

Abstract
Structural perturbations in cytochrome P450cam (CYP101) induced by the soluble fragment of rate cytochrome b5, a non-physiological effector of CYP101, were investigated by NMR spectroscopy and compared with the perturbations induced by the physiological reductant and effector, putidaredoxin (Pdx). Chemical shifts of perdeuterated [U-15N] CYP101 backbone amide (NH) resonances were monitored as a function of cytochrome b5 concentration by 1H, 15N TROSY-HSQC experiments. The association of cytochrome b5 with the reduced CYP101-camphor-carbon monoxide complex (CYP-S-CO) perturbs many of the same resonances that Pdx does, including regions of the CYP101 molecule implicated in substrate access and orientation. The perturbations are smaller in magnitude than those observed with Pdxr due to a lower binding affinity (Kd = 13 ± 3 mM, for reduced cytochrome b5-CYP-S-CO complex compared to Kd = 26 ± 12 μM for the Pdx-CYP-S-CO complex). The results are in accord with our previous suggestion that the observed perturbations are related to effector activity and support the proposal that the primary role of the effector is to populate the active conformation of CYP101 to prevent uncoupling [Pochapsky et al. Biochemistry 42, 5649-5656 (2003)]. A titratable perturbation is observed at the 1H resonance of the 8-CH3 of CYP101-bound camphor upon addition of cytochrome b5, a phenomenon also associated with the formation of CYP101·Pdx complex albeit with larger perturbations [Wei et al., J. Am. Chem. Soc. 127, 6974-6 976 (2005)]. The effector activity of the particular rat cytochrome b5 construct used for NMR studies was confirmed by monitoring the enzymatic turnover to yield 5-exo-hydroxy camphor using gas chromatography/mass spectrometry. Finally, the common features of the perturbations observed in the NMR spectra of the two complexes are discussed and their relevance to effector activity considered.
Keywords: cytochrome P450, cytochrome b5, putidaredoxin, NMR, effector
Introduction
Cytochromes P450 are ubiquitous heme-thiolate enzymes that catalyze oxidative modifications of diverse substrates (1). Due to their central role in drug metabolism and involvement in steroid biosynthesis, cytochromes P450 are one of the most intensively investigated metalloenzymes (2). There are nearly 4000 identified P450 genes and structures for 20 unique P450s on deposit in the RCSB PDB. The structurally and biochemically best-characterized cytochrome P450 is P450cam (CYP101) from Pseudomonas putida. CYP101 has provided a paradigm for P450 structure and function studies (3). CYP101 catalyzes the regio- and stereospecific hydroxylation of camphor by dioxygen to 5-exo-hydroxycamphor. Two redox proteins, Pdx reductase (PdR), a flavoprotein NADH reductase, and putidaredoxin (Pdx), a Cys4Fe2S2 ferredoxin, provide the electrons necessary for camphor hydroxylation (4). Pdx serves as the proximal electron donor for CYP101 and sequentially transfers two electrons from PdR to CYP101 per turnover, the second electron transfer being rate-limiting under physiological conditions (5). In addition, Pdx is an effector for substrate turnover by CYP101, and the complex between CYP101 and Pdx is the catalytically competent species (6).
Elucidation of the mechanism of electron transfer and the origin of the effector activity of the in vivo reductant Pdx is essential for understanding the CYP101 reaction cycle. While the availability of structures for CYP101 (3, 7-9) and Pdx (10-14) has provided insight into mechanistic questions, the details of electron transfer and the origins of effector activity have not yet been determined. A number of kinetic (5, 15), mutational (16-18), and theoretical studies (19, 20) have been conducted to gain insight into the specific interactions between Pdx and CYP101. NMR techniques have emerged as an effective method for gaining insight into molecular recognition (21), and have been applied extensively to investigation of the CYP101·Pdx system (22-24). In particular, mapping of chemical shift perturbations upon complex formation using NMR spectroscopy provides a sensitive tool for identifying the residues that are perturbed by these interactions (21). Our group recently reported the application of multidimensional NMR to the characterization of the complex between the Fe(II)-carbonmonoxy-camphor-CYP101 complex (CYP-S-CO) and reduced Pdx (Pdxr) (22). The CYP-S-CO complex provides a convenient diamagnetic model for the one-electron reduced complex of CYP101 with camphor and O2, an obligate intermediate in the reaction pathway. We found that the binding of Pdxr to CYP-S-CO perturbs not only residues in the proposed Pdx binding site on CYP101, but also residues in regions remote from the binding site that are involved in substrate access to and orientation within the CYP101 active site. These observations suggested to us that the primary role of the effector is to prevent the loss of substrate and/or intermediates from the active site and to insure that the substrate is in the correct orientation for reaction by selecting the active (closed) conformation of CYP101 from a manifold of conformations occupied in the absence of effector.
If the Pdx-induced perturbations in the CYP101 structure are indeed critical for effector activity, other identified effectors of CYP101 should induce spectral perturbations similar to those observed in the presence of Pdx. In our previous study, we found that adrenodoxin, a ferredoxin structurally and functionally similar to Pdx that is not an effector of CYP101 turnover, did not induce any obvious spectral changes in CYP101 in molar excesses up to five-fold. On the other hand, cytochrome b5 has previously been reported to act as an effector for camphor hydroxylation by CYP101 (6), and is a good candidate to establish which structural perturbations observed in the presence of Pdx are related to effector activity. Cytochrome b5 is an integral membrane protein consisting of an amino-terminal hydrophilic domain that contains a single heme b with two histidines as axial ligands for the iron and a carboxyl-terminal membrane-binding region (25). The soluble heme-containing domain of cytochrome b5 is small (11 kDa) and structurally well characterized. Cytochrome b5 participates in electron transfer processes in a number of biological redox pathways, and shows effector activity for some class II cytochrome P450s, including CYP3A4 and CYP2B4 (25, 26). The reduction potentials of cytochrome b5 and ferric CYP101 are approximately +25 mV and −300 mV respectively, making cytochrome b5 unable to donate the first electron to CYP101 (25). Sligar and colleagues showed that cytochrome b5 is a competitive inhibitor of electron transfer from Pdx to CYP101, and proposed that cytochrome b5 and Pdx bind to similar sites on the CYP101 surface (27). In current work, we show via multinuclear NMR methods that cytochrome b5 perturbs many of the same resonances in CYP-S-CO as Pdx, including those for residues involved in substrate access to and orientation within the active site of CYP101. We also confirm that the particular construct of cytochrome b5 used for these studies is an effector for CYP101 enzymatic activity. Finally, we discuss the common features of the two complexes in terms of their implications for the mechanism of effector activity.
MATERIALS AND METHODS
Protein Expression and Purification
Perdeuterated [U-15N] CYP101 was over-expressed, isolated and purified from Escherichia coli NCM533 strain harboring the pDNC334A plasmid that encodes the C334A mutant of CYP101 under control of the lac promotor. The C334A mutant is identical to wild-type monomer in terms of enzymatic activity and spectroscopic properties, but does not dimerize even at millimolar concentrations (28). Cell growth was performed at 37°C with shaking, which was started by inoculating a freshly-transformed colony into 5 ml LB supplemented with 25 μg/ml of chloramphenicol (Cmp) and 50 μg/ml of kanamycin (Kan). When the optical density at 600 nm (OD600) reached 0.6, the starter culture was scaled up to 50 ml in LB medium, which was subsequently spun down at OD 0.6, resuspended in 50 ml M9 medium, and incubated for 2 hrs. M9 medium (per liter) contains 6.8 g Na2HPO4, 3 g KH2PO4, 0.5 g NaCl, 1 g NH4Cl, 2 mM MgSO4, 0.04 mM CaCl2, 0.01 mM FeCl3, 0.5% glycerol, 0.25 mg thiamine, 25 mg Cmp, 50 mg Kan, and 2 ml trace metal mix stock solution containing 34.36 g H3BO3, 4.32 g MnCl2· 4H2O, 0.315 g ZnCl2, 0.03g MoO3, 0.003g CuSO4 · 5H2O, 0.012g CoCl2 · 6H2O per liter. The cell culture was then pelleted with centrifugation and resuspended into 1 liter of fresh M9+ media prepared using 15NH4Cl, fully deuterated (d8)-glycerol, and 99.9% D2O (Cambridge Isotope Laboratories, Inc., Andover, MA). When OD600 reached 1.0, 70 mg of porphyrin precursor δ-aminolevulinic acid (Sigma, St. Louis, MO) were added, followed by the addition of IPTG to a final concentration of 1 mM to induce protein expression. Finally the culture was harvested after 12 hrs of incubation.
