

Abstract
Pharmacologic activation of the heterodimeric HIF transcription factor appears promising as a strategy to treat diseases, such as anemia, myocardial infarction, and stroke, in which tissue hypoxia is a prominent feature. HIF accumulation is normally linked to oxygen availability because an oxygen-dependent posttranslational modification (prolyl hydroxylation) marks the HIFα subunit for polyubiquitination and destruction. Three enzymes (PHD1, PHD2, and PHD3) capable of catalyzing this reaction have been identified, although PHD2 (also called Egln1) appears to be the primary HIF prolyl hydroxylase in cell culture experiments. We found that conditional inactivation of PHD2 in mice is sufficient to activate a subset of HIF target genes, including erythropoietin, leading to striking increases in red blood cell production. Mice lacking PHD2 exhibit premature mortality associated with marked venous congestion and dilated cardiomyopathy. The latter is likely the result of hyperviscosity syndrome and volume overload, although a direct effect of chronic, high-level HIF stimulation on cardiac myocytes cannot be excluded.
Introduction
Inadequate oxygen delivery and cellular hypoxia contributes to the pathophysiology of many diseases, including myocardial infarction, stroke, acute tubular necrosis, peripheral vascular disease, and anemia. Hypoxia causes cellular dysfunction and cell death if sufficiently severe and prolonged. The HIF transcription factor, comprised of an unstable alpha subunit and a stable beta subunit, regulates many genes that promote survival in response to acute or chronic hypoxia. The former group includes genes that preserve ATP levels by promoting anaerobic glycolysis or suppressing energy-requiring processes such as protein translation. The latter group includes angiogenic genes, such as VEGF, or erythropoietic genes, such as Erythropoietin.
There are 3 HIFα genes, although HIF1α and HIF2α have been the most intensively studied. HIF1α is ubiquitously expressed, while the expression of HIF2α is more restricted. Oxygen-dependent posttranslational modifications dictate the stability and transcriptional regulatory activity of the HIFα protein subunits.1,2 Under well-oxygenated conditions, HIFα becomes hydroxylated on 1 (or both) of 2 prolyl residues by members of the PHD (also called Egln) prolyl hydroxylase family. This recruits an E3 ubiquitin ligase complex that contains the von Hippel–Lindau protein (pVHL) tumor suppressor protein, which marks HIFα for proteasomal degradation. In addition, HIFα contains 2 transactivation domains (NTAD and CTAD). The HIFα CTAD is silenced when a conserved asparagine residue is hydroxylated by FIH1. The oxygen Km of FIH1 is lower than that of the PHD family members,3 and there is experimental evidence that dual inactivation of FIH1 and PHD in cells requires more profound oxygen deprivation than does inactivation of PHD function alone.4 In addition, some HIF target genes are more sensitive to FIH1 inhibition than others,5 presumably because they differ with respect to their requirement for the CTAD relative to the NTAD and for their responsiveness to HIF2α, which is a poor FIH1 substrate relative to HIF1α.4,6
All 3 PHD family members can hydroxylate HIFα in vitro. Acute ablation of PHD2 (Egln1), but not PHD1 or PHD3, leads to HIF stabilization in cell culture siRNA experiments, suggesting that PHD2 is the primary HIF prolyl hydroxylase.7 However, PHD3 is a HIF target gene and might compensate for PHD2 loss under some conditions.8–10 The extent to which the 3 PHD family members are redundant in intact mammals is not known, nor is it clear whether PHD2 loss, without concurrent FIH1 inactivation, would suffice to promote erythropoietin production.
In this regard, Percy and coworkers recently described a family in which individuals carrying a hypomorphic PHD2 (Egln1) missense allele exhibited inappropriate erythropoietin production and polycythemia, suggesting that PHD2 is indeed the primary HIF prolyl hydroxylase in man.11 The fact that these individuals were heterozygotes, however, implies that partial reduction of PHD2 function is sufficient to cause disease or that the corresponding missense mutant is dominant-negative with respect to PHD2, and perhaps its paralogs. Moreover, the small size of the kindred did not exclude the formal possibility that the apparent linkage of this allele with the development of polycythemia was due to chance.
We found that inactivation of PHD2 is indeed sufficient to promote red blood cell production in mice but, unexpectedly, leads to the development of dilated cardiomyopathy and premature mortality. These findings, and their underlying mechanisms, have implications for the development of drugs that inhibit PHD activity.
Methods
Homologous recombination
A 6.5-kb EcoRI fragment from a 129Sv-BAC clone spanning the mouse PHD2 locus (RP22-256K16; Children's Hospital Oakland Research Institute, Oakland, CA) was subcloned into pBluescript II SK+ vector (Stratagene, La Jolla, CA). EcoRI/HindIII (830 bp), HindIII/SpeI (2.3 kb), and SpeI/EcoR1 (3.3 kb) genomic fragments were then subcloned into the pKOII plasmid along with a LoxP site 5′ of the SpeI/EcoRI fragment12 (Figure 1A). A pKOII-PHD2 targeting construct was linearized with NotI and electroporated into TC1 embryonic stem (ES) cells (derived from the 129SvEv strain) by using standard techniques. A total of 23 of 288 G418-resistant ES clones were successfully recombined, as determined by polymerase chain reaction (PCR; Fwd, 5′-ATCCCAATGCCCTCTCCCTTAC-3′; Rev, 5′-TGGATGTGGAATGTGTGCGAG-3′) and Southern blot analysis with a probe (Figure 1A) made by PCR of the PHD2 BAC clone (Fwd, 5′-AAGACACGACACACCCTGCTGTTG-3′; Rev, 5′-CCACCCCTAACCCCTTATTTCG-3′).
Figure 1.

