

Abstract
Thymocytes undergoing TCRβ gene rearrangements are maintained in a low or nonproliferating state during early T cell development. This block in cell cycle progression is not released until the expression of a functional pre-TCR, which is composed of a successfully rearranged TCRβ-chain and the Pre-Tα-chain. The regulatory molecules responsible for the coordination of these differentiation and proliferation events are currently unknown. E2A and HEB are structurally and functionally related basic helix-loop-helix transcription factors involved in T cell development. To reveal the function of E2A and HEB through the stage of pre-TCR expression and alleviate functional compensation between E2A and HEB, we use a double-conditional knockout model. The simultaneous deletion of E2A and HEB in developing thymocytes leads to a severe developmental block before pre-TCR expression and a dramatic reduction of Pre-Tα expression. These developmentally arrested thymocytes exhibit increased proliferation in vivo and dramatic expansion ex vivo in response to IL-7 signaling. These results suggest that E2A and HEB are not only critical for T cell differentiation but also necessary to retain developing thymocytes in cell cycle arrest before pre-TCR expression.
T lymphocyte development ensues in the thymus upon the arrival of bone marrow-derived lymphoid precursors. Maturation follows an ordered progression in which these precursor cells begin at the CD4−CD8− double-negative (DN)3 stage and progress through the CD4+CD8+ double-positive (DP) stage, to reach the CD4+ or CD8+ single-positive (SP) stages of development. TCRβ and TCRα gene segments recombine at the DN and DP stage of development, respectively, to form a clonotypic TCR responsible for the recognition of antigenic peptide/MHC complexes.
DN cells can be further delineated based on their expression of CD44 (hyaluronic acid receptor) and CD25 (IL-2Rα) such that they begin at the DN1 (CD44+CD25−), and progress through the DN2 (CD44+CD25+), DN3 (CD44−CD25+), and DN4 (CD44−CD25−) stages (1). Early thymocytes begin commitment to the T cell lineage at the DN2 stage of development, during which thymocytes commence rearrangement of Dβ and Jβ gene segments within the TCRβ locus. At the DN3 stage, Vβ segments recombine with DβJβ gene products to form the TCRβ-chain. Expression of a functional TCRβ-chain constitutes a critical developmental checkpoint during the DN stage of thymocyte development. Only in-frame TCRβ rearrangements are able to produce a functional TCRβ-chain, which is expressed on the cell surface paired with a surrogate α-chain (Pre-Tα) to form the pre-TCR (pre-TCR). Signaling through this receptor complex triggers beta selection, a process that ceases further TCRβ rearrangement, stimulates thymocyte proliferation, and allows for the progression to the DN4 and DP stages of development.
Proliferation and differentiation are coordinately regulated during thymocyte development. During the DN stages of development, cells must balance cell cycle progression with ongoing rearrangement of their TCRβ-chain. As cells reach the DN2 stage, a transient proliferative expansion occurs which is then followed by a slow or nonproliferating phase as they enter the DN3 stage of development (2). Importantly, the majority of DN3 cells are retained within the G1 phase of cell cycle before pre-TCR expression (3). However, upon the successful expression of the pre-TCR and completion of beta selection, this cell cycle arrest is released and proliferation ensues. The molecular mechanism responsible for regulating cell cycle arrest before TCRβ expression is presently unknown.
Basic helix-loop-helix (bHLH) family E-protein transcription factors have been implicated in the regulation of both differentiation and proliferation during T cell development. Members of this family share two important structural similarities: 1) a basic region that allows for DNA binding and 2) an adjacent HLH domain that allows for dimerization with other bHLH proteins (4). Subsequent to DNA binding at sites termed E-boxes (CANNTG), activation domains within the dimerized E-proteins stimulate transcription of their target genes. Id proteins, encoded by four separate Id genes (Id1– 4) in mammals, lack the basic DNA-binding domain and consequently inhibit E-protein activity upon the formation of heterodimers with E-proteins (5). In the thymus, E2A-HEB heterodimers form the predominant E-protein dimer pair (6, 7) and regulate several important lineage- and stage-specific target genes important for lymphocyte development. These include the TCRα and -β enhancers, the Pre-Tα promoter, and various CD4 regulatory elements (6, 8, 9).
Mice lacking a functional E2A gene exhibit multiple lymphopoietic developmental defects including an accumulation of DN1 cells and an accelerated transition from the DP to SP stage of development (10). In addition, E2A-deficient mice display a high frequency of thymic-derived T cell leukemia (11, 12). HEB knockout mice display a developmental arrest at the CD8 immature SP (ISP) stage, a transitional stage between the DN and DP stages of thymocyte development (13). Although the disruption of individual Id genes generally causes little or no T cell developmental defects, ectopic expression of Id1 (14) or Id2 (15) in thymocytes results in severe developmental consequences, including a block in T cell differentiation and enhanced proliferation of thymocytes.
Genetic and biochemical studies have provided evidence of a significant degree of functional redundancy among E-proteins (13, 16, 17). The overlapping functions of E2A-HEB heterodimers and E2A or HEB homodimers in T cell development have been explored in mice carrying a dominant-negative allele of HEB (HEBbm) (17). This variant is able to form dimers with E2A but cannot bind DNA due to the lack of the basic region required for DNA binding. HEBbm mice display a complete block at the DN3 stage of T cell development and are deficient in TCRβ V-DJ rearrangement. Importantly, in contrast to the developmental block induced by the disruption of pre-TCR signaling, the developmental block in HEBbm mice cannot be rescued by the introduction of a functional TCR transgene (17). This finding suggests the existence of currently unappreciated functions of E-proteins in T cell development that are independent of TCRβ gene rearrangement and expression.
Available genetic models have clearly indicated the importance of E2A and HEB before and during the beta selection checkpoint. Unfortunately, functional compensation among different E-proteins in combination with the pleiotropic effect of germline gene disruption has obscured in-depth analysis. To address these issues and facilitate the investigation of E-protein function in T cell development, we have created a T cell-specific E2A and HEB double-conditional knockout (DKO) mouse using a Cre-recombinase system. Our study reveals a crucial role for E-proteins to retain developing T cells in a nonproliferative state before pre-TCR expression.
Materials and Methods
Construction of the HEB-loxP targeting construct
Genomic HEB sequence encoding the bHLH exon was cloned into pSK(+) immediately upstream of a floxed PGK-neomycin positive selection cassette. A partial digest using BamHI was performed to insert an additional loxP site 5′ of the bHLH exon. A 1.1-kb fragment was then cloned into a SpeI site upstream of the floxed bHLH domain to serve as the short arm for homologous recombination. The construct was excised using BglII and inserted into a shuttle vector containing a 7-kb long arm corresponding to the genomic sequence downstream of the bHLH exon, along with a PGK-thymidine kinase negative selection cassette. The complete targeting construct was linearized and transfected into cultured embryonic stem (ES) cells. These cells were grown under dual selection conditions using G418 and gancyclovir, and surviving clones were screened by PCR for homologous recombination using a primer upstream of the short arm (genomic downstream: 5′-CTTAGGACATGCGTTCATAATAAC-3′) and a primer that partially hybridizes to the loxP site upstream of the bHLH region (lox/bHLH upstream: 5′-GTTATTCAGACTGGATCTAATAAC-3′). Following the identification of successful recombinants (~5% of all clones (14 of 270)) and subsequent karyotyping, the ES cell clones were transiently transfected in vitro with a Cre-recombinase expression vector and screened by PCR for the floxed HEB allele (PGK-Neo deleted) and SpeI digest. Although we were unable to detect bHLH deletion alone due to PCR design, percentages for total deletion (bHLH plus PGK-Neo) and PGK-Neo deletion alone were 11.5 and 1%, respectively. Primers used for detection of Cre-deleted products were genomic downstream (see above) and a primer that hybridizes with the long arm of the targeting construct (upstream long arm: 5′-GCCAGAGGGTGGAAGCTCAGG-3′). These clones were then injected into blastocysts and implanted into pseudopregnant foster recipient mice to generate chimeras. Following chimera matings, F1 offspring containing the heterozygous HEBflox/+ allele were produced. HEBflox/+ mice were then crossed with E2Aflox/+lck-Cre+ mice (18) to establish HEBflox/floxE2Aflox/floxCre+ (DKO) and HEBflox/floxCre+ mice. Viability and fertility of the HEBflox/floxCre+ as well as E2Aflox/floxCre+ mice are similar to those of wild-type (WT) mice.
