

Abstract
Hookworms infect nearly a billion people. The Ancylostoma caninum hookworm of canids is a model for studying human infections and information from its genome coupled with functional genomics and proteomics can accelerate progress towards hookworm control. As a step towards a full-scale A. caninum genome project, we generated 104,000 genome survey sequences (GSSs) and determined the genome size of the canine hookworm. GSSs assembled into 57.6 Mb of unique sequence from a genome that we estimate by flow cytometry of isolated nuclei to be 347±1.2 Mb, substantially larger than other Rhabditina species. Gene finding identified 5,538 genes in the GSS assembly, for a total of 9,113 non-redundant A. caninum genes when EST sequences are also considered. Functional classifications of many of the 70% of genes with homology to genes in other species are provided based on Gene Ontology and KEGG associations and secreted and membrane-bound proteins are also identified.
Keywords: hookworm, Ancylostoma caninum, genome survey sequences, expressed sequence tags, genome, comparative genomics
Hookworms are parasitic nematodes that live in the host small intestine and affect mammals including humans, dogs, and cats. Ancylostoma duodenale and Necator americanus infect close to a billion people [1] causing anemia and malnutrition, and also diminished physical and cognitive development in children. A. caninum is a hookworm species that infects canids and is commonly used as a model for studying human infections. Hookworm infections are usually treated with anthelmintic drugs [2]. However, recurrence of infections and lack of immunity has bolstered the need for vaccines to reduce infections.
Tremendous advances towards a hookworm vaccine have been made in the last decade. Ancylostoma Secreted Proteins, ASP-1 and ASP-2 [3, 4] and several hookworm digestive proteases like aspartic haemoglobinase [5] are being studied as potential vaccines against hookworm infections. In addition, hookworm gene products may have application against other diseases. For instance, recombinant hookworm rNAPC2 has anti-coagulant properties and is being studied for the treatment of acute coronary syndrome in the U.S. [6]. A recent study has shown that burns treated with this protein heal more rapidly with reduced scar contracture [7].
Genomic information from hookworms can enhance development of new, safer and sustainable control strategies. Our earlier effort to explore the A. caninum genome involved an EST-based approach [8]. By sampling 9,331 ESTs from three stage-specific cDNA libraries we identified and analyzed 3,840 genes. In this study we sampled the A. caninum genome by whole-genome shotgun. While one would expect that the A. caninum genome size would be in the 53-59 MB range based on other clade V Strongylida species including Haemonchus contortus [9], our recent genome size estimate using flow sorted nuclei surprisingly revealed a genome more than six times this size. Genome size was estimated by prodidium iodide staining and flow cytometry of isolated nuclei following methods described in [10]. To prepare the nuclei, the A. caninum L3s were washed in cold Galbraith buffer pH 7.2 and then pipetted into a plastic petri dish along with 30-50 μl cold Galbraith buffer and chopped (50 times) with a fresh single edge razor blade. The chopped material was washed into one edge of the (tilted) petri dish using an additional 1 ml of cold Galbraith buffer (per liter: 4.26g MgCl2, 8.84 g sodium citrate, 4.2 g [N-morpholino] propane sulfonic acid, 1ml Triton X-100, 1 mg boiled ribonuclease A, pH 7.2-7.4 with 1M KOH) and this solution was pipetted into a 1.5 ml Dounce homogenizer. Standards (similarly chopped C. elegans, the head of a D. melanogaster (Iso-1) and/or D. virilis female) were added to the Dounce at this time and the final amount in the Dounce was adjusted to 1ml adding cold Galbraith buffer as necessary. Each sample, standard, and co-prepared sample was ground using 15 strokes of the “A” pestle at a rate of 3 strokes per two seconds. The ground solution was poured through 20 μm nylon filter into a microfuge tube, brought to a final volume of 1 ml using additional cold Galbraith buffer as needed and then maintained on ice. Propidium Iodide (50 μl of 1 mg/ml H2O) was added to a final concentration of 0.075 mM, the tube capped and inverted several times to mix, and then stored in the dark for 2-8 hours prior to running. Samples were run in a Beckman-Coulter Epics Elite Cytometer using 25 mW of 488 mn (blue) excitation. PI fluorescence was measured after passing the collected fluorescent output across a 610 nm long pass filter. Counting was activated by PI fluorescence (gating: Debris, partial nuclei and nuclei with adhering cytoplasmic “tags” were excluded from the final analysis on the basis of forward and side scatter parameters. Only those nuclei with the lowest level of scatter were counted, as these have been shown by sorting to be the intact, untagged nuclei). A total of five replicates with at least 6,000 scored nuclei in each replicate were used to estimate genome size. C. elegans (1C = 100 Mb), D. melanogaster (1C = 175 Mb) and D. virilis (1C = 333.5 Mb) were used as standards. In every replicate, A. caninum ran just above the Drosophila virilis 2C peak (Fig. 1). The number of nuclei isolated from individual A. caninum was modest (maximum 200). However, the relative mean position of these nuclei compared to the standard was very consistent (Fig 1). The average (± standard error) genome size of A. caninum was 347.2±1.2 Mb. According to our estimate we sampled the A. caninum genome to an anticipated ~17% coverage.
