

Abstract
The actin-related protein 2/3 (Arp2/3) complex mediates the formation of branched actin filaments at the leading edge of motile cells and in the comet tails moving certain intracellular pathogens. Crystal structures of the Arp2/3 complex are available, but the architecture of the junction formed by the Arp2/3 complex at the base of the branch was not known. In this study, we use electron tomography to reconstruct the branch junction with sufficient resolution to show how the Arp2/3 complex interacts with the mother filament. Our analysis reveals conformational changes in both the mother filament and Arp2/3 complex upon branch formation. The Arp2 and Arp3 subunits reorganize into a dimer, providing a short-pitch template for elongation of the daughter filament. Two subunits of the mother filament undergo conformational changes that increase stability of the branch. These data provide a rationale for why branch formation requires cooperative interactions among the Arp2/3 complex, nucleation-promoting factors, an actin monomer, and the mother filament.
Introduction
Dynamic actin networks drive cell locomotion, phagocytosis, and intracellular motility of vesicles, organelles, and certain pathogens (Welch and Mullins, 2002). The actin-related protein 2/3 (Arp2/3) complex is the key component of these networks by virtue of its ability to initiate actin filament branches (daughter filaments) at an angle on the sides of preexisting mother filaments (Mullins et al., 1998). Conserved among eukaryotes, the Arp2/3 complex consists of two Arps, Arp2 and Arp3, and five additional subunits named ARPC1–5. In the presence of ATP and nucleation-promoting factors, the complex binds to the side of a mother filament and initiates a daughter filament, which grows at its free barbed end. Arp2 and Arp3 are proposed to form the first two subunits of the daughter filament. Although actin filaments are flexible, branch junctions appear to be very rigid (Blanchoin et al., 2000).
Crystal structures of the inactive Arp2/3 complex (Robinson et al., 2001; Nolen et al., 2004; Nolen and Pollard, 2007) revealed the architecture of the subunits but not how the complex forms branches, which also requires knowledge of the 3D structure of the branch junction. Current models of the branch junction (Robinson et al., 2001; Volkmann et al., 2001; Beltzner and Pollard, 2004; Aguda et al., 2005; Egile et al., 2005) are based solely on indirect evidence and 2D projection maps. Consequently, they lack detailed 3D information on the interactions and on conformational changes required to assemble a branch. The absence of these details has limited the progress in understanding the mechanism of branching and its contributions to cellular function.
In this study, we describe a 3D reconstruction of the branch at 2.6-nm resolution from electron tomography. Statistics-based docking of the crystal structures of the Arp2/3 complex and models of actin filaments into the reconstruction revealed that conformational changes are required in both the mother filament and Arp2/3 complex to fit the proteins into the density. Our structure includes 12 subunits of the mother filament, seven subunits of the Arp2/3 complex, and four subunits of the daughter filament. This reconstruction provides the structural framework for understanding branch formation during diverse cellular processes.
Results and discussion
Electron microscopy and image processing
We used a combination of electron tomography and single-particle volume processing to make a 3D reconstruction of the actin filament branch junction at 2.6-nm resolution (Figs. 1 and Figs.2). We generated the reconstruction from 279 individual branch junctions from negatively stained filament networks formed by the Acanthamoeba castellanii Arp2/3 complex. Reconstructions of branches generated by bovine and yeast samples were very similar in appearance to the amoeba samples (Fig. 1). The reconstructions are fully consistent with previous observations in two dimensions (Volkmann et al., 2001) and show three bridges of density connecting the two filaments comprising the branch (Fig. 2).
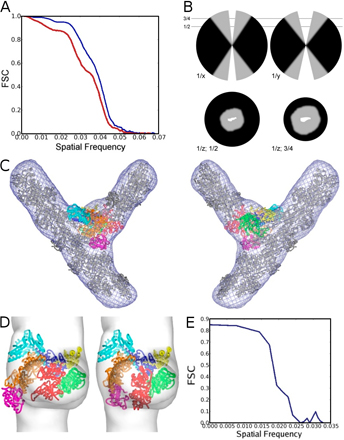
Figure 1.

Electron tomography of actin filament branches mediated by the Arp2/3 complex. (A) Central slice showing several branches (boxes), individual Arp2/3 complexes (circles), and gold markers (arrowheads). (B and C) Enlarged view of individual complexes (B) and branches (C1–3). (D) Automatically segmented branch junctions show individual actin subunits and substructure in the junction. (E–G) Central slices through the averages of branch junctions from A. castellanii (E), cow (F), and budding yeast (G). (H) Composite of three x-y slices through a dual-axis cryotomogram. Because the branches are not contained within a single slice, we concatenated three slices for visualization purposes. (I) Central slices through the three branches marked in H after reorientation to coincide with the x-y plane. (J) Detail of a filament from H showing individual actin subunits after automatic segmentation. Bars: (A, C, D, H, and I) 20 nm; (G and J) 10 nm.
Figure 2.

Reconstruction of filament branch junction formed by the A. castellanii Arp2/3 complex. (A) Three views related by 90° clockwise rotations. The reconstruction was sharpened in Fourier space with 300 Å2 and is displayed at a 100% mass contour level. All other density representations are from the original reconstruction. Numbers indicate three bridges of density between the two branches. (B) Stereo view of the fit of the branch junction model (α carbon trace) into the branch junction. Actin subunits are gray. Inset shows the crystal structure of the Arp2/3 complex with the mother filament position indicated by a gray rectangle. The color code for subunits of the Arp2/3 complex is used throughout: Arp2, red; Arp3, orange; ARPC1, green; ARPC2, cyan; ARPC3, magenta; ARPC4, blue; and ARPC5, yellow.
To control for artifacts associated with negative staining, we made reconstructions from averaged cryotomograms of 82 frozen-hydrated branch junctions suspended over holes (Fig. 1 and Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200709092/DC1). These reconstructions share all of the features of negatively stained specimens, but the resolution was limited to ∼5.5 nm because of the significantly lower signal to noise ratio. Angles between the mother and daughter filaments were measured using only the actin subunits of the mother and daughter filaments resolved in the averaged reconstructions because the actin filaments bend further away from the branch point. This angle was remarkably uniform (±2°) for both types of samples at 78°.
