

Abstract
Over the last decade, cysteine thiolate ligands have been shown to be critical to the Cu(I) (cuprous) binding chemistry of many cytosolic metallochaperone and metalloregulatory proteins involved in copper physiology. More recently, the thioether group of methionine has begun to emerge as an important Cu(I) ligand for trafficking proteins in more oxidizing cellular environments.
Inorganic chemistry poses several paradoxical challenges in the life of a cell. Transition metals such as zinc, copper and iron must be avidlyacquired to reach intracellular concentrationquotas of tens to hundreds of micromolar; however, few if any of these ions are thought to be ‘free’ or readily accessible in terms of theirthermodynamic availability or reaction chemistry. The mechanisms by which cells control metal occupancy of a given metal-binding site–that is, how metalloenzymes acquire the correct metal ion at the right time in cellular growth–are emerging through physiochemical characterization of a myriad of conserved metal trafficking pathways. We now know that regulation of metal ion availability is achieved by the concerted activity of numerous metal receptors, including metal transporters, metallochaperonesand metalloregulatory factors.
Mechanistic and structural insights into the chemistry of cellular copper-trafficking machinery have predominately focused on cysteine (that is, thiol)-rich Cu(I) sites associated with inherited diseases of copper metabolism. In a series of recent studies characterizing methionine-rich domains of copper trafficking proteins, some unexpected copper-thioether chemistry is emerging. As was the case for cysteine thiolate–rich sites, the Cu(I) coordination chemistry of such proteins is quite distinct from that seen in canonical copper enzymes. Furthermore, both classes of copper sites show accessible and flexible coordination chemistry that allows for both facile Cu(I) transfer events and metal ion selectivity1. Two fundamental differences are emerging between the two classes: they each function in chemically distinct subcellular compartments, and they use quite different electrostatic contributions to achieve Cu(I) binding selectivity and transfer. Here we describe a few biological imperatives and chemical principles that underlie this note-worthy division of labor among proteins that share common functions.
Copper coordination chemistry
The chemical identity, number and geometry of the ligating groups of a copper-binding site determine the relative stabilities of the resulting Cu(I) and Cu(II) complexes and thus the selectivity of that site. Studies of small-molecule copper chemistry generally indicate that Cu(I) binding is thermodynamically favored by the inclusion of more sulfur donors, as opposed to oxygen or nitrogen donors, and by lower coordination numbers (Fig. 1). The well-studied small-molecule sulfur-containing macrocycles demonstrate this trend: the inclusion of nitrogen donor atoms generally increases Cu(II) affinity2, and a Cu(II) macrocycle typically adopts a lower coordination number upon reduction to the Cu(I) complex(Fig. 1a). Ready accessibility of two-and three coordinate states is another feature of cuprous chemistry: Cu(I) is notable among first-row transition metal ions in its formation of two coordinate bisthiolate species. Although such Cu(I) centers are frequently constituents of multi-copper(I) clusters, three discrete two coordinate Cu(I)–thiolate complexes have been achieved and structurally characterized, and three-coordinate Cu(I)–thiolate complexes are some what more common (see citations in ref.3) (Fig. 1b). This chemistry can be compared to that of Hg(II), Au(I) and Ag(I), in which linear two-coordinate complexes are more easily isolated.
Figure 1.