Purification of CYP101 followed published procedures (29) with modifications. Briefly, 10 g of cell paste resuspended in 40 ml of buffer L (50 mM Tris·Cl, pH 7.4, 50 mM KCl, and 1 mM camphor) were disrupted by sonication using a Misonix sonicator 3000 (Farmingdale, NY) 8 times for 30 sec spaced by 30 sec rest intervals at power setting of 55%. All sonications were performed with cooling in an ice bath. Following centrifugation at 32,000 × g for 50 min to remove the cell debris, 200 mg of protamine sulfate (Sigma) dissolved in 5 ml buffer L were added dropwise with stirring to the protein solution to precipitate DNA, which was then removed by centrifugation. The resulting supernatant was precipitated by addition of ammonium sulfate to a w/v of 70%. After solution of the pellet, dialysis and concentration, the protein was applied to a DEAE sepharose fast flow anion-exchange column (Amersham Biosciences, Piscataway, NJ) and developed with a linear gradient of 50 mM to 300 mM KCl. Finally, the protein solution was subjected to gel filtration on a P-100 column (Bio-Rad) equilibrated and developed by buffer L. The fractions with A391/A280 value greater than 1.45 were pooled and used for the NMR experiments.
Perdeuterated cytochrome b5 was purified from E. coli BL21(DE3) cells containing the plasmid pETb5 that encodes the gene for the 98-residue soluble core domain of rat cytochrome b5 under the control of the T7 promoter (30). Growth and expression of cytochrome b5 followed a procedure similar to that used for CYP101 with the exception that 0.3% d6-glucose (Cambridge Isotope Laboratories) was used as the carbon source in the D2O-based M9 medium. For purification, cell pellets were resuspended in 4 volumes of 50 mM Tris·Cl, pH 7.5, 1.0 mM EDTA and lysed by sonication, followed by centrifugation to remove the cell debris. Cytochrome b5 is isolated primarily isolated as apoprotein from this overexpression system and was reconstituted with hemin (Sigma) prior to purification. Ten milligrams of hemin (iron protoporphyrin IX) were dissolved in a minimal volume of 0.1 N NaOH, diluted 10-fold with H2O, and add dropwise to the lysate containing apo-cytochrome b5 with stirring. Reconstitution was monitored by periodically removing aliquots for spectroscopic assay and was terminated when the absorbance at 414 nm maximized and a shoulder began to appear on the same peak. Excess hemin was then precipitated by lowering the pH of the protein solution to 6.0 and removed by filtration though 0.45 μm filter paper. The clarified cytochrome b5 holoprotein solution was loaded onto a Whatman DE-52 ion exchange column and eluted with a linear 0 mM to 0.4 mM NaCl gradient at a 1 ml/min flow rate. Fractions with A413/A280 value more than 3.5 were pooled and concentrated using Amicon centrifugal concentrators (Millipore, Bedford, MA) and applied to a Biogel P-30 (Bio-Rad) size exclusion column. Fractions with A413/A280 more than 5.7 were pooled, concentrated, and used for NMR experiments. Protein concentrations and absorbance ratios for both enzymes were estimated by UV-visible spectroscopy using available extinction coefficients (29, 30). Conditions for preparation and purification of isotopically-labeled Pdx have been described previously (22).
NMR sample preparation
Concentrated protein samples were exchanged either into 10% D2O/90% H2O NMR buffer (50 mM Tris-d11·HCl, pH 7.4, 100 mM KCl, and 2 mM d-camphor) for 1H-15N HSQC experiments or into 100% D2O NMR buffer for one-dimensional NMR experiments using spin columns packed with P-2 resin (Bio-Rad). Reduction of CYP101 (0.5 mM), Pdx (3 mM), and cytochrome b5 (9 mM) was achieved by addition of aliquots of 0.25 M and 1 M sodium dithionite (prepared in degassed 1 M Tris·Cl, pH 8.0), respectively, with a slight excess of reductant compared to the amount of protein. All protein samples were placed under a CO atmosphere prior to and after the addition of the reducing agent for 10 min and 2 min, respectively. CYP-S-CO samples thus obtained were then transferred to an NMR sample tube (Shigemi, Inc., Allison Park, PA) inside an anaerobic chamber under a N2 (90%)/H2 (10%) atmosphere, and the NMR tube was sealed with Parafilm (Pechiney Inc., Menasha, WI) before removal from the anaerobic chamber. Aliquot additions of the desired protein (cytochrome b5 or Pdxr) to the deuterated 15N-labeled CYP101 samples for NMR titrations were performed in the anaerobic chamber. For titrations with cytochrome b5, the molar ratio between the two proteins was varied from 0 to 16, in 6 titration points. The Pdxr titration was performed as described previously (22).
NMR experiments
NMR titrations of CYP-S-CO with cytochrome b5 were performed at 298 K on a 14 T Varian UNITY Inova spectrometer operating at 599.702 and 60.774 MHz for 1H and 15N, respectively. The spectrometer is equipped with a pulsed field gradient amplifier and a 5 mm 1H {13C, 15N} triple resonance PFG probe. 15N, 1H TROSY-HSQC spectra (31) were acquired to follow the chemical shift perturbations of NH coupled pairs during titrations. For these experiments, gradient coherence selection was used to select desired coherences and suppress solvent signals, and the Rance-Kay method was used to obtain quadrature detection in the indirectly detected dimension (32, 33). Spectra were acquired with 596 (1H) × 128 (15N) complex points, 1H sweep width of 8000 Hz, and 15N sweep width of 2200 Hz. All 1H and 15N chemical shifts (δ) are reported in parts per million relative to trimethylsilylpropionic acid sodium salt. All of the NMR spectra were processed and analyzed using the Topspin software package (Bruker Biospin). Conditions for the titration of CYP-S-CO with Pdxr were described previously (22).
Estimation of Kd for the cytochrome b5-CYP-S-CO complex
For eighteen 15NH pairs with well-resolved resonances for each titration point, 1H chemical shifts δ measured in Hertz were fit to the following standard equation using the nonlinear regression analysis package of Mathematica 5.2:
| Eq. 1 |
where δ0 and δmax are the chemical shifts of the free and bound forms of CYP101, respectively, and m0 and p0 are the nominal concentrations of CYP101 and cytochrome b5, respectively. The fits yielded values for δmax and Kd.