Creation of conditional PHD2 allele. (A) Targeting strategy for mouse PHD2. NEO cassette flanked by 2 Frt sites (▵) is excised by fliplase to create floxed allele. Exons 2 and 3 flanked by 2 LoxP sites (▴) are excised by Cre recombinase to create a null allele. DT-A indicates diphtheria toxin-A used to select against nonhomologous recombinants; ■, exon; and □, untranslated region (UTR). Selected restriction sites are shown: EcoRI (RI), HindIII (H), BamHI (B), SpeI (Sp), and NotI (N). One-sided arrows = PCR primers used for genotyping. 2-sided arrows indicate BamHI fragment detected by Southern Blot with indicated probe. (B) Southern blot analysis of tail genomic DNA, digested with BamHI, from mice with indicated genotypes using probe shown in panel A. In the bottom panel, mice contained EIIA-Cre transgene where indicated. (C,D) RT-PCR (C) and immunoblot analysis (D) of MEFs with indicated genotypes. *The faint band is probably a background band, as PHD2 mRNA is undetectable in these cells. (E) Immunoblot analysis of PHD2 flox/flox (F/F);Cre-ER MEFs after treatment with 4-hydroxy tamoxifen (4-OHT; 200 nM) for the indicated time period. (F) Immunoblot analysis of kidney and liver from PHD2 flox/flox;Cre-ER mice after tamoxifen treatment.
Mice
A total of 3 ES clones were microinjected into C57BL/6 blastocysts. High-percentage chimeric mice were bred to FVB-actin-flipase mice to eliminate the neomycin resistance cassette. Resulting PHD2+/flox mice were mated with C57BL/6 chicken beta-actin-Cre-ER mice13 to obtain PHD2+/flox;Cre-ER mice, which were then back-crossed 4 times to C57BL/6 mice. VHL flox/flox mice were a generous gift from Volker Haase (University of Pennsylvania, Philadelphia) and were likewise mated with C57BL/6 chicken beta-actin-Cre-ER mice and then backcrossed 4 times to C57BL/6 mice. Male PHD2+/flox;Cre-ER mice were crossed to female PHD2+/flox mice, and pregnant females were given 2 mg of tamoxifen (Sigma-Aldrich, St Louis, MO) at 17.5 days after coitus (dpc) by intraperitoneal injection. PHD2 flox/flox;Cre-ER offspring were again treated with 2 mg of tamoxifen by intraperitoneal injection at 3 weeks of age. Conditional inactivation of VHL was performed by treating 3-week-old VHL flox/flox;Cre-ER mice with 2 mg of tamoxifen by intraperitoneal injection every other day for 2 doses.
To obtain PHD2+/− mice, PHD2+/flox mice were bred to FVB-EIIA-Cre mice (a gift of Dr Ronald DePinho, Dana-Farber Cancer Institute, Boston, MA) and PHD2+/flox;EIIA-Cre offspring were backcrossed to C57BL/6 mice. PHD2+/− pups that did not carry the EIIA-Cre transgene were back-crossed to C57BL/6 mice 4 times. Mice were genotyped by PCR using the following primers: PHD2 Fwd, 5′-CGGGATCCGTTGCTTGTTATCCGGGCAATGGAACG-3′; PHD2 Rev1 (for Wt and floxed allele), 5′-CGGAATTCCTAGAAGACGTCTTTACCGAC-3′; and PHD2 Rev2 (for null allele), 5′-CGGAATTCTAGAGTTCAACCCTCACACCTTTTTCACC-3′.
MEFs
Mouse embryonic fibroblasts (MEFs) were obtained from embryonic day (E) 13.5 offspring using standard techniques and maintained in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum. Cells with the desired genotype, as determined by PCR, were immortalized with a retrovirus encoding the K1 variant of SV40 large-T antigen.14 To activate Cre-ER 4-hydroxy tamoxifen (4OH-TM; Sigma-Aldrich) was added to the media (final concentration, 200 nM).
Isolation of erythroid lineage cells
Bone marrow cells were obtained from 12-week-old PHD2 flox/flox;Cre-ER (−) and PHD2 flox/flox;Cre-ER (+) mice after treatment with tamoxifen at E17.5 and at 3 weeks of age. Ter-119+ cells were purified using rat anti–mouse Ter-119 monoclonal antibody (15-5921-82; eBioscience, San Diego, CA) and sheep anti–rat IgG magnetic beads (110.35; Dynal Biotech, Oslo, Norway) after hemolysis with standard NH4Cl buffer (155 mM NH4Cl, 10 mM KHCO3, and 1 mM EDTA-2Na [pH 7.0]).
mRNA analysis
mRNA was purified from MEFs with RNeasy column (Qiagen, Valencia, CA), or from tissues with Trizol (Invitrogen, Carlsbad, CA) and RNeasy column. Total RNA (1 μg) was then reverse transcribed (StrataScript First Strand cDNA Synthesis Kit; Stratagene). A total of 1 μg cDNA was used for quantitative PCR using QuantiTect SYBR Green kit (Qiagen) and Mx3005 (Stratagene). The following primer and probe sets were used: PHD2 (Fwd, 5′-GCCCAGTTTGCTGACATTGAAC-3′; Rev, 5′-CCCTCACACCTTTCTCACCTGTTAG-3′), PHD3 (Fwd, 5′-TCAACTTCCTCCTGTCCCTCATC-3′; Rev, 5′-GCGAACATAACCTGTCCCATTTC-3′). The primer and probe sets for EPO and VEGF-A were published before.15 QuantiTect Primers Assays (Qiagen) were used for Erythropoietin receptor, Transferrin, Transferrin receptor, DMT1, Dcytb, ALAS-2, PAI-1, and Bnip3.