PCR genotyping
The presence of floxed or WT HEB and E2A alleles as well as the Cre transgene were detected by PCR. Detection of HEB was accomplished using JW1 (5′-CTGGGACAGAAGTTCAGCACTTAGTAC-3′) and JW2 (5′-CATTCCTATACATCAGCTTCTTGGACG-3′). E2A was genotyped using three primers: YZ-104 (5′-ATGTGTGGTGGCCCACACTTG T-3′), YZ-150 (5′-ACATGGCTGAATATCGACGGT-3′), and YZ-164 (5′-AA GAACGAGGCCTTCCGTGTC-3′). Cre transgene detection was accomplished using Cre5 (5′-CGCAGAACCTGAAGATGTTCGCGATTA-3′) and Cre3 (5′-TCTCCCACCGTCAGTACGTGAGATATC-3′).
PCR detection of Cre-mediated deletion
To amplify floxed and deleted bands for HEB or E2A, genomic DNA was harvested from sorted DN2–4 thymocytes or cultured DKO cells and subject to “touchdown” PCR (19) with JW1 and JW2 primers for HEB (see above for sequence), and E2Aflox forward (fw) (5′-CTGCACTCCGAAT TGTGCCTG-3′), PGKNeo fw (5′-GCCCATTCGACCACCAAGCG-3′), and YZ198 (5′-GATCCTCGTCTTCATTGGTACTG-3′) for E2A. Toe DNA from a HEBflox/floxE2Aflox/floxCre− mouse was used as a negative control. To quantify deletion, a standard curve was generated by mixing known numbers of HEB−/−E2A−/− and HEBflox/floxE2Aflox/flox pre-B cells together in the following ratios: 100% deleted (del), 90% del: 10% flox, 75% del: 25% flox, 50% del: 50% flox, 25% del: 75% flox, 10% del: 90% flox, 100% flox (20).
PCR analysis of TCRβ V(D)J rearrangement
Total thymus genomic DNA was harvested from LAT−/−, Rag2−/−, HEBflox/floxE2Aflox/floxCre−, and HEBflox/floxE2Aflox/floxCre+ mice or sorted ex vivo thymocytes and subjected to touchdown PCR (19) using the following primers: Vβ5-5′ consensus (5′-CCCAGCAGATTCTCA GTCCAACAG-3′), Vβ8-5′ consensus (5′-GCATGGGCTGAGGCTGA TCCATTA-3′), and Jβ2.7-3′ (5′-TGAGAGCTGTCTCCTACTATGGA TT-3′). EF1α was amplified as a loading control using YZ-95 (5′-AG TTTGAGAAGGAGGCTGCT-3′) and YZ-96 (5′-CAACAATCAGGA CAGCACAGTC-3′). Platinum Taq polymerase was used for PCR amplification as recommended by the manufacturer (Invitrogen Life Technologies).
Flow cytometry analysis
Single-cell suspensions were harvested from thymus, resuspended in 1×PBS/5% bovine calf serum, and kept on ice throughout the analysis. Cells were stained on ice for 30 min in the dark using the appropriate mAbs and scored using a FACSCalibur (BD Biosciences). FlowJo software (Tree Star) was used for data analysis. All Abs used for staining were purchased from BD Pharmingen, eBioscience, or Caltag Laboratories. BrdU staining was performed using a BrdU Flow Kit (BD Pharmingen) as per the manufacturer’s instructions, where mice were injected i.p. with 1 mg of BrdU and sacrificed 4 h later.
Cell sorting
To purify CD4−CD8− (DN) populations, thymocytes were first stained with anti-CD4-PE/Cy5 (L3T4) and anti-CD8a-PE/Cy5 (Ly-2) Abs. CD4+ and CD8+ cells were depleted with Dynabeads conjugated to sheep anti-rat IgG (Invitrogen Life Technologies). For some experiments, biotin-conjugated Abs were used followed by antibiotin Dynabead depletion. No differences were detected between the two methods. DN-enriched thymocytes were further stained with anti-CD44 (IM7), anti-Thy-1.2 (53-2.1), and anti-CD25 (PC61.5). DN subpopulations were defined and sorted as CD4−CD8−CD44+CD25− (DN1), CD4−CD8−CD44+CD25+ (DN2), CD4−CD8−CD44−CD25+ (DN3), and CD4−CD8−CD44−CD25−Thy-1.2+ (DN4). For purification of HEBflox/floxE2Aflox/floxCre+ DN populations, thymocytes were directly stained with the above Abs for sorting without any magnetic bead separation. Dead cells were excluded from sorting as positively stained cells with propidium iodide. FACS sorting were performed on a FACSVantage with a DiVa option equipped with 488-nm argon, 599-nm dye, and 408-nm krypton lasers (BD Biosciences; Flow Cytometry Systems).
Real-time PCR
mRNA was harvested from sorted DN3 and DN4 thymocytes (see above) using TRI reagent as recommended (Sigma-Aldrich) and reverse transcribed to cDNA using a Moloney murine leukemia virus reverse transcriptase kit per the manufacturer’s instructions (Invitrogen Life Technologies). cDNA was then subjected to SYBR Green real-time PCR (Roche). Relative units for each target were calculated using EF1α as a standard. Primer sequences are as follows: pTa fw (5′-CCATCACACTGCTGGTA GATGGAA-3′), pTa reverse (rev) (5′-GCAGAAGCAGTTTGAAGAG GAGCA-3′); p18: p18F1 (5′-ACACTGTACAGGCTTTGCTGGAGT-3′), p18R1 (5′-CACATTGCAGGCTGTGTGCTTCAT-3′); p21: p21fw (5′-TTGTCGCTGTCTTGCACTCTGGT-3′), p21rev (5′-AGACCAATCTGC GCTTGGAGTGAT-3′); p27: p27fw (5′-CGTGAGAGTGTCTAACGGG AGCC-3′), p27rev (5′-GCTTCTTGGGCGTCTGCTCCA-3′).
Ex vivo proliferation assay
For cell counting experiments, total DN cell culture was performed with 2.4–2.7 × 105 cells per 1 ml of culture medium (containing 10% FBS, 10 ng/ml IL-7, 5 μM 2-ME, and penicillin/streptomycin in RPMI 1640) in a 24-well plate. Cells from single wells were harvested at specified time points for FACS analysis and numeration on a hemacytometer. Trypan blue staining was used to exclude dead cells. [3H]Thymidine incorporation assays on sorted DN cells were done as follows: WT, p18−/−, and p21−/− thymocytes were first depleted of CD4+ and CD8+ cells using Dynabeads as described above. For total DN thymocyte cultures, 1 × 105 purified DN thymocytes were cultured in triplicate wells of a 96-well plate in 100 ml of IMDM containing 5% FCS, with or without IL-7 (10 ng/ml; R&D Systems) for 48 h. [3H]Thymidine (1 mCi) was added into the culture 6 h before harvesting. Cells were harvested with an auto cell harvester (Harvester 96; Tomtec) onto a glass filter. Radioactivity was determined by a liquid scintillation counter (1450 LSC and Luminescence Counter; PerkinElmer). Sorted DN2 and DN3 cultures were done similarly, except 1 × 104 cells were cultured for 7 days and [3H]thymidine was added into the culture 14–16 h before harvesting. Fresh IL-7 was added at day 3 and 6 of culture.
Results
Construction of a HEB-floxed allele
To investigate the lineage- and stage-specific function of HEB in T cell development, we used a loxP-Cre conditional knockout model. The conditional HEB knockout targeting construct was created by flanking both exon 18 of genomic HEB and the PGK-Neo selection cassette with loxP sites (triple loxP system) (Fig. 1A). Exon 18 of HEB encodes the bHLH domain, which is indispensable for DNA binding and dimerization (21). Targeted ES cell clones were transiently transfected with a Cre-recombinase expression vector and screened for those clones in which the floxed bHLH region remained, but the PGK-Neo cassette had been deleted. The ES cell clones carrying the HEB-floxed allele (HEBflox) were introduced into a mouse embryo for germline transmission. Subsequent breeding revealed that the floxed allele segregated with the same ratio as the WT allele (Fig. 1B) and that HEBflox/flox mice were phenotypically indistinguishable from WT littermates in terms of viability and fertility.