Enzymatic substrate turnover with cytochrome b5 as the effector
The turnover of camphor yielding 5-exo-hydroxycamphor by CYP101 with cytochrome b5 as the effector was monitored using gas chromatography/mass spectrometry (GC/MS) (6). CYP101 in 50 mM potassium phosphate buffer (pH 7.4) was added anaerobically to a solution containing 1 mM camphor, 4 μM proflavine, and 10 mM EDTA to a final CYP101 concentration of 4 μM. The CYP101 was then photoreduced by exposure to a 400-watt white light source. The mixture was then oxygenated by injection of air and cytochrome b5 was injected to a final concentration of 64 μM to initiate the hydroxylation reaction. After one hour, the mixture was extracted with dichloromethane and analyzed with an Hewlett-Packard 5890 GC/MS spectrometer equipped with an HP-5MS column (dimension: 30 m × 0.25 mm × 0.25 μm, Agilent Technologies, Inc., Palo Alto, CA), quadrupole mass spectrometer and flame ion detector. The injector and flame ion detector were maintained at 220°C and 230°C respectively, and a split ratio of 3:1 was used. The He carrier flow rate was maintained at 1.0 ml/min. The temperature program was: 60°C for 3 min and 60°C – 210°C at a rate of 5 °C/min. Camphor (m/z = 152) elutes at 9.0 min and hydroxycamphor (m/z = 168) elutes at 12.3 min under these conditions. Pdx added to a similar reaction mixture provided a positive control, and the photo-reduced CYP101 solution without effector added provided the negative control.
RESULTS
Chemical shift perturbations in the Pdxr-CYP-S-CO complex
We have previously described the use of two-dimensional 1H-15N TROSY-HSQC NMR experiments to monitor the titration of perdeuterated [U-15N] CYP-S-CO with Pdxr (22). As a significant number of backbone resonances have been additionally assigned in the CYP-S-CO spectrum since our last report (22), the results of those titrations can now be more completely analyzed. Furthermore, some ambiguities concerning perturbations in crowded regions of the spectrum have been resolved, and a number of resonances that we previously reported as unperturbed are now seen to be perturbed upon addition of Pdxr. Among the newly assigned resonances, perturbations are observed at the backbone NH resonances of Arg 109, Phe 111, Ala 115, and Val 118 in the N-terminal and middle regions of the C helix (residues 106-126), Ser 190 in the F-G loop, Thr 192 and Phe 193 in the N-terminal end of the G helix (residues 192-214), Thr 252 in the center of the I helix (residues 234-267), and residues His 347, Phe 350, Gly 351, Gly 353, Ser 354, Leu 358 and Gly 359 within the Cys ligand loop (residues 350-357). The NH resonance of the axial heme ligand, Cys 357, has not yet been identified, but the NH resonance of Leu 358 is shifted upfield by 2.75 ppm due to the shielding effect of the heme, an unusually large shift, and it is possible that the Cys 357 NH proton resonates near or under the water peak at 4.78 ppm. Residues previously classified as ambiguous or unperturbed by Pdx addition that are now classified as perturbed include Gly 37, Ser 48, Val 50 and Asp 52 in the A helix and A-β1 loop, Ala 92 in the B’ helix, Gln 108 in the C helix, Gly 168 and Glu 173 in the E-F loop, Ala 196 and Ile 208 in the G helix, Val 338, Ile388 and Gln 289 in the β5 sheet. All of these observations support our previous identification of the CYP101 structural features perturbed by binding of Pdx (22). In particular, the first three turns of the C helix (residues 107-118), previously proposed as a primary interaction site for Pdx (19), are now seen, along with the preceding B’ helix, to be among the CYP101 structural features most uniformly affected by Pdx binding.
Previously, we classified the CYP-S-CO resonances perturbed by Pdxr binding as either in the slow/intermediate exchange or fast exchange regimes (22). A non-linear fit of chemical shift as a function of Pdx concentration to Eq. 1 for resonances in the fast exchange regime yields a value of Kd = 26 ± 12 μM for the Pdxr/CYP-S-CO complex (Ref. 22, Fig. 1A). The apparent exchange rate of the complex at half-saturation was estimated from line width measurements for resonances in the fast-exchange regime to be ~300 s−1 at 17 °C (22, 34). As expected, all resonances in the fast exchange regime show 1H shift perturbations with Δδmax < 300 s−1, as determined from the value for Δδmax = δo - δmax obtained from fits of experimental shifts to Eq. 1.
Figure 1.
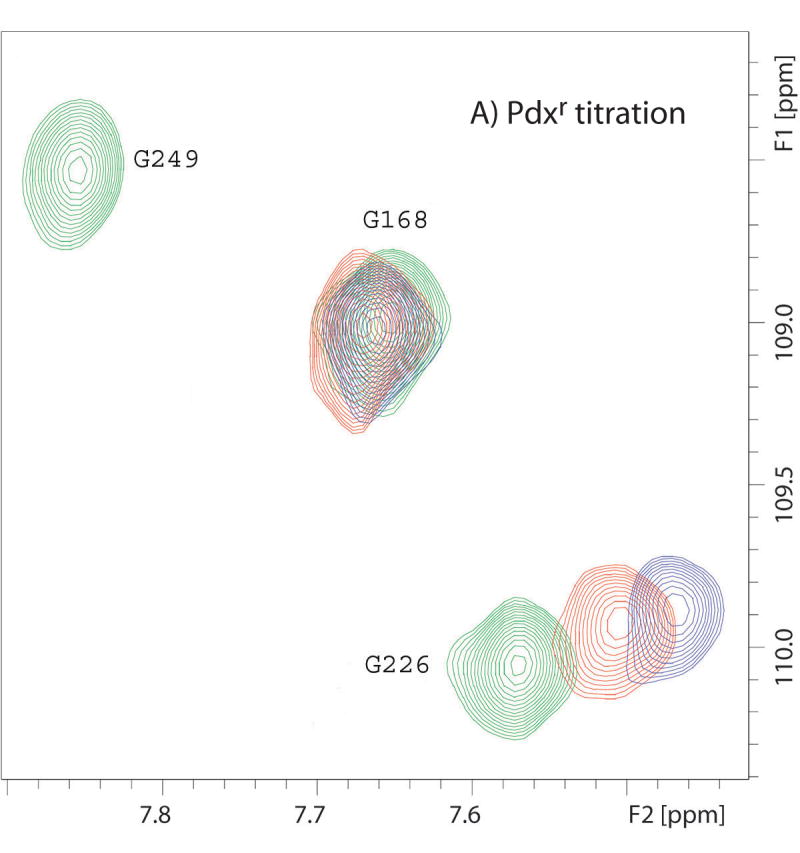
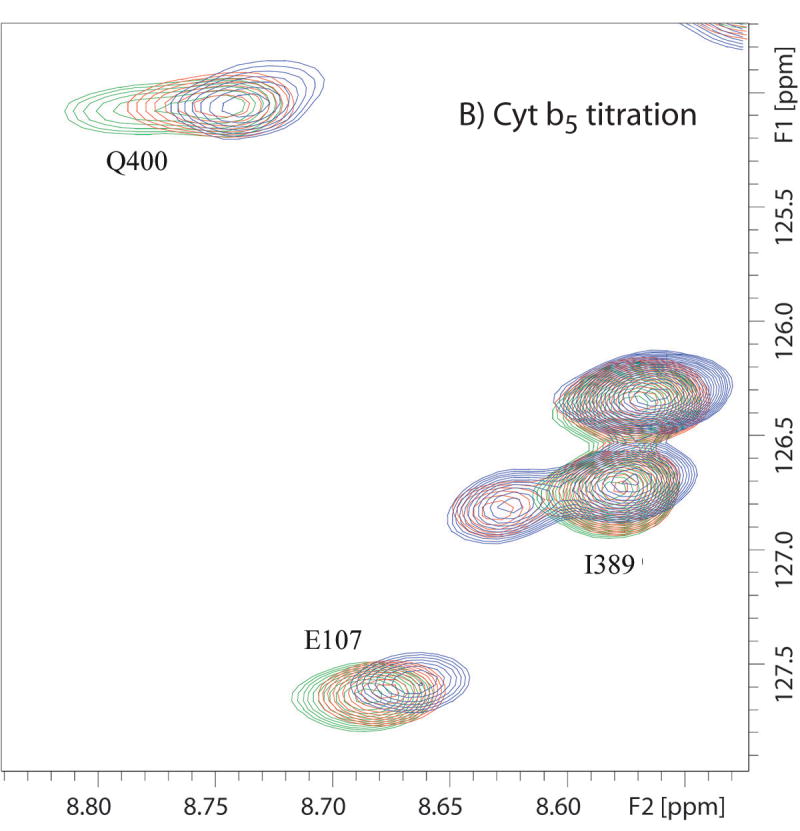
Superimposed 14T (600 MHz 1H) 1H-15N TROSY-HSQC spectra showing different types of effects of titration of CYP-S-CO with Pdxr or reduced cytochrome b5. Color coding of peaks are follows: green corresponds to no Pdx or cytochrome b5, red to ~0.5 equivalent of Pdx or 4 equivalents of b5, and blue to 1 equivalent of Pdx or 16 equivalents of b5. (A) Portions of superposition of the amide region of the 1H-15N TROSY-HSQC spectra (600 MHz 1H, 10% D2O/90% H2O, 50 mM Tris-d11 · HCl, pH 7.4, 100 mM KCl, and 2 mM d-camphor) as a function of added Pdxr. Gly 226 is shifted upon Pdx addition, Gly 168 is unperturbed by Pdx, while Gly 249 is broadened to invisibility at the first addition of Pdx. (B) Portions of superposition of the amide region of the 1H-15N TROSY-HSQC spectra (600 MHz 1H, 10% D2O/90% H2O, 50 mM Tris-d11 · HCl, pH 7.4, 100 mM KCl, and 2 mM d-camphor) as a function of added cytochrome b5. Glu 107 and Gln 400 are shifted upon cytochrome b5 addition, while Ile 389 shows peak splitting (indicating with arrow) that increase in intensity with increasing cytochrome b5 concentration.