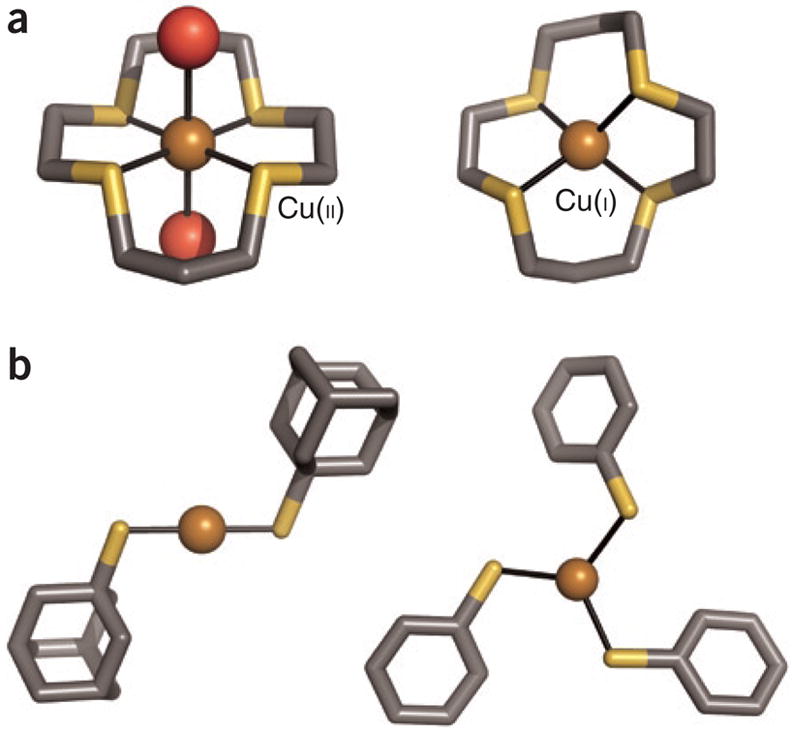
Four crystallographically characterized small-molecule complexes are shown (gray, C; dark yellow, S; red, O; bronze, Cu), labeled with the copper oxidation state. (a) The Cu(II)–thioether macrocycle complex, left, shows a higher coordination number (with two water ligands) than that of the reduced Cu(I) complex (ref. 2 and references therein). (The macrocycle structure has been simplified for the figure.) (b) Discrete two-and three-coordinate Cu(I)-thiolate complexes have been obtained with the use of sterically encumbering ligands and appropriate counterions28,29. The discussionof Cu(I) coordination chemistry is simplified here, with certain nitrogen-donor ligands, such as the phenanthroline-based bathocuproine disulfonic acid (BCS), acting as very tight Cu(I) complexing agents.
As we consider copper-binding sites in proteins from the broader perspective of inorganic chemistry, the array of amino acid functional groups that bind copper ions is rather limited :it generally includes the histidine imidazole, the cysteine thiolate, the carboxylate group of aspartate and glutamate, the methionine thioether and, much less frequently, the tyro-sine or serine hydroxyl groups, the deprotonated peptide amide nitrogen or the N-terminal amine. Although the protein-based ligand set is limited, these biopolymers have complex architectures that can tune coordination sites to achieve exquisite selectivity and precise thermo dynamic affinities. Cysteine thiolate coordination to Cu(I) has been well documented in the chemistry of copper sensing and trafficking proteins, where low-coordination-numberCys2 sites favor Cu(I) and aid in the discrimination against Cu(II) and other divalent metalions. By donating sulfur-containing ligands, methionine-rich sites might also be expected to enhance their selectivity for Cu(I). There are several notable features to the chemical properties of the methionine thioether compared tothe cysteine thiolate that are relevant to metal recognition. Bearing a longer side chain with no ionizable group, methionine is less polar and more hydrophobic than cysteine and has greater conformational flexibility. There is a small but real steric demand of the thioether methyl group in a metal-coordination sphere that is not present in cysteine. Finally, the ability of cysteine thiol sulfur to lose a proton has a major impact on susceptibility to oxidation, and it provides cysteine with an electrostatic component in the coordinate-covalent bonding. As described below, the differential oxidation chemistry and the electrostatic contributions are important determinants affecting which side chain is deployed in a given biological site.
Chemistry of cellular compartments
Copper sensing and trafficking proteins operate in several chemically distinct environments. Gram-negative bacteria (Fig. 2a) have essentially two types of compartments–the cytosol and the periplasmic space, which is part of the cell envelope–whereas the generic eukaryotic cell (Fig. 2b) has several chemically distinct compartments and/or organelles. The cytosols of both types of cells (shown in blue in Fig. 2)are reducing, with an estimated effective reduction potential between –190 and –250 mV with respect to the normal hydrogen electrode4.Although the cytosolic environment may vary with cell metabolism and external stresses, most cells maintain high levels (~3 mM) of the reduced small thiol-containing molecule glutathione, and specific cellular components such as the thioredoxin proteins work to preserve the cytosol of both prokaryotic and eukaryotic cells as overall reducing environment. Thus cytosolic proteins rarely contain stable disulfide bonds, and the cysteine-rich motifs found in a number of cytosolic copper trafficking and sensing proteins are reduced and available for Cu(I) binding. In contrast, the periplasmic space, and the endoplasmic reticula, as well as the extracellular surfaces, are generally more oxidizing in aerobic environments. Specific proteins are translocated into each compartment by cellular machinery, and copper trafficking proteins are no exception.