FIGURE 1.

Generation of the HEBflox allele. A, The targeting strategy for HEBflox mice. Shown are partial representations of the HEB genomic locus, the targeting construct, the HEBflox allele, and the HEBdel allele. Restriction enzyme site designations are as follows: B, BamHI; E, EcoRI; S, SpeI; and X, XhoI. Exons are depicted by black boxes and solid triangles represent loxP sites. The arrow above the WT HEB diagram indicates the direction of transcription. The primers JW1 and JW2 used for genotyping PCR are shown. B, HEBflox genotyping PCR. Mouse toe DNA was harvested and the primers JW1 and JW2 were used to detect WT and HEB-floxed alleles, yielding 1.05-kb and 1.3-kb fragments, respectively. C, Deletion PCR for HEB. The presence of floxed vs deleted HEB alleles in sorted DN2–4 thymocytes from HEBflox/floxCre+ SKO mice. A HEB serial dilution PCR with known ratios of floxed vs deleted alleles is shown for reference to estimate the efficiency of deletion. Nonspecific products are marked by an asterisk.
Conditional deletion of HEB results in a block at the ISP stage of T cell development and a reduction in thymocyte numbers
Mice carrying the HEBflox allele were crossed to lck-Cre transgenic mice. This transgene was previously used in the construction of E2A conditional knockout mice (18) and provides T lineage-specific expression of Cre recombinase under the control of the lck gene proximal promoter. To examine the expression pattern of Cre and the efficiency of HEB deletion in developing thymocytes, we assessed the presence of floxed and deleted bands in DN2–4-sorted thymocytes from HEBflox/floxCre+ mice. Serial dilutions of control DNA with predetermined ratios of floxed to deleted ranging from 100% floxed to 100% deleted were used to estimate the amount of deletion in HEBflox/floxCre+ mice (20). PCR analysis shows that deletion of HEB occurs during the DN stage of thymocyte development, starting as early as the DN2 stage and persisting through the DN4 subset, where deletion appears to be at least 75% complete (Fig. 1C). Flow cytometric analysis of total thymocytes from HEBflox/floxCre+ mice showed an accumulation of CD8 SP cells (Fig. 2A). CD5 expression was then evaluated as a marker to differentiate between mature CD8+ and ISP CD8+ cells, because the latter cell type have lower CD5 expression levels than mature CD8+ cells (13). As such, CD5 staining revealed that the block was at the ISP stage of development. DP cells also show two distinct peaks for CD5 expression, a finding that is consistent with the HEB germline knockout (13). A 2.5-fold average reduction in thymocyte numbers was detected in HEBflox/floxCre+ mice compared with WT or HEBflox/floxCre− controls (39.9 ± 15.4 × 106 vs 133 ± 32.6 × 106, respectively) (Fig. 2D). Importantly, this phenotype closely resembles that of conventional HEB knockout mice, which demonstrate a block at the ISP stage of development and a 5- to 10-fold reduction in thymocyte numbers (13). This finding confirms the importance of HEB in T cell differentiation and suggests that the conditional deletion of HEB is either complete or at least to a sufficient degree such that its function is abrogated by the ISP stage of thymocyte development.
FIGURE 2.

Conditional deletion of HEB alone or HEB and E2A together results in a block in thymocyte development. A, Deletion of HEB results in a block at the ISP stage of thymocyte development. Total thymocytes were harvested from HEBflox/floxCre− and HEBflox/floxCre+ mice and subject to flow cytometric analysis after staining with CD8-FITC, CD5-PE, 7-aminoactinomycin D (7AAD), and CD4-allophycocyanin. Following gating on forward and side scatter, 7AAD staining was used to exclude dead cells from the analysis. Percentage of cells in each quadrant is indicated. CD5 histograms are shown to differentiate between DP and ISP CD5lowCD8+ and mature CD5highCD8+ T cells. Arrows indicate quadrant origin for CD5 plots. Shaded areas represent HEBflox/floxCre− thymocytes, solid lines without shading represent HEBflox/floxCre+ thymocytes. B, Deletion of E2A and HEB results in a block at the DN stage of thymocyte development. Total thymocytes from HEBflox/floxE2Aflox/floxCre−, HEBflox/floxE2Aflox/floxCre+ (DKO), and LAT−/− mice were harvested and subject to flow cytometric analysis after staining with CD8-FITC, CD5-PE, 7AAD, and CD4-allophycocyanin. Following gating on forward and side scatter, 7AAD staining was used to exclude dead cells. Quadrant percentages are indicated. LAT−/− thymocytes are shown as a control and are also blocked at the DN stage of development. C, Normal distribution of DN thymocyte subsets in DKO mice. Total thymocytes were stained with CD44-FITC, DX5-PE, CD4-Tricolor (TC), CD8-TC, B220-TC, Mac1-TC, 7AAD, and CD25-allophycocyanin to examine DN cell subsets. Following gating on forward and side scatter, cells negative for DX5, CD4, CD8, B220, Mac1, and 7AAD were used for analysis. Percentage of cells in each quadrant is indicated. LAT−/− thymocytes are blocked at the DN3 stage of T cell development. D, Decreased cell numbers in SKO and DKO mice. Total thymocytes from WT (n = 17), HEBflox/floxCre+ single conditional knockout (n = 8), and DKO (n =13) mice were harvested and counted using a Coulter Counter as per the manufacturer’s recommendations. Values of p between WT and SKO, and WT and DKO cell numbers are indicated.
Deletion of both HEB and E2A results in a complete block in thymocyte development
Functional compensation between HEB and E2A has been well documented in early studies using HEB and E2A single gene knockouts. Therefore, we attempted to more clearly investigate the function of these E-proteins in T cell development by creating a HEB and E2A DKO. This model is valuable in that it eliminates the compensation issues that have hindered previous studies. HEBflox/flox mice were mated to previously generated E2Aflox/floxlck-Cre (Cre+) mice (18) to obtain HEBflox/floxE2Aflox/floxCre+ mice. Thymocyte development in conditional HEB and E2A double-knockout mice is blocked at the DN stage of differentiation (Fig. 2B). Examination of DN1– 4 cell subsets using surface CD44 and CD25 staining reveals a relatively normal distribution of cells throughout the DN stages of development (Fig. 2C). This result is in contrast to the E2A or HEB single knockouts (SKO), which manifest partial blocks at the DN1 and ISP stages, respectively (11, 13, 22). Linker for activation of T cell (LAT)-deficient thymocytes were used as a control, because these cells are also blocked at the DN stage of development. In contrast to the DKO, however, LAT−/− thymocytes are arrested at the DN3 stage. Very few DP cells are observed in all DKO mice examined (<1% of WT). Total thymic cellularity is significantly reduced (average 20-fold) in HEBflox/floxE2Aflox/floxCre+ mice in comparison to WT or Cre-negative controls (6.7 ± 3.1 × 106 vs 134.4 ± 33.4 × 106, respectively) (Fig. 2D). FACS analysis of DKO and WT thymi consistently reveals that the average proportion of DN cells is ~95% and 3% of the total thymocyte pool, respectively (data not shown). Therefore, although total thymic cellularity is decreased in DKO mice, there is a ~50% greater number of DN thymocytes in DKO mice than WT controls. γδ T cells are modestly reduced (2- to 4-fold) in DKO mice compared with WT controls (data not shown). The increased phenotypic severity of the DKO in comparison to the HEB SKO supports a cooperative role for E2A and HEB in thymocyte development.