Western blot analysis
Cells were rinsed with cold phosphate-buffered saline (PBS), and scraped and lysed with EBC buffer (50 mM Tris [pH 8.0], 120 mM NaCl, and 0.5% NP-40) supplemented with complete protease inhibitor cocktail (Roche Molecular Biochemicals, Indianapolis, IN). Tissues were snap-frozen, thawed, homogenized, and lysed in 8 M urea buffer supplemented with 40 mM Tris-HCl (pH 7.6), 300 mM NaCl, and 0.5% NP-40. Nuclear preps were done with a Ne-Per kit (Pierce, Rockford, IL). Protein concentration was determined by the Bradford method (Bio-Rad, Hercules, CA). Lysates were resolved by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and blotted onto nitrocellulose membranes (Bio-Rad). Membranes were blocked with Tris-buffered saline with 5% nonfat dry milk and probed with the following primary antibodies: anti-HIF1α polyclonal antibody (AG10001; A&G Pharmaceuticals, Columbia, MD, or kind gift from Dr Jacques Pouyssegur, University of Nice, Nice, France), anti-HIF2α polyclonal antibody (NB100-122; Novus, Littleton, CO), anti-Glut1 polyclonal antibody (NB300-666; Novus), anti-PHD3 polyclonal antibody (NB100-333; Novus), anti-PHD2 polyclonal antibody (generous gift from Dr Robert S. Freeman, University of Rochester School of Medicine and Dentistry, Rochester, NY), anti-vinculin monoclonal antibody (V9131; Sigma-Aldrich), and antitubulin mouse monoclonal antibody (B-512; Sigma-Aldrich). Bound antibody was detected with horseradish peroxidase (HRP)–conjugated secondary antibodies (W4021/W4011; Promega, Madison, WI) and Immobilon Western Chemiluminescent HRP Substrate (Millipore, Billerica, MA).
Blood analysis
A total of 70 μL blood was collected from the optical venous plexus of isoflurane-anesthetized mice using heparinized capillary tubes. Complete hematologic profile was obtained using a blood analyzer (Hemavet 850; Drew Scientific, Dallas, TX). Serum erythropoietin was measured by enzyme-linked immunosorbent assay (ELISA; R&D Systems, Minneapolis, MN).
Histology
All tissues except the heart were fixed with Bouin solution. Heart tissues were perfused and fixed with buffered 10% formalin solution. Tissues were embedded in paraffin prior to sectioning and staining. Photomicrographs were obtained with a Leica DMLB microscope (20× objective lens and 10× eyepiece lens; total magnification, 200×) with a Leica DFC320 camera and Leica IM500 acquisition software (Leica, Wetzlar, Germany).
Echocardiography
Murine transthoracic echocardiography was conducted in conscious 6-week-old mice using a Vevo 770 high-resolution microultrasound system (Visualsonics, Toronto, ON) as previously described.16 Briefly, the heart was imaged in the 2-dimensional parasternal short-axis view. M-mode echocardiogram of the midventricle was recorded at the level of the papillary muscle. Heart rate, posterior wall thickness, and end-diastolic and end-systolic internal dimensions of the left ventricle were measured from the M-mode image. Left ventricle (LV) fractional shortening was defined as the end-diastolic dimension minus the end-systolic dimension normalized for the end-diastolic dimension and was used as an index of cardiac contractile function.
Quantitative PCR for genomic DNA
Genomic DNA from multiple tissues of in utero tamoxifen-treated PHD2 flox/flox;Cre-ER females (9 weeks old) or MEFs (PHD2+/+ and PHD2+/−) were extracted with PureGene kit (Qiagen). A total of 100 ng DNA was used for quantitative PCR using the QuantiTect SYBR Green kit (Qiagen) and Mx3005 (Stratagene). Primers were designed to amplify intronic sequences of PHD2 or VHL that are removed upon Cre-mediated recombination. The following primers were used: PHD2 (Fwd, 5′-GCACCGTGGTTGTTTTCCTG-3′; Rev, 5′-TGTTGTAAGCCCTTGCCATAGAC-3′), VHL (Fwd, 5′-ATCTACCCCGAGAAAATGCCC-3′; Rev, 5′-GTAACCCTCCAAGTTGTCATCACC-3′).
Results
To ask whether PHD2 inactivation is sufficient to stabilize HIF and promote red blood cell production in vivo, we used standard techniques to make mice in which PHD2 exons 2 and 3, which encode almost the entire PHD2 catalytic domain, were flanked by LoxP sites (“flox” allele; Figure 1A,B). To create a null PHD2 allele, these mice were bred to transgenic mice expressing Cre recombinase under the control of the EIIA promoter, which is active during very early embryogenesis.17,18 PHD2+/flox;EIIA-Cre mice were backcrossed to wild-type mice to obtain PHD2+/− mice that did not express Cre. Consistent with a recent report,19 no viable PHD2−/− animals were obtained after mating PHD2+/− animals, indicating that PHD2 is essential for embryonic development. Viable PHD2−/− E12.5 embryos were obtained, however, and used to isolate MEFs. PHD2 mRNA and PHD2 protein were, as expected, undetectable in PHD2−/− MEFs, which displayed increased levels of HIF1α and the HIF-responsive gene product Glut1 (Figure 1C,D).
Next we generated, through appropriate matings, PHD2 flox/flox mice that ubiquitously produce a Cre-ER fusion protein, which can be activated by tamoxifen. Treatment of PHD2 flox/flox;Cre-ER MEFs with tamoxifen led, as expected, to the accumulation of HIF and HIF-responsive gene products Glut1 and PHD3 (Figure 1E). Treatment of PHD2+/+;Cre-ER MEFs with tamoxifen had no effect (data not shown). Notably, the induction of PHD3 protein was transient in this system. This might be due, at least partly, to negative feedback of PHD3 on HIF and changes in protein translation after chronic HIF stimulation.20
To inactivate PHD2 in vivo, PHD2 flox/flox;Cre-ER and PHD2+/+;Cre-ER embryos were exposed to tamoxifen near term (17.5 dpc) and 3 weeks after birth. Recombination of the floxed allele occurred in many tissues, albeit with varying efficiencies, based on a quantitative PCR assay (Figure S1, available on the Blood website; see the Supplemental Materials link at the top of the online article). Recombination of PHD2 was, as expected, associated with loss of PHD2 mRNA (Figure S2) and accumulation of HIF1α and HIF2α (Figure 1F).