Resonances identified as being in the slow/intermediate exchange regime are broadened to invisibility upon the first addition of Pdxr (CYP101:Pdx molar ratio of 5:1), and are for the most part observed for residues in proximity to the heme and/or active site, including the heme axial ligand loop containing Cys 357 and portions of the C and I helices (see Figure 5C). Based on their disappearance in the first titration point, they are assumed to have chemical shift differences between the Pdx-bound and free forms Δδ > 300 s−1 at 17°C, although the chemical shifts of these resonances in the bound form have not confirmed. Recently, we detected a high-barrier conformational shift in the active site of CYP101 that occurs upon Pdx binding, and by comparing chemical shift changes for the 8-CH3 (which is at slow exchange) and 9-CH3 (which is at fast exchange) of bound camphor (34), we estimated an exchange rate over this barrier between 150 s-1 and 300 s-1 at 25 °C. Based on the similarities of the rates obtained from two different observations (line widths (22) and camphor 1H shifts (34), as well as the proximity of NH resonances undergoing slow/intermediate exchange to the active site, we find it likely that the same event is responsible for both observations. The precise nature of this high-barrier event is currently under investigation in our laboratory.
Figure 5.




The CYP101 structure (3CPP) color-coded to show distribution of secondary structural features perturbed by Pdx or cytochrome b5 as determined by NMR. Structural features shown in dark blue are not affected by either Pdx or cytochrome b5 binding, those shown in yellow are perturbed (δmax < kex), orange if peak splitting occurs, and in red for resonances that are essentially undetectable after the first addition of Pdx (δmax ~ kex). Features for which insufficient data are available are shown in light blue. The heme and camphor are shown in pink. A) Perturbation caused by Pdxr, updated from Fig. 4 of Ref. 22 to include newly assigned residues. B) Perturbations caused by cytochrome b5. C) Only resonances that are essentially invisible after 1/5 of an equivalent of Pdxr (shown in beige) are localized on the CYP101 structure and shown in red. Complex shown is that proposed in Ref. 19. D) Interior and partially solvent exposed residues in the CYP101 structure affected by both Pdx and cytochrome b5 titration. Perturbed residues are shown in yellow. The figures were generated using MolMol (40).
In addition to these two categories of perturbations, some resonances, most prominently around the region of A-β1 loop (residues 47-51) and β1 strand (residues 52-66), are observed to split into two peaks during the titration and the intensities of the new peaks increase with the increasing Pdxr concentration. Although this splitting represents a slow-exchange event, the lack of any significant line broadening associated with the splitting, as well as the small chemical shift differences between the split peaks (less than 50 Hz, in some cases) indicates that the exchange involved is considerably slower than that associated with the high-barrier event discussed above, and is likely not related to the conformational changes responsible for the slow/intermediate exchange perturbations discussed in the preceding paragraph. As such, it appears that there is more than one slow conformational change induced by Pdx in the CYP101 structure, one that affects the active site, substrate and adjacent structural features (the “high barrier event” (34)), and another, slower event that connects conformations in regions remote from the active site in the A-β1 loop and β1 strand.
Chemical shift perturbations in CYP-S-CO due to cytochrome b5
Two-dimensional 1H-15N TROSY-HSQC NMR experiments were used to monitor the titration of perdeuterated [U-15N] CYP101 with unlabeled cytochrome b5. Chemical shift perturbations were observed in the spectrum of CYP-S-CO upon binding of cytochrome b5 (Fig.1B). Unlike Pdx, we did not observe the disappearance or significant exchange broadening of any amide 1H resonance in the CYP-S-CO complex due to the addition of cytochrome b5, although a slight exchange broadening is observed at high resolution to the camphor 8-methyl peak (Fig. 2). Instead, chemical shifts change in a continuous fashion during the titration, that is, a single resonance peak is observed for each perturbed resonance of CYP-S-CO at any point during the titration. These results indicate that CYP-S-CO·cytochrome b5 complex is in fast exchange on the 1H chemical time scale between bound and unbound forms. In general the chemical shift changes observed in CYP-S-CO upon complexation with cytochrome b5 are smaller in magnitude than those induced by Pdxr binding (Fig. 3). However, the perturbations can be interpreted with confidence and only peaks with Δδmax(obs) > 10 Hz are considered as being perturbed by cytochrome b5 addition. Nonlinear fits of chemical shift changes for eighteen affected resonances to Eq. 1 as of function of cytochrome b5 and CYP-S-CO concentrations yields a Kd of 13 ± 3 mM at 50 mM Tris·Cl buffer at pH 7.4 and 25°C with 100 mM KCl and 2 mM camphor. The uncertainty represents the reproducibility of fitting the 1H chemical shift induced by titration with cytochrome b5 for eighteen residues in CYP-S-CO.
Figure 2.

Superposition of the upfield region of 600 MHz 1H NMR spectra showing titration of perdeuterated CYP-S-CO with perdeuterated cytochrome b5 in 100% D2O, 50 mM potassium phosphate buffer, pH 7.4, 50 mM KCl. The 8-CH3 group of camphor shifts downfield upon cytochrome b5 titration, the 10-CH3 moves very slightly (Δδmax(obs) < 10 Hz) but noticeably downfield. The camphor 9-methyl resonance is not appreciably perturbed (peak not shown).
Figure 3.

Comparison of the magnitudes of the chemical shift changes for the NH correlation of the same residue (Glu 107 in the C helix) of CYP101 upon complexation with Pdxr and cytochrome b5 in superimposed 14 T (600 MHz 1H) 1H-15N TROSY-HSQC spectra. Color-coding of peaks follows the convention in Fig. 1.