TCRβ rearrangement is normal, but Pre-Tα expression is dramatically reduced in DKO thymocytes
Previous studies have indicated a requirement for E-proteins in efficient TCRβ gene rearrangement (17) and Pre-Tα gene transcription (23, 24). The successful completion of V(D)J β-recombination and subsequent pairing of the β-chain with Pre-Tα to form the pre-TCR is absolutely required for the DN to DP stage transition during thymocyte development. We therefore investigated whether the defect in DKO thymocyte development could be due to the loss of proper TCRβ-rearrangement and/or Pre-Tα expression.
Touchdown PCR was used to evaluate Vβ-DβJβ recombination for both the Vβ5 and Vβ8 gene families, which together constitute approximately one-fourth of the entire TCRβ repertoire (25). LAT knockout and RAG2 knockout mice were included as positive and negative controls, respectively, to allow for direct comparison of Vβ-DβJβ rearrangement within the DN compartment. This assay revealed the presence of TCRβ Vβ-DβJβ recombination in DKO mice (Fig. 3A). Using the same assay, we were also unable to detect any differences in recombination between WT and DKO-sorted DN3 and DN4 thymocytes (data not shown). Together, these results indicate that the developmental block in DKO thymocyte development is unlikely due to defective TCRβ gene recombination.
FIGURE 3.
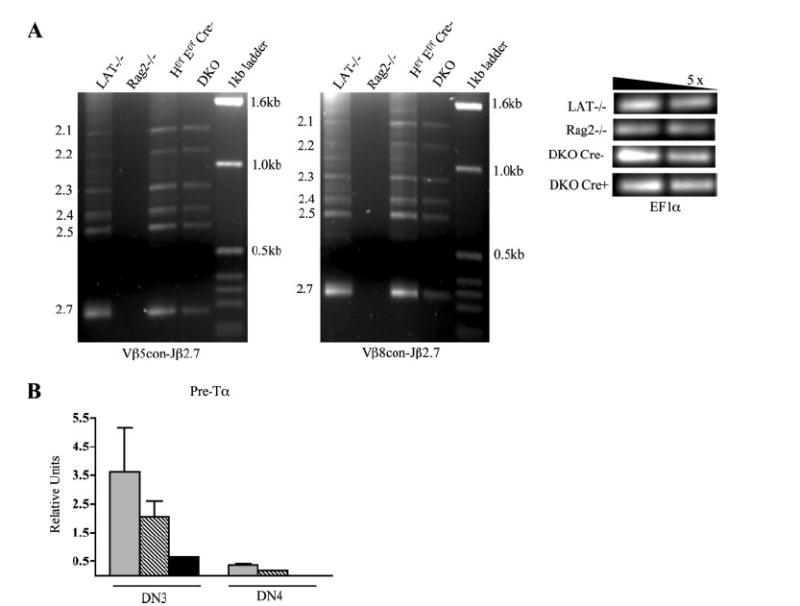
DKO thymocytes appear to have normal V to DJ TCRβ rearrangement but severely reduced levels of Pre-Tα expression. A, Analysis of TCRβ recombination involving Vβ5 and Vβ8 gene families. Genomic DNA from total thymocytes was harvested and used for touchdown PCR analysis with the indicated primers for the amplification of V-DJ recombination products. Rearrangement products involving Jβ2.1–7 (marked on the left of each gel) are detected in LAT−/−, HEBflox/floxE2Aflox/floxCre−, and DKO mice but not Rag2−/− mice. A serial 5-fold dilution of EF1α was used as a loading control. B, Real-time RT-PCR analysis of Pre-Tα expression. mRNA was harvested from sorted WT, SKO, and DKO DN3 and DN4 thymocytes, and cDNA was reverse transcribed and amplified by real-time PCR.
 represents WT samples;
represents WT samples;
 represents HEB SKO; and ■ represent DKO samples. Relative units were calculated in reference to a standard curve generated by four 5-fold serial dilutions of WT thymic cDNA. Error bars represent the SD of samples done in duplicate. Results are representative of three independent experiments.
represents HEB SKO; and ■ represent DKO samples. Relative units were calculated in reference to a standard curve generated by four 5-fold serial dilutions of WT thymic cDNA. Error bars represent the SD of samples done in duplicate. Results are representative of three independent experiments.
We then evaluated Pre-Tα expression using real-time RT-PCR on sorted DN3 and DN4 thymocytes (Fig. 3B). Consistent with previous reports, Pre-Tα transcription is highly expressed in DN3 cells and down-regulated in DN4 cells (26). We detected a dramatic down-regulation of Pre-Tα expression in DKO thymocytes that was most evident in DN3 stage cells, the same developmental population in which thymocyte development is blocked in Pre-Tα knockout mice (27). This result suggests that loss of Pre-Tα expression may partially contribute to the developmental defect of DKO mice. An E-protein dose-related effect was also seen, because HEB single conditional knockouts displayed intermediate levels of Pre-Tα expression. These observations are consistent with reports that Pre-Tα is directly regulated by E-proteins (23) and further confirms that E-protein activity has been dramatically down-regulated as early as the DN3 stage of development in DKO mice.
Double-knockout thymocytes undergo enhanced proliferation in vivo and display deregulation of cell cycle regulatory genes
An increase in the total number of DN cells in DKO mice suggests the presence of enhanced cell proliferation resulting from the loss of E-proteins. Accordingly, we chose to investigate in vivo thymocyte proliferation in DKO mice by measuring BrdU incorporation. In agreement with previous studies that indicate normal DN2 cells have a greater proliferative capacity than DN3 cells (2), WT DN2 thymocytes proliferate more robustly than DN3 cells (Fig. 4A). However, although DN2 thymocytes from both WT and DKO mice appear to undergo a similar degree of proliferation, DKO DN3 thymocytes proliferate >1.5-fold the amount of their WT counterparts (12.7% vs 20.1%, respectively). This result suggests that the loss of E-proteins during T cell development releases DN3 thymocytes from the G1 phase retention and allows for cell cycle progression. In contrast, DN4 cells from DKO mice display a reduction in proliferation compared with WT cells (Fig. 4A). Because proliferation of DN4 cells is dependent on pre-TCR signaling, this finding is likely due to the severely compromised Pre-Tα expression in DKO thymocytes.
FIGURE 4.
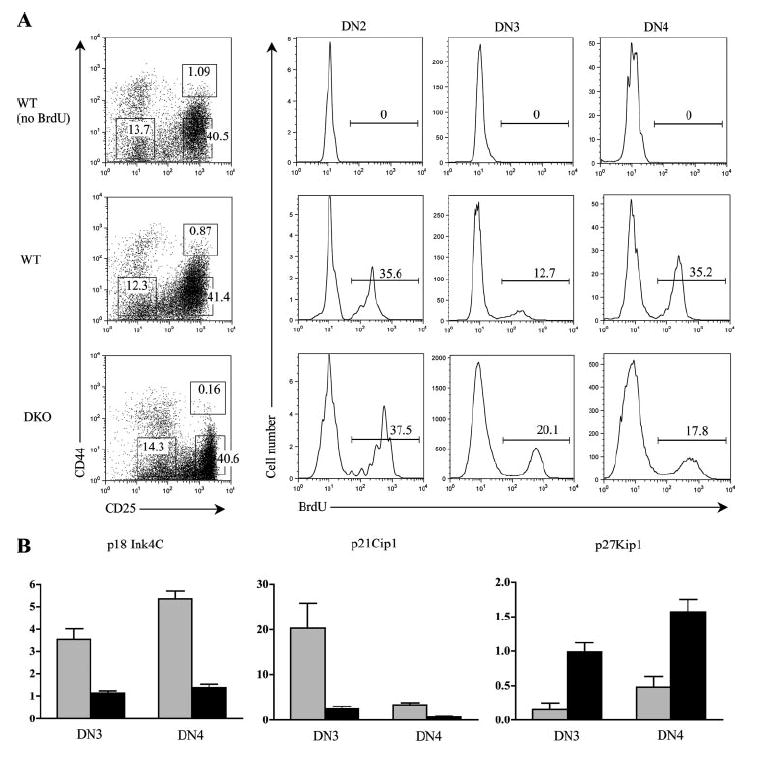
Thymocytes from DKO mice hyperproliferate in vivo and display altered levels of CDKI expression. A, Proliferation analysis of WT and DKO thymocytes. Mice were injected with 1 mg of BrdU and sacrificed 4 h later. Total thymocytes were stained with BrdU-FITC, CD44-PE, CD4-TC, CD8-TC, B220-TC, and CD25-allophycocyanin. Following gating on forward and side scatter, cells staining negative for CD4, CD8, and B220 were analyzed for CD44/25 expression. The boxed regions on the CD44/25 plot corresponding to DN2, DN3, and DN4 cells were analyzed for BrdU incorporation. Cell percentages in the boxed regions are indicated. Histograms indicating the percentage of BrdU-positive cells for each DN subset examined are shown. Results are representative of two experiments. B, Real-time RT-PCR analysis of CDKIs p18Ink4c, p21Cip1, and p27Kip1 in sorted DN3 and DN4 thymocytes.
