A association with the IgH intronic (Eμ) and hs3b enhancers. D, no detectable E2A interaction with the 3′ IgH enhancers hs3a and hs4.
Although E2A was previously shown to regulate transcription and rearrangement within the IgH locus our original ChIP study generated ambiguous results regarding E2A interaction with the IgH enhancers (16). It was therefore important to clarify the potential for E2A interaction with these regulatory regions using the two-step purification strategy. One intronic enhancer (Eμ) and several 3′ enhancers, including hs3a, hs3b, and hs4, contribute to the regulation of the IgH locus during B-lymphocyte development (Fig. 2A) (24-26). We first screened for E2A binding within the IgH intronic enhancer, which had previously shown only slight enrichment due to a high background signal (16). Significant enrichment of Eμ was observed after dual epitope purification, whereas immunoprecipitated E2AGFP chromatin extract generated minimal background signal across this regulatory region (Fig. 2C). The new two-step purification procedure dramatically improved the purity of the isolated DNA fragments, thereby enhancing our ability to discern E2A association with target regulatory regions. Similar results were obtained for the IgH 3′ enhancer hs3b, which was also significantly enriched in sequentially purified E2AFH1B DNA (Fig. 2C).
Consistent with the results obtained in the original ChIP study, the IgH 3′ enhancer hs3a was not enriched after dual epitope purification (Fig. 2D). However, the two-step purification approach eliminated the high background signal observed in the first study and therefore established with greater certainty the lack of detectable E2A binding at this enhancer. This finding indicated that either E2A interaction with the hs3a enhancer was below the threshold of sensitivity for our dualepitope ChIP assay or that E2A does not interact with this IgH enhancer region in the pre-B-cell lines. Interestingly, the IgH 3′ enhancer hs4 also showed no detectable enrichment after dual epitope purification of E2A-bound DNA fragments (Fig. 2D). Although this enhancer appeared to be slightly enriched in the original ChIP study, the high background signal again raised doubt as to whether this minimal enrichment was actually indicative of association with E2A (16). The lack of detectable hs4 sequence enrichment and clean background further emphasized the utility of the dual purification strategy in elucidating true genomic targets for physiological E2A interaction.
Few studies thus far have investigated the role of E2A in suppression of gene transcription. Once we established the efficacy of the dual-epitope ChIP, we used this method to evaluate the role of E2A in mediating gene repression. ChIP analysis was performed on several genes shown to be up-regulated in E2A-deficient pre-B-cells in order to determine whether they were direct targets for physiological regulation by E2A proteins. Nfil3 (also known as E4BP4) and FGFR2 (fibroblast growth factor receptor 2) were chosen for further characterization based on their functional relevance to B-cell development. Transcription of the basic region/leucine zipper transcription factor Nfil3 was up-regulated in each of three E2A-deficient pre-B-cell lines, although this probe set on the microarray failed to meet the stringent criteria used to create Table I. The Nfil3 promoter region contains five canonical E-box sequences (CANNTG), which could represent potential E2A-binding sites, including two adjacent E-boxes within 100 bp of the transcription start site (Fig. 3A). We therefore sought to determine whether E2A was directly involved in Nfil3 repression in pre-B-cells. Single or dual epitope purifications were carried out on E2AFH1B and E2AGFP pre-B-cell chromatin extracts to evaluate the potential for E2A association with the Nfil3 locus. Significant E2A interaction with the Nfil3 promoter region was detected by dual epitope ChIP analysis (Fig. 3B). As seen with the IgH enhancers, the dual epitope purification significantly reduced the background signals seen after only one round of purification. In fact, significant enrichment was only detectable at the Nfil3 promoter after the dual epitope purification.
FIG. 3. Chromatin immunoprecipitation analysis of genomic regulatory elements within potential E2A-repressed genes.
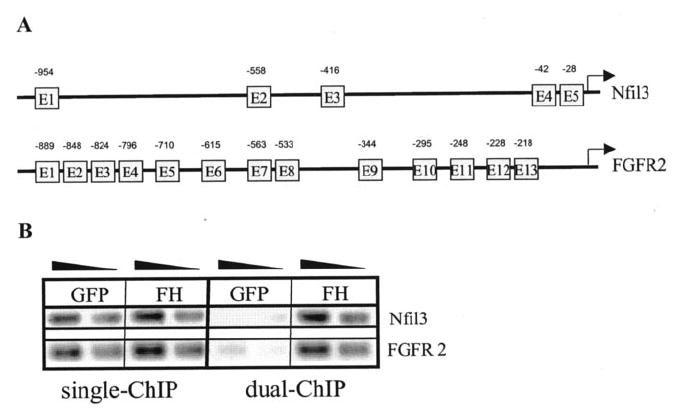
A, promoters and 5′ regulatory regions of Nfil3 and FGFR2. E-box motifs (CANNTG) representing potential E2A binding sites are indicated, including approximate positions with respect to transcription start sites or the 5′ end of published cDNA. B, ChIP-PCR screen on 3-fold dilutions of E2A-bound DNA fragments from single (FLAG only) and dual epitope purifications. E2A interacts with the Nfil3 promoter and the 5′ upstream regions of the FGFR-2.
