

Abstract
Mutations in the extracellular signal-regulated kinase (ERK) pathway, particularly in the mitogen-activated protein kinase/ERK kinase (MEK) activator B-Raf, are associated with human tumorigenesis and genetic disorders. Hence, B-Raf is a prime target for molecule-based therapies, and understanding its essential biological functions is crucial for their success. B-Raf is expressed preferentially in cells of neuronal origin. Here, we show that in mice, conditional ablation of B-Raf in neuronal precursors leads to severe dysmyelination, defective oligodendrocyte differentiation, and reduced ERK activation in brain. Both B-Raf ablation and chemical inhibition of MEK impair oligodendrocyte differentiation in vitro. In glial cell cultures, we find B-Raf in a complex with MEK, Raf-1, and kinase suppressor of Ras. In B-Raf–deficient cells, more Raf-1 is recruited to MEK, yet MEK/ERK phosphorylation is impaired. These data define B-Raf as the rate-limiting MEK/ERK activator in oligodendrocyte differentiation and myelination and have implications for the design and use of Raf inhibitors.
Introduction
The Raf kinases (A-Raf, B-Raf, and Raf-1) relay extracellular signals to the MAPK/extracellular signal-regulated kinase (ERK) kinase (MEK)/ERK signaling module. Among the three Raf kinases, B-Raf binds best to MEK and has the highest basal MEK kinase activity. Growth factor–stimulated ERK activation is reduced (∼60%) in cells lacking B-Raf but not in A-Raf– or Raf-1–deficient cells (Wojnowski et al., 2000; Huser et al., 2001; Mikula et al., 2001; Mercer et al., 2002; Pritchard et al., 2004). Finally, Raf kinases from lower organisms (lin-45 in Caenorhabditis elegans and D-Raf in Drosophila melanogaster) are more similar to B-Raf than to A-Raf or Raf-1 (Wellbrock et al., 2004). All these observations hint at B-Raf as the essential mammalian MEK kinase, whereas A-Raf and Raf-1 may have a subordinate role or have diverged to perform other functions.
B-Raf is mutated in a high percentage of certain human cancers (Davies et al., 2002) and congenital progressing conditions like cardio-facio-cutaneous syndrome (Rodriguez-Viciana et al., 2006). This discovery has generated huge industrial interests and inhibitors are currently being tested in preclinical and clinical trials. However, our fundamental understanding of B-Raf biology is still scanty, and embryonic lethality associated with germline B-Raf ablation (Wojnowski et al., 1997; Galabova-Kovacs et al., 2006) has precluded its investigation. B-Raf is highly expressed in cells of neuronal origin (Wojnowski et al., 2000) and reportedly plays a prosurvival role in cultured primary embryonic neurons (Wiese et al., 2001). Therefore, the analysis of animals in which B-Raf is selectively ablated in neuronal cells is of particularly interest. Indeed, conditional ablation in forebrain excitatory neurons has shown an essential role for B-Raf in hippocampus-dependent learning (Chen et al., 2006). The more generalized ablation of b-raf in neuronal precursors leads to growth retardation, disorders of hypothalamic-pituitary function, and premature death. b-raf ablation does not interfere with neurogenesis or neuron survival, and the only molecular defect investigated to date is the reduced expression of the glial cell line–derived neurotrophic factor receptor Ret in dorsal root ganglion neurons at postnatal stages, a rather mild phenotype. Concomitant elimination of b-raf and c-raf-1 strongly reduced axon growth in vitro and cutaneous axon terminal arborization in vivo, which suggests that Raf-1 can compensate for the loss of B-Raf function in this sytem (Zhong et al., 2007).
We show that mice with epiblast-restricted (Mox2cre/+;b-raf f/f) and neuronal precursor–restricted b-raf ablation (b-rafΔ/Δneu) develop progressive loss of coordination, tremors, and severe muscular weakness and die around postnatal day 21 (P21). In line with this phenotype, we detect a major defect in central nervous system (CNS) myelination and reduced oligodendrocyte differentiation. B-Raf knockout (KO) oligodendrocytes in situ and in culture fail to activate MEK/ERK and to differentiate, despite the increased recruitment of Raf-1 to MEK in KO cells. Blunting ERK activation in wild-type (WT) oligodendrocytes with a MEK inhibitor similarly impairs differentiation. Together, the data indicate an essential role of B-Raf and its downstream effector ERK in oligodendrocyte differentiation and myelination.
Results
Ablation of B-Raf in the epiblast and in neuronal precursors causes severe neuromuscular defects
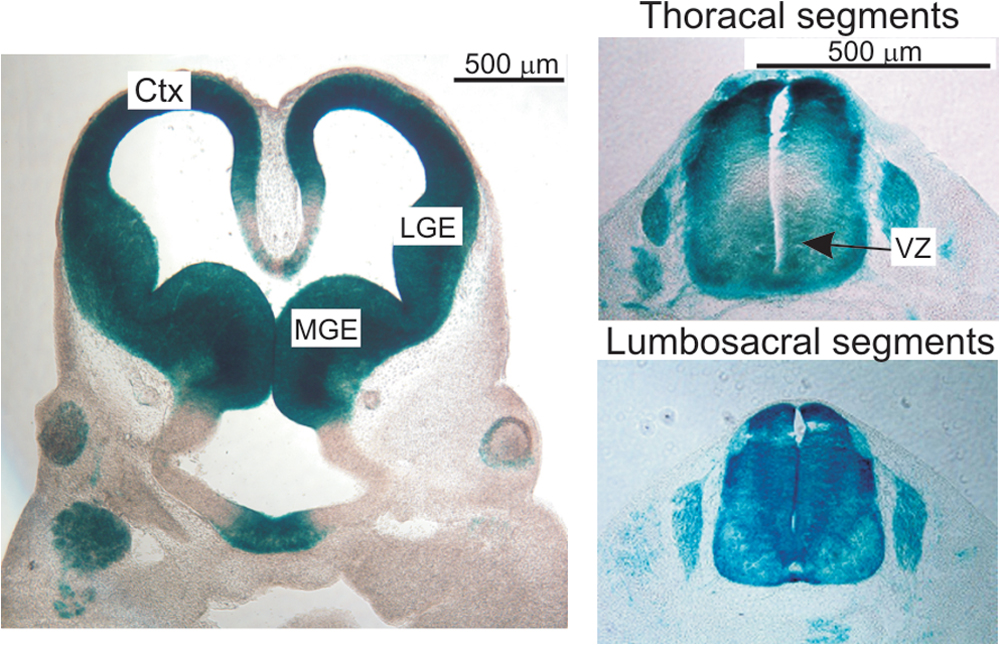
b-raf was inactivated by Cre-loxP–mediated deletion of exon 11, which encodes the kinase domain (Chen et al., 2006). Deletion leads to a shift in the open reading frame and completely abrogates B-Raf protein expression (Fig. 1, C–D). Epiblast-restricted ablation in the 129/Sv background yielded live offspring. Mox2cre/+;b-raf f/f were indistinguishable from littermate controls at birth but showed growth retardation starting around P10. This phenotype was followed by loss of coordination, the onset of tremors, ataxia, and muscle weakness (at P15). P10–21, B-Raf–deficient animals suspended by the tail clasped their limbs to their trunks in a dystonic fashion, a diagnostic sign of neurological impairment (Fig. 1 A). After P18, the Mox2cre/+;b-raf f/f mice deteriorated rapidly, showing increasing difficulties in ambulating and finally in breathing (Videos 1–3, available at http://www.jcb.org/cgi/content/full/jcb.200709069/DC1). Spleen size was markedly decreased (unpublished data), likely because of the previously reported essential role of B-Raf in B cell development (Brummer et al., 2002). With the exception of the latter, all phenotypes were phenocopied in NestinCre;b-raf f/f (b-raf Δ/Δneu) animals, which express the Cre recombinase in CNS neural precursor cells that give rise to both neurons and glia (Tronche et al., 1999; Haigh et al., 2003). NestinCre mediated efficient recombination in brain and spinal cord as early as embryonic day 11.5 (Fig. S1). Complete conversion of the b-raf f to the b-raf Δ allele was evident in brain and spinal cord (not depicted) but not in other tissues derived from b-raf Δ/Δneu mice (Fig. 1 B). Accordingly, B-Raf could not be detected by immunoblotting in b-raf Δ/Δneu brain (Fig. 1, C and D), spinal cord (Fig. 1 D), and glial cell cultures derived from P0 animals (see Fig. 6, A–C). In B-Raf–deficient brains, A-Raf expression was unchanged, whereas Raf-1 was slightly up-regulated (Fig. 1 D). Thus, the pathology (growth retardation, muscle weakness, tremors, and ataxia) observed in Mox2cre/+;b-raf f/f was caused by a defect of neural precursor cells. Histological examination revealed severe atrophy of skeletal muscle fibers (Fig. S2 C) but axon retraction/degeneration was not detected, and both the morphology and innervation of the neuromuscular junctions were normal in the b-raf Δ/Δneu mice (Fig. S2 D).