Figure 1.
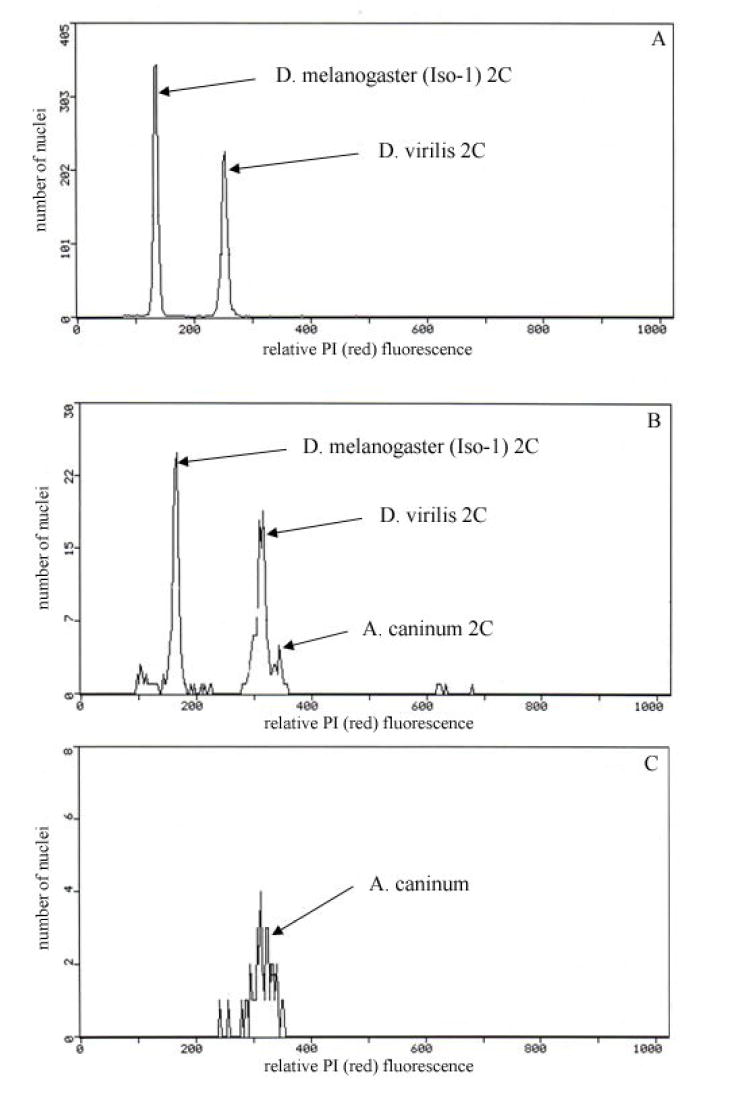
The number of nuclei scored at differing levels of red fluorescence. The red fluorescence corresponds to binding of propidium iodide to the DNA of 2C nuclei in: A. Co-prepared heads of female D. melanogaster Iso-1 (1C = 175 Mb) and female D. virilis (1C = 333.5 Mb); B. Co-prepared heads of female D. melanogaser, and D. virilis with A. caninum. C. A single female A. caninum (1C = 347.2 Mb). Small differences in PMT voltage shift means to slightly higher or lower channels in the three histograms. The DNA estimate is based on the ratio of co-prepared sample and standard as in 1B and 1C.
Genomic DNA from adult A. caninum derived from experimental infection (strain Baltimore was isolated by G.A. Schad [11] from natural infection in the Baltimore area in the 1960s) was randomly sheared, end-repaired and size fractioned to enrich for 2-4 kb fragments. A total of 104,000 genome survey sequences (GSSs) were generated (72.6 Mb)(http://genome.wustl.edu/platforms_index.cgi), 95% passed quality screening and were submitted to dbGSS division of GenBank. Eighty nine per cent of clones had sequences from both ends. The 94,602 GSSs were assembled using PCAP [12]. Thirty five percent of the sequences assembled in 14,208 contigs and subsequently in 12,430 supercontigs (largest supercontig 9,358 bp). Total length of the contigs was 14.5 Mb and the 60,172 singletons contributed to 43.1 Mb of additional unique sequence. The low redundancy in the GSSs is consistent with the larger genome size estimate and rules out the possibility of a ~60 Mb genome like the Strongylid H. contortus.