 and ■ represent HEBflox/floxE2Aflox/floxCre− and DKO samples, respectively. Error bars represent the SD of samples done in duplicate. Results are representative of two to three independent experiments with independently sorted thymocytes from different litter mice. Relative units were calculated in reference to a standard curve generated by four 5-fold serial dilutions of WT thymic cDNA.
and ■ represent HEBflox/floxE2Aflox/floxCre− and DKO samples, respectively. Error bars represent the SD of samples done in duplicate. Results are representative of two to three independent experiments with independently sorted thymocytes from different litter mice. Relative units were calculated in reference to a standard curve generated by four 5-fold serial dilutions of WT thymic cDNA.
We next investigated potential mechanisms contributing to the aberrant cellular proliferation. Progression through the cell cycle is tightly regulated by the balance of cyclins and cyclin-dependent kinase inhibitors (CDKIs) (28, 29). E-proteins have been implicated as being positive transcriptional regulators of the CDKI p21Cip1 in non-T cells and thus act to inhibit cell cycle (30-32). Evidence for E-proteins regulating p21Cip1 expression in T lineage cells has also been reported in which E2A-deficient thymoma cells display an up-regulation of p21Cip1 when transduced with E47 (33). In addition, it has been shown that mice lacking the CDKIs p18Ink4c (34) or p27Kip1 (35) but not p21Cip1 (36) have increased thymic cellularity. Therefore, we evaluated the effect of E-protein deletion on the expression of these CDKIs using real-time RT-PCR on sorted DN3 and DN4 thymocytes (Figs. 4B). In WT mice, p21Cip1 is highly expressed at the DN3 stage and down-regulated at the DN4, whereas both p18Ink4c and p27Kip1 are slightly increased from the DN3 to DN4 stage of development. Analysis of DKO thymocytes showed a dramatic down-regulation of p18Ink4c and p21Cip1expression in both the DN3 and DN4 stages, suggesting a positive role for E-proteins in regulating these two CDKI genes. In contrast, p27Kip1 is up-regulated in DKO mice, indicating that it is negatively regulated by E-proteins. Thus, E-proteins appear to function in a complex manner to influence the expression of cell cycle regulatory genes.
IL-7-dependent proliferation of DKO thymocytes ex vivo
IL-7 is an essential cytokine for normal DN thymocyte expansion and survival (reviewed in Ref. 37). The increased proliferation of DKO DN3 cells in vivo prompted us to investigate the responsiveness of DKO thymocytes to IL-7 in an ex vivo culture system. Total DN cells were isolated from WT or DKO mice and placed in culture in the presence or absence of IL-7 and pulsed with [3H]thymidine. Although both WT and DKO DN cells exhibit IL-7-dependent proliferation, at 48 h DKO thymocytes display ~3- to 4-fold higher [3H]thymidine incorporation than WT cells (Fig. 5A). Next, we investigated the kinetics of proliferation. In these experiments, we used LAT−/− DN thymocytes which respond similarly to WT DN cells in our culture system (data not shown). In contrast to LAT−/− DN cells which fail to proliferate, we observed a significant expansion of DKO thymocytes following 7 days of culture in the presence of IL-7 (Fig. 5B). This expansion succeeds a transient decrease in DKO cell numbers, suggesting that cell death occurs in the majority of DN cells and only a subset is responsible for the increased proliferation. Consistent with an increase in cell number, the proliferating DKO cells show a blasting phenotype, whereas the nonproliferating LAT−/− cells show a decrease in cell size by both FACS (Fig. 5C) and microscopic analysis (data not shown).
FIGURE 5.

DKO thymocytes are hyperproliferative ex vivo in response to IL-7 signaling. A, DKO thymocytes undergo enhanced proliferation in the presence of IL-7. A total of 1 × 105 purified DN thymocytes from WT or DKO mice was cultured in the presence or absence of 10 ng/ml IL-7 for 48 h and pulsed with [3H]thymidine 6 h before harvesting and determining radioactive incorporation. Error bars represent the SD of samples done in triplicate. Results are representative of three independent experiments. B, Increase in cell number of DKO but not LAT−/− DN thymocytes following a 7-day culture in the presence of 10 ng/ml IL-7. Plot shown is representative of three independent experiments. Values of p between WT and DKO samples are indicated. C, DKO but not LAT−/− thymocytes display a blasting phenotype following ex vivo culture in the presence of 10 ng/ml IL-7. Total thymocytes from DKO and LAT−/− culture were harvested at day 0 (preculture), day 3, and day 5 and analyzed by flow cytometry. Cells were stained with CD44-FITC, c-kit-PE, CD4-TC, CD8-TC, Mac-1-TC, B220-TC, 7AAD, and CD25-allophycocyanin. Cells negative for CD4, CD8, Mac-1, B220, and 7AAD were gated and used for forward and side scatter analysis. D, The emergence of DN2 phenotype cells in DKO but not LAT−/− thymocyte culture. Cells used in C were further analyzed for CD44 and CD25 expression following gating on forward and side scatter and exclusion of CD4, CD8, Mac-1, B220, and 7AAD signals. Quadrant percentages are indicated. E, HEB and E2A deletion analysis of cultured cells. An E2A serial dilution PCR with known ratios of floxed vs deleted alleles is shown for reference to estimate the efficiency of deletion (analogous to assay used in Fig. 1C for HEB deletion). Toe (T) DNA from HEBflox/floxE2Aflox/floxCre− mice and DNA from cultured (C) DKO cells were assayed for HEB or E2A deletion. Floxed and deleted alleles for HEB and E2A are indicated.
Interestingly, over the course of culture we observe the gradual emergence of DN2-like cells in DKO but not LAT−/− cultures (Fig. 5D). These cells appear to be T lineage cells as evidenced by positive Thy1 staining and were confirmed to have a DN2 phenotype by expressing CD44, CD25, and CD117 (c-kit) but not non-DN2 markers including CD4, CD8, CD5, DX5, γδ TCR, and Mac1 (data not shown). Also, there was no change in either DX5+ NK cells or γδ T cells during the culture period (data not shown). PCR analysis was used to quantify the deletion of E2A and HEB in the cultured cells. We show a significant, yet incomplete deletion of E2A and HEB in cultured DKO cells (Fig. 5E).
Because DKO DN3 cells hyperproliferate in vivo yet we detect the expansion of a DN2-like population following ex vivo culture, we sought to determine which specific subset of DN thymocytes was responsible for the observed increase in proliferation. To address this question, DN2 and DN3 thymocytes from DKO mice were sorted and placed in culture independently in the presence or absence of IL-7. Cells were pulsed with [3H]thymidine on day 6 of culture and harvested at day 7. In agreement with results obtained from in vivo BrdU-labeling experiments, DN3 cells from DKO mice hyperproliferated in response to IL-7 signaling (Fig. 6A). Specifically, DN3 cell proliferation was ~10-fold that of DN2 cells from DKO mice. This increase in proliferation is accompanied by a DN2-like phenotypical shift over the course of the culture resembling that previously seen in total DN cell culture (Fig. 6B). In addition, examination of the V(D)J rearrangement status of sorted DKO DN3 cells under these culture conditions revealed the presence of V-DJ recombination (Fig. 6C). Together, these results imply that DN3 cells are the primary cell subset responsible for the increased proliferation of DKO thymocytes in IL-7 culture.
FIGURE 6.