As was the case with Pdx, we observe that some NH resonances of CYP101 split in the presence of cytochrome b5, indicating that a minor conformation is populated under these conditions at slow exchange with the major ensemble. Splittings are observed primarily in the A-β1 loop, the N terminal of β1 strand, the C and G helices, with scattered resonances splitting in the A helix, B helix, E helix, and β2 sheet. As was the case with Pdx, the structural origin(s) of this phenomenon are unknown, but it is apparently not related to the high-barrier conformational shift that occurs when Pdx binds to CYP101 (see reference 34), and no phenomenon corresponding to the high-barrier event is observed in the course of the cytochrome b5 titration.
Perturbations induced in CYP101 by cytochrome b5 binding were mapped and the distribution of the perturbations related to both the sequence and the three-dimensional structure of CYP-S-CO (Figs. 4 and 5A). As can be seen from these figures, cytochrome b5 binding perturbs many of the same regions that Pdx binding does (22), including the C helix, portions of the F helix, F-G loop and G helix as well as portions of the Cys 357 loop. Other regions perturbed by both Pdx and cytochrome b5 include both ends of the E-helix and portions of β3 and β5 sheets. A number of regions are affected only by cytochrome b5 and not by Pdx, including several residues in the F-G loop (Arg 187 and Gly 190), one strand of the β5 sheet (Gly 146-Asn 149), and residues near the C-terminal of CYP101, Ala 409, Thr 410 and Val 414.
Figure 4.

The amino acid sequence of CYP101, color-coded to show the structural perturbations due to Pdxr and cytochrome b5 mapped via 1H, 15N-HSQC experiments. Wavy red lines indicate helices, and boat-shaped lines in black indicate β-sheets. Secondary structural features are named above the sequence using the nomenclature of Raag and Poulos (8). Residues whose amide NH groups are perturbed in CYP-S-CO upon addition of either cytochrome b5 or Pdxr are backgrounded in beige, in green if perturbed only by Pdxr, and in purple if perturbed only by cytochrome b5. Residues bordered in red are broadened to invisibility or shifted to unknown positions upon the first addition of Pdxr, those bordered in dark blue indicate resonances that are unperturbed by either Pdxr or cytochrome b5 addition, those bordered in light blue are residues whose NH correlations have been assigned but for which perturbations due to effector addition are ambiguous due to spectral overlap.
Perturbation of bound camphor by cytochrome b5
We titrated samples of perdeuterated CYP-CO-S in D2O with perdeuterated cytochrome b5 in order to observe any effects the addition of cytochrome b5 might have on the orientation of camphor bound in the active site. Unlike Pdx, reduced cytochrome b5 has a large number of resonances in the same region of the 1H NMR spectrum as the methyl groups of bound camphor. Although perdeuteration of cytochrome b5 significantly reduces the interference of the cytochrome b5 signals with the camphor methyl 1H signals, at higher ratios of cytochrome b5 to CYP-S-CO, residual protonation of cytochrome b5 results in some complexity in this region of the spectrum. Still, the three methyl signals of CYP101-bound camphor, 10-CH3, 8-CH3, and 9-CH3 in the upfield region of the 1H spectrum of perdeuterated CYP-S-CO can be observed in the absence and presence of perdeuterated cytochrome b5 and based on the assignments reported recently by our group (34) can be followed with confidence during the titration. Of the three camphor methyl signals, the 9-CH3 resonance did not show significant perturbation, the 10-CH3 1H resonance showed some minimal titratable movement with Δδmax(obs) less than 10 Hz, and the 8-CH3 1H resonance is perturbed slightly (Δδmax = 30 Hz) by the addition of cytochrome b5 (Fig. 2). The 1H resonance of 8-CH3 moves in a continuous fashion during the titration with only minimal line broadening, again indicating that a fast exchange event is being monitored.
Confirmation of cytochrome b5 effector activity
The original reports concerning the effector activity of cytochrome b5 in the reconstituted CYP101 turnover assay were made using a trypsin-solubilized form of cytochrome b5 obtained from liver homogenates (6). As a bacterially-expressed cytochrome b5 construct was used in our experiments, we needed to confirm that this construct also exhibits effector activity in the reconstituted CYP101 system. We used GC-MS to detect CYP101 substrate turnover in the presence of cytochrome b5 and have confirmed that the current construct used in the NMR studies is capable of supporting turnover under the conditions reported by Lipscomb and Gunsalus (6). Addition of either cytochrome b5 or Pdx to photoreduced and oxygenated camphor-bound CYP101 resulted in the formation of 5-hydroxycamphor (Fig. 6) under the reported conditions (6), while in the absence of effector, hydroxycamphor formation was not detected. The experiment was done in triplicates and the results were consistent.
Figure 6.

Elution profile of substrate turnover by CYP101 using cytochrome b5 as effector monitored by GC-MS. MS data for substrate (camphor, eluted at 8.97 min with m/z 152) and product (5-hydroxy-camphor, eluted at 12.26 min with m/z 168) are shown in the inserts. Reaction conditions are detailed in Materials and Methods.
DISCUSSION
A model for cytochrome b5-CYP101 interactions
As was previously observed with Pdx, the titration of CYP-S-CO with cytochrome b5 affects structural features throughout the CYP101 molecule. However, the perturbations observed with cytochrome b5 are generally smaller than the corresponding perturbations in the Pdxr-CYP-S-CO complex. The calculated dissociation constant for the CYP-S-CO complex with cytochrome b5 (13 ± 3 mM at 25 °C) is three orders of magnitude higher than that of CYP-S-CO · Pdxr complex calculated previously at 17°C, 26 ± 12 μM (22) (Fig. 7). As it takes less than three equivalents of Pdx to saturate CYP101 in the complex, the experimental Δδmax(obs) obtained from the last reported titration point with Pdx (2:1 Pdx/CYP101) are close to the Δδmax calculated from non-linear fits to Eq. 1 (22). It would require approximately 30 equivalents of cytochrome b5 to saturate CYP-S-CO based on the calculated Kd for that complex. However, such a large excess of cytochrome b5 is not practical in multidimensional NMR experiments. Consequently, we chose 16 equivalents of cytochrome b5 as the final titration point and under these conditions, observed chemical shift perturbations are not at a maximum.
Figure 7.

Normalized chemical shift perturbations of amide proton resonances in CYP101 as a function of titrant concentration (Pdxr or cytochrome b5).. Vertical axis values are Δδ at each titration point normalized to (δmax − δ0), where δmax was calculated by fitting titration data to Eq. 1 using the nonlinear regression analysis package of Mathematica 5.2. Chemical shifts of five residues in CYP101 monitored throughout the Pdxr titration and ten residues monitored through the cytochrome b5 titration were fit to the binding curves. The curves represent the expected normalized chemical shift value given the calculated Kd and starting CYP101 concentration (see Experimental section for further details). For a CYP-S-CO starting concentration of 0.5 mM in both cases, the Pdxr titration shows almost complete saturation at 1 mM Pdx, while the curve for the cytochrome b5 titration is still nearly linear at much higher cytochrome b5 concentrations. Error bars represent the maximum deviation of the experimental chemical shift values for the resonances used in the analysis. The larger error for cytochrome b5 versus Pdx titration points is due to the increased uncertainty of the value of δmax obtained from the fit of the cytochrome b5 data to Eq. 1.