As a first step to explore the available A. caninum genome sequence we identified the repetitive elements and masked them prior to gene identification. We evaluated repeat content in the assembly and singletons by masking simple repeats, low-complexity repeats and repeats identified by generating a custom library of repeat sequences. The custom library was built using RECON [13] and default parameters. This library was screened for non-coding RNA and protein-coding genes using Rfam [14] and Non-Redundant GenBank (built 03/23/2005) respectively, yielding 1,141 repeat families (Suppl. File S1). RepeatMasker (RepeatMasker Open-3.0.) was used to estimate the percentage of repeats. The total repeat content is estimated to be around 26.9 % (C. elegans repeat content is 16.5%) out of which 0.4% are simple repeats, 0.2% are low complexity repeats, and 26.3% are repeats identified by the custom library (The list of the ten most abundant repeats is available as on line Suppl. Table S1a). The GC content of the repeats was 44.6%, similar to the genome GC (43.2%) but lower than the protein coding exons GC (47.3%). Twenty-six repeat families had hits to repeats from various organisms in RepBase [15] (1e-05 cut-off; Suppl. Table S1b). Further analysis, especially on its resident mobile genetic elements, identified a novel mariner-like element [16], a non-long terminal repeat (LTR) retrotransposons [17] and transib transposon [18].
The masked assembly was used to call genes. In total, 5,538 genes were identified through a 6-tier gene-calling pipeline [19]. In addition, of the 3,840 genes that were identified by our previous A. caninum EST approach [8] 3,589 (represented by 4,816 contig consensus sequences) were not incorporated in calling genes, and therefore added to the list of identified unique genes. Similarly, 258 out of 498 contigs from external EST projects ([20] and Datu B., Loukas A., and Gasser, R., personal communication), were also non-overlapping genes and added to the non-redundant list of genes, making the total number of identified genes 9,385. The 9,385 genes are likely an overestimate of gene discovery, as one gene could be represented by multiple non-overlapping EST clusters or gene fragments identified from the GSS assembly. Such ‘fragmentation’ was estimated at 20% using C. elegans as a reference genome. As expected the fragmentation of the GSS derived genes was higher than the EST derived genes (21% vs 4%), and the overall fragmentation of 20% partially explains the low overlap between our EST and GSS gene collections. While the gene calling pipeline provides us with translations, we used prot4EST, a translational prediction pipeline optimized for EST datasets [21], to generate protein prediction for the EST clusters. The GFF and fasta files for the final set of 9,113 confident translations used in our analysis can be found at http://www.nematode.net [22]. Our approach indicates that the moderate number of ESTs and low-coverage obtained by GSS approach is a powerful combination which could allow an initial cost-effective identification of genes in neglected genomes. By this approach we identified 62 out of 77 A. caninum proteins previously deposited in the non-redundant GenBank (cut-off 1e-30) and increased the number of available genes from this species greater than 100-fold. The data contained 2.5 Mb of sequence from predicted genes, of which 1.9 Mb was exonic sequence. Direct comparison of the A. caninum genes to the homologous C. elegans counterparts indicated a smaller median exon size in A. caninum than C. elegans (128 bp vs. 174bp), and very similar median intron size of 72bp vs. 76bp (Suppl. Table S2). However, exons per genes and length of genes cannot be adequately estimated due to the fragmented nature of the assembly. At this coverage of the genome, it is premature to determine whether A. caninum’s larger genome will also contain an increase in the number or size of protein-coding genes compared to C. elegans. The GC content for A. caninum differed from C. elegans for the genome as a whole (43.2% for A. caninum versus 35.4% for C. elegans) and for protein coding exons (47.3% versus 42.7%).
