

SUMMARY
Sulfonylurea receptor 1 (SUR1) is a molecule with more diverse and critically important functions than previously recognized. Long viewed simply as a subunit involved in formation of subset of K(ATP) channels, accumulating evidence indicates that SUR1 is newly upregulated in CNS ischemia and injury and is surprisingly promiscuous in its association with different pore-forming subunits, which endow it with new roles not previously envisioned. In this review, we focus on the SUR1-regulated NC(Ca-ATP) channel, its emerging role in CNS ischemia and trauma, and the growing evidence from preclinical and clinical studies demonstrating the potential importance of block of SUR1 by sulfonylureas such as glibenclamide (glyburide) in conditions as seemingly diverse as stroke and spinal cord injury.
Keywords: SUR1, NCCa-ATP channel, cerebral ischemia, stroke, CNS trauma, spinal cord injury
Introduction
CNS ischemia and trauma are leading causes of serious, long-term disability and death worldwide. Each year in the U.S. alone, 700,000 people suffer a stroke, with 20% dying within four weeks, 1.5 million people suffer traumatic brain injuries, with 52,000 resulting in death, and 11,000 people suffer spinal cord injuries. Despite major recent disappointments [1], the continuing search for pharmacological agents that could impact CNS ischemia and trauma remains a top priority, and any success in this endeavor will likely have an immeasurable impact on the human condition.
Numerous molecular mechanisms have been implicated in the pathophysiological responses to CNS ischemia and trauma. Here, we examine emerging evidence for the role of sulfonylurea receptor 1 (SUR1) and for the SUR1-regulated NCCa-ATP channel in these pathological processes. Involvement of this channel in capillary dysfunction induced by CNS ischemia / hypoxia has recently been reviewed [2].
Sulfonylurea receptor 1 (SUR1)
SUR1 is a member of the ATP-binding cassette (ABC) superfamily and is best known for its role in formation of KATP channels [3–7]. Recently, SUR1 has also come to be recognized for its role in formation of SUR1-regulated NCCa-ATP channels [8;9]. SUR1 contains two nucleotide-binding folds plus high affinity binding sites for therapeutic sulfonylurea drugs and related compounds. Drugs such as glibenclamide (glyburide) and repaglinide bind with subnanomolar or nanomolar affinity and are potent inhibitors of SUR1-regulated channel activity (Table 1). SUR1-regulated channels are exquisitely sensitive to changes in the metabolic state of the cell, responding to physiologically meaningful changes in intracellular ATP concentration by modulating channel open probability. For SUR1-regulated channels, sensitivity to ATP is conferred not by SUR1 but by the pore-forming subunit of the channel complex [10].
Table 1.
Sulfonylurea receptors (SURx) — their principal constitutive locations and blockers.
| SUR1 | SUR2A | SUR2B | ||
|---|---|---|---|---|
| principal constitutive locationsa | pancreatic β cell hippocampus neocortex olfactory bulb cerebellum hypothalamus substantia nigra | cardiac muscle skeletal muscle | smooth muscle vascular smooth muscle | |
| blockersb | glibenclamide | 0.13–4.2 | 27–45 | 42–166 |
| glimepiride | 3.0 | 5.4 | 7.3 | |
| repaglinide | 5.6–21 | 2.2 | 2 |
A notable feature of SUR1 is its apparent promiscuity. SUR1 forms heteromultimeric assemblies with several pore-forming subunits to create channels with different biophysical properties but similar pharmacological profiles. The best known native association is with Kir6.2 to form pancreatic and neuronal KATP channels [3;4;11]. SUR1 also forms stable heteromeric assemblies with Kir6.1 and Kir1.1a [12], resulting in formation of functional inward rectifier channels. The most recent addition to this list is the SUR1-regulated NCCa-ATP channel, the only channel with pore forming subunits not selective for K+, but that act instead as a non-selective cation channel [8]. The molecular identity of the pore-forming subunit is not known with certainty, but speculation has arisen that TRPM4 may be involved [13]. All of these channels are sensitive to the metabolic state of the cell, with channel opening promoted by a decrease in intracellular [ATP]. However, because of their different pore-selectivities, metabolic stress has opposite effects on cells expressing KATP channels compared to those expressing NCCa-ATP channels – ATP depletion causes hyperpolarization via KATP but depolarization via NCCa-ATP.