We also investigated the potential for E2A interaction with the regulatory elements of FGFR2. Microarray analysis indicated consistent up-regulation of FGFR2 in the absence of E2A expression (Table I). Sequence analysis identified 13 E-box motifs within the 1-kb region upstream of the FGFR2 gene transcription start site (Fig. 3A). Significant E2A interaction was detected within the FGFR2 promoter region by dual epitope ChIP analysis, suggesting that E2A may be directly involved in FGFR2 repression in the pre-B-cell lines (Fig. 3B).
Gene expression studies using E2A−/− pre-B-cells reconstituted with an inducible human E47-estrogen receptor ligand binding domain fusion protein (E47R) were used to complement the ChIP assays in order to gain a clearer picture of which genes are actively suppressed by E2A (23). A retroviral vector encoding E47R-IRES-GFP was stably transduced into one of our E2A-deficient pre-B-cell clones (Fig. 4A). The E47R fusion protein exhibits very low activity due to the fused estrogen repressor domain, but E2A DNA binding activity can be strongly induced in the presence of the drug tamoxifen (Fig. 4B). Semiquantitative RT-PCR was conducted on RNA isolated from cells reconstituted with E47R and cultured in the presence or absence of tamoxifen. These samples were compared with control cells (E2A+/+) and the untransduced E2A-deficient (E2A−/−) pre-B-cell clone to determine whether E2A activity influences repression of the putative target genes analyzed by ChIP. E47R mediated the transcriptional suppression of the Nfil3 and FGFR2 genes in a tamoxifen-dependent manner (Fig. 4C, E47R(+) and E47R(−) compared with E2A−/−). Tamoxifen alone is not sufficient to suppress the expression of Nfil3 and FGFR2, since the treatment of E2A−/− cells with tamoxifen does not decrease the expression of these genes (Fig. 4D). FGFR2 and Nfil3 each exhibited E2A-dependent suppression of transcript levels and were shown to have physiologic interaction of E2A with their promoter regions, suggesting that these genes may be true targets of E2A-mediated gene suppression. We have also identified additional genes that exhibit physiologic E2A interaction with their promoter regions and increased expression in E2A−/− cells but are not suppressed by activated E47R (data not shown). This illustrates that expression profiling or ChIP alone may not be completely reliable for predicting true targets of E2A. However, when these two assays are combined together, we believe that the probability of identifying true E2A target genes is dramatically improved.
FIG. 4. Modulated expression of E2A-repressed genes in an inducible E2A reconstitution system.