By 10 weeks of age, the PHD2 flox/flox;Cre-ER mice were smaller than their wild-type counterparts (Figure 2A), were erythematous (Figure 2B), and began dying suddenly (Figure 2C). At autopsy, some of these animals had evidence of retroperitoneal hemorrhage (data not shown; see also Figure 5B,D).
Figure 2.

Gross phenotypes of mice after conditional inactivation of PHD2. (A) Body weights of 10-week-old female mice with indicated genotypes after treatment with tamoxifen at E17.5 and at 3 weeks of age. (B) Appearance of ears (top row) and skin (bottom row). Note vascular dilation and erythema in PHD2 flox/flox;Cre-ER mice (right column). (C) Kaplan-Meier survival curves for mice with indicated genotypes. Error bars indicate 1 SD.
Figure 5.

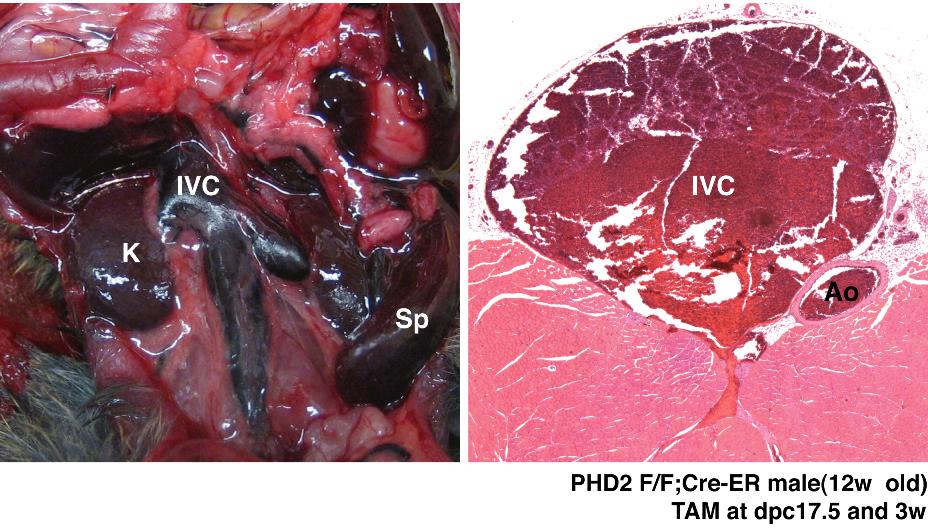
Histologic and cardiac alterations after conditional inactivation of PHD2. (A,B) Gross appearance of abdomen in PHD2+/+;Cre-ER (A) and PHD2 flox/flox;Cre-ER (B) mice treated with tamoxifen at E17.5 and at 3 weeks of age. Note large retroperitoneal hematoma (asterisk) in PHD2 flox/flox;Cre-ER mouse (B). (C-L) Hematoxylin and eosin staining of PHD2+/+;Cre-ER (C,E,G,I,K) and PHD2 flox/flox;Cre-ER (D,F,H,J,L) treated with tamoxifen as in panels A and B. Sections are from spine (C,D), liver (E,F), kidney (G,H), lung (I,J), and heart (K,L). Note large retroperitoneal hematoma (arrowheads), severely dilated inferior vena cava (IVC; see also Figure S3), as well as evidence of hemorrhage in spinal canal (arrows). SC indicates spinal cord; V, vertebral body. *Aorta.
The prominent erythema, together with the knowledge that HIF regulates erythropoiesis, suggested that PHD2 inactivation was causing polycythemia. Polycythemia, in turn, can cause hyperviscosity and attendant complications such as thrombosis and cardiac failure.21 Indeed, PHD2 flox/flox;Cre-ER mice treated with tamoxifen exhibited profound increases in red blood cell production (Figure 3A), which was associated with increased serum erythropoietin levels (Figure 3B). In contrast, white blood and platelet counts were normal (data not shown). Similar results were obtained when PHD2 flox/flox;Cre-ER mice were treated twice with tamoxifen at 3 weeks of age and not in utero (data not shown).
Figure 3.

PHD2 loss causes severe polycythemia. (A) Hematologic parameters and (B) serum erythropoietin (Epo) levels in 10-week-old PHD2+/+;Cre-ER (+/+) and PHD2 flox/flox;Cre-ER (F/F) mice after treatment with tamoxifen at E17.5 and at 3 weeks of age. (C) Real-time RT-PCR analysis for the indicated mRNAs from PHD2 conditional knockout (F/F) mice treated as in panels A and B. *P < .05; **P < .01. Error bars indicate 1 SD.
The liver and kidneys are the major sources of erythropoietin during fetal and adult life, respectively. Erythropoietin mRNA levels were markedly increased in the kidneys, but not the livers, of PHD2 flox/flox;Cre-ER mice treated with tamoxifen compared with wild-type counterparts (Figure 3C). In contrast, mRNAs for the HIF targets VEGF and PHD3 were not increased in either the kidneys or the livers from PHD2 flox/flox;Cre-ER mice treated with tamoxifen (Figure 3C), and circulating VEGF was not increased (data not shown). As a control, we did detect an increase in Erythropoietin, VEGF, and PHD3 mRNA in both the kidneys and livers of adult VHL flox/flox;Cre-ER mice treated with tamoxifen (Figures S1,3C). This indicates that some HIF target genes are down-regulated by pVHL in the absence of PHD2.