Figure 1.

Neurological defects and growth retardation in B-Raf–deficient mice. (A) Limb clasping reflex in P18 Mox2cre/+;b-raf f/f and b-raf Δ/Δneu mice suspended by the tail. (B) Complete conversion of the b-raf f/f to b-raf Δ/Δ alleles in brain but not in other tissues of P18 b-raf Δ/Δneu mice. PCR analysis of: T, tail; B, brain; Liv, liver; L, lung; Sp, spleen; Th, thymus; H, heart; and K, kidney. N, negative control (H2O); f/− and +/+, positive controls. (C) Immunoblots of brain lysates from P18 b-raf f/f (WT) and b-raf Δ/Δneu (KO) mice probed with antibodies against N- or C-terminal B-Raf epitopes demonstrate the complete absence of B-Raf protein. The position of the molecular weight markers is shown between the autoradiograms. The arrow indicates B-Raf. Actin immunoblot, loading control. (D) Immunoblot of brain and spinal cord lysates from P18 b-raf f/f (WT) and b-raf Δ/Δneu (KO) mice. MEK2 immunoblot, loading control.
Figure 6.

B-Raf is required for MEK/ERK phosphorylation and ERK activation is required for differentiation in oligodendrocyte-enriched glial cell cultures. Immunoblot analysis of whole cell lysates (40 μg) from WT and B-Raf KO oligodendrocyte-enriched (A and C) or oligodendrocyte-depleted mixed glial cell cultures (B) left untreated or stimulated for 10 min with 100 ng/ml PDGF (A and B; 10 min), 20 ng/ml EGF (C), or 20 ng/ml FGF (C) before lysis. MEK/ERK phosphorylation is impaired in oligodendrocyte-containing but not oligodendrocyte-depleted cultures. In B, the first lane from the left is slightly underloaded. (D) ERK phosphorylation in oligodendrocyte cultures. Cells allowed to differentiate for 2 d and either left untreated or stimulated with 20 ng/ml FGF for the indicate times. Cells were colabeled with antibodies against pERK (green) and pan-tubulin (red). (E) MEK1 immunoprecipitates were prepared from 400 μg WT and B-Raf null (KO) oligodendrocyte-enriched glial cell cultures, and left untreated or stimulated with 100 ng/ml PDGF (for 10 min) before lysis and immunoblotting. C, isotype-matched irrelevant Ab control; MEK1 KO, MEK1 ips prepared from whole cell lysate of MEK1 KO MEFs. (F) ERK phosphorylation in cells of the oligodendrocyte lineage in situ. Brain sections were colabeled with antibodies against pERK (green) and βIV-tubulin (red). Cells were classified on the basis of their morphology as premyelinating oligodendrocytes (left and middle) and mature oligodendrocytes (right). Asterisks indicate autofluorescent erythrocytes. Bars, 30 μm. (G) A working model Raf/MEK/ERK complexes in differentiating oligodendrocytes. Soluble or axonal signals activate the ERK pathway in oligodendrocyte precursors. In WT cells, three complexes may be formed, two of which are B-Raf dependent: complex I, which comprises B-Raf, KSR, and MEK/ERK; and complex II, which consists of a B-Raf–Raf-1 heterodimer bound to KSR and MEK/ERK. In complex I, MEK is phosphorylated by B-Raf, whereas in complex II, the Raf heterodimer is the MEK kinase. Both complexes produce a strong, sustained ERK signal ultimately leading to oligodendrocyte differentiation. Complex III comprises Raf-1 and MEK/ERK, is B-Raf independent, and is the only complex found in B-Raf–deficient oligodendrocytes. This complex gives rise to a much weaker ERK signal, leading to defective oligodendrocyte differentiation.
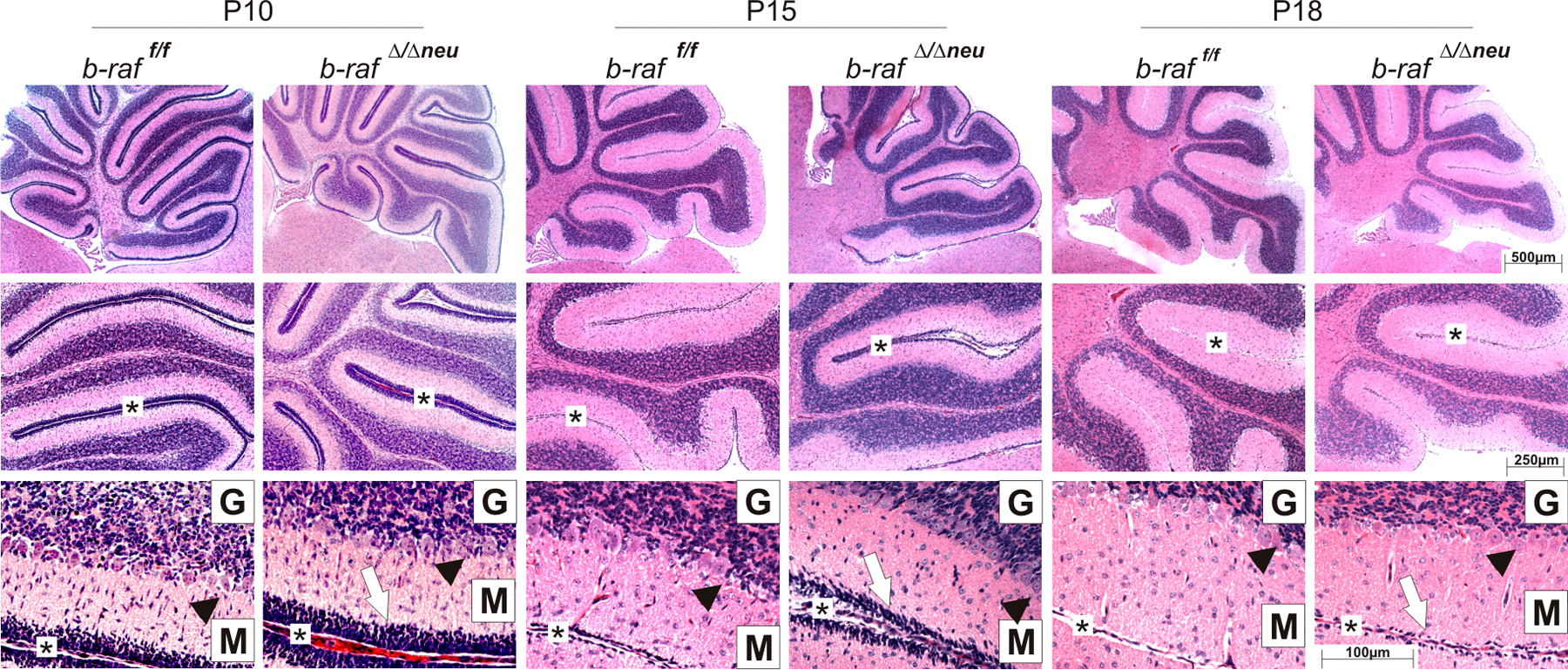
Macroscopically and histologically, the brains of Mox2cre/+;b-raf f/f and b-raf Δ/Δneu did not show gross anomalies. P18 B-Raf–deficient brains were slightly smaller than those of WT littermates and the molecular layer of their brain cortex was thinner (Fig. S2, A and B; Zhong et al., 2007). In the cerebellum, B-Raf ablation caused a slight accumulation of granular cells at the cerebellar surface, which could be caused by a delay in postnatal migration or a mild increase in precursor proliferation (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200709069/DC1). These defects were observed with a 100% penetrance.