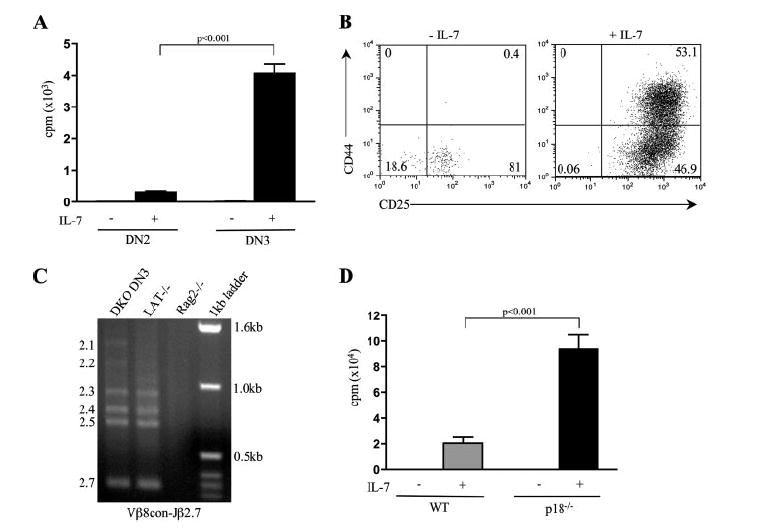
DN3 thymocytes are responsible for the increased proliferation of DKO cells in IL-7 culture. A, DN3 cells undergo enhanced proliferation in the presence of IL-7. Thymocytes from DKO mice were sorted into DN2 and DN3 populations based on CD44, CD25, and c-kit staining. DN2 cells were identified as CD44+CD25+c-kit+ whereas DN3 cells were CD44−CD25+c-kit−. A total of 1 × 104 cells was cultured in the presence or absence of 10 ng/ml IL-7 for 7 days and pulsed with [3H]thymidine 14 h before harvesting and determining radioactive incorporation. Error bars represent the SD of three to five samples. Results are representative of two independent experiments. Values of p between DKO DN2 and DN3 samples are indicated. B, DKO DN3 thymocytes undergo a phenotypical shift to DN2-like cells following 7 days of culture in the presence of 10 ng/ml IL-7. Cells were stained with CD44-FITC, c-kit-PE, CD4-TC, CD8-TC, Mac-1-TC, B220-TC, 7AAD, and CD25-allophycocyanin. Following gating on forward and side scatter, cells negative for CD4, CD8, Mac-1, B220, and 7AAD were used in the analysis. Quadrant percentages are indicated. C, Cultured DN3 cells show normal V(D)J recombination. Genomic DNA from day-7 cultured thymocytes was harvested and used for touchdown PCR analysis with the indicated primers for the amplification of Vβ8-Jβ2.1–7 recombination products (marked on the left of each gel) are as indicated. D, p18−/− thymocytes hyperproliferate ex vivo in the presence of IL-7. A total of 1 × 105 purified DN thymocytes from WT and p18 knockout mice was cultured in the presence or absence of 10 ng/ml IL-7 for 48 h and pulsed with [3H]thymidine 6 h before harvesting and determining radioactive incorporation. Results are representative of three independent experiments. Values of p between WT and p18−/− samples are indicated.
We next chose to investigate the mechanism(s) responsible for the increased proliferation in DKO thymocytes. Because our real-time RT-PCR data indicate that p18Ink4c and p21Cip1 expression is down-regulated in both DN3 and DN4 DKO thymocytes, we sought to determine whether the elimination of p18Ink4c or p21Cip1 alone is sufficient to cause cell cycle deregulation in DN cells. Using a proliferation assay of cultured DN thymocytes from p18Ink4c or p21Cip1 knockout mice, we show that loss of p18Ink4c results in ~a 4- to 5-fold increase in proliferation (Fig. 6D), suggesting that p18Ink4c is indeed an important CDKI for the suppression of proliferation in DN thymocytes. In contrast, no change in proliferation was detected for p21Cip1 (data not shown). These observations indicate a novel role for E-proteins in antagonizing IL-7-dependent proliferation during early thymocyte development.
Discussion
We have described herein a HEB/E2A DKO system and directly examined the T cell intrinsic contribution of E-proteins to thymocyte development. Simultaneous disruption of HEB and E2A alleviates concerns of functional compensation that have influenced the results of earlier studies on single E-protein knockouts (11, 13). Although a third E-protein (E2–2) exists in mammals, its involvement in thymocyte development is uncertain. Although some authors have suggested that E2–2 plays a minor role in very early thymocyte development, others have reported that E2–2 mRNA is undetectable in the thymus (38). Although the possibility of E2–2 compensating for the loss of HEB and E2A exists, the severe block in thymocyte development in the DKO model suggests that E2–2 is unlikely to be a significant contributor to E-protein function in early thymocytes or is unable to substitute for the loss of HEB and/or E2A.
DKO mice exhibited a complete block in thymocyte development before the DN to DP transition, much like that of previously described HEBbm mice (17). However, two important phenotypical discrepancies were observed between these models: First, DKO thymi contained a substantial number of DN4 cells, which were nearly absent in HEBbm mice. Second, recombination at the TCRβ locus was found to be normal in DKO thymocytes, whereas TCRβ V-DJ recombination was severely disrupted in HEBbm mice. Both of these differences can most likely be attributed to incomplete Cre-mediated E2A and/or HEB deletion at the onset of V(D)J rearrangement. However, caution must be exercised in regards to interpreting the results of the TCRβ recombination status of DKO mice. Although touchdown PCR is a sensitive assay capable of detecting TCR rearrangement, it is not a quantitative assay and subtle defects in recombination may pass undetected.
Previous work has shown that anti-CD3ε stimulation is unable to drive the differentiation of HEBko (13) or HEBbm thymocytes from the DN to DP stage of development (Y. Zhuang, unpublished results). Similarly, expression of a TCR transgene is not sufficient to rescue T cell development in HEBko (13) or HEBbm mice (17). These results suggest that the phenotype caused by the disruption of E-proteins cannot simply be explained by defects in the expression and/or signaling of either the pre-TCR or mature TCR. Accordingly, the loss of Pre-Tα expression revealed in our study of DKO mice is unlikely to be the sole defect responsible for the developmental block at the beta selection checkpoint. As such, other currently unidentified functions of E-proteins exist that are required for proper T cell development.
In vivo BrdU labeling assays revealed an increased proliferation of DN3 thymocytes in DKO mice. This increased proliferative capacity was further supported by the ex vivo culture assay, in which DN3 thymocytes from DKO mice were found to expand in the presence of IL-7 more vigorously than controls. Curiously, most proliferating cells in our ex vivo culture assay acquire canonical DN2 markers including CD44, CD25, and CD117 (c-kit) but not other lineage markers including Mac1, γδ TCR, or DX5 (to identify myeloid cells, γδ T cells, and NK cells, respectively). Importantly, these cells display TCRβ V(D)J gene rearrangement, suggesting that the emergence of DN2 cells in our culture represents a phenotypic shift from the more mature DN3 subset. This developmental reversion suggests a novel and pivotal role for E-proteins in promoting normal maturational progression while simultaneously inhibiting the backwards differentiation of DN3 to DN2 thymocytes. Notably, this phenomenon seems to be IL-7 dependent, because we do not see the phenotypical reversion in non-IL-7-containing cultures nor in vivo, which may be attributed to IL-7 availability or an unidentified role for thymic stroma in regulating T cell differentiation.