Docking of crystal structures into the reconstruction
We used a statistics-based docking algorithm (Volkmann and Hanein, 1999) to fit models of actin filaments (Holmes et al., 2003; Volkmann et al., 2005) and substructures of the bovine Arp2/3 complex (Robinson et al., 2001; Nolen et al., 2004) into the reconstruction. Docking confirmed that the Arp2/3 complex binds to the side of the mother filament oriented with Arp2 and Arp3 at the pointed end of the daughter filament (Egile et al., 2005). However, as anticipated from both the crystal structure of inactive bovine Arp2/3 complex (Robinson et al., 2001) and 2D reconstructions of the branch junction (Volkmann et al., 2001), the inactive Arp2/3 complex does not fit into the density at the branch junction as a result of mismatches at Arp3-ARPC3 and Arp2 (Fig. 3 and Video 2, available at http://www.jcb.org/cgi/content/full/jcb.200709092/DC1). A filament model with conventional symmetry docks faithfully into the reconstruction of the daughter filament (Fig. 2 B) but not into that of the mother filament (Fig. 3 C). Therefore, we used the docking algorithm to rebuild the Arp2/3 complex and the mother filament so that the proteins fit into the reconstruction.
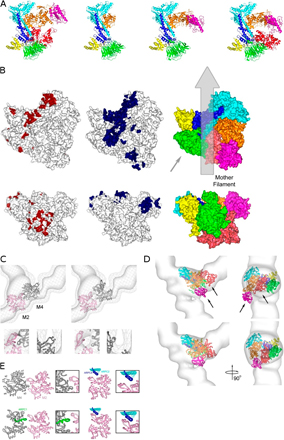
Figure 3.

Fits of unmodified and modified crystal structures in the reconstruction of branch junctions. (A) Best fit of the Arp2/3 complex crystal structure. Arrows indicate mismatches with the reconstruction. The magenta and red arrows indicate the movement of ARPC3 and Arp2 upon remodeling. (B) Best fit of the remodeled Arp2/3 complex. (C and D) Comparisons between mother filament density in the branch reconstruction and a filament model viewed from the side opposite to the origin of the daughter filament. (C) Superimposition of the models fit to optimize the top portion of the reconstruction (orange) and the bottom portion of the reconstruction (dark pink). The gray arrow points to the area where a break in symmetry occurs at subunit M4. The pink and orange arrows indicate the movement necessary to fit the density. (D) Fit after replacing M4 with a monomer conformation. (E and F) Surface contours of the reconstruction at ∼70% of the mass. (E) Fit of the subdomains 2 of M2 (light pink) and M4 (dark gray) in the original filament conformation. The arrow points at subdomain 2 of actin subunit M4. (F) Improved fit with modified subdomains 2. (G) Close-up stereo view of the branch junction model inside the reconstruction displayed at the 100% mass contour level. The tethered helix of APC1 (green helix in the foreground) is near the groove between subdomains 1 and 3 of mother filament subunit M4.
The docking algorithm allows an objective estimation of precision and the definition of confidence levels for conformational changes. We cross-validated all results by repeating each docking experiment multiple times using different crystal structures, reconstructions, and resolution cutoffs (see Materials and methods). All reported changes are significant at a 99.5% confidence level (P < 0.005). The analysis indicates that placement of rigid body elements is reproducible within ∼0.4 nm. The final model (Fig. 2 and Video 1, available at http://www.jcb.org/cgi/content/full/jcb.200709092/DC1) contains all seven subunits of the Arp2/3 complex as well as 16 actin subunits (a total molecular mass of ∼900 kD).
Docking and remodeling of the Arp2/3 complex and daughter filament
We used a series of conservative steps to fit the Arp2/3 complex into the reconstruction (Fig. S3 A, available at http://www.jcb.org/cgi/content/full/jcb.200709092/DC1). The core subunits of the complex consisting of ARPC1, ARPC2, ARPC4, and ARPC5 fit into the reconstruction without modification. We left intact the contacts of subdomain 1 of Arp3 with these subunits observed in crystal structures because they are stable even when the rigid unit of subdomains 3 and 4 of Arp3 with ARPC3 changes conformation in response to nucleotide binding (Nolen et al., 2004; Nolen and Pollard, 2007). The short-pitch helix of the daughter filament continues into the mass occupied by the Arp2/3 complex, evidence that Arp2 and Arp3 are arranged next to each other like two subunits at the pointed end of a filament. To generate this conformation of the Arps, to move the Arp2/3 complex into the density, and to avoid steric clashes with the rest of the complex, we made the following changes: (1) we moved Arp2 next to Arp3 in a short-pitch helix dimer; (2) we closed the nucleotide-binding clefts of both Arps; and (3) we twisted subdomains 3 and 4 of both Arps by ∼15° relative to subdomains 1 and 2 (Figs. 2 and S2 A). The reconstruction has density for subdomains 1 and 2 of Arp2, which are disordered in most crystals. Arp2 has a completely different set of contacts with ARPC1, ARPC2, and Arp3 in the branch model than in the crystal structures. The new contacts bury a surface area that is 1,700 Å2 larger than the contacts in the inactive complex. The rest of the intersubunit contacts do not need to be modified to fit into the reconstruction. We are confident that Arp2 is located next to Arp3 in the branch, but the resolution of the reconstruction does not reveal conformational changes in other parts of the complex that may occur as part of this repositioning of Arp2.
Docking and remodeling of the mother filament
The reconstruction includes 12 actin subunits of the mother filament. When we docked a filament model into the density, we found that the fit was poor (Fig. 3 C). We could not correct this misfit by varying the global twist of the filament model. This established that the misfit arises from a local difference between the model and the reconstruction. Because of its local nature, the mismatch is not evident in projection (Volkmann et al., 2001). We found that modifying the conformation of actin subunit M4 from a filamentous to monomeric conformation untwists the filament locally so that it fits into the density (Fig. 3 D). The major difference between monomeric actin structures and filament models is a twist of ∼15° between subdomains 1/2 and 3/4 (Fig. S2 A). This modification of M4 disrupts its interactions in both directions along the long-pitch helix where subdomain 2 of M2 interacts with the groove between subdomains 1 and 3 of M4 and subdomain 2 of M4 interacts with the groove of M6.