Homology search of the 9,113 A. caninum genes with WU-BLAST (www://blast.wustl.edu) versus three specific phylogenetic databases (Fig. 2) revealed that 70% (6,335/9,113) had homologs among known and predicted proteins from other species. The majority of those with homology (63% or 3,996/6,335) matched all three databases. Of all proteins with homology, 92.5% shared similarity to Caenorhabditis elegans and/or C. briggsae, a higher percentage than the 80% match rate obtained using EST clusters alone [8]. Reasons for the increase in observed homology include the addition of non-abundant genes sampled by the GSS method, increased sequence length and quality contributing towards more significant hits, and not the effect of increased database size on score cut-off. This was confirmed by BLAST searching the EST clusters from Mitreva et al., [8] against the newly built databases used for this study (data not shown). The 7% of genes with homology only to Caenorhabditis species genes are potential nematode Clade V lineage-specific genes. Furthermore, 59% (3,330/5,641) of the hits to C. elegans were best reciprocal hits, therefore putative orthologs between A. caninum and C. elegans. RNA interference (RNAi) has not been reported in hookworms, therefore extrapolation from the C. elegans orthologs/homologs with observed phenotype by RNAi can be very informative for functional analysis of the orthologous counterparts in A. caninum. Of all orthologous C. elegans genes, 98% (3,253/3,330) had available RNAi information (RNAi data used from Wormpep v. 156), and of these 46% have observable phenotypes by RNAi knockdown. Of the genes with observable phenotypes, 60% (897/1,494) had severe phenotypes including embryonic, larval, or adult lethal, sterile, sterile progeny, and larval or adult growth arrest. A list of all RNAi information for C. elegans genes with A. caninum homolog is available as online Suppl. Table S3.
Figure 2.
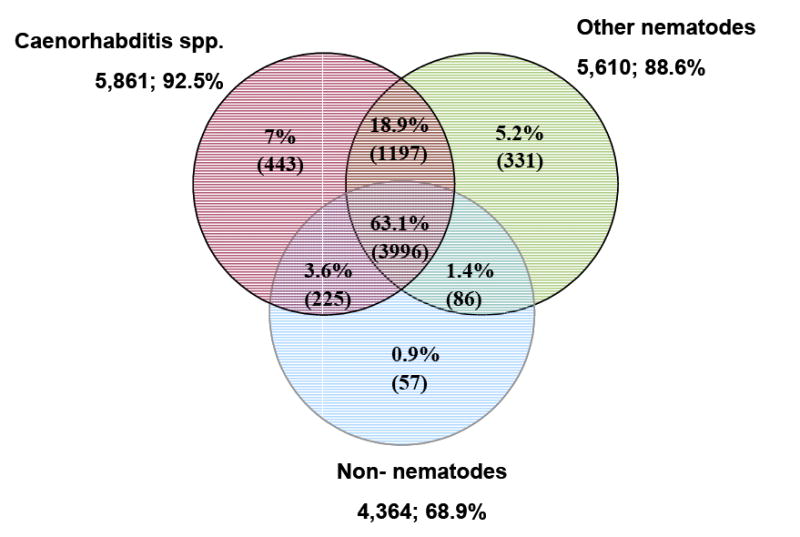
Distribution of Ancylostoma caninum BLAST matches by database. Caenorhabditis spp., represents C. elegans, C. briggsae and C. remanei; Other nematodes, all nematode beyond Caenorhabditis and Ancylostoma; and Non-nematoda, all species beyond nematodes. Databases were built 06/09/06, e-value scores ≤ 1e-05 are considered.
To functionally classify and categorize the genes, homologies to Kyoto Encyclopedia of Genes and Genomes (KEGG; [23]) and Interpro [24] database members were analyzed. The KEGG database contains information on metabolic pathways and interactions. A. caninum genes were mapped to the metabolic pathways using the highest-scoring WU-BLAST match and corresponding enzyme commission (EC) numbers of the homologous KEGG representative. Twenty-four percent of the A. caninum genes were mapped to 124 KEGG metabolic pathways classified into 11 major metabolic categories. Relative to mappings from the complete C. elegans genome, 70% of potential mappings were represented by A. caninum sequences. Biosynthesis of Secondary Metabolites and Energy metabolism were among the better represented categories using C. elegans as a reference (Table 1a). The complete listing of all mapping is available as online Suppl. Table S4. Metabolic pathways reconstruction is a productive approach for identifying novel anthelmintic drug targets. To facilitate this research, we have made the A. caninum KEGG associations to the 132 represented metabolic pathways graphically viewable on our web site (www.nematode.net; [22]). The viewer provides associations to specific enzyme commission (EC) numbers, strength of the match and associated KO identifiers. The A. caninum KEGG viewer coupled with our viewer of C. elegans metabolic mappings with incorporated RNAi information (http://www.nematode.net/KEGGscan/cgi-bin/KEGGscan_hit_distribution.cgi?species_selection=Caenorhabditis%20elegans) enables identification of potential ‘chokepoints’ in biochemical pathways and corresponding phenotypes. The combination of information from multiple sources can bolster the case for selection of new anti parasitic drug targets.