In addition to the Erythropoietin mRNA, a number of mRNAs that promote erythropoiesis through changes in iron metabolism and heme synthesis, including some that are regulated by hypoxia or HIF,22–24 were increased in erythroid precursors, kidneys, and small intestines after PHD2 inactivation in vivo (Figure 4). Whether the induction of these mRNAs was due primarily to increased circulating erythropoietin or due to a cell-autonomous effect of PHD2 loss is uncertain. In this regard, these mRNAs were not increased in erythroid precursors after PHD2 inactivation ex vivo (data not shown), possibly due to the presence of erythropoietin added to the cell culture media, and marrow hypoxia is not sufficient to stimulate erythropoiesis.25,26 Likewise, we have not observed polycythemia in PHD2 flox/flox mice expressing Cre under the control of the erythropoietin receptor promoter27 (data not shown).
Figure 4.

Induction of erythropoietic mRNAs in mice lacking PHD2. Real-time RT-PCR analysis for indicated mRNAs in erythroid lineage cells (A), kidneys (B), and small intestines (C) from 12-week-old PHD2 flox/flox;Cre-ER (−) and PHD2 flox/flox;Cre-ER (+) mice after treatment with tamoxifen at E17.5 and at 3 weeks of age. *P < .05; **P < .01. Error bars indicate 1 SD.
Gross and histologic examination of 8- to 10-week-old PHD2 flox/flox;Cre-ER mice that were treated with tamoxifen consistently revealed marked venous congestion and occasional retroperitoneal hemorrhages (Figures 5 and S3). To further delineate the cause of the congestion, we considered the possibility of cardiac dysfunction. Indeed, echocardiography of tamoxifen-treated PHD2 flox/flox;Cre-ER mice consistently revealed evidence of a dilated cardiomyopathy (Figure 6). Upon histologic examination the cardiomyocytes were enlarged, with occasional large nuclei (Figure 5K,L). The space between myocardial fibers was also increased (Figure 5K,L). Interestingly, there was increased contraction band necrosis indicative of early cardiac ischemia (Figure S4). Immunoblot analysis confirmed that HIF2α and Glut1 protein levels were modestly increased in the heart after PHD2 inactivation (Figure 7A), while HIF1α was below the limit of detection (Figure 7B). This was associated with an increase in a subset of HIF-responsive mRNAs, including mRNAs encoding PAI-1, Bnip3, PGK1, and PHD3 (Figure 7C).
Figure 6.

In vivo transthoracic echocardiography. Representative echocardiograms of 6 week old PHD2+/+;Cre-ER (A) and PHD2 flox/flox;Cre-ER (B) mice after Tamoxifen treatment as in Figure 4A,B. Short axis views (left panels) show evidence of left ventricular dilatation in PHD2 flox/flox mice. Representative M-mode views (right panels), in which a cross section of the short axis view is monitored over time, shows dilatation as well a depressed left ventricular function. Inactivation of PHD2 did not affect heart rate (C) or wall thickness (D), but increased end diastolic left ventricular diameter (E) and decreased fractional shortening (F). *P < .05; **P < .01. Error bars indicate 1 SD.
Figure 7.

Cardiac HIF activity after PHD2 inactivation. (A,B) Immunoblot analysis of heart whole-cell extracts from 6-week-old (VHL flox/flox) or 8-week-old (PHD2 flox/flox) mice that did (+) or did not (−) express Cre-ER. Mice were treated with tamoxifen at E17.5 and at 3 weeks of age. (C) Real-time RT-PCR analysis for the indicated mRNAs in hearts from 26- to 33-day old PHD2 flox/flox;CreER (−) and PHD2 flox/flox;Cre-ER (+) mice after treatment with tamoxifen at E17.5 and at 3 weeks of age. *P < .05; **P < .01. Error bars indicate 1 SD.
The observed cardiac changes might be indirect consequences of severe polycythemia and hyperviscosity. On the other hand, mice lacking VHL specifically in the heart also develop cardiomyopathy.28 In addition, cardiac ventricular development is abnormal in PHD2−/− embryos.19 These 2 observations raise the intriguing possibility that the development of cardiomyopathy in PHD2-deficient mice is a direct consequence of chronic, high-level HIF activation on cardiac myocytes.
Discussion
In Caenorhabditis and Drosophila there is a single PHD family member (Egl9), and Egl9−/− organisms display increased HIF levels.29,30 We found that germ-line deletion of PHD2 (Egln1) in mice causes embryonic lethality and that somatic PHD2 inactivation promotes HIF accumulation and activation of HIF target genes, including Erythropoietin, leading to profound polycythemia. The marked polycythemia was associated with venous congestion and premature death, which is consistent with findings in individuals who are homozygous for a hypomorphic VHL allele (Chuvash polycythemia)31 or carry the PHD2 variant described by Percy and coworkers.11 In contrast, PHD1−/− and PHD3−/− mice are grossly normal and do not develop polycythemia19 (Y.A.M. and W.G.K., unpublished data, August 2007). These studies establish that PHD2 is the primary HIF regulatory prolyl hydroxylase in higher metazoans in vivo. The requirement for PHD2 for embryonic development is consistent with earlier studies in which perturbations of HIF, such as HIF1α or VHL inactivation, likewise caused embryonic lethality.32–35
We found that some HIF target genes are repressed by pVHL despite the absence of PHD2. This might reflect residual hydroxylation of HIFα by other PHD family members or pVHL's ability to cooperate with FIH1 to repress HIF transactivation function.36 With respect to the latter, both VEGF and PHD3 transcription are inhibited by FIH15 and were not induced upon PHD2 inactivation in the liver or kidney. It is conceivable that the degree to which PHD2 loss is compensated by FIH1 and the other PHD family members is both cell-type and gene specific. Finally, it is also possible that there are subtle differences between prolyl hydroxylated HIFα, such as would accumulate in pVHL-defective cells, and non–prolyl hydroxylated HIFα, such as might accumulate in PHD2-defective cells, with respect to transcriptional competence in vivo.