Sligar and Stayton demonstrated that the same cytochrome b5 construct used in our study is a competitive inhibitor of Pdx binding to CYP101 and proposed that portions of the Pdx binding site and cytochrome b5 binding site(s) overlap on the surface of CYP101 (27). Specifically, they implicated Arg 72 (B helix), Arg 112 (C helix), Lys 344, and Arg 364 (in the L helix following the loop containing the heme axial ligand Cys 357) as possibly interacting with cytochrome b5 on the proximal surface of CYP101. Based on the perturbations we observe, it is likely that cytochrome b5 interacts with CYP101 directly at the C helix and the Cys ligand loop. Also, residues near to Arg 72 in the B helix and near Lys 344 are perturbed, although neither of those residues are assigned as yet. However, none of the assigned resonances in the L helix are perturbed by cytochrome b5, suggesting that Arg 364 is not involved in the interaction. Furthermore, the possibility of direct interaction between cytochrome b5 and CYP101 away from the proximal surface is suggested by doubling of resonances corresponding to residues in the G helix and residues in the A-β1 loop and β1 sheet in the presence of cytochrome b5.
Comparison of complexes of cytochrome b5 and Pdx with CYP101
Comparison of perturbation patterns in the CYP101 structure induced by cytochrome b5 and Pdx suggest similarities between the two complexes. Although the extent and precise patterns of affected residues differ, most regions of the CYP101 structure that are affected by Pdx binding are perturbed to some extent by cytochrome b5 as well (Fig. 4). The secondary structural features that are most uniformly perturbed by both cytochrome b5 and Pdx include the N-terminal regions of the A (residues 36-39) and C helices (residues 107-113), the A-β1 loop (residues 48-52) and the loop containing the heme axial ligand cysteine (residues 351-354). Other regions that are perturbed in both complexes include the N-terminal of the B helix, portions of the B’, G and I helices. The B’ helix is uniformly perturbed by Pdxr, although only two residues, Glu 91 and Ala 95, are noticeably perturbed by cytochrome b5.
In many cases, the differential perturbation patterns due to Pdx and cytochrome b5 within affected secondary structural features may simply be a reflection of the weaker interaction of cytochrome b5 with CYP101 relative to Pdx, and perturbations that are apparent with Pdx are too small to be detected in the absence of saturating cytochrome b5. Nevertheless, as seen in the comparison in Fig. 5, there are differences between the complexes besides the differences in binding affinity. Parts of the of β2 sheet (residues 226-233) are perturbed by Pdx association, but not by cytochrome b5; in contrast, binding of cytochrome b5 perturbs the β5 strand (residues 146-150) preceding the E helix as well as some C-terminal and N-terminal residues that are not affected by Pdxr. Taken together, these observations suggest that the binding sites on CYP101 for Pdx and cytochrome b5 are not identical but overlapping, and that cytochrome b5 is less specific in its interactions with CYP101 than is Pdx.
The most obvious difference between the two complexes is the apparent absence in the cytochrome b5 complex of the high barrier conformational change observed upon binding of Pdxr to CYP-S-CO. This conformational change results in a reorientation of the camphor molecule within the active site of CYP101 and is detected as a broadening to essential invisibility of many backbone amide resonances for residues adjacent to the heme and camphor binding site (vide supra and Ref .34). As a result of this conformational shift, most identified residues in the central I helix are heavily perturbed by Pdx binding, especially the central part of the helix containing residues bracketing the heme and active site. The event likely involves reorganization of side chains in the active site and may be responsible for the large chemical shift changes that occur for many functionally important residues (Leu 244, Leu 245, Leu 246, Gly 248, Gly 249, leu358, and Gly359) upon binding of Pdxr. Fig. 6 shows the localization of the residues that are affected by the high-barrier event as reflected by the fact that they are undergoing perturbations that are slow in the 1H chemical shift time scale upon binding of Pdx. The change is profound and appears to be forced with almost 100% efficiency by the binding of Pdxr (that is, the equilibrium constant for the conformational change lies far to the right when Pdx is bound). However, upon cytochrome b5 binding (Fig. 5B), perturbations to the I helix are much smaller in magnitude and only Leu 250 and Thr 252 are appreciably perturbed among the eight assigned residues in this region (Figs. 4 and 5). Likewise, although both the C helix and Cys 357 heme ligand loop are affected by cytochrome b5, evidence for the high-barrier conformational change is absent. The camphor 8-CH3 resonance is perturbed upon cytochrome b5 binding (Fig. 2), and the shift is in the same down-field direction as observed upon titration with Pdx (34). However, the magnitude of the effect is much smaller and fast on the chemical shift time scale in the case of the cytochrome b5- CYP-S-CO complex.
Implications for effector activity
It was demonstrated some years ago that trypsin-solublized bovine cytochrome b5 is an effector for camphor hydroxylation by CYP101, albeit at much higher concentrations for full activity than the physiological effector and reductant Pdx. The effector concentrations necessary for the maximum yield of 5-exo-hydroxycamphor from photo-reduced camphor-bound O2-CYP101 were 0.16 μM and 2.6 μM for Pdx and cytochrome b5, respectively (6). The current work indicates that, like Pdx, cytochrome b5 binding perturbs many of the regions involved in substrate access and orientation (1, 7, 8), and supports our previous suggestion that the primary role of the effector is to select the catalytically active conformation from an ensemble of conformations that are occupied in solution by the CYP101 molecule. In this conformation, active site access from solution is blocked, preventing loss of substrate/intermediate, and the substrate orientation is appropriate for the observed chemistry. Note that this model does not preclude the possibility that effector binding may also make the rate-limiting second electron transfer more favorable, but currently, direct evidence for this is lacking.
For insight into the conformational changes involved in effector activity, one can look at perturbations that occur upon effector binding to CYP101 structural features with minimum surface exposure. Structural perturbations mapped by NMR may be caused by direct contact with a bound molecule or indirectly via conformational changes induced by the binding event (34). Fig. 5D shows perturbations that occur due to binding of both effectors at residues that are partially or completely buried in the CYP101 structure. “Hot spots” for such changes are contact points between secondary structural features. The region of contact between helices G, H and I is one such place, as are points of contact between helices F, G, B’, the F-G loop and the β5 sheet. Again, many of these structural features have been implicated in substrate access and orientation (7, 35, 36). In a recent crystallographic study, it was found that binding to CYP101 of a modified camphor linked to an external ruthenium sensitizer exhibits a more open substrate access channel that involves displacements in the distal B’, F and G helices as well as conformational changes in the I helix relative to the positions of these features in closed-channel CYP101 structures (37). It was suggested by the authors of this work that the F and G helices translate relative to the protein core in order to open the access channel and cause the N-terminus of the I helix to rotate in order to conserve interhelical contacts. Based on the localization of internal changes to contact points between secondary structures detected in the current work, it is likely that internal effects on the CYP101 structure detected by NMR upon binding of effector are transmitted in a similar fashion.
It is worth considering how the internal perturbations discussed above might be transmitted from the effector binding site(s) on the proximal face of the protein (to the left in Fig. 7) to the affected structural features on the distal side (the F, G and B’ helices, F-G loop and β5 sheet). It has been proposed that Pdx binds at the proximal surface of CYP101 and that electrostatic interactions involving Arg 109 and Arg 112 on the C helix contribute to the specificity of the association between these physiological redox partners (18, 19). It has also been proposed that Arg 112 forms part of the electron transfer pathway in the CYP101·Pdx complex (18, 20). We observe that the C helix and Cys 357 loop are affected by both Pdx and cytochrome b5 and are likely sites for interaction with both proteins. One possible route for mechanical transmission of effector-induced perturbations from the proximal to distal side is via contacts between the C helix and the N-terminal region of the long I helix. A second route is via the irregular structure connecting the B’ and C helices. This connection is also close to the Cys 357 loop. Together, the two pathways bracket the heme, and appear ideally situated to provide a “pincer” closure of the active site. Unfortunately, both the N-terminal of the I helix and the connection between B’ and C have proven to be difficult to assign, and until backbone resonances assignments are completed in these regions, these transmission pathways remain conjectural. As discussed above, the possibility that cytochrome b5 may interact directly with the G helix cannot be discounted. However, such a binding mode would not seem to provide the mechanical stability needed to force closure or rearrangement of the substrate access channel.