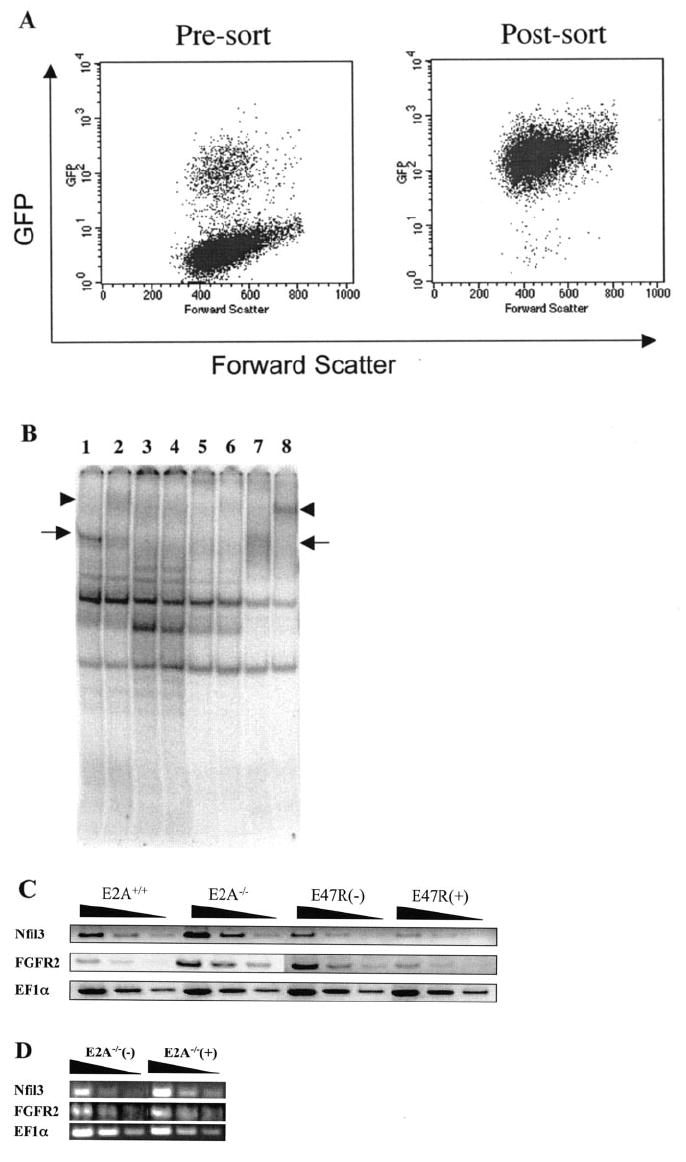
A, E2A−/− pre-B-cells were infected with E47R-IRES-GFP retrovirus (left plot), and GFP+ cells were sorted and cultured (right plot). B, tamoxifen treatment of E47R reconstituted B-cells restores inducible E2A DNA binding activity. Nuclear extracts from E2A+/+, E2A−/−, and E47R reconstituted pre-B-cells were incubated with labeled μE5 oligonucleotide probe in the presence (even-numbered lanes) or absence (odd-numbered lanes) of a pan-E2A monoclonal antibody (YAE). Specific DNA binding is indicated by the arrows, and the supershifted band is shown by arrowheads. E2A DNA binding activity was detected in extracts from E2A+/+ (lanes 1 and 2) and E47R reconstituted cells treated with 1 μm tamoxifen (lanes 7 and 8) but not E2A−/− (lanes 3 and 4) or 0.1% Me2SO-treated E47R-reconstituted cells (lanes 5 and 6). C, RT-PCR analysis of total RNA isolated from pre-B-cell lines with either functional E2A (E2A+/+), deleted E2A (E2A−/−), or E2A−/− cell line reconstituted with E47R fusion protein. Reconstituted cells were treated with either 0.1% Me2SO (E47R(−)) or 1 μm tamoxifen in Me2SO (E47R(+)) for 5 h to activate the E47R protein. Samples are presented as a series of serial 3-fold dilutions. D, tamoxifen treatment in the absence of E47R does not suppress gene expression. RT-PCR analysis of total RNA from E2A−/− pre-B-cells mock-treated with 0.1% Me2SO (−) or treated with 1 μm tamoxifen in Me2SO (+) for 5 h. Each sample is presented as a series of 3-fold serial dilutions.
DISCUSSION
We have successfully generated a stable E2A-deficient pre-B-cell line to allow us for the first time to directly evaluate the function of E2A-mediated gene regulation in the context of developing B lymphocytes. Previous work has identified genes such as EBF, Pax-5, mb-1, TdT, RAG1, immunoglobulin heavy chain, and immunoglobulin light chain as likely downstream targets of E2A regulation (8, 16). Each of these genes plays an important role either to regulate B lymphocyte development or produce a functional B-cell antigen receptor. Microarray analysis helped confirm that immunoglobulin gene expression was indeed perturbed by E2A deficiency. Surprisingly, many previously defined E2A target genes, although slightly decreased, were still expressed at relatively high levels compared with wild type controls, indicating that sustained expression of these genes is not dependent on E2A. These results suggest that E2A is not absolutely required for maintaining the expression of these B-lineage-specific genes once B-cell development has progressed to pre-B-cell stage.
The two-tiered approach of gene expression analysis and chromatin immunoprecipitation analysis provides a highly effective system for characterizing numerous E2A target genes. Here we have verified E2A-mediated regulation of the immunoglobulin heavy chain locus by combining gene expression analysis with ChIP-based chromatin binding studies. The dual epitope purification approach significantly enhances our ability to determine target sequence enrichment and thus E2A interaction with the IgH locus as well as numerous other lymphoid regulatory regions. Clarification of E2A association with the IgH enhancers will facilitate future studies on the regulation of immunoglobulin gene transcription and rearrangement by E2A and other transcription factors. E2A protein association with these enhancers might contribute to the stage-specific activation of IgH locus accessibility, transcription, and rearrangement during B-lymphocyte differentiation. The absence of even the earliest IgH gene rearrangements in bone marrow E2A-deficient mice may be explained in part by the lack of E2A binding within the IgH enhancers. These findings may also have mechanistic significance in the context of earlier observations regarding E2A-induced IgH transcription and rearrangement in non-B-cells (9, 15).