Table 1a.
KEGG biochemical pathway category mappingsa for A. caninum and C. elegans genes
| KEGG PATHWAY | A. caninum | C. elegans | ||
|---|---|---|---|---|
| Genes | Enzymes | Genes | Enzymes | |
| Carbohydrate Metabolism | 422 | 127 | 1485 | 169 |
| Energy Metabolism | 276 | 43 | 362 | 56 |
| Lipid Metabolism | 267 | 79 | 1098 | 118 |
| Nucleotide Metabolism | 207 | 49 | 676 | 73 |
| Amino Acid Metabolism | 348 | 135 | 929 | 179 |
| Metabolism of Other Amino Acids | 93 | 35 | 289 | 46 |
| Glycan Biosynthesis and Metabolism | 147 | 44 | 598 | 65 |
| Biosynthesis of Polyketides and Nonribosomal Peptides | 45 | 6 | 307 | 11 |
| Metabolism of Cofactors and Vitamins | 249 | 51 | 839 | 83 |
| Biosynthesis of Secondary Metabolites | 89 | 24 | 344 | 34 |
| Xenobiotics Biodegradation and Metabolism | 154 | 37 | 825 | 48 |
WU-BLAST (www://blast.wustl.edu) with top hits of each gene; KEGG v. 40 was used.
As an alternative method for categorizing predicted proteins we used Interpro [25], an integrated database of known protein domains from well characterized data sources, to identify signatures/domains in our sequences. InterproScan [26] was used to locate homology to the Interpro database. 73% of the A. caninum genes mapped to one or more of 1,826 unique Interpro domains. The protein kinase (IPR000719), Protein kinase-like (IPR011009), Transposase, type 1 (IPR001888) and allergen V5/Tpx-1 related domains (IPR001283) were among the most frequently identified (Table 1b). Intriguingly, there were no C. elegans genes mapped to the IPR001888. Blasting of the 65 A. caninum genes against the C. elegans proteome did not yield any hits (cutoff 1e-10). We also looked at the Pfam entries for PF01359 (seed and full alignments)[27] and the corresponding Interpro id IPR001888 and we found 4 C. elegans proteins included in this entry. However all of these proteins were superseded or retired from Wormbase. Among the 65 A. caninum genes mapped to this IPR was the novel mariner-like element, bandit-transposon, that we characterized from the genome of A. caninum [16]. The phylogenetic analysis of Tc1/mariner superfamily of transposons indicated that the closest relative to bandit was the human HSmar1 rather than nematodes or arthropods originated transposons.
Table 1b.
The top 10 most abundant Interpro identifiers of A. caninum in C. elegans
| Interpro | Description | A. caninum | C. elegans |
|---|---|---|---|
| IPR000719 | Protein kinase | 106 | 435 |
| IPR011009 | Protein kinase-like | 101 | 494 |
| IPR001888 | Transposase, type 1 | 65 | 0 |
| IPR001283 | Allergen V5/Tpx-1 related | 55 | 36 |
| IPR012337 | Polynucleotidyl transferase | 51 | 52 |
| IPR006201 | Neurotransmitter-gated ion-channel | 46 | 101 |
| IPR013032 | EGF-like region | 42 | 193 |
| IPR001506 | Peptidase M12A, astacin | 42 | 42 |
| IPR002048 | Calcium-binding EF-hand | 41 | 128 |
Furthermore, based on A. caninum InterPro protein domain matches, 32% of the gene products mapped to one or more organizing principles of the Gene Ontology (GO; [28]), representing 789 unique GO identifiers. Among the most common GO categories were ATP binding (361 genes mapped to GO:0005524), membrane (355 to GO:0016020), and intracellular (213 to GO:0005622). The GO associations are available on line through the Amigo viewer - (http://www.nematode.net/cgi-bin/amigo/go_AC/go.cgi).
Most of the nematode vaccine targets studied to date are excretory/secretory (ES) or intestinal antigens that are secreted or membrane-bound. Many chemotherapeutic targets are receptors or channels also found in membranes. We processed the A. caninum sequences through the Phobius server [29] which predicts secreted proteins (SP) and transmembranes containing domains (TM) by comparing the Hidden Markov Models of different sequence regions. There were 490 genes with predicted SPs for secretion and 1,437 with predicted transmembrane domains. Of the SP, 17% had associations to 48 unique IPR entries and 25% of the TM to 206 unique IPR entries (on line Suppl. Table S5).