Both before and after remodeling M4, the docked model of the Arp2/3 complex left unoccupied some densities near subdomains 2 of M2 and M4, where both of these subdomains are outside the density in the mother filament region (Fig. 3 E). Thus, we modified both subunits to fit the density by opening their nucleotide-binding clefts (Fig. 2, F and G). These changes are consistent with the fact that the change in actin subunit M4 releases both subdomains 2 from filament interactions. The modified cleft of M4 is similar to that of apo-Arp3 (Robinson et al., 2001), and the cleft of M2 resembles that of monomeric actin crystal structures in the typical closed monomeric conformation (Fig. S2; Otterbein et al., 2001; Klenchin et al., 2006).
Model of the entire branch junction
With the changes in the Arp2/3 complex and the mother filament, the entire model of the branch junction fits into the reconstruction, leaving no substantial regions of density unoccupied (Figs. 2 B and 3 G). Although the resolution of the reconstruction is limited to 2.6 nm, the statistical docking analysis indicates that the placement of crystal structures can be reproduced within ∼0.4 nm. Therefore, the model provides reliable information about the relative positions of all 23 protein subunits, but side chain interactions cannot be deduced. The model reveals an extensive interface between the Arp2/3 complex and the mother filament (Fig. 4) burying a surface area of 9,100 Å2, which is consistent with the rigidity of the branch junction (Blanchoin et al., 2000).
Figure 4.

Low resolution surface representation of the branch junction model. Actin subunits are shown in white or gray. M1–6 are subunits in the mother filament. D1 and D2 are the first two subunits in the daughter filament. Some of the actin and Arp subunits are indicated by numbers. (A) Three different views related by 90° clockwise rotations. (B) The three views of A showing the mother filament with areas of contact with subunits of the Arp2/3 complex color coded according to the subunit coloring in A. In the middle view, the Arp2/3 complex is viewed from the aspect of the mother filament (turned 180° clockwise from A) with points of mother filament contact marked in gray (subunits M2 and M4) and white (subunits M1, M3, M5, and M6). Bar, 5 nm.
Five mother filament subunits, M1–M4 and M6, participate in the interactions (Fig. 4 B). Unexpectedly, all seven subunits of the Arp2/3 complex contact the mother filament. M2 and M4 contribute the largest patch of interactions (Fig. 4 B, gray), contacting all subunits of the Arp2/3 complex except for Arp3. The distortion around M4 and the altered conformations of subdomains 2 of M2 and M4 provide surfaces for several of these interactions (Fig. S3 E), including those with Arp2 and ARPC3. Both of these interactions may stabilize the filamentlike conformation of the Arps. Distortion of the mother filament creates a groove that appears to be occupied by the C-terminal helices of ARPC2 and ARPC4. Many of the residues of the Arp2/3 complex that are close enough to interact with the mother filament are highly conserved between species (Fig. S3 B; Beltzner and Pollard, 2004).
Arp2 and Arp3 form the first two subunits of the daughter filament
Our reconstruction shows unambiguously that Arp2 and Arp3 form the first two subunits of the daughter filament. This rules out hypotheses suggesting that one of the Arps is incorporated into the mother filament (Pantaloni et al., 2000). Having the Arps at the pointed end of the daughter filament requires a substantial conformational change from the inactive Arp2/3 complex, in which the Arps are arranged back to back like two successive subunits along the short-pitch helix of an actin filament. We expect that Arp3 maintains its contacts with ARPC2 and ARPC4 during this conformational change because they are stable even when other parts of the complex change conformation after ATP binding and because it is possible to isolate a complex from fission yeast that lacks Arp2 but has all of the other subunits in their normal positions (Nolen, B., personal communication). Therefore, Arp2 must shift the longitudinal distance of 2.5 nm, or the whole complex must bend to place Arp2 next to Arp3. In the active branch-junction conformation, the first actin subunit of the daughter filament interacts with the barbed end of Arp3 and the side of Arp2. In keeping with this interpretation, fission yeast Arp2/3 complex lacking Arp2 has no nucleation activity (Nolen, B., personal communication).
The binding site of the Arp2/3 complex overlaps the tropomyosin-binding site on actin
The location of the Arp2/3 complex on the side of the mother filament partially overlaps the site where skeletal muscle tropomyosin in the active conformation binds to muscle thin filaments (Pirani et al., 2006). This provides a structural explanation for the finding that microinjection of skeletal muscle tropomyosin displaces the Arp2/3 complex from lamellipodia of epithelial cells (Gupton et al., 2005) and that skeletal muscle tropomyosin inhibits branch formation (Blanchoin et al., 2001). The position of the tropomyosin is not known in nonmuscle cells, but a similar overlap between tropomyosin and the Arp2/3 complex may explain why nonmuscle tropomyosin isoforms also inhibit branch formation (Blanchoin et al., 2001). In the cell, the presence of the Arp2/3 complex on freshly formed filaments may inhibit tropomyosin binding and explain why tropomyosin binds to filaments behind the leading edge, where many branches have dissociated (DesMarais et al., 2002). This structural incompatibility may also contribute to the stable segregation of dynamic and functionally distinct actin structures that contain either the Arp2/3 complex (actin patches) or tropomyosin (actin cables) at sites of polarized growth in yeast (Moseley and Goode, 2006).
The Arp2/3 complex firmly anchors the daughter filament on the mother filament
Our reconstruction also shows that formation of the interface between the Arp2/3 complex and the mother filament requires substantial conformational changes of both partners. The large interface thus created anchors the daughter filament to the mother filament, providing a mechanical reinforcement for the growth of the daughter filament to exert force on the inside of the plasma membrane or the surface of a bacterium. On the other hand, formation of the branch junction requires conformations far from equilibrium. Assuming that these conformations are poorly populated at equilibrium, rarely will either a segment of a filament or an Arp2/3 complex be in conformations favorable for binding. This situation may explain why the Arp2/3 complex binds to filaments extremely slowly with an association rate constant on the order of 10−3 μM−1s−1 (Beltzner and Pollard, 2008). Once formed, the large interface is stable, and the dissociation rate constant is 10−3 s−1. The cell can afford an unfavorable binding reaction to build this complicated structure because the concentration of actin filaments at the leading end of a cell is in the millimolar range (Abraham et al., 1999), allowing branches to form on a time scale of seconds.