Ablation of B-Raf in neuronal precursors causes CNS dysmyelination
The mild anomaly in cerebellar development may contribute to the ataxic behavior of the b-raf Δ/Δneu mice (Grusser-Cornehls and Baurle, 2001). Overall, however, the neurological symptoms of the b-raf Δ/Δneu mice and their onset on P14 are reminiscent of those observed in myelin mutants occurring naturally, like shiverer or jimpy (Sturrock, 1980; Popko et al., 1987; Nave, 1994), or generated by gene targeting (Olig1 null mice; Xin et al., 2005), which suggests a defect in myelination. Indeed, at P18, significantly fewer myelinated fibers could be observed in different areas of P18 b-raf Δ/Δneu brains (Fig. 2, A–C; Fig. S4 A; and Fig. S5, A and B, available at http://www.jcb.org/cgi/content/full/jcb.200709069/DC1), and immunoblotting confirmed a reduction in the total amount of myelin basic protein (MBP; Figs. 2 D and S4, B and C). In the spinal cord and peripheral nerves, MBP staining was very strong and the differences between WT and b-raf Δ/Δneu were not as clear (unpublished data). Immunohistochemical analysis and quantitative immunoblotting of P0–18 brains showed that myelination was reduced significantly in b-raf Δ/Δneu animals throughout postnatal development (Fig. S4 D). The decrease in MBP expression was also evident at the mRNA level (Fig. S4 E). In contrast to MBP, the expression of other major myelin components (MAG, MOG, and PLP/DM20) was unchanged. Transmission electron microscopy of selected CNS areas showed that B-Raf ablation did not prevent axon recognition and ensheathment; however, the number of unmyelinated fibers in the optic nerve, corpus callosum, and, less obviously, in the spinal cord (Fig. 3 and not depicted) of b-raf Δ/Δneu mice was clearly increased. In addition, the g ratio (the numerical ratio between the diameter of the axon proper and the outer diameter of the myelinated fiber) was higher than in WT CNS. Signs of gliosis were observed in the optic nerve but we did not observe axonal swelling or degeneration, fragmented organelles, myelin debris, or macrophage infiltration. In addition, the total number of axons in WT and b-raf Δ/Δneu optic nerves did not differ. All these data are consistent with a delay in CNS myelination (dysmyelination) rather than with demyelination.
Figure 2.

b-raf ablation in neuronal precursors leads to hypomyelination of the brain. (A–C) Hypomyelination of different regions of the P18 b-rafΔ/Δneu brain revealed by immunohistochemistry (IHC) using an α-MBP antibody on paraffin sections. Brown staining indicates MBP+ fibers and cells. The sections were counterstained with hematoxylin. (A) Brain cortex. (B) Detail of the cortex. (C) Hippocampus and the dentate gyrus (arrows). (D) MBP expression determined by immunoblotting of brain lysates. The reduced MBP expression in the b-rafΔ/Δneu lysates correlates with the results obtained in the IHC.
Figure 3.

Hypomyelination in the CNS of the of b-rafΔ/Δneu mice. Electron micrographs of P18 optic nerves (A) and spinal cords (B) in cross section. Note the high proportion of unmyelinated axons (plotted on the right as mean ± SD for the optic nerve; *, P < 0.05 comparing b-raf f/f and b-raf Δ/Δneu mice) and the thinner myelin sheaths in b-raf Δ/Δneu sections. The scatter plots show g ratios (diameter of the axon proper:outer diameter of the myelinated fiber) as a function of axon diameter for optic nerve and spinal cord (100 axons/mouse, three b-raf f/f, and three b-raf Δ/Δneu mice). White symbols indicate WT and gray symbols indicate b-raf Δ/Δneu mice. g ratios are higher in the b-raf Δ/Δneu mice (P < 0.0005), which indicates hypomyelination. Bars, 2.5 μm.
Oligodendrocytes are the myelin-forming macroglia of the vertebrate CNS. They originate mostly from ventral but also dorsal sources in the spinal cord and forebrain (Richardson et al., 2006). Oligodendrocyte precursors with proliferative capacity express the NG2 proteoglycan and PDGF receptor α (PDGFRα) and migrate from their sites of origin to their final position in the white matter, where terminal differentiation starts at the end of embryogenesis. Terminal differentiation is characterized by loss of NG2 and PDGFRα, the up-regulation of the cytoskeletal marker βIV-tubulin (expressed by premyelinating, early myelinating, and myelinating oligodendrocytes; Terada et al., 2005), and the induction of myelin genes like MBP. At P18, B-Raf–deficient brains contained more undifferentiated PDGFRα+ oligodendrocytes than WT brains in all regions investigated (Fig. 4 A and Fig. S5, C and D). In parallel, the amount of βIV-tubulin+ cells was reduced. Together with the reduced expression of MBP and the dysmyelination observed in B-Raf KO CNS, this suggested that B-Raf ablation caused a delay in oligodendrocyte differentiation. To confirm this, we established oligodendrocyte precursor cultures (Zezula et al., 2001) from WT and B-Raf KO brains. The initial purity (88 ± 12% NG2+ cells) and the total number of cells present in these cultures were comparable. After differentiation, however, B-Raf KO cultures contained significantly higher numbers of NG2+ prooligodendrocytes, whereas the amount of premyelinating oligodendrocytes (NG2+/O4+ with radial processes and limited arborization, and O4+ cells with a complex process network) was correspondingly decreased (Fig. 4 B). MBP+ cells were hardly present in B-Raf–deficient cultures and the few detected expressed much less MBP than WT cells (Fig. 4 C). WT oligodendrocyte precursor cultures differentiating in the presence of the MEK inhibitor U0126 also failed to produce MBP+ cells (Fig. 4 C). This shows a causal link between ERK phosphorylation and oligodendrocyte differentiation and suggests that B-Raf is required for efficient ERK phosphorylation in this system.
Figure 4.

Oligodendrocytes maturation is defective in b-raf Δ/Δneu mice. Immature, PDGFRα+ oligodendrocytes, stained in brown, are present in several areas of the P18 b-raf Δ/Δneu but not b-raf f/f brain. Subjects were counterstained with hematoxylin. (A) Brain cortex (inset and bottom) and the corresponding quantification of positive cells/brain area. The plot shows means ± SD; *, P < 0.02; **, P < 0.005; and ***, P < 0.0005 comparing three b-raf f/f and three b-raf Δ/Δneu mice. A quantification of the βIV-tubulin+ cells (premyelinating, early myelinating, and myelinating oligodendrocytes) from the same areas is also shown. (B and C) b-raf Δ/Δneu oligodendrocytes fail to differentiate in vitro. Oligodendrocyte precursors from P0 b-raf f/f and b-raf Δ/Δneu pups were allowed to differentiate for 5 (B) or 6 d (C). Differentiation was analyzed by immunofluorescence staining with α-NG2, α-O4, and α-MBP antibodies. Examples of single+ and double+ cells from a WT culture are shown below in B. In C, cell morphology was visualized by staining with an α-tubulin antibody. The percentage of NG2+ cells (oligodendrocyte progenitors), O4+ cells (pro-oligodendrocytes), and MBP+ cells (terminally differentiated oligodendrocytes) present in the b-raf f/f and b-raf Δ/Δneu cultures were compared. Each culture consisted of a pool of two WT or KO mice; for the chemical inhibition of MEK, oligodendrocyte cultures were established from pools of four WT mice and kept for 6 d in differentiation medium in the absence (untreated [UT]) or presence of MEK inhibitor (U0126; 10 μM). In all cases, a minimum of 100 cells/culture were counted independently by two investigators. The experiment was repeated three times, the plots show the mean ± SD of the results obtained by assessing three separate experiments (*, P < 0.05 for b-raf f/f vs. b-raf Δ/Δneu). Bars, 30 μm.