Figure 2.

Characterized Cu(I) trafficking and sensing coordination sites classified by cellular localization and function. (a) Protein structures depicted in theprokaryotic cell are the periplasmic putative trafficking proteins, which all form positively charged Cu(I) complexes (colored blue): Cu(I)-DR1885 (ref. 25),Cu(I)-CusF26 and Cu(I)2CopC224); and the cytoplasmic proteins, which all form negatively charged Cu(I) complexes (colored red): the chaperone Cu(I)-CopZ11and the metalloregulatory DNA-binding proteins Cu(I)2CsoR2 (ref. 15) and Cu(I)2CueR2 (ref. 6). (b) In the cytosol of the eukaryotic cell (not drawn to scale),two chaperone structures are shown, the yeast Cu(I)-Atx1 (ref. 7) and the human Cu(I)-Hah12 (ref. 12). A number of mitochondrial Cco assembly proteinshave suspected Cu(I) transport function, and the Cu(I)-Sco1 (ref. 17) and Cu(I)-Cox17 (ref. 20) structures are shown. The coordination sites are shown indetail outside of the cell diagrams, and have been characterized by a combination of crystallographic, XAS and NMR structural studies. In addition to theprotein sites listed above, a Met3His site of CopC23 and a Met2His site of PcoC22 are included with the methionine-rich cationic sites of the prokaryoticperiplasmic proteins. The anionic Cys2Cu(I) sites of the eukaryotic cytosolic proteins Hah1 (ref. 30) and Atx1 (ref. 7) and the cytosolic metal-bindingdomains of the Ccc2, Menkes (Mnk)9 and Wilson (Wnd)10 disease Cu(I) transporting P-type ATPases are also included.
Copper in the eukaryotic cytosol is largely trafficked via a Cu(I) (cuprous) binding proteins. In contrast, the metal-binding sites of many of the ‘clients’ of the trafficking proteins–that is, copper-containing enzymes–typically accommodate both Cu(II) (cupric)and Cu(I) oxidation states. Based on thermodynamic and kinetic studies of copper trafficking proteins, it has been proposed that essentially no ‘free’ (that is, aquo or uncomplexed) copper is tolerated in the cytosol of either eukaryotic5 or prokaryotic cells6 under normal physiological conditions. Several copper-specific metallochaperones ferry Cu(I) through the cytosol to specific proteins or compartments where it is inserted into target proteins. In prokaryotes, copper management occurs outside the cytoplasm. In fact, to date, there are no known copper-containing enzymes in the cytosol of E. coli: the half-dozen copper enzymes so far identified, as well as the copper trafficking machinery, are found in the cell envelope, which includes the periplasmic space.
Whether it be in the cell envelope of bacteria or the cytosol of eukaryotes, copper availability in cellular compartments is tightly regulated by specific metal-binding sites in trafficking proteins. The structures of a number of copper trafficking and sensing protein domains from different cellular compartments have been characterized (Fig. 2), and below we discuss emerging themes of the Cu(I) chemistry found in different cellular environments.
Cysteine-based Cu(I) sensing andtrafficking
The first recognized copper sensing and trafficking proteins contain Cys-X-X-C (CXXC)motifs and function in the reducing environment of the cytosol7. Biscysteinate Cu(I) coordination, a hallmark of cytosolic coordination chemistry, results in a [Cu(I)(Cys)2]1– moietythat bears a net negative change. Examples include the buried Cu(I) site in the E. coli copper-responsive transcription factor CueR6,8 and in the more solvent-exposed CXXC metal binding sites of the eukaryotic copper chaperones and their target metal-binding domains of the copper P-type ATPases7,9,10. The protein conformation in the latter case allows for flexible coordination chemistry: a third coordinated ligand is sometimes detected7,9,11, and intermediates in the copper transfer process, such as homo-and heterodimeric complexes of chaperones and their targets, show higher coordinate, Cu(I)-bridged complexes12,13.