The complexity of the interactions at branch junctions also offers an explanation for why the Arp2/3 complex, nucleation-promoting factors, the mother filament, and actin monomers must cooperate to assemble a branch. Each interaction may contribute free energy to overcome barriers along the assembly pathway. Knowledge of the arrangement of the protein subunits in the branch junction together with crystal structures of the Arp2/3 complex provide the framework required to design and interpret the experiments that will define the detailed pathway of branch formation.
The structural roles of ARPC1–5
The organization of subunits in our branch model suggests some hypotheses regarding details of the protein interactions, which we consider in the closing paragraphs (Fig. 5). Some of these details are not resolved in the reconstruction but are reasonable based on the relative positions of the protein subunits.
Figure 5.

Schematic representations of the precursors and assembled components of the branch junction. (A) Drawing of the inactive Arp2/3 complex and a standard actin filament. (B) Model of the branch junction. The following conformational changes are proposed to occur: (1) opening the nucleotide-binding clefts of mother filament subunits M2 and M4; (2) converting subunit M4 from a filament to a monomeric conformation; (3) converting Arp3 into filament conformation; (4) moving Arp2 tethered by ARPC5 next to Arp3 to form the first two subunits of the daughter filament; and (5) converting Arp2 into filament conformation.
The ARPC2–ARPC4 heterodimer provides the main surface for interaction of the complex with the mother filament and anchors Arp3, the first subunit of the daughter filament (Figs. 3 G and 4 B). These interactions explain why the ARPC2–ARPC4 dimer is crucial for branch formation and why ARPC2–ARPC4 and the Arp2/3 complex have similar affinities for filamentous actin (Gournier et al., 2001).
ARPC3 forms a bridge between mother filament subunits M4 and M6 and Arp3 (Figs. 3 G and 4 B), putting it in a position to stabilize the closed conformation of Arp3 and the open conformation of actin subunit M4 in the mother filament. This reinforcing function is not absolutely essential because subcomplexes lacking ARPC3 show only relatively minor functional defects (Winter et al., 1999; Gournier et al., 2001).
ARPC5 appears to contribute to the branch in two ways. First, its helical domain provides some minor contacts with mother filament subunit M2 (Fig. 4 B). Second, the extended N terminus of ARPC5 wraps around Arp2, so it may help to tether Arp2 to the rest of the complex during the conformational changes that reposition Arp2 as the second subunit of the daughter filament. Consistent with this idea, Arp2 and ARPC1 bind weakly to a budding yeast subcomplex lacking ARPC5 (Winter et al., 1999).
The β propeller domain of ARPC1 makes only minor contacts with the mother filament (Fig. 4), but budding yeast Arp2/3 complex lacking ARPC1 is completely inactive for actin nucleation (Kelly et al., 2006). Thus, ARPC1 may have additional roles in Arp2/3 complex function. One possibility is binding nucleation-promoting factors (Pan et al., 2004; Kelly et al., 2006). Another involves a short helix tethered to the β propeller domain by disordered loops. In the crystal structure (Robinson et al., 2001), this helix packs between subdomains 1 and 3 of Arp3 in a neighboring complex. In our model of the branch, ARPC1 is close enough to mother filament subunit M4 (Fig. S2 B) for this helix to bind M4 in analogous fashion (Fig. 3 G). Like profilin (Chik et al., 1996), the ARPC1 helix might stabilize the open conformation of M4 observed in the branch junction and promote nucleotide exchange by M4. Also in analogy with profilin, the ARPC1 helix might then dissociate after M4 hydrolyzes ATP and dissociates phosphate. Dissociation of the ARPC1 helix might destabilize the open conformation of M4, weaken the contact of M4 subdomain 2 with ARPC2–ARPC4, and destabilize the closed filamentlike conformation of Arp2 (directly) and Arp3 (through ARPC3). These reactions would help to explain why the Arp2/3 complex has nearly the same affinity for ADP- and ATP-actin filaments (Beltzner and Pollard, 2008) but why branches dissociate 10-fold faster from ADP-actin filaments than ATP– or ADP–inorganic phosphate filaments (Mahaffy and Pollard, 2006).
Materials and methods
Proteins
Actin was purified from rabbit skeletal muscle as described previously (Volkmann et al., 2005). Purification of A. castellanii, the bovine and budding yeast Arp2/3 complex, as well as the nucleation-promoting factors was described previously (Volkmann et al., 2001). Samples for tomography were prepared as previously described for 2D analysis (Egile et al., 2005). In brief, reactions were performed by mixing 2 μM Mg2+-ATP–G-actin (10% pyrene labeled) with the Arp2/3 complex and nucleation-promoting factor (100 nM amoeba Arp2/3 complex with 200 nM Scar-VCA, 25 nM bovine Arp2/3 complex with 500 nM Scar-VCA, and 50 nM budding yeast Arp2/3 complex with 100 nM VCA-Bee1p). Actin polymerization was initiated by adding KMEI buffer (50 mM KCl, 2 mM MgCl2, 1 mM EGTA, 0.2 mM DTT, 0.1 mM ATP, 0.02% azide, and 10 mM imidazole, pH 7.0). Polymerization was followed using a fluorescence spectrophotometer (MOS-250 spectrofluorimeter; Bio-Logic) equipped with BioKine 32 software (BioLogic) using 365 nm as the excitation wavelength and 407 nm as the emission wavelength. When the reaction reached the plateau, 2 μM phalloidin was added, and the samples were applied to electron microscopy grids as described in the next section.