Apart from its role in channel formation, SUR1 also serves a critical role as chaperone, which is required for trafficking of functional channels to the cell membrane [14–16]. For KATP channels at least, correct trafficking and cell surface expression are under the control of a tripeptide endoplasmic reticulum (ER)-retention signal, RKR, present in both the SUR1 and Kir6.2 subunits. When expressed independently, the two proteins are retained in the ER because of exposure of the RKR signal. Under normal conditions, SUR1 and Kir6.2 associate with one another, which results in shielding of the ER-retention signal and permits the channel complex to traffic to the cell surface. Notably, drugs that bind to SUR1 can affect trafficking to the cell surface [16]. Prolonged exposure to glibenclamide results in desensitization that has been attributed to reduced surface expression of KATP channels [17;18], presumably due to abnormal trafficking induced by drug. It is possible that SUR1 plays a similar role in surface expression of SUR1-regulated NCCa-ATP channels, and that prolonged exposure to glibenclamide may also reduce surface expression of these channels.
SUR1-regulated KATP channel
The SUR1-regulated KATP channel has been extensively reviewed [3–7]. Its potential role in CNS ischemia and trauma will only be briefly mentioned here. The first potential role for KATP channels relates to their function in regulating insulin release in pancreatic β-cells. It is well established that hypoglycemia is associated with neurological abnormalities and seizures, and that hyperglycemia is associated with significantly poorer outcomes in stroke [19;20] and traumatic brain injury [21]. The second potential role for KATP channels regards direct neuroprotection. Neuronal KATP channels become activated in ATP-depleted metabolic states such as hypoxia and may contribute to protection against energy-consuming generalized seizures [22;23]. Sulfonylureas such as glibenclamide can depress hypoxia-induced hyperpolarization in hippocampal and neocortical neurons, thereby antagonizing the neuroprotective function of these channels. However, KATP channels are believed to fully exert their protective role only in very limited severities of hypoxic challenge just above the critical transition, apparently delaying but ultimately not preventing major depolarization, which likely limits their overall contribution to neuroprotection [22;23].
The SUR1-regulated KATP channel is not considered for direct targeting in CNS ischemia and injury, since serum glucose is better regulated by insulin infusion and direct neuroprotection via KATP channels is considered to be of limited value. However, recognizing these roles is important due to potential adverse effects that could be associated with sulfonylurea administration intended to target SUR1-regulated NCCa-ATP channels.
SUR1-regulated NCCa-ATP channel – biophysical and pharmacological properties
The SUR1-regulated NCCa-ATP channel is a 35 pS cation channel that conducts all inorganic monovalent cations (Na+, K+, Cs+, Li+, Rb+), but is impermeable to Ca2+ and Mg2+ [24]. The fact that it conducts Cs+ readily distinguishes it from KATP channels (Fig. 1). Studies using a variety of organic monovalent cations indicate that the channel has an equivalent pore radius of 0.41 nm. Channel opening requires nanomolar concentrations of Ca2+ on the cytoplasmic side. Channel opening is blocked by intracellular ATP (EC50, 0.79 µM), but is unaffected by ADP or AMP.
Figure 1.

The SUR1-regulated NCCa-ATP channel and the SUR1-regulated KATP channel have similar pharmacological profiles but very different channel properties. Like KATP, NCCa-ATP is inhibited by intracellular ATP (A) and is blocked by glibenclamide in a pH-dependent manner (D,E). However, unlike KATP, NCCa-ATP requires intracellular Ca2+ (B) and it conducts all monovalent cations (C). Panels A, B, D from Simard et al., 2006 [9], with permission; panel C from Chen and Simard, 2001 [24], with permission; panel E, from Chen et al., 2003 [8], with permission (○), and unpublished observations (●) (J. M. Simard and M. Chen).
The SUR1-regulated NC<sub>Ca-ATP</sub> channel has a pharmacological profile similar to SUR1-regulated K<sub>ATP</sub> channels [8]. Opening of SUR1-regulated NCCa-ATP channels is blocked by first and second generation sulfonylureas, tolbutamide (EC50, 16.1 µM at pH 7.4) and glibenclamide (EC50, 48 nM at pH 7.4), respectively. Block by sulfonylurea is due to prolongation of and an increase in the probability of long closed states, with no effect on open channel dwell times or channel conductance. In the presence of ATP, channel opening is increased by diazoxide, but not pinacidil or cromakalim, as expected for SUR1 but not SUR2. The inhibitory effect of glibenclamide on opening of the SUR1-regulated NCCa-ATP channel is prevented by antibody directed against one of the cytoplasmic loops of SUR1 [8]. Knockdown of SUR1 using antisense-oligodeoxynucleotide reduces SUR1 expression and prevents expression of functional SUR1-regulated NCCa-ATP channels [25].