The PHD family members and FIH1 are dioxygenases that require iron and 2-oxoglutarate for catalytic function. Small-molecule PHD inhibitors have been developed that activate HIF.2 It is not yet clear to what extent these small molecules discriminate among the PHD family members, and between PHD members and FIH1, in intact cells. Pharmacologic studies with such agents, as well as genetic studies, suggest that HIF contributes to tissue preservation in preclinical models of myocardial infarction,37,38 stroke,39–41 and acute tubular necrosis.42 In some cases it is unclear whether these beneficial effects were due to cell autonomous changes or due to hormonal effects mediated by survival factors such as VEGF and erythropoietin. In addition, PHD inhibitors have been used to correct anemia in mice43 and macques,44 and are now being tested in humans for this indication.
Our study has several implications for the development these agents. First, further studies will be required to determine their safety during pregnancy. Second, they suggest that inhibition of PHD2 alone will suffice to promote erythropoietin production in patients with functional kidneys. In functionally anephric patients, it might be necessary to target additional PHD family members based on the observation that pVHL loss, unlike PHD2 loss, promotes hepatic erythropoietin production,45,46 as do some small-molecule PHD inhibitors.43 The mice described in this report should be useful for addressing whether PHD2 inhibition is also sufficient to promote tissue preservation following regional ischemia. It is conceivable that the salutary effects of HIF in this setting, for example, requires activation of targets that are critically dependent on FIH1 inactivation in addition to PHD2 inactivation. Finally, they suggest that overzealous correction of anemia with these agents might lead to thrombotic complications and premature mortality, as has been observed with recombinant erythropoietin.47–49 Teleologically, stabilization of HIF, which regulates an entire suite of genes involved in hypoxic adaptation, might be safer and more effective than pharmacologic doses of erythropoietin. Similarly, it will be important to determine whether pharmacologic inhibition of PHD2, leading to a partial (rather than complete) loss of PHD2 function, will lead to cardiac dysfunction over time. Resolving these issues will require additional preclinical and clinical studies.
Supplementary Material
Acknowledgments
The authors thank Ronglih Liao's laboratory for assistance with mouse echocardiography, Rafael Bejar and Benjamin Ebert for help with bone marrow assays, and Ronald A. DePinho and James Horner for helping with gene targeting.
J.M. was supported by a National Institutes of Health (NIH) Clinical and Research Training Program for Academic Vascular Medicine Specialists (K12 HL083786). W.G.K. was supported by NIH and the Murray Foundation, and is a Howard Hughes Medical Institute (HHMI) Investigator.
Note added in proof:
Polycythemia and premature mortality in transgenic mice engineered to overproduce erythropoietin was reported by Wagner et al (Blood. 2001;97:536-542).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: Y.A.M. and J.M. designed and performed research and analyzed data. N.B. provided technical guidance. D.C. performed research. R.T.B. performed research and analyzed data. W.G.K. designed research, analyzed data, and wrote the paper.
Conflict-of-interest disclosure: W.G.K. owns equity in Fibrogen, which is developing prolyl hydroxylase inhibitors as potential therapeutic agents. All other authors declare no competing financial interests.
Correspondence: William G. Kaelin Jr, Department of Medical Oncology, Brigham and Women's Hospital and Dana-Farber Cancer Institute, Harvard Medical School, 44 Binney St., Boston, MA 02115; e-mail: william_kaelin@dfci.harvard.edu.
References
- 1.Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol. 2004;5:343–354. doi: 10.1038/nrm1366. [DOI] [PubMed] [Google Scholar]
- 2.Kaelin WG., Jr The von Hippel-Lindau protein, HIF hydroxylation, and oxygen sensing. Biochem Biophys Res Commun. 2005;338:627–638. doi: 10.1016/j.bbrc.2005.08.165. [DOI] [PubMed] [Google Scholar]
- 3.Koivunen P, Hirsila M, Gunzler V, Kivirikko KI, Myllyharju J. Catalytic properties of the asparaginyl hydroxylase (FIH) in the oxygen sensing pathway are distinct from those of its prolyl 4-hydroxylases. J Biol Chem. 2004;279:9899–9904. doi: 10.1074/jbc.M312254200. [DOI] [PubMed] [Google Scholar]
- 4.Bracken CP, Fedele AO, Linke S, et al. Cell-specific regulation of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha stabilization and transactivation in a graded oxygen environment. J Biol Chem. 2006;281:22575–22585. doi: 10.1074/jbc.M600288200. [DOI] [PubMed] [Google Scholar]
- 5.Dayan F, Roux D, Brahimi-Horn MC, Pouyssegur J, Mazure NM. The oxygen sensor factor-inhibiting hypoxia-inducible factor-1 controls expression of distinct genes through the bifunctional transcriptional character of hypoxia-inducible factor-1alpha. Cancer Res. 2006;66:3688–3698. doi: 10.1158/0008-5472.CAN-05-4564. [DOI] [PubMed] [Google Scholar]