Molecular correlates of B-Raf ablation in the brain
We next determined the expression of the Raf kinases and the activation status of their downstream targets MEK/ERK during postnatal brain development (Fig. 5 A). A-Raf and MEK2 were stably expressed in both WT and B-Raf–deficient brains. In contrast, B-Raf and MEK1 were expressed at low levels in newborn brains and increased steadily thereafter. Raf-1 showed an opposite pattern, being expressed at higher levels in P0 and P5 animals and decreasing in WT animals older than 10 d. Raf-1 expression was slightly up-regulated in P10–18 B-Raf–deficient brains. ERK phosphorylation paralleled B-Raf/MEK1 expression, being very low in WT P0–5 brains and rising steadily from P10 on. B-Raf ablation strongly decreased ERK phosphorylation between P10 and 18. In contrast, the kinetics and extent of phosphorylation of p38 and JNK were similar in WT and KO brains (unpublished data). Lack of ERK phosphorylation correlated with a dramatic reduction in the expression of EGR1, a downstream target of the ERK cascade (Harada et al., 2001) expressed in oligodendrocyte progenitors and induced upon differentiation (Fig. 5 A; Sock et al., 1997). In addition, the B-Raf–dependent rise in ERK phosphorylation coincided with an increase in βIV-tubulin expression, which is indicative of oligodendrocyte maturation (Terada et al., 2005). Consistent with a defect in oligodendrocyte maturation, less βIV-tubulin was detected in B-Raf KO lysates (Fig. 5 A). Thus, increased B-Raf expression and B-Raf–dependent ERK phosphorylation correlated temporally with the developmental window during which myelination of the upper CNS takes place (Foran and Peterson, 1992) and with the onset of the neurological symptoms in the b-raf Δ/Δneu mice.
Figure 5.

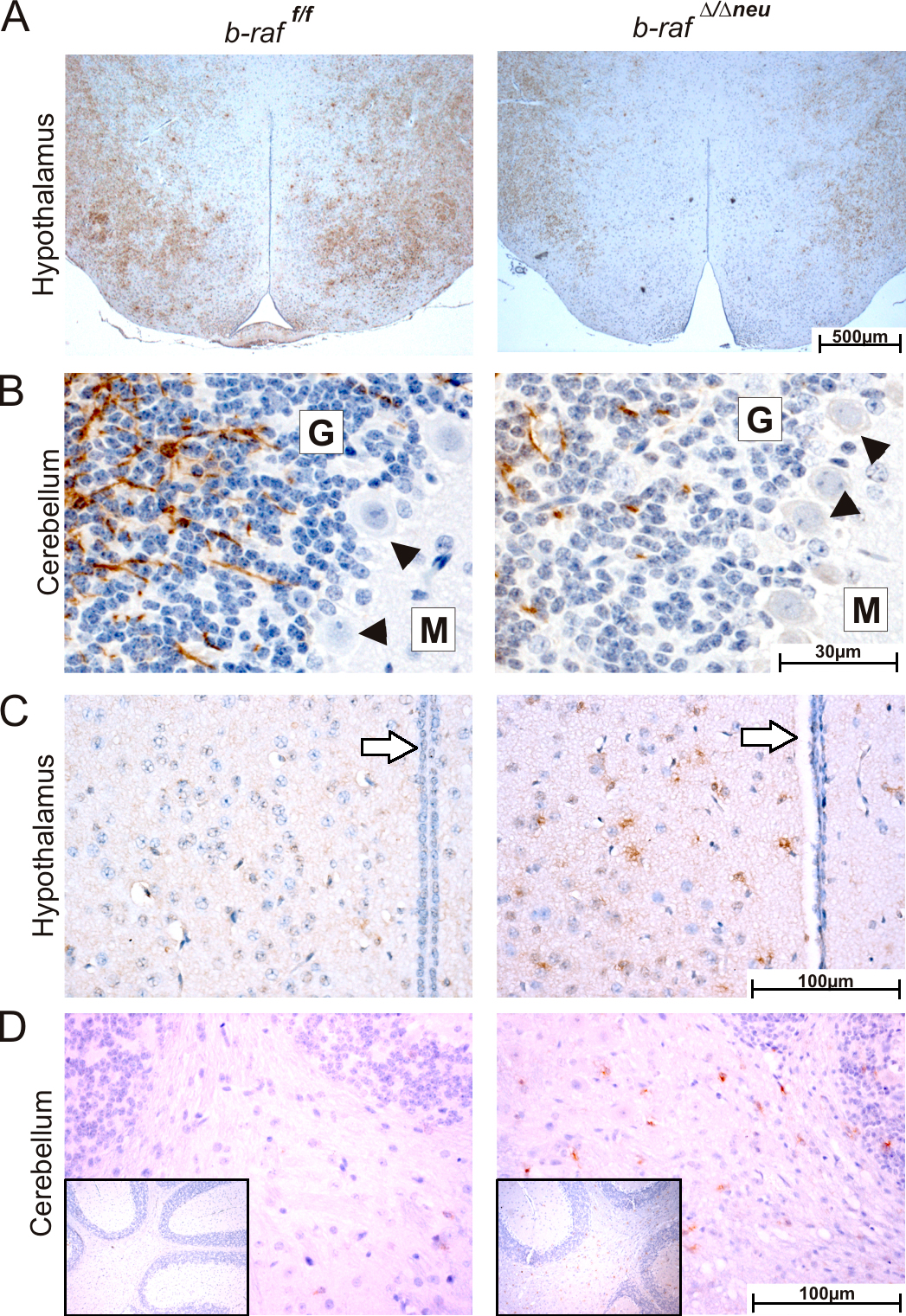
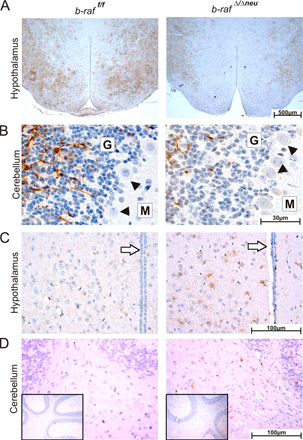
B-Raf is required for ERK activation during postnatal brain development. (A) Expression/phosphorylation of components and targets of the ERK pathway in b-raf f/f (WT) and b-raf Δ/Δneu (KO) brains during postnatal development. (right) A quantification of B-Raf, Raf-1, pMEK, and pERK levels from at least three animals (mean ± SD). Levels are expressed as arbitrary units. Variation among experiments was minimized by normalizing the levels of the proteins of interest to loading controls that showed no change over time (actin). The dividing line in the βIV-tubulin panel indicates that the P18 samples were taken from a different gel. (B) Immunohistochemical analysis of ERK phosphorylation in B-Raf KO and WT brains. The brown staining indicates pERK+ cells. The sections were counterstained with hematoxylin. The vertical bar spans the neuropil-rich molecular layer of the cerebral cortex. G indicates the granular and M the molecular layer of the cerebellar cortex. Arrowheads indicate Purkinje cells.