Finally, the role of the high-barrier conformational shift observed in the active site of CYP101 upon addition of Pdx (34) in effector activity, if any, must be considered. While there is no direct evidence linking this conformational change to effector activity, we have recently found that in a mutant of CYP101, L358P, that exhibits many of the same spectral shifts as the Pdx-CYP-S-CO complex and has some constitutive camphor hydroxylation activity in the absence of Pdx (38), the camphor orientation is similar to that found in the Pdxr-CYP-S-CO complex (that is, the conformation forced by Pdx binding) rather than that observed in wild-type CYP-S-CO alone (B. OuYang and S. Pochapsky, unpublished results). This indicates that the conformation forced by binding of Pdxr to wild-type CYP-S-CO is stabilized in the absence of Pdx in the L358P mutant, and suggests that the constitutive activity of the L358P mutant may linked to this conformational change. If the high-barrier conformational change observed upon addition of Pdx in WT CYP101 is essential for camphor hydroxylation (that is, if it is essential to effector activity) it is possible that a small fraction of the CYP101·cytochrome b5 complex exists in this conformation, but the equilibrium constant for the conformational change is smaller than in the Pdxr-CYP-S-CO complex, and so is present to such a small extent that it is not readily detectable by NMR methods. A more complete analysis of NMR relaxation behavior of the CYP101-effector complex may provide insight into this possibility (39).
Acknowledgments
The authors are grateful to Luet-Lok Wong of Oxford University, Oxford, UK. for the plasmid pDNC334A that expresses CYP101 C334A and Prof. J. T. L. LeComte (Pennsylvania State University) for the plasmid pETb5 that expresses the soluble domain of cytochrome b5. This work was supported in part by a grant from the U.S. Public Health Service, R01-GM44191 (T.C.P.).
ABBREVIATIONS
- Cmp
chloramphenicol
- CYP101
cytochrome P450cam
- CYP-S-CO
carbon monoxide and camphor-bound reduced cytochrome P450cam
- GC/MS
gas chromatography/mass spectroscopy
- IPTG
isopropyl β-D-thiogalactoside
- Kan
kanamycin
- LB
Luria-Bertani medium
- NMR
nuclear magnetic resonance
- HSQC
heteronuclear single-quantum correlation
- M9
minimal growth medium
- OD600
optical density at 600 nm
- PdR
putidaredoxin reductase
- Pdx
putidaredoxin
- Pdxr
reduced putidaredoxin
- TROSY
transverse relaxation optimized spectroscopy
Footnotes
This work was supported in part by a grant from the US Public Health Service R01-GM44191 (T.C.P)
References
- 1.Mueller EJ, Loida PG, Sligar SG. Twenty-five Years of P450cam Research. In: Ortiz de Montellano PR, editor. Cytochrome P450: Structure, Function and Biochemistry. Plenum Press; New York: 1995. pp. 83–124. [Google Scholar]
- 2.Denisov IG, Makris TM, Sligar SG, Schlichting Ilme. Structure and Chemistry of Cytochrome P450. Chem Rev. 2005;105:2253–2277. doi: 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]
- 3.Poulos TL, Finzel BC, Howard AJ. High-resolution crystal structure of cytochrome P450cam. J Mol Biol. 1987;195:687–700. doi: 10.1016/0022-2836(87)90190-2. [DOI] [PubMed] [Google Scholar]
- 4.Peterson JA, Graham-Lorence SE. Bacterial P450s: Structural similarity and Functional Differences. In: Ortiz de Montellano PR, editor. Cytocrome P450: Structure, Mechanism, and Biochemistry. Plenum Press; New York: 1995. pp. 151–179. [Google Scholar]
- 5.Brewer CB, Peterson JA. Single Turnover Kinetics of the Reaction between Oxycytochrome P-450cam and Reduced Putidaredoxin. J Biol Chem. 1988;263:791–798. [PubMed] [Google Scholar]
- 6.Lipscomb JD, Sugar SG, Namtvedt MJ, Gunsalus IC. Autooxidation and Hydroxylation Reactions of Oxygenated Cytochrome P-450cam. J Biol Chem. 1976;251:1116–1124. [PubMed] [Google Scholar]
- 7.Raag R, Li H, Jones BC, Poulos TL. Inhibitor-induced conformational change in cytochrome P-450CAM. Biochemistry. 1993;32:4571–4578. doi: 10.1021/bi00068a013. [DOI] [PubMed] [Google Scholar]
- 8.Raag R, Poulos TL. Crystal Structure of the Carbon Monoxide-Substrate-Cytochrome P-450CAM Ternary Complex. Biochemistry. 1989;28:7586–7592. doi: 10.1021/bi00445a013. [DOI] [PubMed] [Google Scholar]
- 9.Schlichting I, Berendzen J, Chu K, Stock AM, Maves SA, Benson DE, Sweet RM, Ringe D, Petsko GA, Sligar SG. The Catalytic Pathway of Cytochrome P450cam at Atomic Resolution. Science. 2000;287:1615–1622. doi: 10.1126/science.287.5458.1615. [DOI] [PubMed] [Google Scholar]
- 10.Jain NU, Tjioe E, Savidor A, Boulie J. Redox-Dependent Structural Differences in Putidaredoxin Derived from Homologous Structure Refinement via Residual Dipolar Couplings. Biochemistry. 2005;44:9067–9078. doi: 10.1021/bi050152c. [DOI] [PubMed] [Google Scholar]
- 11.Pochapsky TC, Jain NU, Kuti M, Lyons TA, Heymont J. A Refined Model for the Solution Structure of Oxidized Putidaredoxin. Biochemistry. 1999;38:4681–4690. doi: 10.1021/bi983030b. [DOI] [PubMed] [Google Scholar]
- 12.Pochapsky TC, Ye XM, Ratnaswamy G, Lyons TA. An NMR-Derived Model for the Solution Structure of Oxidized Putidaredoxin, a 2-Fe, 2-S Ferredoxin from Pseudomonas. Biochemistry. 1994;33:6424–6432. doi: 10.1021/bi00187a006. [DOI] [PubMed] [Google Scholar]
- 13.Sevrioukova IF. Redox-dependent Structural Reorganization in Putidaredoxin, a Vertebrate-type [2Fe-2S] Ferredoxin from Pseudomonas putida. J Mol Biol. 2005;347:607–621. doi: 10.1016/j.jmb.2005.01.047. [DOI] [PubMed] [Google Scholar]
- 14.Sevrioukova IF, Garcia C, Li H, Bhaskar B, Poulos TL. Crystal structure of putidaredoxin, the [2Fe-2S] component of the P450cam monooxygenase system from Pseudomonas putida. J Mol Biol. 2003;333:377–392. doi: 10.1016/j.jmb.2003.08.028. [DOI] [PubMed] [Google Scholar]
- 15.Hintz MJ, Mock DM, Peterson LL, Tuttle K, Peterson JA. Equilibrium and kinetic studies of the interaction of cytochrome P- 450cam and putidaredoxin. J Biol Chem. 1982;257:14324– 14332. [PubMed] [Google Scholar]
- 16.Holden M, Mayhew M, Bunk D, Roitberg A, Vilker V. Probing the Interactions of Putidaredoxin with Redox Partners in Camphor P450 5-Monooxygenase by Mutagenesis of Surface Residues. J Biol Chem. 1997;272:21720–21725. doi: 10.1074/jbc.272.35.21720. [DOI] [PubMed] [Google Scholar]