Electron microscopy and data collection
For the negatively stained samples, the preparations were applied to glow discharged electron microscopy carbon-coated grids and partially blotted, and a 10-nm gold colloid solution (Ted Pella, Inc.) was applied and blotted. The sample was stained with 1 or 2% uranyl acetate and air dried. Tilt series of negatively stained samples were acquired at room temperature and 4-μm defocus at 31,000× (pixel size of 0.75 nm) from approximately −70 to 70° every 3° on a microscope (Tecnai TF30; FEI) operated at 300 keV using a 2,048 × 2,048-pixel CCD camera (Multiscan 894; Gatan), the serialEM package (Mastronarde, 2005), and an advanced tomography holder (model 2020; Fischione Instruments) for data acquisition. For frozen-hydrated samples, protein solutions were applied to copper grids coated with holey carbon films (Quantifoil Micro Tools GmbH) and partially blotted, a 10-nm gold colloid solution was applied, and the sample was blotted again and plunged in liquid ethane. Tilt series of frozen-hydrated specimens were taken over holes and collected using a microscope (Tecnai F20; FEI) equipped with a 2,048 × 2,048-pixel slow scan CCD camera (TemCam-F224HD; Tietz Video and Image Processing Systems). Electron micrographs were recorded at a magnification of 26,500× (pixel size of 0.54 nm) from approximately −65 to 65° every 4° at 4-μm defocus using the TOM package (Nickell et al., 2005) for data collection. The microscope was operated at liquid nitrogen temperature. Before data collection, conditions for all samples were refined using a microscope (Tecnai T12; FEI) at 120 keV.
We recorded a total of 18 tomographic tilt series of negatively stained and 27 tilt series of frozen-hydrated A. castellanii Arp2/3-mediated actin networks. For both the negatively stained and frozen-hydrated samples, several tomographic tilt series were repeated after a 90° sample rotation (double-tilt tomograms) to provide the best possible initial template models for the alignment (with minimal artifacts caused by missing data). We collected 10 tilt series of negatively stained filaments with yeast Arp2/3 complex and five tilt series with bovine Arp2/3 complex. To minimize beam damage, all data, including the negatively stained samples, were collected under strict low-dose conditions with a cumulative dose per tomogram not exceeding 100–300 e−/Å2.
Tomographic reconstruction
Reconstructions were generated using IMOD (Kremer et al., 1996). A minimum of 10 fiducial markers (10-nm gold beads) per reconstruction were used. 54, 161, and 279 branches were selected and boxed out from the tomograms of specimens with negatively stained bovine, yeast, and A. castellanii Arp2/3 complex, respectively. Although the orientation of most branch junctions in these negative stain datasets are roughly perpendicular to the beam direction, different in-plane orientations as well as slight differences in the out-of-plane orientation increased data coverage in the averages substantially (Fig. S1 B). We selected 82 branches from the A. castellanii cryotomograms. For tomograms of both negatively stained and frozen-hydrated specimen, we restricted the averaging and alignment of branches to a small neighborhood because the filaments are so flexible that they can bend close to the branch junctions.
Single-particle volume analysis
For each of the three negatively stained samples, we selected a well-defined double-tilt branch as a reference volume. For the frozen-hydrated samples, we used the average of the reconstruction of negatively stained A. castellanii Arp2/3 complex branches as an initial reference. We aligned all branches to the respective references using a modified version of the density alignment algorithm in CoAn (Volkmann and Hanein, 1999, 2003). The modification explicitly accounts for the missing data caused by tomographic data collection during the alignment step. The aligned branches were then averaged using the averaging module of CoAn, which contains a weighting function that accounts for the data coverage of the individual contributors. We applied a spherical Butterworth filter to the individual volumes to deemphasize features distal from the junction. The same filter was used for all boxed-out volumes. The boxed-out branches from cryotomograms were denoised using three rounds of iterative median filtering (van der Heide et al., 2007) to improve alignment accuracy. The resulting alignment transforms were then applied to the original boxes that were then used for generating the average. For negatively stained samples, the 10% worst matching branches were excluded from the average; for frozen-hydrated samples, 25% were excluded. Then, all branches (including the ones removed from the average) were realigned to the average. For negatively stained samples, this procedure was repeated until there were no significant changes between subsequent averages (approximately five rounds). The refinement of the cryo data was restricted to two rounds because the resolution significantly worsened after the second round of refinement. To test for biasing toward the starting volume in the negatively stained samples, the complete process was repeated, choosing as the reference the (double tilt) branch that matched worst to the initial reference in the first round. The resulting averages were virtually identical to those using the initial references. For the data from frozen-hydrated samples, the alignment and averaging were repeated with the handedness of the reference inverted (by mirroring along the short axis). The resulting average was also virtually identical to the one with the initial reference.
We calculated resolution by dividing the final aligned volume sets into two random halves, averaging these halves separately, and calculating the 0.5 cut-off of the Fourier shell correlation between the two (Figs. S1 A and S2 C). To improve resolution, we repeated the processing of the negatively stained samples using spherical Butterworth filters with different cut-off radii. If the radius is chosen too large, the resolution deteriorates as a result of the flexibility of the actin filaments distal from the branch point. The largest cut-off radius that still gave the highest achievable resolution according to the Fourier shell correlation was used for the final reconstruction. A hierarchical descendant cluster analysis indicated that the dataset from negatively stained A. castellanii samples is homogeneous. However, we detected two subpopulations for negatively stained samples made with bovine and yeast Arp2/3 complex by image processing. These subpopulations correlate well with the two possible orientations (up or down) a branch can adopt on the sample support. The existence of these subpopulations could suggest that these structures were distorted by interaction with the substrate. As a consequence, we restricted the docking analysis to the samples made with A. castellanii Arp2/3 complex.
Docking and modeling
We docked atomic models of structural domains into the reconstructions using the CoAn software (Volkmann and Hanein, 1999, 2003). All analyses were run on the original reconstructions without sharpening. The algorithm used in CoAn is based on a global evaluation of the 6D fitting space followed by statistical analysis. The target function used in the analysis is the real-space density correlation coefficient between the experimental density and the density calculated from the search model. In contrast to other approaches that return only a single best fit based on a single scoring function value, the CoAn algorithm relies on the identification of solution sets, which are small regions in fitting space (confidence intervals) that are compatible with the data and its associated error level at a predefined confidence level (we used 99.5%; P < 0.005). This concept efficiently captures variations caused by random errors, low reconstruction quality, and insufficient local resolution. Differences in fit using alternative models can then be tested for significance using standard statistical tests with these solution sets. All proposed conformational changes were tested using this concept and were found to be statistically significant at the 99.5% confidence level.