A role for E-proteins in the regulation of proliferation has been indicated in studies of both nonlymphoid and lymphoid systems (30, 39, 40). In particular, a role for E2A in suppressing DN3 cell proliferation has been observed by Engel and Murre (41) in the analysis of E47-deficient mice. However, the signaling events responsible for cell cycle control that are mediated by E-proteins at this early stage of T cell development are unknown. Previous studies have implicated E2A in cell cycle regulation by activating CDKIs such as p21Cip1 in cultured fibroblasts and thymoma cells (30, 32, 33). Although we observed a decrease in p21Cip1 expression in DKO thymocytes, no changes in the proliferative status of DN thymocytes from p21Cip1-deficient mice were detected in vivo or ex vivo in the presence of IL-7 (data not shown). In agreement with these findings, defects in T cell development have not been reported in the study of p21Cip1-deficient mice (36). In contrast, gene knockout studies have indicated that p18Ink4c and p27Kip1 appear to play important roles in thymocyte development, because disruption of either p18Ink4c (34) or p27Kip1 (35) leads to increased numbers of thymocytes. Our analysis showed that p18Ink4c is down-regulated whereas p27Kip1 is up-regulated upon the deletion of E2A and HEB. Transcriptional up-regulation of p27Kip1 is somewhat surprising because we observed an overall increase of cell cycle proliferation in DKO mice. It is possible that even though p27Kip1 expression was increased, it was not sufficient to overcome the dramatic decreases of p18Ink4c and p21Cip1, because the decision to proliferate or arrest is ultimately regulated by the balance of factors that inhibit vs those that promote cell cycle. Also possible is the fact that enhanced p27Kip1 expression can activate cyclin D-dependent kinases (28) and therefore promote cell cycle. Finally, the observed increase p27Kip1 in expression may not be representative of protein levels, because p27Kip1 activity is regulated at the posttranslational level (42).
Our analysis of p18Ink4c knockout mice provides an important clue as to how E-proteins may participate in cell cycle regulation during early T cell development. The increased proliferation seen in p18Ink4c-deficient DN thymocytes even in the presence of E-protein function suggests that p18Ink4c may act downstream of E-proteins. In fact, the observed decrease of p18Ink4c expression in DKO thymocytes further implicates p18Ink4c as a downstream effector molecule of E-protein signaling that bridges their function to cell cycle regulation.
The ex vivo culture of DN cells was conducted in the presence of IL-7 as the sole cytokine and in the absence of stromal cells. IL-7 has been shown to provide both survival and proliferation signals during early T cell development (reviewed in Ref. 37). Thus, the observed increase in proliferation of DKO and p18Ink4c-deficient thymocytes suggests that E-proteins provide a critical counterbalance to suppress IL-7-mediated proliferation in developing T cells undergoing TCRβ gene rearrangement. In addition to IL-7 signaling, Notch has been shown to provide an essential signal for early thymocyte development (43). However, the absence of stromal cells in our culture system suggests that proliferation of DKO thymocytes occurs independent of Notch signaling.
In summary, our findings suggest that E-proteins are important regulators for coordinating differentiation and proliferation events before the beta selection checkpoint. Our studies further suggest a role for E-proteins in suppressing IL-7-mediated proliferation before pre-TCR expression. The exact mechanism(s) responsible for cell cycle regulation before pre-TCR expression still requires further investigation, and p18 is most likely only one of many targets regulated by E-proteins. The genetic system reported herein provides a valuable experimental model for identifying and testing additional E-protein targets responsible for cell cycle regulation. Work reported here is also relevant to the observation that E2A-deficient mice frequently develop T cell leukemia (11, 12). Studies have suggested that leukemiogenesis is linked to the loss of E2A in the early stage of T cell development (18). Therefore, future research on E-protein function in cell cycle progression should impact our understanding of both basic mechanisms of T cell development and leukemiogenesis.
Acknowledgments
We thank Adam Lazorchak, Beth Jones, Caron Jia, Ikuko Hayakawa, and Stephen Greenbaum for critical reading of this manuscript. We thank Cheryl Bock, Dave Schnider, and Mei Lang Flowers of the Duke Transgenic Facility for assistance in the generation of the HEBflox mice. We thank Dr. Yue Xiong for providing the p18 mice and thoughtful discussion of the project and Matthew Smith for assistance in the p18 mouse work.
Footnotes
This work was supported by National Institutes of Health Grant R01 GM059638 (to Y.Z.).
Abbreviations used in this paper: DN, double negative; DP, double positive; SP, single positive; bHLH, basic helix-loop-helix; ISP, immature SP; DKO, double-conditional knockout; ES, embryonic stem; WT, wild type; fw, forward; rev, reverse; SKO, single knockout; LAT, linker for activation of T cell; CDKI, cyclin-dependent kinase inhibitor; 7AAD, 7-aminoactinomycin D; TC, Tricolor; del, deleted.
Disclosures The authors have no financial conflict of interest.
References
- 1.Godfrey DI, Kennedy J, Suda T, Zlotnik A. A developmental pathway involving four phenotypically and functionally distinct subsets of CD3−CD4−CD8− triple-negative adult mouse thymocytes defined by CD44 and CD25 expression. J Immunol. 1993;150:4244–4252. [PubMed] [Google Scholar]
- 2.Tourigny MR, Mazel S, Burtrum DB, Petrie HT. T cell receptor (TCR)-β gene recombination: dissociation from cell cycle regulation and developmental progression during T cell ontogeny. J Exp Med. 1997;185:1549–1556. doi: 10.1084/jem.185.9.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoffman ES, Passoni L, Crompton T, Leu TM, Schatz DG, Koff A, Owen MJ, Hayday AC. Productive T-cell receptor β-chain gene rearrangement: coincident regulation of cell cycle and clonality during development in vivo. Genes Dev. 1996;10:948–962. doi: 10.1101/gad.10.8.948. [DOI] [PubMed] [Google Scholar]
- 4.Murre C, McCaw PS, Vaessin H, Caudy M, Jan LY, Jan YN, Cabrera CV, Buskin JN, Hauschka SD, Lassar AB, et al. Interactions between heterologous helix-loop-helix proteins generate complexes that bind specifically to a common DNA sequence. Cell. 1989;58:537–544. doi: 10.1016/0092-8674(89)90434-0. [DOI] [PubMed] [Google Scholar]
- 5.Benezra R, Davis RL, Lockshon D, Turner DL, Weintraub H. The protein Id: a negative regulator of helix-loop-helix DNA binding proteins. Cell. 1990;61:49–59. doi: 10.1016/0092-8674(90)90214-y. [DOI] [PubMed] [Google Scholar]
- 6.Sawada S, Littman DR. A heterodimer of HEB and an E12-related protein interacts with the CD4 enhancer and regulates its activity in T-cell lines. Mol Cell Biol. 1993;13:5620–5628. doi: 10.1128/mcb.13.9.5620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shen CP, Kadesch T. B-cell-specific DNA binding by an E47 homodimer. Mol Cell Biol. 1995;15:4518–4524. doi: 10.1128/mcb.15.8.4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Greenbaum S, Zhuang Y. Identification of E2A target genes in B lymphocyte development by using a gene tagging-based chromatin immunoprecipitation system. Proc Natl Acad Sci USA. 2002;99:15030–15035. doi: 10.1073/pnas.232299999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takeuchi A, Yamasaki S, Takase K, Nakatsu F, Arase H, Onodera M, Saito T. E2A and HEB activate the pre-TCRα promoter during immature T cell development. J Immunol. 2001;167:2157–2163. doi: 10.4049/jimmunol.167.4.2157. [DOI] [PubMed] [Google Scholar]