Binding sites for sulfonylurea and related drugs are accessed via the lipid layer. For weak acids such as tolbutamide, glibenclamide and repaglinide, it is thus the un-ionized form that is active [26]. As a result, both drug binding [27] and drug-induced channel block are increased at acidic pH. An increase in potency at low pH is observed not only with KATP channels [26] but also with SUR1-regulated NCCa-ATP channels. In neurons from the core of ischemic brain, the magnitude of channel block by 50 nM glibenclamide is doubled by decreasing the pH from 7.4 to 6.8 [9]. In astrocytes from the ischemic inner zone of the gliotic capsule, decreasing the pH from 7.4 to 6.8 shifts the EC50 for block from 54 nM to 6 nM (Fig. 1). This effect of pH on apparent drug potency is a feature that is believed to enhance drug effect at the acidic pH characteristic of ischemic or injured CNS tissues [28].
SUR1-regulated NCCa-ATP channel – expression
The SUR1-regulated NCCa-ATP channel is not constitutively expressed, but is expressed in the CNS under conditions of hypoxia or injury. The channel was first discovered in freshly isolated reactive astrocytes obtained from the hypoxic inner zone of the gliotic capsule [8;24]. Since then, the channel has also been identified using patch clamp electrophysiology in neurons from the core of an ischemic stroke [9] and in cultured human and mouse endothelial cells subjected to hypoxia [25].
Apart from patch clamp recordings to demonstrate presence of the channel, CNS tissues have been analyzed to detect the regulatory subunit of the channel, SUR1, at both the protein and mRNA levels. In controls, baseline expression of SUR1 is attributable to constitutively expressed KATP channels in a subset of neurons [11], but SUR1 is not normally expressed in astrocytes or capillaries.
SUR1 is strongly upregulated in several rodent models of CNS injury, including models of cerebral ischemia [9], penetrating brain injury with foreign body [8], as well as severe cervical spinal cord injury [25]. Upregulation of SUR1 is found in all members of the neurovascular unit, i.e., neurons, astrocytes and capillary endothelial cells. Notably, SUR1 upregulation is not accompanied by upregulation of Kir6.x, at least in ischemia [9], the only one of these conditions in which this question has been evaluated.
SUR1 has also been identified in human brain tissues where it would not normally be expected. Fresh biopsy specimens were obtained from patients undergoing brain surgery for resection of a metastatic tumor surrounded by a gliotic capsule or for removal of an intracerebral hematoma secondary to rupture of an arteriovenous malformation. Immunolabeling for SUR1 was prominent in these cases (Fig. 2). In the gliotic capsule, astrocytes were labeled exclusively, whereas in the specimen from the patient with intracerebral hemorrhage, immunolabeling for SUR1 as well as in situ hybridization for Abcc8 mRNA, which encodes SUR1, was most prominent in neurons and microvessels.
Figure 2.

SUR1 is upregulated in human brain tissues with injury. Freshly isolated human gliotic capsule shows widespread SUR1 expression in astrocytes co-labeled for GFAP (A). Freshly isolated tissue adjacent to intracerebral hemorrhage (*) shows widespread SUR1 protein (B, left panel) and mRNA (C, left panel) in large neuron-like cells and microvessels; post-mortem human brain tissue (B, right panel) and tissue adjacent to intracerebral hemorrhage labeled with sense probe (C, right panel) showed minimal signal; methods for immunohistochemistry and in situ hybridization as in Simard et al., 2006 [9].