To allow for cross-validation and estimation of precision of the fit and confidence intervals, we divided the data in two random halves and generated two averages that were treated as two independent experimental realizations of the underlying structure. We repeated all docking and modeling experiments described in the following paragraphs for both averages as well as for the apo-Arp2/3, ADP, and ATP crystal structures (Robinson et al., 2001; Nolen et al., 2004) to compile the statistics and precision estimates as described previously (Volkmann and Hanein, 2003). Thus, each modular fit is cross-validated by six different docking experiments. We used the 3D watershed transform (Volkmann, 2002) to generate three segments for each reconstruction: one corresponding to the mother filament, one corresponding to the daughter filament, and one corresponding to the Arp2/3 complex. First, we performed docking of the Arp2/3 complex portion. We docked the unmodified, inactive crystal structures into the corresponding segments. The orientation of the complex was very similar to that obtained independently using labeling information (Egile et al., 2005). Several areas showed severe mismatches between density and model (Figs. 2 A and Video 2), clearly implicating the need for conformational changes even in the cryoreconstruction with its limited resolution (Fig. S2).
The resulting docking precision, which was compiled from the six independent docking experiments, was ∼0.4 nm. The docking precision is related to the reproducibility of the fit within the error margin given by the data. This value is only a good estimate for the actual coordinate error (accuracy) if the model is near correct and no conformational changes occur that were not included in the modeling. Because the accuracy of the actin model used is limited (it was built using low resolution fiber diffraction and EM data), the docking precision value should be used as a lower limit of accuracy. An accuracy of 0.4–0.6 nm at an interface is high enough to make reliable predictions about residue patches being likely involved in interactions. Individual residues in the center regions of these patches are almost guaranteed to be close enough to interact; residues at the periphery are less certain. Because we have no knowledge about the side chain conformations at the protein surfaces, especially at the new interfaces, the data do not allow reliable predictions of specific residue–residue interactions.
Next, we modeled a filamentlike conformation of the Arps by superimposing individual subdomains of each Arp on an atomic model (Volkmann et al., 2005) of the corresponding filamentous actin subunit subdomain. Disordered portions of Arp2 were replaced by the corresponding portions of filamentous actin. We then arranged the modified Arps using the actin filament symmetry to provide a short-pitch template for filament growth. We inserted this short-pitch Arp2–Arp3 heterodimer into the complex by superimposing subdomains 1 of the modified and unmodified Arp3. We performed an energy minimization with REFMAC (Murshudov et al., 1997) to relieve minor clashes and strains. We then redocked this modified complex into the reconstructions, giving a significantly higher (P < 0.005) correlation with the density than the unmodified one and an improved visual fit (Figs. 2 and S2).
Then, we added four filamentous actin subunits to the modified Arp2/3 complex model using the actin filament symmetry. We docked this whole entity (corresponding to the modified Arp2/3 complex and the four subunits of the daughter filament) into the merged density segments corresponding to the Arp2/3 complex and the part of the daughter filament visible in the reconstruction. The root mean square deviation between the position of the Arp2/3 complex subunits except for Arp2 and Arp3 (which were modified) deduced from the unmodified crystal structure fitting and that for the branch model fitting are 0.45 nm, which is close to the cross-validated uncertainty of ∼0.4 nm for the individual model fits. This close correspondence indicates that the absence or presence of the daughter filament and the remodeling of Arp2 and Arp3 does not introduce significant bias into the positioning of the rest of the Arp2/3 complex. In the configuration with the four daughter filament subunits added to the remodeled Arp2 and Arp3, all members of the Arp2/3 complex and the daughter filament are within the high density region of the reconstructions, indicating that there is no need for further large-scale conformational changes. The precision estimate of the fit in this region was 0.35 nm.
A crystal structure for filamentous actin is not yet available. Current models were obtained by refining monomeric actin against x-ray fiber diffraction data (0.8-nm resolution; Holmes et al., 1990, 2003; Lorenz et al., 1993; Tirion et al., 1995) and by docking monomeric actin into electron microscopic reconstructions of actin filaments (∼2-nm resolution; Volkmann et al., 2005). We fitted an actin-filament model (Volkmann et al., 2005) with 12 subunits into the density segment corresponding to the mother filament. A symmetry mismatch was visually apparent, but global changes of the filament symmetry parameters did not lead to improvements, indicating the existence of a local effect. A division of the filament model in two halves (breaking it between M3 and M4) and redocking of these individual subfilaments leads to a statistically significant increase in density correlation (P < 0.005). The accompanying change in the mother filament suggests an untwisting close to the central density bridge with the Arp2/3 complex. Insertion of a monomeric actin in the presence of ATP (PDB accession no. 1ATN; Kabsch et al., 1990) instead of a filamentous actin at this location accommodates the change. Replacement of this subunit with other monomeric actin structures also accommodates the change. Compared with the filamentous conformation, monomeric actin is bent ∼15° between subdomains 1 and 2 and between subdomains 3 and 4 (Fig. S2 A). If the interactions of subdomains 3 and 4 of subunit M4 with its long-pitch neighbors M6 and M2 are maintained, this monomeric conformation of subunit M4 reproduces the observed symmetry mismatch within the estimated precision of the docking (∼0.3 nm). We further modified the monomeric actin subunit (M4) and its barbed end neighbor (M2) to move the protruding subdomains 2 (Fig. 3 E) into the density. We performed a global search subject to simultaneously maximize the correlation coefficient with the density, maintaining main-chain connectivity in the actin subunits and minimizing steric clashes. The resulting models were subjected to energy minimization to relieve minor strains and clashes. The precision estimate for the fit in the mother filament region is ∼0.4 nm.