At P18, ERK phosphorylation was readily detectable by immunohistochemistry in various areas of the WT brain, including cortex, cerebellum, and hypothalamus (Fig. 5 B and not depicted). In particular, a strong positive signal was observed in the neuropil of the forebrain and cerebellar cortical plate, which consists of axons, dendrites and glial processes, and in the Purkinje cells of the cerebellar cortex. In the forebrain cortex, the pERK signal was more rarely associated with cell bodies, and if it was, it was mainly observed in cells featuring the small heterochromatic nuclei typical of glial cells. pERK was dramatically reduced in all cells and areas of b-raf Δ/Δneu brains (Fig. 5 B).
B-Raf is required for MEK/ERK activation in oligodendrocyte-enriched cultures and in oligodendrocytes in vivo
To assess whether B-Raf was required for MEK/ERK activation in cells of the oligodendrocytic lineage, we stimulated oligodendrocyte-enriched primary mixed glial cultures with PDGF, which reportedly regulates oligodendrocyte development (Fruttiger et al., 1999) and activates ERK in differentiating oligodendrocytes (Bhat and Zhang, 1996). PDGF-induced phosphorylation of MEK and ERK was impaired in the B-Raf–deficient cells (Fig. 6 A). In contrast, B-Raf ablation had hardly any effect on basal and PDGF-induced MEK/ERK phosphorylation in oligodendrocyte-depleted glial cell cultures (≥90% astrocytes; Fig. 6 B), which supports the idea that oligodendrocytes are the glial cell type in which ERK phosphorylation is most affected by the lack of B-Raf. A decrease in MEK/ERK phosphorylation was also apparent in B-Raf–deficient, oligodendrocyte-enriched cultures treated with other stimuli involved in oligodendrocyte differentiation, namely FGF (Kessaris et al., 2004; Oh et al., 2003) and EGF (Fig. 6 C; Aguirre et al., 2007). Consistent with the decrease in ERK phosphorylation, nuclear translocation of ERK was impaired in B-Raf–deficient oligodendrocytes (Fig. 6 D).
To gain more insight into the wiring of the ERK pathway, we prepared MEK1 immunoprecipitates from mixed glial cultures and monitored the association of MEK with other ERK pathway components (Fig. 6 E). Although, as described previously (Kortum and Lewis, 2004; Fujioka et al., 2006), the amounts of coprecipitating proteins were very low, WT MEK immunoprecipitates contained kinase suppressor of Ras 1 (KSR1), a scaffold protein with high similarity to Raf-1 that can bind to all members of the ERK pathway and facilitate signal propagation (Morrison, 2001; Nguyen et al., 2002), B-Raf, and Raf-1. B-Raf ablation decreased the association of the MEK with KSR, whereas the amount of coprecipitating Raf-1 was unchanged or increased (Fig. 6 E and not depicted). Thus, in this oligodendrocyte-enriched glial cell population, Raf-1 can bind to MEK1 in the absence of B-Raf but cannot take its place as a MEK kinase or stimulate MEK's association with KSR, which ultimately results in impaired ERK phosphorylation (Fig. 6, A, C, and D).
Finally, to confirm that B-Raf is the ERK activator in differentiating oligodendrocytes in vivo, we colabeled P18 brain sections with antibodies against pERK and βIV-tubulin. On the basis of their morphology (Zhang, 2001), pERK+ cells were classified as premyelinating oligodendrocytes, featuring processes with a low degree of arborization (Fig. 6 F, left) or more elaborate process networks (middle); and mature myelinating cells extending their processes toward myelin sheets (Fig. 6 F, right). In the latter case, a very strong pERK signal was associated with the myelinated fibers. ERK phosphorylation was undetectable in βIV-tubulin+ cells from b-raf Δ/Δneu brains (Fig. 6 F).
Discussion
Our data allow several important conclusions. The fact that the deletion of b-raf in neuronal precursors almost completely phenocopies the deletion in the embryo proper (with the exception of the reduced spleen) implies that in vivo, at least during the first 3 wk of life, B-Raf is not absolutely required for proliferation, survival, or differentiation of other cell types. As has been previously suggested (Zhong et al., 2007), the lack of major changes in the cellularity and architecture of the nervous system itself further implies that, in the context of the whole animal, B-Raf is not essential for the proliferation or survival of neurons. Previous studies have predicted a role for B-Raf in neuronal survival (Wiese et al., 2001), differentiation (Marshall, 1995; Kao et al., 2001), and axon growth (Markus et al., 2002). With the caveat that the level of analysis presented in this paper is not sufficient to draw conclusions on subtle differences in neuronal differentiation, the simplest explanation for the absence of major defects is that the in vivo environment provides a variety of signals that promote neuronal survival and sustains differentiation independently of B-Raf, possibly via other Raf kinases. In line with this hypothesis, the marginal defects in dorsal root ganglia differentiation caused by ablation of b-raf alone become progressively more dramatic when one or both c-raf-1 alleles are eliminated (Zhong et al., 2007). Compound tissue-restricted KOs of the Raf genes will be required to circumvent redundancy and clarify the biological role of Raf in neuronal tissues.
In brain lysates, MEK and ERK phosphorylation are not synchronized. MEK phosphorylation is highest at early stages of postnatal development, when B-Raf is hardly expressed, whereas ERK phosphorylation coincides with B-Raf expression and is evident at later stages. One trivial but not likely explanation is that MEK and ERK may not be expressed concomitantly in the same cell types during early postnatal development. Alternatively, if MEK and ERK are expressed concomitantly, the discrepancy between MEK and ERK phosphorylation could be explained by a stronger negative feedback (e.g., phosphatase activity) targeting ERK at these early stages. In PC12 cells, modular response analysis has recently confirmed that ERKs negatively feed back on MEKs (Santos et al., 2007). If this were true in the developing brain in vivo, less sustained ERK activity could explain the stronger MEK phosphorylation.
Be that as it may, in postnatal brain development, the highest levels of ERK phosphorylation were observed in coincidence with the increased expression of B-Raf and with the onset and progress of myelination, which was critically dependent on B-Raf. B-Raf–deficient oligodendrocyte precursors show impaired MEK/ERK activation and differentiation in vitro and in vivo, and chemical inhibition of MEK prevents the development of MBP-expressing cells in WT cultures (Figs. 4 and Figs.6), demonstrating a causal relationship between ERK activation and terminal oligodendrocyte differentiation. The latter result is in agreement with previous reports demonstrating that FGF-induced ERK activation cooperates with Sonic hedgehog to promote oligodendrocyte maturation in culture (Kessaris et al., 2004) and that MEK/ERK inhibition reduces MBP production by basal forebrain oligodendrocytes (Du et al., 2006).
Thus, in the oligodendrocyte lineage, B-Raf is the critical ERK activator. This is underscored by the inability of Raf-1 to compensate for B-Raf ablation, notwithstanding its binding to MEK in B-Raf–deficient oligodendrocytes (complex III; Fig. 6 G). The molecular basis for this may be simply that the higher intrinsic kinase activity of B-Raf is directly required for MEK activation in these cells (complex I; Fig. 6 G); alternatively, the presence of B-Raf may be required to regulate the kinase activity of Raf-1 in the context of the recently described B-Raf–Raf-1 heterodimer, which represent the Raf kinase with the highest activity (Garnett et al., 2005; Rushworth et al., 2006). In the latter case (complex II; Fig. 6 G), the physical presence of B-Raf may be more important than its kinase activity (Garnett et al., 2005). Consistent with the latter hypothesis, mice expressing a kinase-dead form of B-Raf do not have a neurological phenotype (Pritchard, C., personal communication).