SUR1-regulated NCCa-ATP channel – cellular function
The consequences of opening the SUR1-regulated NCCa-ATP channel have been studied in isolated cells that express the channel by depleting ATP using Na azide or Na cyanide plus 2-deoxyglucose, or by using diazoxide. These treatments induce a strong inward current that depolarizes the cell completely to 0 mV. Morphological studies demonstrate that cells subsequently undergo changes consistent with cytotoxic edema (oncotic cell swelling), with formation of membrane blebs [24]. Bleb formation is reproduced without ATP depletion by diazoxide [8], and is prevented by increasing the osmolarity of the bath solution [24], consistent with bleb formation being a manifestation of oncotic cell swelling. ATP-depletion leads not only to oncotic cell swelling, but eventually to cell death, predominantly by non-apoptotic, propidium iodide-positive oncotic (necrotic) cell death [9]. These processes leading to cell death may involve neurons, astrocytes or capillary endothelial cells, with involvement of capillaries leading to formation of ionic edema, vasogenic edema and, eventually, to catastrophic failure of capillary integrity with formation of petechial hemorrhages (Fig. 3) [2].
Figure 3.
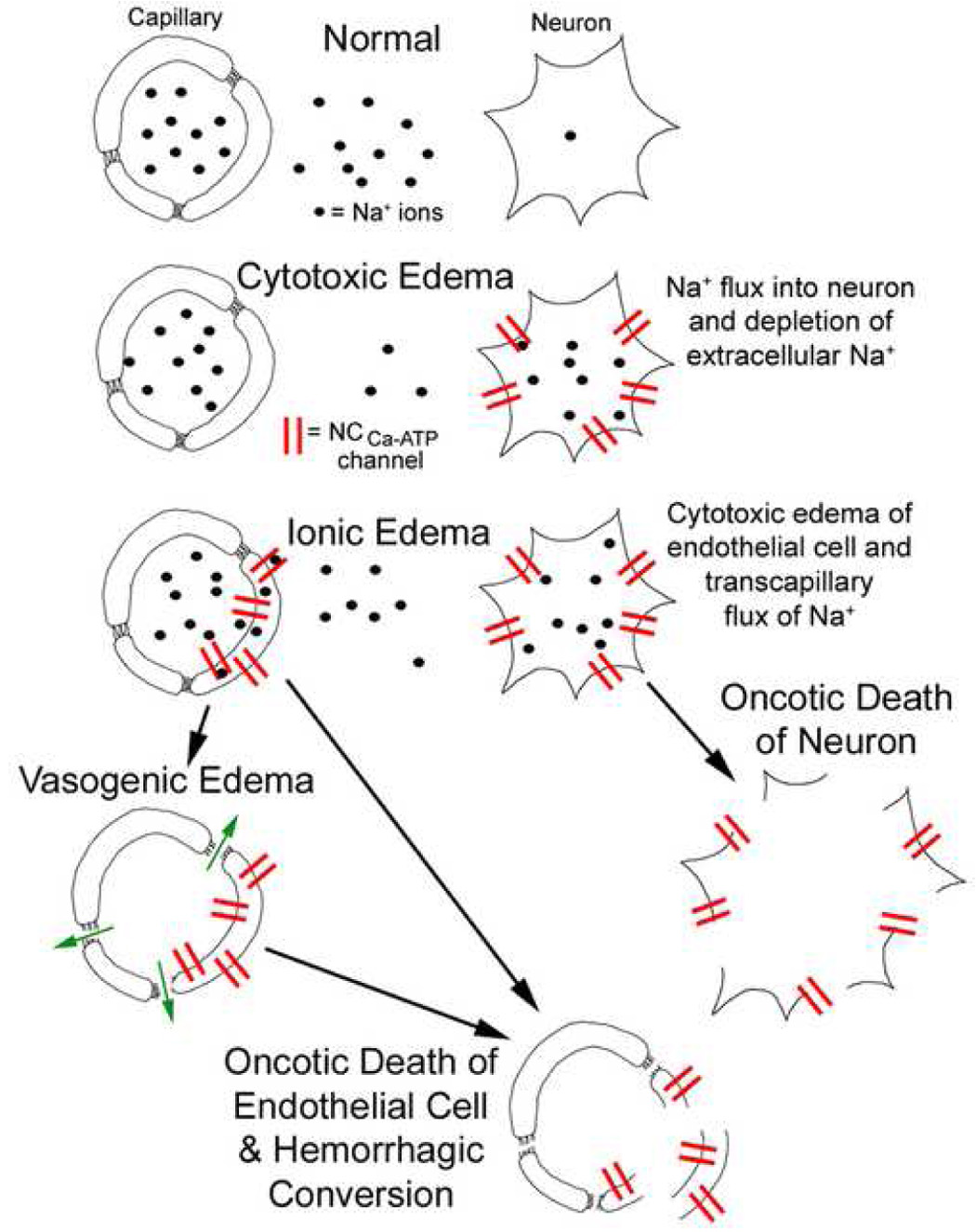
Schematic diagram illustrating various types of edema progressing to hemorrhagic conversion in the CNS. Normally, Na+ concentrations in serum and in extracellular space are the same, and much higher than inside the neuron. Cytotoxic edema of neurons is due to entry of Na+ into ischemic neurons via pathways such as SUR1-regulated NCCa-ATP channels, depleting extracellular Na+ and thereby setting up a concentration gradient between intravascular and extracellular compartments. Ionic edema results from cytotoxic edema of endothelial cells, due to expression of cation channels on both the luminal and abluminal side, allowing Na+ from the intravascular compartment to traverse the capillary wall and replenish Na+ in the extracellular space. Vasogenic edema results from degradation of tight junctions between endothelial cells, transforming capillaries into “fenestrated” capillaries that allow extravasation (outward filtration) of proteinatious fluid. Oncotic death of neuron is the ultimate consequence of cytotoxic edema. Oncotic death of endothelial cells results in complete loss of capillary integrity and in extravasation of blood, i.e., hemorrhagic conversion. From Simard et al. [2], with permission.