- 6.Yan Q, Bartz S, Mao M, Li L, Kaelin WG., Jr The hypoxia-inducible factor 2alpha N-terminal and C-terminal transactivation domains cooperate to promote renal tumorigenesis in vivo. Mol Cell Biol. 2007;27:2092–2102. doi: 10.1128/MCB.01514-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berra E, Benizri E, Ginouves A, Volmat V, Roux D, Pouyssegur J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1alpha in normoxia. EMBO J. 2003;22:4082–4090. doi: 10.1093/emboj/cdg392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marxsen JH, Stengel P, Doege K, et al. Hypoxia-inducible factor-1 (HIF-1) promotes its degradation by induction of HIF-alpha-prolyl-4-hydroxylases. Biochem J. 2004;381:761–767. doi: 10.1042/BJ20040620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aprelikova O, Chandramouli GV, Wood M, et al. Regulation of HIF prolyl hydroxylases by hypoxia-inducible factors. J Cell Biochem. 2004;92:491–501. doi: 10.1002/jcb.20067. [DOI] [PubMed] [Google Scholar]
- 10.Appelhoff RJ, Tian YM, Raval RR, et al. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J Biol Chem. 2004;279:38458–38465. doi: 10.1074/jbc.M406026200. [DOI] [PubMed] [Google Scholar]
- 11.Percy MJ, Zhao Q, Flores A, et al. A family with erythrocytosis establishes a role for prolyl hydroxylase domain protein 2 in oxygen homeostasis. Proc Natl Acad Sci U S A. 2006;103:654–659. doi: 10.1073/pnas.0508423103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bardeesy N, Sinha M, Hezel AF, et al. Loss of the Lkb1 tumour suppressor provokes intestinal polyposis but resistance to transformation. Nature. 2002;419:162–167. doi: 10.1038/nature01045. [DOI] [PubMed] [Google Scholar]
- 13.Hayashi S, McMahon A. Abstract efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol. 2002;244:305–318. doi: 10.1006/dbio.2002.0597. [DOI] [PubMed] [Google Scholar]
- 14.Gjoerup O, Chao H, DeCaprio JA, Roberts TM. pRB-dependent, J domain-independent function of simian virus 40 large T antigen in override of p53 growth suppression. J Virol. 2000;74:864–874. doi: 10.1128/jvi.74.2.864-874.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rankin EB, Higgins DF, Walisser JA, Johnson RS, Bradfield CA, Haase VH. Inactivation of the arylhydrocarbon receptor nuclear translocator (Arnt) suppresses von Hippel-Lindau disease-associated vascular tumors in mice. Mol Cell Biol. 2005;25:3163–3172. doi: 10.1128/MCB.25.8.3163-3172.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liao R, Jain M, Cui L, et al. Cardiac-specific overexpression of GLUT1 prevents the development of heart failure attributable to pressure overload in mice. Circulation. 2002;106:2125–2131. doi: 10.1161/01.cir.0000034049.61181.f3. [DOI] [PubMed] [Google Scholar]
- 17.Lakso M, Pichel JG, Gorman JR, et al. Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc Natl Acad Sci U S A. 1996;93:5860–5865. doi: 10.1073/pnas.93.12.5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dooley TP, Miranda M, Jones NC, DePamphilis ML. Transactivation of the adenovirus EIIa promoter in the absence of adenovirus E1A protein is restricted to mouse oocytes and preimplantation embryos. Development. 1989;107:945–956. doi: 10.1242/dev.107.4.945. [DOI] [PubMed] [Google Scholar]
- 19.Takeda K, Ho VC, Takeda H, Duan LJ, Nagy A, Fong GH. Placental but not heart defects are associated with elevated hypoxia-inducible factor alpha levels in mice lacking prolyl hydroxylase domain protein 2. Mol Cell Biol. 2006;26:8336–8346. doi: 10.1128/MCB.00425-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu L, Simon MC. Regulation of transcription and translation by hypoxia. Cancer Biol Ther. 2004;3:492–497. doi: 10.4161/cbt.3.6.1010. [DOI] [PubMed] [Google Scholar]
- 21.Schafer AI. The hypercoagulable states. Ann Int Med. 1985;102:814–828. doi: 10.7326/0003-4819-102-6-814. [DOI] [PubMed] [Google Scholar]
- 22.Wenger RH. Cellular adaptation to hypoxia: O2-sensing protein hydroxylases, hypoxia-inducible transcription factors, and O2-regulated gene expression. FASEB J. 2002;16:1151–1162. doi: 10.1096/fj.01-0944rev. [DOI] [PubMed] [Google Scholar]
- 23.Hofer T, Wenger RH, Kramer MF, Ferreira GC, Gassmann M. Hypoxic up-regulation of erythroid 5-aminolevulinate synthase. Blood. 2003;101:348–350. doi: 10.1182/blood-2002-03-0773. [DOI] [PubMed] [Google Scholar]
- 24.Lis A, Paradkar PN, Singleton S, Kuo HC, Garrick MD, Roth JA. Hypoxia induces changes in expression of isoforms of the divalent metal transporter (DMT1) in rat pheochromocytoma (PC12) cells. Biochem Pharmacol. 2005;69:1647–1655. doi: 10.1016/j.bcp.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 25.Stohlman F, Rath CE, Rose JC. Evidence for a humoral regulation of erythropoiesis: studies on a patient with polycythemia secondary to regional hypoxia. Blood. 1954;9:721–733. [PubMed] [Google Scholar]
- 26.Schmid R, Gilbertsen AS. Fundamental observations on the production of compensatory polycythemia in a case of patent ductus arteriosus with reversed blood flow. Blood. 1955;10:247–251. [PubMed] [Google Scholar]
- 27.Heinrich A, Pelanda R, Klingmüller U. A mouse model for visualization and conditional mutations in the erythroid lineage. Blood. 2004;104:659–666. doi: 10.1182/blood-2003-05-1442. [DOI] [PubMed] [Google Scholar]
- 28.Giordano FJ. Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Invest. 2005;115:500–508. doi: 10.1172/JCI200524408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bruick R, McKnight S. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001;294:1337–1340. doi: 10.1126/science.1066373. [DOI] [PubMed] [Google Scholar]