Some variations on the cysteine-rich sites are emerging, including the copper chaperone from the cyanobacterium Synechocystis PCC6803 where the trigonal Cu(I) is bound in aCys2His site14. A similar Cys2His site was also reported for the copper-sensing transcription factor CsoR from Mycobacterium tuberculosis15, a homolog of CueR. Inclusion of a histidine ligand in these proteins may affect their affinity for Cu(I) as well as their recognition of, and metal-transfer chemistry with, partner proteins.
In addition, a number of cytochrome c oxidase (Cco) assembly proteins with mitochondrial and cytosolic localization have beenhypothesized to transfer Cu to Cco16, and conflicting evidence points to a role for Cys-Cu(I)chemistry in these systems. A [Cu(Cys)2His]1–site has been identified in the human Sco1 andSco2 (refs. 17–19) proteins, whereas biscysteine Cu(I) coordination is implicated for yeastCox17 (ref. 20); all three of these proteins are Cco assembly proteins localized in the mitochondrial inter membrane space. Notably, some of the uncertainty about the function of the Cco assembly proteins concerns their disulfide status in vivo, and it has been suggested that Cox17 may access several conformational forms with differing Cu(I)-binding properties depending on its disulfide status21. In these systems the disulfide redox potential and mechanism may have important bearing on copper binding and transfer events.
Methionine-rich copper trafficking sites
Methionine-rich sites are emerging as a dominant inorganic motif in less redox-balanced spaces that are outside the protective cytosolic environment, including the periplasm of prokaryotic organisms and the cell-surface environment of eukaryotic cells. Methionine is certainly not immune to oxidative modification, but it is more difficult to oxidize than copper-binding CXXC motifs, which readily form disulfides catalyzed by disulfide bond isomerase enzymes (Dsb family) in the prokaryotic periplasm and protein disulfide isomerase proteins (PDI family) in the eukaryotic endo-plasmic reticulum. As a ligand, the methionine thioether group lacks the electrostatic component of the cysteine thiolate ligand and is therefore expected to have weaker Cu(I) affinities. In the absence of anionic ligands, the resulting [Cu(I)(Met)n]+ centers bear a net positive charge. Furthermore, the thioether sulfur atoms bound to Cu(I) sites show very few H-bonding interactions (if any) in a methionine-rich site but can have extensive H-bonding interactions in the cysteine-rich sites. These differences are expected to significantly influence copper transfer reactions and may factor into protein-protein partnerships that are important to specific trafficking pathways.
Thus far the best-defined Cu(I)-Metn coordination chemistry is found among soluble prokaryotic periplasmic proteins, which generally feature higher-coordinate Cu(I) complexes(three or four coordinate) than the cysteine-rich Cu(I) handling proteins of the cytosol, perhaps to compensate for the lower Cu(I) affinity of thioethers compared to thiolates. These periplasmic proteins are generally associated with copper tolerance systems, which function to export copper and protect the cell from metal-induced toxicity. Much like the cytosolic trafficking components, they feature surface-exposed sites, showing some coordination flexibility. For example, a surface exposed Met2 His has been characterized by X ray absorption spectroscopy (XAS) for the E.coli Pco C protein22, whereas its Pseudomonas syringae homologs (CopC) shows Met3 His coordination by EXAFS23 in addition to crystallographically characterized Cu(I)Met4 and[MetHisCu(I)]2X bridged dimers24. The cyanobacterial protein DR1885, a putative Cco Cu(I) accessory protein, is localized to the periplasm and also shows Met3 His coordination as determined by EXAFS25. It is intriguing to consider that the Cco Cu(I) assembly chemistry, although still unclear, may differ between prokaryotic and eukaryotic organisms, based on the localization of the components, as the Cco components of prokaryotes are localized in the cellular envelope with some assembly factors found in the periplasm.