The generation and analysis presented here is a valuable addition to resources for the study of hookworms. The A. caninum sequencing data have been submitted to public databases and the functional classifications and sequence characterizations are available on line, and are therefore accessible to researchers working on hookworms and other parasites. The generated data can also serve as a resource for more complete microarrays, RT-PCR, RNA interference and proteomic experiments and analysis. Such studies will aid in the identification of genes involved in host recognition, infection, migration and immune evasion as well as the characterization of targets for vaccines and anthelmintic drugs.
Furthermore, the importance of generating as complete a genome sequence as possible from both Necator and Ancylostoma species is recognized. Complete genome sequences from these species will serve as ongoing references for improved methods of parasite control, not only for drug and vaccine target identification, but also for development of diagnostic tools, drug resistance monitoring, vaccine response tracking, and parasite population surveys. Therefore, among several Strongylida species proposed for genome sequencing in 2006, N. americanus was considered because it is the most prevalent hookworm species infecting humans, and therefore most important from a public health standpoint. A. caninum was recommended as the Ancylostoma species to be sequenced based on its use as a model and the preliminary sequencing data (ESTs and GSSs generated here and in [8]). This genus is made up of very closely related species, which should make comparisons between them relatively easy. In October 2006, the National Human Genome Research Institute announced that the Large-Scale Sequencing Research Network received financial support for sequencing the two hookworm genomes (http://www.genome.gov/10002154). We anticipate that complete annotated genomes for these species are approximately 3-4 years away. Based on the size estimate of the A. caninum genome reported here, nearly 2.7 million ABI3730 reads (average length of 800 bp) would be required for 6X coverage of the genome. The greater genome size increases the need for a physical map to enable sequence assembly. The larger than expected A. caninum genome size together with changing sequencing technology will also lead to a reconsideration of sequencing strategy. For example, one such ‘massively parallel’ sequencing platform is the FLX sequencer from 454 Life Sciences. This is a sequencing system that offers a 100-fold increase in throughput over the current state-of-the-art Sanger sequencing technology on capillary electrophoresis instruments [30]. The apparatus uses a novel fiber-optic slide (PicoTiterPlates) of individual 40 μm wells in which beads containing individual DNA fragments amplified by an emulsion PCR step are subjected to sequencing by synthesis using a pyrosequencing protocol optimized for solid support and picoliter-scale volumes. The FLX Genome Sequencer is capable of generating up to 100 Mb of data in a single run (7 hours) as 200-210 bp reads. No cloning is involved in the sample preparation; therefore no cloning bias can be introduced.
The extended genomic studies can offer a growing and fundamental base of information, which when coupled with downstream functional genomics and proteomics, could shorten the route towards developing more efficient and sustainable control programs.
Supplementary Material
The following data are available on line:
File S1. Consensus sequences of the 1,141 A. caninum repeat families.
Table S1a. The ten most abundant repeats in A. caninum.
Table S1b. Repeat characterization in A. caninum.
Table S2. Ancylostoma caninum genes identified by whole genome shotgun approach.
Table S3. C. elegans RNAi information for genes with A. caninum homologs.
Table S4. KEGG associations for A. caninum and C. elegans.
Table S5. A. caninum genes with predicted signal peptide for secretion and transmembrane domain(s) having InterPro matches.
Acknowledgments
Ancylostoma EST sequencing at Washington University was supported by NIH-NIAID research grant AI 46593 to RKW. JPM is employee and equity holder of Divergence Inc; this research was not company funded.