- 17.Stayton PS, Sligar SG. The Cytochrome P-450Cam, Binding Surface As Defined by Site-Directed Mutagenesis and Electrostatic Modeling. Biochemistry. 1990;29:7381–7386. doi: 10.1021/bi00484a005. [DOI] [PubMed] [Google Scholar]
- 18.Unno M, Shimada H, Toba Y, Makino R, Ishimura Y. Role of Arg112 of Cytochrome P450cam in the Electron Transfer from Reduced Putidaredoxin. ANALYSES WITH SITE-DIRECTED MUTANTS. J Biol Chem. 1996;271:17869–17874. doi: 10.1074/jbc.271.30.17869. [DOI] [PubMed] [Google Scholar]
- 19.Pochapsky T, Lyons T, Kazanis S, Arakaki T, Ratnaswamy G. A structure-based model for cytochrome P450cam-pufidaredoxin interactions. Biochimie. 1996;78:723–733. doi: 10.1016/s0300-9084(97)82530-8. [DOI] [PubMed] [Google Scholar]
- 20.Roitberg AE, Holden MJ, Mayhew MP, Kurnikov IV, Beratan DN, Vilker VL. Binding and Electron Transfer between Putidaredoxin and Cytochrome P450cam. Theory and Experiments. J Am Chem Soc. 1998;120:8927–8932. [Google Scholar]
- 21.Simonneaux G, Bondon A. Mechanism of Electron Transfer in Heme Proteins and Models: The NMR Approach. Chem Rev. 2005;105:2627–2646. doi: 10.1021/cr030731s. [DOI] [PubMed] [Google Scholar]
- 22.Pochapsky SS, Pochapsky TC, Wei JW. A Model for Effector Activity in a Highly Specific Biological Electron Transfer Complex: The Cytochrome P450cam-Putidaredoxin Couple. Biochemistry. 2003;42:5649–5656. doi: 10.1021/bi034263s. [DOI] [PubMed] [Google Scholar]
- 23.Shire Y, Iizuka T, Makino R, Ishimura Y, Morishima I. 15N NMR Study on Cyanide (C15N-) Complex of Cytochrome P-450cam. Effects of d-Camphor and Putidaredoxin on the Iron-Ligand Structure. J Am Chem Soc. 1989;111:7707–7711. [Google Scholar]
- 24.Tosha T, Yoshioka S, Takahashi S, Ishimori K, Shimada H, Morishima I. NMR Study on the Structural Changes of Cytochrome P450cam upon the Complex Formation with Putidaredoxin: Functional Significance of the Putidaredoxin-Induced Structural Changes. J Biol Chem. 2003;278:39809–39821. doi: 10.1074/jbc.M304265200. [DOI] [PubMed] [Google Scholar]
- 25.Paine MJI, Scrutton NS, Munro AW, Gutierrez A, Roberts GCK, Wolf CR. Electron Transfer Partners of Cytochrome P450. In: Ortiz de Montellano PR, editor. Cytochrome P450: Structure, Mechanism, and Biochemistry. Kluwer Academic/Plenum Publishers; New York: 2005. pp. 115–148. [Google Scholar]
- 26.Bridges A, Gruenke L, Chang Y-T, Vakser IA, Loew G, Waskell L. Identification of the Binding Site on Cytochrome P450 2B4 for Cytochrome b5 and Cytochrome P450 Reductase. J Biol Chem. 1998;273:17036–17049. doi: 10.1074/jbc.273.27.17036. [DOI] [PubMed] [Google Scholar]
- 27.Stayton PS, Poulos TL, Sligar SG. Putidaredoxin Competitively Inhibits Cytochrome b5-Cytochrome P-450cam Association: A Proposed Molecular Model for a Cytochrome P-45Ocam Electron-Transfer Complex. Biochemistry. 1989;28:8201–8205. doi: 10.1021/bi00446a035. [DOI] [PubMed] [Google Scholar]
- 28.Nickerson DP, Wong L-L. The dimerization of Pseudomonas putida cytochrome P450cam: practical consequences and engineering of a monomeric enzyme. Protein Eng. 1997;10:1357–1361. doi: 10.1093/protein/10.12.1357. [DOI] [PubMed] [Google Scholar]
- 29.Gunsalus IC, Wagner GC. Bacterial P-450cam Methylene Monooxygenase Components: Cytochrome m, Putidaredoxin, and Putidaredoxin Reductase. Methods Enzymol. 1978;52:166–187. doi: 10.1016/s0076-6879(78)52019-3. [DOI] [PubMed] [Google Scholar]
- 30.Von Bodman SB, Schuler MA, Jollie DR, Sligar SG. Synthesis, bacterial expression, and mutagenesis of the gene coding for mammalian cytochrome b5. Proc Natl Acad Sci USA. 1986;83:9443–9447. doi: 10.1073/pnas.83.24.9443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pervushin K, Riek R, Wider G, Wüthrich K. Attenuated T2 relaxation by mutual cancellation of dipole–dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc Natl Acad Sci USA. 1997;94:12366–12371. doi: 10.1073/pnas.94.23.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kay LE, Keifer P, Saarinen T. Pure Absorption Gradient Enhanced Heteronuclear Single Quantum Correlation Spectroscopy with Improved Sensitivity. J Am Chem Soc. 1992;114:10663–10665. [Google Scholar]
- 33.Palmer AG, III, Cavanagh J, Wright PE, Rance M. Sensitivity Improvement in Proton-Detected Two-Dimensional Heteronuclear Correlation NMR Spectroscopy. J Magn Reson. 1991;93:151–170. [Google Scholar]
- 34.Wei JY, Pochapsky TC, Pochapsky SS. Detection of a High-Barrier Conformational Change in the Active Site of Cytochrome P450cam upon Binding of Putidaredoxin. J Am Chem Soc. 2005;127:6974–6976. doi: 10.1021/ja051195j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lüdemann SK, Lounnas V, Wade RC. How do substrates enter and products exit the buried active site of cytochrome P450cam? 2. Steered molecular dynamics and adiabatic mapping of substrate pathways. J Mol Biol. 2000;303:813–830. doi: 10.1006/jmbi.2000.4155. [DOI] [PubMed] [Google Scholar]
- 36.Loida PJ, Sligar SG. Molecular recognition in cytochrome P-450: Mechanism for the control of uncoupling reactions. Biochemistry. 1993;32:11530–11538. doi: 10.1021/bi00094a009. [DOI] [PubMed] [Google Scholar]
- 37.Dunn AR, Dmochowski IJ, Bilwes AM, Gray HB, Crane BR. Probing the open state of cytochrome P450cam with ruthenium-linker substrates. Proc Natl Acad Sci USA. 2001;98:12420–12425. doi: 10.1073/pnas.221297998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tosha T, Yoshioka S, Ishimori K, Morishima I. L358P Mutation on Cytochrome P450cam Simulates Structural Changes upon Putidaredoxin Binding: The Structural Changes Trigger Electron Transfer to Oxy-P450cam from Electron Donors. J Biol Chem. 2004;279:42836–42843. doi: 10.1074/jbc.M404216200. [DOI] [PubMed] [Google Scholar]
- 39.Millet O, Loria JP, Kroenke CD, Pons M, Palmer AG. The Static Magnetic Field Dependence of Chemical Exchange Linebroadening Defines the NMR Chemical Shift Time Scale. J Am Chem Soc. 2000;122:2867–2877. [Google Scholar]
- 40.Koradi R, Billeter M, Wüthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graphics. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]