In oligodendrocyte-enriched cultures, B-Raf promotes the recruitment of KSR1 to the Raf-MEK-ERK module (Fig. 6 C and complex I and II in Fig. 6 E). Currently, KSR is thought to bind constitutively to MEK/ERK and present it to Raf, thereby both insulating and enhancing ERK signaling. Our data instead imply that B-Raf regulates the scaffold function of KSR. It is conceivable that B-Raf binds to KSR1 and brings it to the MEK/ERK module in the context of a B-Raf–KSR heterodimer. Alternatively, B-Raf may activate KSR, enabling it to bind to MEK1. Considering the strong similarity between Raf-1 and KSR, activation may occur allosterically, in analogy to the recently described activation of Raf-1 by B-Raf (Garnett et al., 2005). Finally, the association of KSR with the MEK/ERK module might be regulated by KSR phosphorylation, mediated directly or indirectly by B-Raf.
Both soluble and axonal signals could stimulate B-Raf activation and oligodendrocyte differentiation in vivo. Candidates are FGF, PDGF, and ErbB receptor ligands, including the membrane-tethered neuregulin-1 type III, which has been implicated as a major axonal signal regulating myelination in Schwann cells (Nave and Salzer, 2006). Whatever the signals in vivo may be, the impaired differentiation observed in b-raf Δ/Δneu oligodendrocyte cultures suggests that B-Raf is either an essential effector of the crucial signal or a point of convergence of multiple factors promoting oligodendrocyte differentiation.
Histological examination shows that ERK phosphorylation is strongly reduced from P15 on in the brain of b-raf Δ/Δneu mice, and we couldn't identify any specific cell type or location that would be spared. With the exception of Purkinje cells, pERK staining was seldom associated with the soma of neurons but was rather found in smaller cells with heterochromatic nuclei characteristic of glia. Specifically, pERK was clearly detectable in premyelinating and myelinating oligodendrocytes of WT but not b-raf Δ/Δneu brains (Figs. 5 and Figs.6). Given the extensive cross-talk between glia and neurons, it is possible that the lack of ERK phosphorylation in B-Raf–deficient neurons may not, or not exclusively, be cell autonomous but rather due to the lack of support by the oligodendrocytes. It is, for instance, conceivable that the defect may result from the decreased action potentials generated as a consequence of delayed/decreased myelination. Besides myelination, other oligodendrocyte functions may impact on neurons. Among these are the secretion of factors that promote neuronal survival (Sherman and Brophy, 2005) and the recently reported physical connection between NG2+ glial progenitors present in different brain regions with surrounding neurons via direct synapses. Particularly relevant to the topic of this work, the NG2+ cells of the cerebellum are directly innervated by the climbing fibers from the inferior olive (Lin et al., 2005) and actually share climbing fibers with the surrounding Purkinje cells. Therefore, they are ideally positioned to modulate Purkinkje cells excitation by climbing fibers and cerebellar functions (Lin et al., 2005).
The involvement of the macroglia is being increasingly recognized in an expanding number of neurological diseases, including Alzheimer's disease and schizophrenia (Rowitch, 2004; Georgieva et al., 2006). We have shown that B-Raf ablation and the consequent impairment of ERK activation in oligodendrocytes lead to delayed differentiation and dysmyelination and are ultimately incompatible with extrauterine life. With the development of safe B-Raf inhibitors for clinical use in mind, it will be important to assess whether B-Raf is essential in this lineage during early development exclusively or whether its ablation later in life will lead to harmful disturbances of the macroglia including demyelination.
Materials and methods
Mice
Generation of mice carrying floxed b-raf alleles (b-raf f / f) has been described previously (Chen et al., 2006). b-raf f/f mice were maintained on a 129/Sv background and crossed to transgenic mice expressing Cre from the Mox2 locus (Tallquist and Soriano, 2000) for epiblast-restricted Raf ablation or to transgenic mice expressing Cre under the control of the Nestin promoter (Tronche et al., 1999) for neuronal precursor-restricted ablation. Animal care was in accordance with the guidelines of the Max F. Perutz Laboratories.
PCR analysis of offspring and tissues
Tail and tissue DNA preparation and b-raf PCR were performed as described previously (Galabova-Kovacs et al., 2006).
Histology, immunohistochemistry, and electron microscopy
Unless stated otherwise, serial sections of 4% paraformaldehyde-fixed and paraffin-embedded tissues were cut at 3-μm thickness and placed on precoated slides (0.5% VECTABOND reagent; Vector Laboratories) and routinely stained with hematoxylin and eosin.
The following primary antibodies were used for immunohistochemistry: α-pERK (Cell Signaling Technology), α-βIV-tubulin (Sigma-Aldrich), α-MBP (Santa Cruz Biotechnology, Inc.), α-PDGFRα (Thermo Fisher Scientific), or unrelated, isotyped-matched controls. Immunocomplexes were visualized using appropriate secondary antibodies conjugated with Alexa 488 or 594 (Invitrogen), the ultrasensitive avidin biotinylated enzyme complex staining kit (ABC; Vector Laboratories), or the EnVision peroxidase system (Dako) according to the manufacturer's instructions, followed by incubation with 0.01% diaminobenzidine (Sigma-Aldrich). Epifluorescence was performed at room temperature using Immersol 518 (Carl Zeiss, Inc.) as an imaging medium and a microscope (Axioplan 2; Carl Zeiss, Inc.) equipped with a Plan Neofluar 40× 1.30 oil objective (Carl Zeiss, Inc.) and a charge-coupled device (CCD) camera (Spot2; Diagnostic Instruments, Inc.). Images were acquired using the MetaVue 5.0r6 software (MDS Analytical Technologies). Light microscopy was performed at room temperature using Immersol 518 as an imaging medium and a microscope (AxioImager M1) equipped with a CCD camera (AxioCam Mrc5; both from Carl Zeiss, Inc.). Images were acquired using the Axiovision 4.6 software (Carl Zeiss, Inc.)
For transmission electron microscopy, tissue was collected and fixed in 3% glutaraldehyde in 0.1 M Sorensen phosphate buffer, pH 7.4, at 4°C. After fixation, specimens were contrasted with osmium tetraoxide and embedded in epoxy resin. Ultrathin sections were cut at 70 nm, stained with alkaline-lead citrate and methanolic uranyl acetate, and viewed with a transmission electron microscope (EM 900; Carl Zeiss, Inc.).
Cell culture
Mixed glial cell cultures were obtained from newborn animals (P0), plated in NM15 media (DME containing 15% fetal calf serum) for at least 7 d. Oligodendrocyte precursors were obtained by differential shaking of mixed glial cultures. Immediately after shaking, progenitors were plated on poly-d-lysine–coated dishes in NM15 medium. Differentiation was induced 24 h later by changing the medium to differentiation medium (DME supplemented with 0.5% FCS, 25 μg/ml insulin, 5 ng/ml selenium, 50 μg/ml transferrin, and 20 ng/ml tri-iodothyronine; all from Sigma-Aldrich) and was assessed by immunofluorescence, staining the cells with α-NG2 (Millipore), α-O4 (R&D Systems), and α-MBP (Santa Cruz Biotechnology, Inc.) followed by Alexa 488– or 594–conjugated secondary antibodies (Invitrogen). The overall purity of the cultures used in the experiments shown in Figs. 4 and Figs.6 was between 80 and 90%, as assessed by immunofluorescence using a combination of antibodies against GFAP, NG2, O4, MBP, pan-tubulin, and βIV-tubulin. For biochemistry, oligodendrocyte-enriched cultures were generated by plating the mixed glial cell cultures in differentiation medium supplemented with 10 ng/ml rat recombinant ciliary neurotrophic factor for 2 d to allow oligodendrocyte precursors to differentiate. Oligodendrocyte-depleted cultures were obtained after repeated differential shaking of mixed glial cell cultures to get rid of oligodendrocyte precursors and consisted of >90% astrocytes.