The study identified a novel group of genes whose expression is negatively affected by E2A expression. Few studies thus far have evaluated the potential for E2A-mediated gene repression. One such study has demonstrated the potential for E2A proteins to function as repressors of E-cadherin expression through direct interactions with E-box-containing promoter sequences (27). E-cadherin down-regulation is thought to be a critical event during embryonic development and in the acquisition of invasive properties in carcinoma tumor progression. E2A was isolated from a yeast one-hybrid screen to identify factors directly involved in mediating E-cadherin repression. Subsequent studies on E2A and the zinc finger protein Snail suggested that these two transcription factors might coordinate E-cadherin down-regulation both during embryonic epithelial-mesenchymal transitions and at the onset of tumor metastasis. E2A has also been implicated in regulating the activity of a cell type-specific repressor within the CR2/CD21 proximal promoter (28). CR2 functions as the receptor for complement C3 activation fragments and is expressed on immature and mature B-cells, epithelial cells, and select subsets of thymocytes, peripheral T cells, and dendritic cells. Mutagenesis experiments have shown that the CR2 proximal promoter contains a cell type-specific repressor element involved in controlling CR2 expression. Interestingly, the promoter contains two functional E-box sites: one of these (E box 1) is required for transcriptional activation, whereas the other (E box 2) is essential for transcriptional repression (28). E2A has been shown to bind E box 2 within the repressor element of the CR2 promoter, suggesting that E2A might be involved in CR2 repression during early B-lymphocyte development. The expression patterns of E2A and CR2 correlate well with this model, since E2A protein activity is high in pro- and pre-B-cells and is then down-regulated at the onset of CR2 expression in immature B-cells (18).
Here we have established a potential direct role for E2A-mediated suppression of Nfil3 and FGFR2. The observation that E2A interacts with genomic sequences within these loci is intriguing; however, it is still unclear how these interactions may influence gene repression. The E47R reconstitution system further demonstrates that E2A-mediated gene repression is reversible for Nfil3 and FGFR2. Previously published studies on the functions of proteins encoded by these genes may provide clues as to the significance of the gene expression and ChIP data. Nfil3 has been implicated in the cytokine-mediated survival of pro-B lymphocytes, and IL-3 signaling has been shown to control Nfil3 expression in murine pro-B-cell lines (29-31). Interestingly, Nfil3 has also been identified as one of four basic region/leucine zipper transcription factors that can competitively bind the consensus sequence recognized by the E2A-HLF oncoprotein. It is likely that the v-Abl oncogene provides sufficient survival and proliferation signals to preclude the need for significant Nfil3 expression in Abelson pre-B-cell lines. However, E2A has been previously shown to be a potent proapoptotic transcription factor (32). It is temping to speculate that active suppression of the Nfil3 gene by E2A may be one mechanism through which E2A mediates its proapoptotic activity.
FGFR2 has been implicated in the differentiation and activation of multiple hematopoietic cell lineages, including B-lymphocytes and myeloid cells (33-35). Basic fibroblast growth factor functions as the ligand for FGFR2 and is highly expressed in the bone marrow (33). Interestingly, FGFR2 expression is not detectable on bone marrow stem cells but is significantly up-regulated in myeloid progenitor populations (33, 34). Thus FGFR2 induction may occur as a consequence of myeloid lineage commitment to regulate subsequent differentiation events. FGFR2 up-regulation in E2A-deficient pre-B-cells may provide insight into how E2A controls B-cell lineage commitment not only through the activation of B-cell specific genes but possibly through the suppression of alternate developmental programs. Interestingly, E2A-deficient hematopoietic progenitors retain the ability to differentiate into multiple lineages that include myeloid cells, raising the possibility that suppression of alternate cell fate decisions may be a mechanism by which E2A mediates B-cell lineage commitment (36).