- 30.Epstein A, Gleadle J, McNeill L, et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107:43–54. doi: 10.1016/s0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- 31.Gordeuk VR, Sergueeva AI, Miasnikova GY, et al. Congenital disorder of oxygen sensing: association of the homozygous Chuvash polycythemia VHL mutation with thrombosis and vascular abnormalities but not tumors. Blood. 2004;103:3924–3932. doi: 10.1182/blood-2003-07-2535. [DOI] [PubMed] [Google Scholar]
- 32.Gnarra J, Ward J, Porter F, et al. Defective placental vasculogenesis causes embryonic lethality in VHL-deficient mice. Proc Natl Acad Sci U S A. 1997;94:9102–9107. doi: 10.1073/pnas.94.17.9102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iyer NV, Kotch LE, Agani F, et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998;12:149–162. doi: 10.1101/gad.12.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ryan H, Lo J, Johnson R. HIF-1α is required for solid tumor formation and embryonic vascularization. EMBO J. 1998;17:3005–3015. doi: 10.1093/emboj/17.11.3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ma W, Tessarollo L, Hong SB, et al. Hepatic vascular tumors, angiectasis in multiple organs, and impaired spermatogenesis in mice with conditional inactivation of the VHL gene. Cancer Res. 2003;63:5320–5328. [PubMed] [Google Scholar]
- 36.Mahon P, Hirota K, Semenza G. FIH-1: a novel protein that interacts with HIF-1α and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001;15:2675–2686. doi: 10.1101/gad.924501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kido M, Du L, Sullivan CC, et al. Hypoxia-inducible factor 1-alpha reduces infarction and attenuates progression of cardiac dysfunction after myocardial infarction in the mouse. J Am Coll Cardiol. 2005;46:2116–2124. doi: 10.1016/j.jacc.2005.08.045. [DOI] [PubMed] [Google Scholar]
- 38.Natarajan R, Salloum FN, Fisher BJ, Kukreja RC, Fowler AA., 3rd Hypoxia inducible factor-1 activation by prolyl 4-hydroxylase-2 gene silencing attenuates myocardial ischemia reperfusion injury. Circ Res. 2006;98:133–140. doi: 10.1161/01.RES.0000197816.63513.27. [DOI] [PubMed] [Google Scholar]
- 39.Ratan RR, Siddiq A, Aminova L, et al. Translation of ischemic preconditioning to the patient: prolyl hydroxylase inhibition and hypoxia inducible factor-1 as novel targets for stroke therapy. Stroke. 2004;35:2687–2689. doi: 10.1161/01.STR.0000143216.85349.9e. [DOI] [PubMed] [Google Scholar]
- 40.Baranova O, Miranda LF, Pichiule P, Dragatsis I, Johnson RS, Chavez JC. Neuron-specific inactivation of the hypoxia inducible factor 1 alpha increases brain injury in a mouse model of transient focal cerebral ischemia. J Neurosci. 2007;27:6320–6332. doi: 10.1523/JNEUROSCI.0449-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bernaudin M, Nedelec AS, Divoux D, MacKenzie ET, Petit E, Schumann-Bard P. Normobaric hypoxia induces tolerance to focal permanent cerebral ischemia in association with an increased expression of hypoxia-inducible factor-1 and its target genes, erythropoietin and VEGF, in the adult mouse brain. J Cereb Blood Flow Metab. 2002;22:393–403. doi: 10.1097/00004647-200204000-00003. [DOI] [PubMed] [Google Scholar]
- 42.Tanaka T, Kojima I, Ohse T, et al. Hypoxia-inducible factor modulates tubular cell survival in cisplatin nephrotoxicity. Am J Physiol Renal Physiol. 2005;289:F1123–F1133. doi: 10.1152/ajprenal.00081.2005. [DOI] [PubMed] [Google Scholar]
- 43.Ivan M, Haberberger T, Gervasi DC, et al. Biochemical purification and pharmacological inhibition of a mammalian prolyl hydroxylase acting on hypoxia-inducible factor. Proc Natl Acad Sci U S A. 2002;99:13459–13464. doi: 10.1073/pnas.192342099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hsieh MM, Linde NS, Wynter A, et al. HIF-prolyl hydroxylase inhibition results in endogenous erythropoietin induction, erythrocytosis, and fetal hemoglobin expression in rhesus macaques. Blood. 2007;110:2140–2147. doi: 10.1182/blood-2007-02-073254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haase V, Glickman J, Socolovsky M, Jaenisch R. Vascular tumors in livers with targeted inactivation of the von Hippel-Lindau tumor suppressor. Proc Natl Acad Sci U S A. 2001;98:1583–1588. doi: 10.1073/pnas.98.4.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rankin EB, Biju MP, Liu Q, et al. Hypoxia-inducible factor-2 (HIF-2) regulates hepatic erythropoietin in vivo. J Clin Invest. 2007;117:1068–1077. doi: 10.1172/JCI30117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Singh AK, Szczech L, Tang KL, et al. Correction of anemia with epoetin alfa in chronic kidney disease. N Engl J Med. 2006;355:2085–2098. doi: 10.1056/NEJMoa065485. [DOI] [PubMed] [Google Scholar]
- 48.Drueke TB, Locatelli F, Clyne N, et al. Normalization of hemoglobin level in patients with chronic kidney disease and anemia. N Engl J Med. 2006;355:2071–2084. doi: 10.1056/NEJMoa062276. [DOI] [PubMed] [Google Scholar]
- 49.Wu WC, Schifftner TL, Henderson WG, et al. Preoperative hematocrit levels and postoperative outcomes in older patients undergoing noncardiac surgery. JAMA. 2007;297:2481–2488. doi: 10.1001/jama.297.22.2481. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}