More recently, we have characterized an unusual feature of the Met2His site in the periplasmic CusF protein (encoded by a component of the cus copper-tolerance gene cluster): a strong cation-pi interaction with a tryptophan residue located some 2.7–2.8 Å from the metal ion is involved in copper recognition26. The Cu(I) ion is in fact displaced 0.5 Å toward this tryptophan from the S2N ligand plane. This cationic [CuMet2His]1+ site shows some of the strongest UV resonance Raman spectroscopic signatures yet described for a cation ion–pi interaction and is a new feature in biological Cu(I) recognition chemistry. In the homologs from about ten organisms, tryptophan is replaced by the more recognizable Cu(I)ligand methionine, bolstering the interpretation that the tryptophan is involved in Cu(I)recognition and stabilization. EXAFS study of the membrane-anchored periplasmic CusB, a possible partner protein of CusF, revealed Met3Cu(I) ligation, providing a hint to possible periplasmic metal-transfer events27. In addition to providing Cu(I) coordinating ligands in less reducing environments, methionine ligands may also be important for facile copper transfer in cellular spaces where weaker copper coordination is required.
The thioether coordination chemistry of these prokaryotic proteins may aid in the elucidation of other methionine-containing trafficking agents, such as the extracellular domains of the eukaryotic Ctr1 copper importer and the methionine-containing regions of the P-type copper ATPases such as the Wilson’s disease protein. A rich variety of methionine-rich sites can be found in genomic databases, but additional structure-function studies are required before it will be possible to define concrete motifs.
Cu(I) coordination motifs:compartmentalization
Most of the cysteine-rich copper sites characterized to date are found in reducing intracellular compartments, whereas an abundance of methionine-rich copper sites are found in the oxidizing compartments and the extracellular milieu (Fig. 2). The functional implications of this dichotomy are still emerging. One important feature of the methionine-rich sites and motifs is their selectivity against the two most abundant intracellular transition metal ions: zinc and iron. Notably, the +2 ions of these metals are not favored for binding in the methionine-rich sites, but hundreds of examples of zinc and iron ions bound to CXXC motifs in higher-coordinate or metal-cluster sites can be found in the metalloprotein literature (for example, in zinc finger proteins and iron-sulfur proteins).
As our understanding of the methionine-rich chemistry31 expands to include extracytosolic proteins, a broad picture is emerging of biological cuprous coordination and ligand-exchange chemistry. Different coordination motifs seem to be tailored to different pathways and cellular environments in order to preserve copper-specific recognition within a robust metal-binding site. A corollary to this idea, however, is the consideration that metal-trafficking sites may also be responsive to changing cellular environments, as oxidative bursts may release copper or zinc ions from cysteine-rich (or methionine-rich) metal-binding sites as the ligating groups are oxidized and the metal-binding sites are disrupted. The interplay between the intimate coordination chemistry of each family of sites and the changing metabolic needs of the cell may well prove to be a key to unlocking the roles of metal ions in intracellular signaling processes. Notably, copper-trafficking proteins are being implicated as critical components in cancer cell proliferation, host-pathogen interactions in infectious disease and neurological disorders. Thus it is increasingly important to understand how healthy cells control this toxic and reactive cofactor, how it is used in signaling processes and how to intervene when disturbances of copper homoeostasis become pathological.
Acknowledgments
This research was supported by US National Institutesof Health grant GM 38784 (to T.V.O.) a nd by NationalResearch Service Award GM 071129 (to A.V.D.)