E2A is likely to play complex roles in the stage- and lineage-specific repression of numerous other genes during cell differentiation. Previous studies on E2F transcription factors have demonstrated the utility of combining ChIP-based studies with microarray-based gene expression analysis (37). We now show that this is also an effective approach to define E2A targets. E2A-mediated repression of certain genes is likely to be required for lineage restriction during lymphocyte differentiation. Other genes are probably repressed by E2A in a stage-specific manner to coordinate dynamic protein expression patterns at different stages of lymphocyte development and activation. Thus, a detailed and comprehensive characterization of E2A-repressed genes should contribute to a more complete understanding of E-protein function in lymphocyte development.
Acknowledgments
We thank Dr. John Choi (University of Pennsylvania) for generously providing the E47R fusion construct used in the inducible E2A reconstitution experiments and Dr. Simon Lin (Department of Biostatistics and Bioinformatics, Duke University) for assistance in statistic analysis of microarray data.
Footnotes
The abbreviations used are: IgH, immunoglobulin heavy; ChIP, chromatin immunoprecipitation; RT, reverse transcription; FACS, fluorescence-activated cell sorting; GFP, green fluorescent protein.
This work was supported by research Grant R01 CA72433 from the National Institute of Health and a scholarship from the Leukemia and Lymphoma Society Ref. 1485-99 (to Y. Z.).
References
- 1.Kondo M, Wagers AJ, Manz MG, Prohaska SS, Scherer DC, Beilhack GF, Shizuru JA, Weissman IL. Annu Rev Immunol. 2003;21:759–806. doi: 10.1146/annurev.immunol.21.120601.141007. [DOI] [PubMed] [Google Scholar]
- 2.Bassing CH, Swat W, Alt FW. Cell. 2002;109(suppl):45–55. doi: 10.1016/s0092-8674(02)00675-x. [DOI] [PubMed] [Google Scholar]
- 3.Greenbaum S, Zhuang Y. Semin Immunol. 2002;14:405–414. doi: 10.1016/s1044532302000751. [DOI] [PubMed] [Google Scholar]
- 4.Murre C, McCaw PS, Vaessin H, Caudy M, Jan LY, Jan YN, Cabrera CV, Buskin JN, Hauschka SD, Lassar AB, Weintraub H, Baltimore D. Cell. 1989;58:537–544. doi: 10.1016/0092-8674(89)90434-0. [DOI] [PubMed] [Google Scholar]
- 5.Murre C, McCaw PS, Baltimore D. Cell. 1989;56:777–783. doi: 10.1016/0092-8674(89)90682-x. [DOI] [PubMed] [Google Scholar]
- 6.Xin XQ, Nelson C, Collins L, Dorshkind K. J Immunol. 1993;151:5398–5407. [PubMed] [Google Scholar]
- 7.Zhuang Y, Soriano P, Weintraub H. Cell. 1994;79:875–884. doi: 10.1016/0092-8674(94)90076-0. [DOI] [PubMed] [Google Scholar]
- 8.Bain G, Maandag EC, Izon DJ, Amsen D, Kruisbeek AM, Weintraub BC, Krop I, Schlissel MS, Feeney AJ, van Roon M, van der Valk M, te Riele HPJ, Berns A, Murre C. Cell. 1994;79:885–892. doi: 10.1016/0092-8674(94)90077-9. [DOI] [PubMed] [Google Scholar]
- 9.Schlissel M, Voronova A, Baltimore D. Genes Dev. 1991;5:1367–1376. doi: 10.1101/gad.5.8.1367. [DOI] [PubMed] [Google Scholar]
- 10.Choi JK, Shen CP, Radomska HS, Eckhardt LA, Kadesch T. EMBO J. 1996;15:5014–5021. [PMC free article] [PubMed] [Google Scholar]
- 11.Kee BL, Murre C. J Exp Med. 1998;188:699–713. doi: 10.1084/jem.188.4.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.O’Riordan M, Grosschedl R. Immunity. 1999;11:21–31. doi: 10.1016/s1074-7613(00)80078-3. [DOI] [PubMed] [Google Scholar]