- 10.Kee BL, Bain G, Murre C. IL-7Rα and E47: independent pathways required for development of multipotent lymphoid progenitors. EMBO J. 2002;21:103–113. doi: 10.1093/emboj/21.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bain G, Engel I, Robanus Maandag EC, te Riele HP, Voland JR, Sharp LL, Chun J, Huey B, Pinkel D, Murre C. E2A deficiency leads to abnormalities in αβ T-cell development and to rapid development of T-cell lymphomas. Mol Cell Biol. 1997;17:4782–4791. doi: 10.1128/mcb.17.8.4782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yan W, Young AZ, Soares VC, Kelley R, Benezra R, Zhuang Y. High incidence of T-cell tumors in E2A-null mice and E2A/Id1 double-knockout mice. Mol Cell Biol. 1997;17:7317–7327. doi: 10.1128/mcb.17.12.7317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barndt R, Dai MF, Zhuang Y. A novel role for HEB downstream or parallel to the pre-TCR signaling pathway during αβ thymopoiesis. J Immunol. 1999;163:3331–3343. [PubMed] [Google Scholar]
- 14.Kim D, Peng XC, Sun XH. Massive apoptosis of thymocytes in T-cell-deficient Id1 transgenic mice. Mol Cell Biol. 1999;19:8240–8253. doi: 10.1128/mcb.19.12.8240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morrow MA, Mayer EW, Perez CA, Adlam M, Siu G. Over-expression of the helix-loop-helix protein Id2 blocks T cell development at multiple stages. Mol Immunol. 1999;36:491–503. doi: 10.1016/s0161-5890(99)00071-1. [DOI] [PubMed] [Google Scholar]
- 16.Zhuang Y, Barndt RJ, Pan L, Kelley R, Dai M. Functional replacement of the mouse E2A gene with a human HEB cDNA. Mol Cell Biol. 1998;18:3340–3349. doi: 10.1128/mcb.18.6.3340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barndt RJ, Dai M, Zhuang Y. Functions of E2A-HEB heterodimers in T-cell development revealed by a dominant negative mutation of HEB. Mol Cell Biol. 2000;20:6677–6685. doi: 10.1128/mcb.20.18.6677-6685.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pan L, Hanrahan J, Li J, Hale LP, Zhuang Y. An analysis of T cell intrinsic roles of E2A by conditional gene disruption in the thymus. J Immunol. 2002;168:3923–3932. doi: 10.4049/jimmunol.168.8.3923. [DOI] [PubMed] [Google Scholar]
- 19.Jackson A, Kondilis HD, Khor B, Sleckman BP, Krangel MS. Regulation of T cell receptor β allelic exclusion at a level beyond accessibility. Nat Immunol. 2005;6:189–197. doi: 10.1038/ni1157. [DOI] [PubMed] [Google Scholar]
- 20.Lazorchak AS, Wojciechowski J, Dai MF, Zhuang Y. E2A promotes the survival of precursor and mature B lymphocytes. J Immunol. 2006;177:2495–2504. doi: 10.4049/jimmunol.177.4.2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murre C, McCaw PS, Baltimore D. A new DNA binding and dimerization motif in immunoglobulin enhancer binding, daughterless, MyoD, and myc proteins. Cell. 1989;56:777–783. doi: 10.1016/0092-8674(89)90682-x. [DOI] [PubMed] [Google Scholar]
- 22.Barndt RJ, Zhuang Y. Controlling lymphopoiesis with a combinatorial E-protein code. Cold Spring Harbor Symp Quant Biol. 1999;64:45–50. doi: 10.1101/sqb.1999.64.45. [DOI] [PubMed] [Google Scholar]
- 23.Petersson K, Ivars F, Sigvardsson M. The pTα promoter and enhancer are direct targets for transactivation by E box-binding proteins. Eur J Immunol. 2002;32:911–920. doi: 10.1002/1521-4141(200203)32:3<911::AID-IMMU911>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 24.Tremblay M, Herblot S, Lecuyer E, Hoang T. Regulation of pTα gene expression by a dosage of E2A, HEB, and SCL. J Biol Chem. 2003;278:12680–12687. doi: 10.1074/jbc.M209870200. [DOI] [PubMed] [Google Scholar]
- 25.Wilson A, Marechal C, MacDonald HR. Biased Vβ usage in immature thymocytes is independent of DJβ proximity and pTα pairing. J Immunol. 2001;166:51–57. doi: 10.4049/jimmunol.166.1.51. [DOI] [PubMed] [Google Scholar]
- 26.Taghon T, Yui MA, Pant R, Diamond RA, Rothenberg EV. Developmental and molecular characterization of emerging β- and γδ-selected pre-T cells in the adult mouse thymus. Immunity. 2006;24:53–64. doi: 10.1016/j.immuni.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 27.Fehling HJ, Krotkova A, Saint-Ruf C, von Boehmer H. Crucial role of the pre-T-cell receptorα gene in development of αβ but not γδ T cells. Nature. 1995;375:795–798. doi: 10.1038/375795a0. [DOI] [PubMed] [Google Scholar]
- 28.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 29.Kozar K, Ciemerych MA, Rebel VI, Shigematsu H, Zagozdzon A, Sicinska E, Geng Y, Yu Q, Bhattacharya S, Bronson RT, et al. Mouse development and cell proliferation in the absence of D-cyclins. Cell. 2004;118:477–491. doi: 10.1016/j.cell.2004.07.025. [DOI] [PubMed] [Google Scholar]
- 30.Prabhu S, Ignatova A, Park ST, Sun XH. Regulation of the expression of cyclin-dependent kinase inhibitor p21 by E2A and Id proteins. Mol Cell Biol. 1997;17:5888–5896. doi: 10.1128/mcb.17.10.5888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Herblot S, Aplan PD, Hoang T. Gradient of E2A activity in B-cell development. Mol Cell Biol. 2002;22:886–900. doi: 10.1128/MCB.22.3.886-900.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pagliuca A, Gallo P, De Luca P, Lania L. Class A helix-loop-helix proteins are positive regulators of several cyclin-dependent kinase inhibitors’ promoter activity and negatively affect cell growth. Cancer Res. 2000;60:1376–1382. [PubMed] [Google Scholar]
- 33.Schwartz R, Engel I, Fallahi-Sichani M, Petrie HT, Murre C. Gene expression patterns define novel roles for E47 in cell cycle progression, cytokine-mediated signaling, and T lineage development. Proc Natl Acad Sci USA. 2006;103:9976–9981. doi: 10.1073/pnas.0603728103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Franklin DS, Godfrey VL, Lee H, Kovalev GI, Schoonhoven R, Chen-Kiang S, Su L, Xiong Y. CDK inhibitors p18INK4c and p27Kip1 mediate two separate pathways to collaboratively suppress pituitary tumorigenesis. Genes Dev. 1998;12:2899–2911. doi: 10.1101/gad.12.18.2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nakayama K, Ishida N, Shirane M, Inomata A, Inoue T, Shishido N, Horii I, Loh DY, Nakayama K. Mice lacking p27Kip1 display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell. 1996;85:707–720. doi: 10.1016/s0092-8674(00)81237-4. [DOI] [PubMed] [Google Scholar]
- 36.Deng C, Zhang P, Harper JW, Elledge SJ, Leder P. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell. 1995;82:675–684. doi: 10.1016/0092-8674(95)90039-x. [DOI] [PubMed] [Google Scholar]
- 37.Jiang Q, Li WQ, Aiello FB, Mazzucchelli R, Asefa B, Khaled AR, Durum SK. Cell biology of IL-7, a key lymphotrophin. Cytokine Growth Factor Rev. 2005;16:513–533. doi: 10.1016/j.cytogfr.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 38.Zhuang Y, Cheng P, Weintraub H. B-lymphocyte development is regulated by the combined dosage of three basic helix-loop-helix genes, E2A, E2–2, and HEB. Mol Cell Biol. 1996;16:2898–2905. doi: 10.1128/mcb.16.6.2898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li L, Olson EN. Regulation of muscle cell growth and differentiation by the MyoD family of helix-loop-helix proteins. Adv Cancer Res. 1992;58:95–119. doi: 10.1016/s0065-230x(08)60292-4. [DOI] [PubMed] [Google Scholar]
- 40.Engel I, Murre C. The function of E- and Id proteins in lymphocyte development. Nat Rev Immunol. 2001;1:193–199. doi: 10.1038/35105060. [DOI] [PubMed] [Google Scholar]
- 41.Engel I, Murre C. E2A proteins enforce a proliferation checkpoint in developing thymocytes. EMBO J. 2004;23:202–211. doi: 10.1038/sj.emboj.7600017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pagano M, Tam SW, Theodoras AM, Beer-Romero P, Del Sal G, Chau V, Yew PR, Draetta GF, Rolfe M. Role of the ubiquitin-proteasome pathway in regulating abundance of the cyclin-dependent kinase inhibitor p27. Science. 1995;269:682–685. doi: 10.1126/science.7624798. [DOI] [PubMed] [Google Scholar]
- 43.Robey EA, Bluestone JA. Notch signaling in lymphocyte development and function. Curr Opin Immunol. 2004;16:360–366. doi: 10.1016/j.coi.2004.03.009. [DOI] [PubMed] [Google Scholar]