The Fourier shell correlation used to estimate the resolution of the reconstructions technically only measures the correlation between two noisy volumes in Fourier space. The resulting value may not correspond to the actual resolution, as it would occur in x-ray crystallography, where the Fourier space signal caused by the order of the underlying structure can easily be identified (diffraction spots). As a consequence, structural interpretations relying on resolution estimates may lead to overinterpretation, especially if Fourier space sharpening is used to boost high resolution features. In this study, no sharpening was used except in Fig. 2 A for visualization purposes. Furthermore, in the CoAn algorithm, Fourier amplitude effects such as those caused by the contrast transfer function or the resolution are explicitly accounted for by decorrecting the Fourier amplitudes of the search model density to (on average) match those of the experimental density. To further guard against resolution-related overinterpretation, we repeated the complete analysis after the reconstructions were truncated to a resolution of 4 nm. The need for changes in the conformation of Arp2 and Arp3 as well as in subdomain 2 of M2 and M4 are still apparent by visual inspection and are significant at a 99.5% confidence level. The untwisting of the mother filament is still highly significant, with a maximum confidence level of 99.2%. The cross-validated precision estimate drops to 0.59 nm, indicating that the higher resolution terms in the nontruncated reconstruction provide significant signal.
Online supplemental material
Fig. S1 shows quality criteria for the negative stain and ice averaged reconstructions. Fig. S2 contains information about remodeling of the mother filament and ARPC1. Fig. S3 contains information about remodeling of the Arp2/3 complex. Video1 shows a180° rotation of the atomic model of the branch junction inside the reconstruction density, and Video 2 shows an illustration of the conformational changes. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200709092/DC1.
Supplemental Material
Acknowledgments
We thank Robert C. Liddington, Alan R. Horwitz, Brad Nolen, and Chris Beltzner for valuable comments on the manuscript. We thank Wolfgang Baumeister for use of his microscopes and Ohad Medalia for help in refining the conditions for image acquisition in Munich.
This work was supported by National Institutes of Health grants to R. Li, T.D. Pollard, N. Volkmann, and D. Hanein. The National Institute of General Medical Sciences Cell Migration Consortium supported feasibility tests for cryotomography (grant to D. Hanein) and aspects of the volume processing (grant to N. Volkmann).
I. Rouiller's present address is Department of Anatomy and Cell Biology, McGill University, Montreal, Quebec H3A 2B2, Canada.
C. Egile's present address is Experimental Therapeutics and Translational Research Oncology, 94403 Vitry Sur Seine, France.
D. Nicastro's present address is Brandeis University, Rosenstiel Center, Waltham, MA 02454.
Abbreviation used in this paper: Arp, actin-related protein.
References
- Abraham, V.C., V. Krishnamurthi, D.L. Taylor, and F. Lanni. 1999. The actin-based nanomachine at the leading edge of migrating cells. Biophys. J. 77:1721–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguda, A.H., L.D. Burtnick, and R.C. Robinson. 2005. The state of the filament. EMBO Rep. 6:220–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltzner, C.C., and T.D. Pollard. 2004. Identification of functionally important residues of Arp2/3 complex by analysis of homology models from diverse species. J. Mol. Biol. 336:551–565. [DOI] [PubMed] [Google Scholar]
- Beltzner, C.C., and T.D. Pollard. 2008. Pathway of actin filament branch formation by Arp2/3 complex. J. Biol. Chem. In press. [DOI] [PubMed]
- Blanchoin, L., K.J. Amann, H.N. Higgs, J.B. Marchand, D.A. Kaiser, and T.D. Pollard. 2000. Direct observation of dendritic actin filament networks nucleated by Arp2/3 complex and WASP/Scar proteins. Nature. 404:1007–1011. [DOI] [PubMed] [Google Scholar]
- Blanchoin, L., T.D. Pollard, and S.E. Hitchcock-DeGregori. 2001. Inhibition of the Arp2/3 complex-nucleated actin polymerization and branch formation by tropomyosin. Curr. Biol. 11:1300–1304. [DOI] [PubMed] [Google Scholar]
- Chik, J.K., U. Lindberg, and C.E. Schutt. 1996. The structure of an open state of beta-actin at 2.65 A resolution. J. Mol. Biol. 263:607–623. [DOI] [PubMed] [Google Scholar]
- DesMarais, V., I. Ichetovkin, J. Condeelis, and S.E. Hitchcock-DeGregori. 2002. Spatial regulation of actin dynamics: a tropomyosin-free, actin-rich compartment at the leading edge. J. Cell Sci. 115:4649–4660. [DOI] [PubMed] [Google Scholar]
- Egile, C., I. Rouiller, X.P. Xu, N. Volkmann, R. Li, and D. Hanein. 2005. Mechanism of filament nucleation and branch stability revealed by the structure of the Arp2/3 complex at actin branch junctions. PLoS Biol. 3:e383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gournier, H., E.D. Goley, H. Niederstrasser, T. Trinh, and M.D. Welch. 2001. Reconstitution of human Arp2/3 complex reveals critical roles of individual subunits in complex structure and activity. Mol. Cell. 8:1041–1052. [DOI] [PubMed] [Google Scholar]
- Gupton, S.L., K.L. Anderson, T.P. Kole, R.S. Fischer, A. Ponti, S.E. Hitchcock-DeGregori, G. Danuser, V.M. Fowler, D. Wirtz, D. Hanein, and C.M. Waterman-Storer. 2005. Cell migration without a lamellipodium: translation of actin dynamics into cell movement mediated by tropomyosin. J. Cell Biol. 168:619–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes, K.C., D. Popp, W. Gebhard, and W. Kabsch. 1990. Atomic model of the actin filament. Nature. 347:44–49. [DOI] [PubMed] [Google Scholar]