- 13.Sigvardsson M. Mol Cell Biol. 2000;20:3640–3654. doi: 10.1128/mcb.20.10.3640-3654.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sigvardsson M, Clark DR, Fitzsimmons D, Doyle M, Akerblad P, Breslin T, Bilke S, Li R, Yeamans C, Zhang G, Hagman J. Mol Cell Biol. 2002;22:8539–8551. doi: 10.1128/MCB.22.24.8539-8551.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Romanow WJ, Langerak AW, Goebel P, Wolvers-Tettero IL, van Dongen JJ, Feeney AJ, Murre C. Mol Cell. 2000;5:343–353. doi: 10.1016/s1097-2765(00)80429-3. [DOI] [PubMed] [Google Scholar]
- 16.Greenbaum S, Zhuang Y. Proc Natl Acad Sci U S A. 2002;99:15030–15035. doi: 10.1073/pnas.232299999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pan L, Hanrahan J, Li J, Hale LP, Zhuang Y. J Immunol. 2002;168:3923–3932. doi: 10.4049/jimmunol.168.8.3923. [DOI] [PubMed] [Google Scholar]
- 18.Zhuang Y, Jackson A, Pan L, Shen K, Dai M. Mol Immunol. 2004;40:1165–1177. doi: 10.1016/j.molimm.2003.11.031. [DOI] [PubMed] [Google Scholar]
- 19.Bain G, Gruenwald S, Murre C. Mol Cell Biol. 1993;13:3522–3529. doi: 10.1128/mcb.13.6.3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dudoit S, Gentleman RC, Quackenbush J. BioTechniques. 2003;(suppl):45–51. [PubMed] [Google Scholar]
- 21.Tsai CA, Hsueh HM, Chen JJ. Biometrics. 2003;59:1071–1081. doi: 10.1111/j.0006-341x.2003.00123.x. [DOI] [PubMed] [Google Scholar]
- 22.Fernandez LA, Winkler M, Grosschedl R. Mol Cell Biol. 2001;21:196–208. doi: 10.1128/MCB.21.1.196-208.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao F, Vilardi A, Neely RJ, Choi JK. Mol Cell Biol. 2001;21:6346–6357. doi: 10.1128/MCB.21.18.6346-6357.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fernex C, Capone M, Ferrier P. Mol Cell Biol. 1995;15:3217–3226. doi: 10.1128/mcb.15.6.3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Giannini SL, Singh M, Calvo CF, Ding G, Birshtein BK. J Immunol. 1993;150:1772–1780. [PubMed] [Google Scholar]
- 26.Chauveau C, Pinaud E, Cogne M. Eur J Immunol. 1998;28:3048–3056. doi: 10.1002/(SICI)1521-4141(199810)28:10<3048::AID-IMMU3048>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 27.Perez-Moreno MA, Locascio A, Rodrigo I, Dhondt G, Portillo F, Nieto MA, Cano A. J Biol Chem. 2001;276:27424–27431. doi: 10.1074/jbc.M100827200. [DOI] [PubMed] [Google Scholar]
- 28.Ulgiati D, Holers VM. J Immunol. 2001;167:6912–6919. doi: 10.4049/jimmunol.167.12.6912. [DOI] [PubMed] [Google Scholar]
- 29.Ikushima S, Inukai T, Inaba T, Nimer SD, Cleveland JL, Look AT. Proc Natl Acad Sci U S A. 1997;94:2609–2614. doi: 10.1073/pnas.94.6.2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuribara R, Kinoshita T, Miyajima A, Shinjyo T, Yoshihara T, Inukai T, Ozawa K, Look AT, Inaba T. Mol Cell Biol. 1999;19:2754–2762. doi: 10.1128/mcb.19.4.2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu YL, Chiang YJ, Yen JJ. J Biol Chem. 2002;277:27144–27153. doi: 10.1074/jbc.M200924200. [DOI] [PubMed] [Google Scholar]
- 32.Park ST, Nolan GP, Sun XH. J Exp Med. 1999;189:501–508. doi: 10.1084/jem.189.3.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berardi AC, Wang A, Abraham J, Scadden DT. Blood. 1995;86:2123–2129. [PubMed] [Google Scholar]
- 34.Allouche M, Bayard F, Clamens S, Fillola G, Sie P, Amalric F. Leukemia. 1995;9:77–86. [PubMed] [Google Scholar]
- 35.Genot E, Petit-Koskas E, Sensenbrenner M, Labourdette G, Kolb JP. Cell Immunol. 1989;122:424–439. doi: 10.1016/0008-8749(89)90089-0. [DOI] [PubMed] [Google Scholar]
- 36.Ikawa T, Kawamoto H, Wright LY, Murre C. Immunity. 2004;20:349–360. doi: 10.1016/s1074-7613(04)00049-4. [DOI] [PubMed] [Google Scholar]
- 37.Wells J, Graveel CR, Bartley SM, Madore SJ, Farnham PJ. Proc Natl Acad Sci U S A. 2002;99:3890–3895. doi: 10.1073/pnas.062047499. [DOI] [PMC free article] [PubMed] [Google Scholar]