References
- 1.Finney LA, O’Halloran TV. Science. 2003;300:931–936. doi: 10.1126/science.1085049. [DOI] [PubMed] [Google Scholar]
- 2.Rorabacher DB. Chem Rev. 2004;104:651–697. doi: 10.1021/cr020630e. [DOI] [PubMed] [Google Scholar]
- 3.Zeevi S, Tshuva EY. Eur J Inorg Chem. 2007:5369–5376. [Google Scholar]
- 4.Schafer FQ, Buettner GR. Free Radic Biol Med. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 5.Rae TD, Schmidt PJ, Pufahl RA, Culotta VC, O'Halloran TV. Science. 1999;284:805–808. doi: 10.1126/science.284.5415.805. [DOI] [PubMed] [Google Scholar]
- 6.Changela A, et al. Science. 2003;301:1383–1387. doi: 10.1126/science.1085950. [DOI] [PubMed] [Google Scholar]
- 7.Pufahl RA, et al. Science. 1997;278:853–856. doi: 10.1126/science.278.5339.853. [DOI] [PubMed] [Google Scholar]
- 8.Chen K, Yuldasheva S, Penner-Hahn JE, O'Halloran TV. J Am Chem Soc. 2003;125:12088–12089. doi: 10.1021/ja036070y. [DOI] [PubMed] [Google Scholar]
- 9.Ralle M, Cooper MJ, Lutsenko S, Blackburn NJ. J Am Chem Soc. 1998;120:13525–13526. [Google Scholar]
- 10.DiDonato M, Hsu HF, Narindrasorasak S, Que L, Sarkar B. Biochemistry. 2000;39:1890–1896. doi: 10.1021/bi992222j. [DOI] [PubMed] [Google Scholar]
- 11.Banci L, Bertini I, Del Conte R, Mangani S, Meyer-Klaucke W. Biochemistry. 2003;42:2467–2474. doi: 10.1021/bi0205810. [DOI] [PubMed] [Google Scholar]
- 12.Wernimont AK, Huffman DL, Lamb AL, O’Halloran TV, Rosenzweig AC. Nat Struct Biol. 2000;7:766–771. doi: 10.1038/78999. [DOI] [PubMed] [Google Scholar]
- 13.Arnesano F, et al. J Biol Chem. 2001;276:41365–41376. doi: 10.1074/jbc.M104807200. [DOI] [PubMed] [Google Scholar]
- 14.Borrelly GPM, et al. Biochem J. 2004;378:293–297. doi: 10.1042/BJ20031669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu T, et al. Nat Chem Biol. 2007;3:60–68. doi: 10.1038/nchembio844. [DOI] [PubMed] [Google Scholar]
- 16.Cobine PA, Pieffel F, Winge DR. BBA Mol CellRes. 2006;1763:759–772. doi: 10.1016/j.bbamcr.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 17.Balatri E, Banci L, Bertini I, Cantini F, Ciofi-Baffoni S. Structure. 2003;11:1431–1443. doi: 10.1016/j.str.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 18.Nittis T, George GN, Winge DR. J Biol Chem. 2001;276:42520–42526. doi: 10.1074/jbc.M107077200. [DOI] [PubMed] [Google Scholar]
- 19.Banci L, et al. Structure. 2007;15:1132–1140. doi: 10.1016/j.str.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 20.Abajian C, Yatsunyk LA, Ramirez BE, Rosenzweig AC. J Biol Chem. 2004;279:53584–53592. doi: 10.1074/jbc.M408099200. [DOI] [PubMed] [Google Scholar]
- 21.Arnesano F, Balatri E, Banci L, Bertini I, Winge DR. Structure. 2005;13:713–722. doi: 10.1016/j.str.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 22.Peariso K, Huffman DL, Penner-Hahn JE, O'Halloran TV. J Am Chem Soc. 2003;125:342–343. doi: 10.1021/ja028935y. [DOI] [PubMed] [Google Scholar]
- 23.Arnesano F, Banci L, Bertini I, Mangani S, Thompsett AR. Proc Natl Acad Sci USA. 2003;100:3814–3819. doi: 10.1073/pnas.0636904100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang L, Koay M, Mahert MJ, Xiao Z, Wedd AG. J Am Chem Soc. 2006;128:5834–5850. doi: 10.1021/ja058528x. [DOI] [PubMed] [Google Scholar]
- 25.Banci L, et al. Proc Natl Acad Sci USA. 2005;102:3994–3999. doi: 10.1073/pnas.0406150102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xue Y, et al. Nat Chem Biol. 2008;4:107–109. doi: 10.1038/nchembio.2007.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bagai I, Liu W, Rensing C, Blackburn NJ, McEvoy MM. J Biol Chem. 2007;282:35695–35702. doi: 10.1074/jbc.M703937200. [DOI] [PubMed] [Google Scholar]
- 28.Fujisawa K, Imai S, Kitajima N, Moro-oka Y. Inorg Chem. 1998;37:168–169. [Google Scholar]
- 29.Garner CD, Nicholson JR, Clegg W. Inorg Chem. 1984;23:2148–2150. [Google Scholar]
- 30.Ralle M, Lutsenko S, Blackburn NJ. J Biol Chem. 2003;278:23163–23170. doi: 10.1074/jbc.M303474200. [DOI] [PubMed] [Google Scholar]
- 31.Jiang J, Nadas IA, Kim MA, Franz KJ. Inorg Chem. 2005;44:9787–9794. doi: 10.1021/ic051180m. [DOI] [PubMed] [Google Scholar]