- Holmes, K.C., I. Angert, F.J. Kull, W. Jahn, and R.R. Schroder. 2003. Electron cryo-microscopy shows how strong binding of myosin to actin releases nucleotide. Nature. 425:423–427. [DOI] [PubMed] [Google Scholar]
- Kabsch, W., H.G. Mannherz, D. Suck, E.F. Pai, and K.C. Holmes. 1990. Atomic structure of the actin:DNase I complex. Nature. 347:37–44. [DOI] [PubMed] [Google Scholar]
- Kelly, A.E., H. Kranitz, V. Dotsch, and R.D. Mullins. 2006. Actin binding to the central domain of WASP/Scar proteins plays a critical role in the activation of the Arp2/3 complex. J. Biol. Chem. 281:10589–10597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klenchin, V.A., S.Y. Khaitlina, and I. Rayment. 2006. Crystal structure of polymerization-competent actin. J. Mol. Biol. 362:140–150. [DOI] [PubMed] [Google Scholar]
- Kremer, J.R., D.N. Mastronarde, and J.R. McIntosh. 1996. Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol. 116:71–76. [DOI] [PubMed] [Google Scholar]
- Lorenz, M., D. Popp, and K.C. Holmes. 1993. Refinement of the F-actin model against X-ray fiber diffraction data by the use of a directed mutation algorithm. J. Mol. Biol. 234:826–836. [DOI] [PubMed] [Google Scholar]
- Mahaffy, R.E., and T.D. Pollard. 2006. Kinetics of the formation and dissociation of actin filament branches mediated by arp2/3 complex. Biophys. J. 91:3519–3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronarde, D.N. 2005. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152:36–51. [DOI] [PubMed] [Google Scholar]
- Moseley, J.B., and B.L. Goode. 2006. The yeast actin cytoskeleton: from cellular function to biochemical mechanism. Microbiol. Mol. Biol. Rev. 70:605–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins, R.D., J.A. Heuser, and T.D. Pollard. 1998. The interaction of Arp2/3 complex with actin: nucleation, high affinity pointed end capping, and formation of branching networks of filaments. Proc. Natl. Acad. Sci. USA. 95:6181–6186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov, G.N., A.A. Vagin, and E.J. Dodson. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53:240–255. [DOI] [PubMed] [Google Scholar]
- Nickell, S., F. Forster, A. Linaroudis, W.D. Net, F. Beck, R. Hegerl, W. Baumeister, and J.M. Plitzko. 2005. TOM software toolbox: acquisition and analysis for electron tomography. J. Struct. Biol. 149:227–234. [DOI] [PubMed] [Google Scholar]
- Nolen, B.J., and T.D. Pollard. 2007. Insights into the influence of nucleotides on actin family proteins from seven structures of Arp2/3 complex. Mol. Cell. 26:449–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolen, B.J., R.S. Littlefield, and T.D. Pollard. 2004. Crystal structures of actin-related protein 2/3 complex with bound ATP or ADP. Proc. Natl. Acad. Sci. USA. 101:15627–15632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otterbein, L.R., P. Graceffa, and R. Dominguez. 2001. The crystal structure of uncomplexed actin in the ADP state. Science. 293:708–711. [DOI] [PubMed] [Google Scholar]
- Pan, F., C. Egile, T. Lipkin, and R. Li. 2004. ARPC1/Arc40 mediates the interaction of the actin-related protein 2 and 3 complex with Wiskott-Aldrich syndrome protein family activators. J. Biol. Chem. 279:54629–54636. [DOI] [PubMed] [Google Scholar]
- Pantaloni, D., R. Boujemaa, D. Didry, P. Gounon, and M.F. Carlier. 2000. The Arp2/3 complex branches filament barbed ends: functional antagonism with capping proteins. Nat. Cell Biol. 2:385–391. [DOI] [PubMed] [Google Scholar]
- Pirani, A., M.V. Vinogradova, P.M. Curmi, W.A. King, R.J. Fletterick, R. Craig, L.S. Tobacman, C. Xu, V. Hatch, and W. Lehman. 2006. An atomic model of the thin filament in the relaxed and Ca2+-activated states. J. Mol. Biol. 357:707–717. [DOI] [PubMed] [Google Scholar]
- Robinson, R.C., K. Turbedsky, D.A. Kaiser, J.B. Marchand, H.N. Higgs, S. Choe, and T.D. Pollard. 2001. Crystal structure of Arp2/3 complex. Science. 294:1679–1684. [DOI] [PubMed] [Google Scholar]
- Tirion, M.M., D. ben-Avraham, M. Lorenz, and K.C. Holmes. 1995. Normal modes as refinement parameters for the F-actin model. Biophys. J. 68:5–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Heide, P., X.P. Xu, B.J. Marsh, D. Hanein, and N. Volkmann. 2007. Efficient automatic noise reduction of electron tomographic reconstructions based on iterative median filtering. J. Struct. Biol. 158:196–204. [DOI] [PubMed] [Google Scholar]
- Volkmann, N. 2002. A novel three-dimensional variant of the watershed transform for segmentation of electron density maps. J. Struct. Biol. 138:123–129. [DOI] [PubMed] [Google Scholar]
- Volkmann, N., and D. Hanein. 1999. Quantitative fitting of atomic models into observed densities derived by electron microscopy. J. Struct. Biol. 125:176–184. [DOI] [PubMed] [Google Scholar]
- Volkmann, N., and D. Hanein. 2003. Docking of atomic models into reconstructions from electron microscopy. Methods Enzymol. 374:204–225. [DOI] [PubMed] [Google Scholar]
- Volkmann, N., K.J. Amann, S. Stoilova-McPhie, C. Egile, D.C. Winter, L. Hazelwood, J.E. Heuser, R. Li, T.D. Pollard, and D. Hanein. 2001. Structure of Arp2/3 complex in its activated state and in actin filament branch junctions. Science. 293:2456–2459. [DOI] [PubMed] [Google Scholar]
- Volkmann, N., H. Liu, L. Hazelwood, E.B. Krementsova, S. Lowey, K.M. Trybus, and D. Hanein. 2005. The structural basis of myosin V processive movement as revealed by electron cryomicroscopy. Mol. Cell. 19:595–605. [DOI] [PubMed] [Google Scholar]
- Welch, M.D., and R.D. Mullins. 2002. Cellular control of actin nucleation. Annu. Rev. Cell Dev. Biol. 18:247–288. [DOI] [PubMed] [Google Scholar]
- Winter, D., T. Lechler, and R. Li. 1999. Activation of the yeast Arp2/3 complex by Bee1p, a WASP-family protein. Curr. Biol. 9:501–504. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}