Immunoblot analysis and immunoprecipitation
Preparation of brain lysates has been described previously (Morice et al., 1999). MEK1 immunoprecipitates were prepared using antibodies from Transduction Laboratories and washed four times in brain lysate buffer. Immunoprecipitates and lysates were subjected to immunoblotting as described previously (Mikula et al., 2001). The antibodies used were B-Raf C-19 (against a C-terminal epitope), H145 (against an N-Terminal epitope), A-Raf C-20, actin, MBP (Santa Cruz Biotechnology, Inc.), Raf-1, MEK1, MEK2, KSR1, panERK (Transduction Laboratories), pMEK1/2, p44/42 MAPK, EGR1 (Cell Signaling Technology), βIV-tubulin, and tubulin (Sigma-Aldrich).
Online supplemental material
Fig. S1 shows the NestinCre activity pattern in an embryonic day 11.5 embryo. Figs S2 and S3 show the morphological defects of b-raf–deficient mouse brain and cerebellum. Fig. S4 shows the myelination defect of b-raf Δ/Δneu throughout postnatal development. Fig. S5 shows hypomyelination and supernumerary oligodendrocyte precursor in different regions of b-raf Δ/Δneu brains. Videos 1–3 show the neurological defects of b-raf Δ/Δneu mice. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200709069/DC1.
Supplemental Material
Acknowledgments
We thank H. Lassman for helpful discussions; J. Zhong and W. Snider for sharing the data in Fig. S1; T. Wirthensohn for cutting the videos; and W. Tschulenk, C. Khrla, and M. Hamerl for outstanding technical assistance.
This work was supported by the Austrian Science Fund (FWF; grants P15784-MOB and P-18712-B09 to M. Baccarini), the FWF Erwin-Schroedinger Return Program (grant R10 to R. Herbst), and an APART grant of the Austrian Academy of Sciences (to R. Herbst).
F. Catalanotti and D. Matzen contributed equally to this paper.
Abbreviations used in this paper: CNS, central nervous system; ERK, extracellular signal-regulated kinase; KO, knockout; KSR, kinase suppressor of Ras; MBP, myelin basic protein; MEK, MAPK/ERK kinase; P, postnatal day; PDGFRα, PDGF receptor α; WT, wild type.
References
- Aguirre, A., J.L. Dupree, J.M. Mangin, and V. Gallo. 2007. A functional role for EGFR signaling in myelination and remyelination. Nat. Neurosci. 10:990–1002. [DOI] [PubMed] [Google Scholar]
- Bhat, N.R., and P. Zhang. 1996. Activation of mitogen-activated protein kinases in oligodendrocytes. J. Neurochem. 66:1986–1994. [DOI] [PubMed] [Google Scholar]
- Brummer, T., P.E. Shaw, M. Reth, and Y. Misawa. 2002. Inducible gene deletion reveals different roles for B-Raf and Raf-1 in B-cell antigen receptor signalling. EMBO J. 21:5611–5622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, A.P., M. Ohno, K.P. Giese, R. Kuhn, R.L. Chen, and A.J. Silva. 2006. Forebrain-specific knockout of B-raf kinase leads to deficits in hippocampal long-term potentiation, learning, and memory. J. Neurosci. Res. 83:28–38. [DOI] [PubMed] [Google Scholar]
- Davies, H., G.R. Bignell, C. Cox, P. Stephens, S. Edkins, S. Clegg, J. Teague, H. Woffendin, M.J. Garnett, W. Bottomley, et al. 2002. Mutations of the BRAF gene in human cancer. Nature. 417:949–954. [DOI] [PubMed] [Google Scholar]
- Du, Y., L.D. Lercher, R. Zhou, and C.F. Dreyfus. 2006. Mitogen-activated protein kinase pathway mediates effects of brain-derived neurotrophic factor on differentiation of basal forebrain oligodendrocytes. J. Neurosci. Res. 84:1692–1702. [DOI] [PubMed] [Google Scholar]
- Foran, D.R., and A.C. Peterson. 1992. Myelin acquisition in the central nervous system of the mouse revealed by an MBP-Lac Z transgene. J. Neurosci. 12:4890–4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruttiger, M., L. Karlsson, A.C. Hall, A. Abramsson, A.R. Calver, H. Bostrom, K. Willetts, C.H. Bertold, J.K. Heath, C. Betsholtz, and W.D. Richardson. 1999. Defective oligodendrocyte development and severe hypomyelination in PDGF-A knockout mice. Development. 126:457–467. [DOI] [PubMed] [Google Scholar]
- Fujioka, A., K. Terai, R.E. Itoh, K. Aoki, T. Nakamura, S. Kuroda, E. Nishida, and M. Matsuda. 2006. Dynamics of the Ras/ERK MAPK cascade as monitored by fluorescent probes. J. Biol. Chem. 281:8917–8926. [DOI] [PubMed] [Google Scholar]
- Galabova-Kovacs, G., D. Matzen, D. Piazzolla, K. Meissl, T. Plyushch, A.P. Chen, A. Silva, and M. Baccarini. 2006. Essential role of B-Raf in ERK activation during extraembryonic development. Proc. Natl. Acad. Sci. USA. 103:1325–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnett, M.J., S. Rana, H. Paterson, D. Barford, and R. Marais. 2005. Wild-type and mutant B-RAF activate C-RAF through distinct mechanisms involving heterodimerization. Mol. Cell. 20:963–969. [DOI] [PubMed] [Google Scholar]
- Georgieva, L., V. Moskvina, T. Peirce, N. Norton, N.J. Bray, L. Jones, P. Holmans, S. Macgregor, S. Zammit, J. Wilkinson, et al. 2006. Convergent evidence that oligodendrocyte lineage transcription factor 2 (OLIG2) and interacting genes influence susceptibility to schizophrenia. Proc. Natl. Acad. Sci. USA. 103:12469–12474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grusser-Cornehls, U., and J. Baurle. 2001. Mutant mice as a model for cerebellar ataxia. Prog. Neurobiol. 63:489–540. [DOI] [PubMed] [Google Scholar]
- Haigh, J.J., P.I. Morelli, H. Gerhardt, K. Haigh, J. Tsien, A. Damert, L. Miquerol, U. Muhlner, R. Klein, N. Ferrara, et al. 2003. Cortical and retinal defects caused by dosage-dependent reductions in VEGF-A paracrine signaling. Dev. Biol. 262:225–241. [DOI] [PubMed] [Google Scholar]
- Harada, T., T. Morooka, S. Ogawa, and E. Nishida. 2001. ERK induces p35, a neuron-specific activator of Cdk5, through induction of Egr1. Nat. Cell Biol. 3:453–459. [DOI] [PubMed] [Google Scholar]
- Huser, M., J. Luckett, A. Chiloeches, K. Mercer, M. Iwobi, S. Giblett, X.M. Sun, J. Brown, R. Marais, and C. Pritchard. 2001. MEK kinase activity is not necessary for Raf-1 function. EMBO J. 20:1940–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao, S., R.K. Jaiswal, W. Kolch, and G.E. Landreth. 2001. Identification of the mechanisms regulating the differential activation of the mapk cascade by epidermal growth factor and nerve growth factor in PC12 cells. J. Biol. Chem. 276:18169–18177. [DOI] [PubMed] [Google Scholar]
- Kessaris, N., F. Jamen, L.L. Rubin, and W.D. Richardson. 2004. Cooperation between sonic hedgehog and fibroblast growth factor/MAPK signalling pathways in neocortical precursors. Development. 131:1289–1298. [DOI] [PubMed] [Google Scholar]
- Kortum, R.L., and R.E. Lewis. 2004. The molecular scaffold KSR1 regulates the proliferative and oncogenic potential of cells. Mol. Cell. Biol. 24:4407–4416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, S.C., J.H.J. Huck, J.D.B. Roberts, W.B. Macklin, P. Somogyi, and D.E. Bergles. 2005. Climbing fiber innervation of NG2-expressing glia in the mammalian cerebellum. Neuron. 46:773–785. [DOI] [PubMed] [Google Scholar]