Abbreviations
- BLAST
basic local alignment search tool
- EST
expressed sequence tags
- GSS
genome survey sequence
- GO
gene ontology
- KEGG
Kyoto encyclopedia of genes and genomes
Footnotes
Note: Nucleotide sequences data reported in this paper are available in the GenBank, EMBL and DDBJ databases under the accession numbers CW698017 - CW717115, CW958972 - CW978588, CZ194948 - CZ250652.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.de Silva NR, Brooker S, Hotez PJ, Montresor A, Engels D, Savioli L. Soil-transmitted helminth infections: updating the global picture. Trends Parasitol. 2003;12:547–51. doi: 10.1016/j.pt.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 2.Bethony J, Brooker S, Albonico M, Geiger SM, Loukas A, Diemert D, Hotez PJ. Soil-transmitted helminth infections: ascariasis, trichuriasis, and hookworm. The Lancet. 2006;367:1521–32. doi: 10.1016/S0140-6736(06)68653-4. [DOI] [PubMed] [Google Scholar]
- 3.Hawdon JM, Jones BF, Hoffman DR, Hotez PJ. Cloning and characterization of Ancylostoma-secreted protein. A novel protein associated with the transition to parasitism by infective hookworm larvae. J Biol Chem. 1996;271:6672–8. doi: 10.1074/jbc.271.12.6672. [DOI] [PubMed] [Google Scholar]
- 4.Hawdon JM, Narasimhan S, Hotez PJ. Ancylostoma secreted protein 2: cloning and characterization of a second member of a family of nematode secreted proteins from Ancylostoma caninum. Mol Biochem Parasitol. 1999;99:149–65. doi: 10.1016/s0166-6851(99)00011-0. [DOI] [PubMed] [Google Scholar]
- 5.Loukas A, Bethony JM, Mendez S, Fujiwara RT, Goud GN, Ranjit N, Zhan B, Jones K, Bottazzi ME, Hotez PJ. Vaccination with recombinant aspartic hemoglobinase reduces parasite load and blood loss after hookworm infection in dogs. PLoS Med. 2005;2:e296. doi: 10.1371/journal.pmed.0020295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Pont AC, Moons AH, de Jonge E, Meijers JC, Vlasuk GP, Rote WE, Buller HR, van der Poll T, Levi M. Recombinant nematode anticoagulant protein c2, an inhibitor of tissue factor/factor VIIa, attenuates coagulation and the interleukin-10 response in human endotoxemia. J Thromb Haemost. 2004;2:65–70. doi: 10.1111/j.1538-7836.2004.00526.x. [DOI] [PubMed] [Google Scholar]
- 7.Mahajan AL, Tenorio X, Pepper MS, Baetens D, Montandon D, Schlaudraff K-U, Pittet B. Progressive tissue injury in burns is reduced by rNAPc2. Burns. 2006;32:957–63. doi: 10.1016/j.burns.2006.02.010. [DOI] [PubMed] [Google Scholar]
- 8.Mitreva M, McCarter JP, Arasu P, Hawdon J, Martin J, Dante M, Wylie T, Xu J, Stajich JE, Kapulkin W, Clifton SW, Waterston RH, Wilson RK. Investigating hookworm genomes by comparative analysis of two Ancylostoma species. BMC Genomics. 2005;6:58. doi: 10.1186/1471-2164-6-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leroy S, Duperray C, Morand S. Flow cytometry for parasite nematode genome size measurement. Mol Biochem Parasitol. 2003;128:91–93. doi: 10.1016/s0166-6851(03)00023-9. [DOI] [PubMed] [Google Scholar]
- 10.Bennett MD, Leitch IJ, Price HJ, Johnston JS. Comparisons with Caenorhabditis (~100 Mb) and Drosophila (~175 Mb) using flow cytometry show genome size in Arabidopsis to be ~157 Mb and thus ~25 % larger than the Arabidopsis initiative estimate of ~125 Mb. Ann Botany. 2003:547–557. doi: 10.1093/aob/mcg057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schad GA. Arrested development of Ancylostoma caninum in dogs: influence of photoperiod and temperature on induction of a potential to arrest. In: Meerovitch E, editor. Aspects of Parasitology: a festschrift dedicated to the fiftieth anniversary of the Institute of Parasitology of McGill University. Montreal: McGill University; 1982. pp. 361–91. [Google Scholar]
- 12.Huang X, Wang J, Aluru S, Yang S-P, Hillier L. PCAP: A Whole-Genome Assembly Program. Genome Res. 2003;13:2164–70. doi: 10.1101/gr.1390403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bao Z, Eddy SR. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 2002;12:1152–5. doi: 10.1101/gr.88502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Griffiths-Jones S, Bateman A, Marshall M, Khanna A, Eddy SR. Rfam: an RNA family database. Nucl Acids Res. 2003;31:439–41. doi: 10.1093/nar/gkg006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;110:462–67. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- 16.Laha T, Loukas A, Wattanasatitarpa S, Somprakhon J, Kewgrai N, Sithithaworn P, Kaewkes S, Mitreva M, Brindley PJ. The bandit, a new DNA transposon from a hookworm - possible horizontal genetic transfer between host and parasite. PloS Neglected Tropical Diseases. 2007;1:e35. doi: 10.1371/journal.pntd.0000035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Laha T, Kewgrai N, Loukas A, Brindley PJ. The dingo non-long terminal repeat retrotransposons from the genome of the hookworm, Ancylostoma caninum. Exp Parasitol. 2006;113:142–53. doi: 10.1016/j.exppara.2005.12.018. [DOI] [PubMed] [Google Scholar]