- Markus, A., J. Zhong, and W.D. Snider. 2002. Raf and akt mediate distinct aspects of sensory axon growth. Neuron. 35:65–76. [DOI] [PubMed] [Google Scholar]
- Marshall, C.J. 1995. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 80:179–185. [DOI] [PubMed] [Google Scholar]
- Mercer, K., A. Chiloeches, M. Huser, M. Kiernan, R. Marais, and C. Pritchard. 2002. ERK signalling and oncogene transformation are not impaired in cells lacking A-Raf. Oncogene. 21:347–355. [DOI] [PubMed] [Google Scholar]
- Mikula, M., M. Schreiber, Z. Husak, L. Kucerova, J. Ruth, R. Wieser, K. Zatloukal, H. Beug, E.F. Wagner, and M. Baccarini. 2001. Embryonic lethality and fetal liver apoptosis in mice lacking the c-raf-1 gene. EMBO J. 20:1952–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morice, C., F. Nothias, S. Konig, P. Vernier, M. Baccarini, J.D. Vincent, and J.V. Barnier. 1999. Raf-1 and B-Raf proteins have similar regional distributions but differential subcellular localization in adult rat brain. Eur. J. Neurosci. 11:1995–2006. [DOI] [PubMed] [Google Scholar]
- Morrison, D.K. 2001. KSR: a MAPK scaffold of the Ras pathway? J. Cell Sci. 114:1609–1612. [DOI] [PubMed] [Google Scholar]
- Nave, K.A. 1994. Neurological mouse mutants and the genes of myelin. J. Neurosci. Res. 38:607–612. [DOI] [PubMed] [Google Scholar]
- Nave, K.A., and J.L. Salzer. 2006. Axonal regulation of myelination by neuregulin 1. Curr. Opin. Neurobiol. 16:492–500. [DOI] [PubMed] [Google Scholar]
- Nguyen, A., W.R. Burack, J.L. Stock, R. Kortum, O.V. Chaika, M. Afkarian, W.J. Muller, K.M. Murphy, D.K. Morrison, R.E. Lewis, et al. 2002. Kinase suppressor of Ras (KSR) is a scaffold which facilitates mitogen-activated protein kinase activation in vivo. Mol. Cell. Biol. 22:3035–3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh, L.Y., A. Denninger, J.S. Colvin, A. Vyas, S. Tole, D.M. Ornitz, and R. Bansal. 2003. Fibroblast growth factor receptor 3 signaling regulates the onset of oligodendrocyte terminal differentiation. J. Neurosci. 23:883–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popko, B., C. Puckett, E. Lai, H.D. Shine, C. Readhead, N. Takahashi, S.W. Hunt III, R.L. Sidman, and L. Hood. 1987. Myelin deficient mice: expression of myelin basic protein and generation of mice with varying levels of myelin. Cell. 48:713–721. [DOI] [PubMed] [Google Scholar]
- Pritchard, C.A., L. Hayes, L. Wojnowski, A. Zimmer, R.M. Marais, and J.C. Norman. 2004. B-Raf acts via the ROCKII/LIMK/cofilin pathway to maintain actin stress fibers in fibroblasts. Mol. Cell. Biol. 24:5937–5952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson, W.D., N. Kessaris, and N. Pringle. 2006. Oligodendrocyte wars. Nat. Rev. Neurosci. 7:11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Viciana, P., O. Tetsu, W.E. Tidyman, A.L. Estep, B.A. Conger, M.S. Cruz, F. McCormick, and K.A. Rauen. 2006. Germline mutations in genes within the MAPK pathway cause cardio-facio-cutaneous syndrome. Science. 311:1287–1290. [DOI] [PubMed] [Google Scholar]
- Rowitch, D.H. 2004. Glial specification in the vertebrate neural tube. Nat. Rev. Neurosci. 5:409–419. [DOI] [PubMed] [Google Scholar]
- Rushworth, L.K., A.D. Hindley, E. O'Neill, and W. Kolch. 2006. Regulation and role of Raf-1/B-Raf heterodimerization. Mol. Cell. Biol. 26:2262–2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos, S.D., P.J. Verveer, and P.I. Bastiaens. 2007. Growth factor-induced MAPK network topology shapes Erk response determining PC-12 cell fate. Nat. Cell Biol. 9:324–330. [DOI] [PubMed] [Google Scholar]
- Sherman, D.L., and P.J. Brophy. 2005. Mechanisms of axon ensheathment and myelin growth. Nat. Rev. Neurosci. 6:683–690. [DOI] [PubMed] [Google Scholar]
- Sock, E., H. Leger, K. Kuhlbrodt, J. Schreiber, J. Enderich, C. Richter-Landsberg, and M. Wegner. 1997. Expression of Krox proteins during differentiation of the O-2A progenitor cell line CG-4. J. Neurochem. 68:1911–1919. [DOI] [PubMed] [Google Scholar]
- Sturrock, R.R. 1980. Myelination of the mouse corpus callosum. Neuropathol. Appl. Neurobiol. 6:415–420. [DOI] [PubMed] [Google Scholar]
- Tallquist, M.D., and P. Soriano. 2000. Epiblast-restricted Cre expression in MORE mice: a tool to distinguish embryonic vs. extra-embryonic gene function. Genesis. 26:113–115. [DOI] [PubMed] [Google Scholar]
- Terada, N., G.J. Kidd, M. Kinter, C. Bjartmar, K. Moran-Jones, and B.D. Trapp. 2005. Beta IV tubulin is selectively expressed by oligodendrocytes in the central nervous system. Glia. 50:212–222. [DOI] [PubMed] [Google Scholar]
- Tronche, F., C. Kellendonk, O. Kretz, P. Gass, K. Anlag, P.C. Orban, R. Bock, R. Klein, and G. Schutz. 1999. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat. Genet. 23:99–103. [DOI] [PubMed] [Google Scholar]
- Wellbrock, C., M. Karasarides, and R. Marais. 2004. The RAF proteins take centre stage. Nat. Rev. Mol. Cell Biol. 5:875–885. [DOI] [PubMed] [Google Scholar]
- Wiese, S., G. Pei, C. Karch, J. Troppmair, B. Holtmann, U.R. Rapp, and M. Sendtner. 2001. Specific function of B-Raf in mediating survival of embryonic motoneurons and sensory neurons. Nat. Neurosci. 4:137–142. [DOI] [PubMed] [Google Scholar]
- Wojnowski, L., A.M. Zimmer, T.W. Beck, H. Hahn, R. Bernal, U.R. Rapp, and A. Zimmer. 1997. Endothelial apoptosis in Braf-deficient mice. Nat. Genet. 16:293–297. [DOI] [PubMed] [Google Scholar]
- Wojnowski, L., L.F. Stancato, A.C. Larner, U.R. Rapp, and A. Zimmer. 2000. Overlapping and specific functions of Braf and Craf-1 proto-oncogenes during mouse embryogenesis. Mech. Dev. 91:97–104. [DOI] [PubMed] [Google Scholar]
- Xin, M., T. Yue, Z. Ma, F.F. Wu, A. Gow, and Q.R. Lu. 2005. Myelinogenesis and axonal recognition by oligodendrocytes in brain are uncoupled in Olig1 null mice. J. Neurosci. 25:1354–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zezula, J., P. Casaccia-Bonnefil, S.A. Ezhevsky, D.J. Osterhout, J.M. Levine, S.F. Dowdy, M.V. Chao, and A. Koff. 2001. p21cip1 is required for the differentiation of oligodendrocytes independently of cell cycle withdrawal. EMBO Rep. 2:27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, S.C. 2001. Defining glial cells during CNS development. Nat. Rev. Neurosci. 2:840–843. [DOI] [PubMed] [Google Scholar]
- Zhong, J., X. Li, C. McNamee, A.P. Chen, M. Baccarini, and W.D. Snider. 2007. Raf kinase signaling functions in sensory neuron differentiation and axon growth in vivo. Nat. Neurosci. 10:598–607. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}