- 18.Kapitonov VV, Jurka J. RAG1 Core and V(D)J Recombination Signal Sequences Were Derived from Transib Transposons. PLoS Biology. 2005;3:e181. doi: 10.1371/journal.pbio.0030181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ding L, Sabo A, Berkowicz N, Meyer RR, Shotland Y, Johnson MR, Pepin KH, Wilson RK, Spieth J. EAnnot: a genome annotation tool using experimental evidence. Genome Res. 2004;14:2503–9. doi: 10.1101/gr.3152604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ranjit N, Jones MK, Stenzel DJ, Gasser RB, Loukas A. A survey of the intestinal transcriptomes of the hookworms, Necator americanus and Ancylostoma caninum, using tissues isolated by laser microdissection microscopy. Inter Journal Parasitol. 2006;36:701–10. doi: 10.1016/j.ijpara.2006.01.015. [DOI] [PubMed] [Google Scholar]
- 21.Wasmuth JD, Blaxter ML. prot4EST: translating expressed sequence tags from neglected genomes. BMC Bioinformatics. 2004;5:187. doi: 10.1186/1471-2105-5-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wylie T, Martin J, Dante M, Mitreva M, Clifton SW, Chinwalla A, Waterston RH, Wilson RK, McCarter JP. Nematode.net: A Tool for Navigating Sequences from Parasitic and Free-Living Nematodes. Nucleic Acids Res. 2004;32:D423–D26. doi: 10.1093/nar/gkh010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucl Acids Res. 2006;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Apweiler R, Attwood TK, Bairoch A, Bateman A, Birney E, Biswas M, Bucher P, Cerutti L, Corpet F, Croning MD, Durbin R, Falquet L, Fleischmann W, Gouzy J, Hermjakob H, Hulo N, et al. The InterPro database, an integrated documentation resource for protein families, domains and functional sites. Nucl Acids Res. 2001;29:37–40. doi: 10.1093/nar/29.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mulder NJ, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D, Bork P, Buillard V, Cerutti L, Copley R, Courcelle E, Das U, Daugherty L, Dibley M, Finn R, Fleischmann W, Gough J, Haft D, Hulo N, Hunter S, Kahn D, Kanapin A, Kejariwal A, Labarga A, Langendijk-Genevaux PS, Lonsdale D, Lopez R, Letunic I, Madera M, Maslen J, McAnulla C, McDowall J, Mistry J, Mitchell A, Nikolskaya AN, Orchard S, Orengo C, Petryszak R, Selengut JD, Sigrist CJA, Thomas PD, Valentin F, Wilson D, Wu CH, Yeats C. New developments in the InterPro database. Nucl Acids Res. 2007;35:D224–28. doi: 10.1093/nar/gkl841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zdobnov EM, Apweiler R. InterProScan - an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 2001;17:847–8. doi: 10.1093/bioinformatics/17.9.847. [DOI] [PubMed] [Google Scholar]
- 27.Finn RD, Mistry J, Schuster-Bockler B, Griffiths-Jones S, Hollich V, Lassmann T, Moxon S, Marshall M, Khanna A, Durbin R, Eddy SR, Sonnhammer ELL, Bateman A. Pfam: clans, web tools and services. Nucl Acids Res. 2006;34:D247–51. doi: 10.1093/nar/gkj149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.The Gene Ontology Consortium. Gene ontology: tool for the unification of biology. Nat Genet. 2000;25:25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kall L, Krogh A, Sonnhammer ELL. A Combined Transmembrane Topology and Signal Peptide Prediction Method. Journal of Molecular Biology. 2004;338:1027–36. doi: 10.1016/j.jmb.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 30.Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen Y-J, Chen Z, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Irzyk GP, Jando SC, Alenquer MLI, Jarvie TP, Jirage KB, Kim J-B, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, Li J, Lohman KL, Lu H, Makhijani VB, McDade KE, McKenna MP, Myers EW, Nickerson E, Nobile JR, Plant R, Puc BP, Ronan MT, Roth GT, Sarkis GJ, Simons JF, Simpson JW, Srinivasan M, Tartaro KR, Tomasz A, Vogt KA, Volkmer GA, Wang SH, Wang Y, Weiner MP, Yu P, Begley RF, Rothberg JM. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–80. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following data are available on line:
File S1. Consensus sequences of the 1,141 A. caninum repeat families.
Table S1a. The ten most abundant repeats in A. caninum.
Table S1b. Repeat characterization in A. caninum.
Table S2. Ancylostoma caninum genes identified by whole genome shotgun approach.
Table S3. C. elegans RNAi information for genes with A. caninum homologs.
Table S4. KEGG associations for A. caninum and C. elegans.
Table S5. A. caninum genes with predicted signal peptide for secretion and transmembrane domain(s) having InterPro matches.