Block of SUR1 - Cerebral ischemia
Rodent models of stroke
The effect of glibenclamide has been studied in different rodent models of ischemic stroke (Table 2). In a rodent model of massive ischemic stroke with malignant cerebral edema associated with high mortality (68%), glibenclamide reduced mortality and cerebral edema (excess water) (Fig. 3) by half [9]. In a rodent model of stroke induced by thromboemboli (with presumed but undocumented delayed spontaneous reperfusion), glibenclamide reduced lesion volume by half, and its use was associated with cortical sparing that was attributed to improved leptomeningeal collateral blood flow due to reduced mass effect from edema [9]. The effect of glibenclamide has also been studied in a rodent model of focal ischemia / reperfusion (105 min / 48 hr) with ischemia induced by 75% or more reduction in relative cerebral blood flow. In this model, glibenclamide reduced cortical lesion volume (corrected for swelling) by 40% (J. M. Simard, in preparation).
Table 2.
Preclinical and clinical studies showing beneficial effects of inhibiting sulfonylurea receptor 1 in central nervous system ischemia or trauma.
| DISORDER | OUTCOME MEASURE | TREATMENT USED IN PRECLINICAL STUDY | TREATMENT USED IN CLINICAL STUDY |
|---|---|---|---|
| malignant cerebral edema | edema | glibenclamidea | |
| mortality | glibenclamidea | ||
| cerebral thromboembolism | severity of presentation | sulfonylureasb glibenclamidec | |
| lesion volume | glibenclamidea | ||
| neurological function | sulfonylureasb | ||
| spinal cord contusion | hemorrhage | glibenclamide, repaglinide, and antisense oligodeoxynucleotided | |
| lesion volume | glibenclamided | ||
| neurological function | glibenclamide, repaglinide, and antisense oligodeoxynucleotided |
Human stroke
The effect of sulfonylureas on stroke in humans has also been examined (Table 2). An outcome analysis was carried out of medical records of patients with diabetes mellitus (DM) hospitalized within 24 hr of onset of acute ischemic stroke in the Neurology Clinic, Charité Hospital, Berlin, Germany, during 1994–2000 [29]. After exclusions, the cohort comprised 33 patients taking a sulfonylurea [glibenclamide (glyburide), glimepiride, or glibornuride] at admission through discharge (treatment group) and 28 patients not on a sulfonylurea (control group). The primary outcome was a decrease in National Institutes of Health Stroke Scale (NIHSS) of 4 points or more from admission to discharge or a discharge NIHSS score = 0, which is considered a “major neurological improvement”. The secondary outcome was a discharge modified Rankin Scale (mRS) score of 2 or less, which signifies functional independence. No significant differences, other than stroke subtype, were observed among baseline variables between control and treatment groups. The primary outcome was reached by 36.4% of patients in the treatment group and 7.1% in the control group (odds ratio=7.5 in favor of sulfonylurea; P=0.007). The secondary outcome was reached by 81.8% vs. 57.1% (odds ratio=3.4 in favor of sulfonylurea; P=0.035). Subgroup analyses showed that improvements occurred only in patients with non-lacunar strokes, and were independent of gender, previous transient ischemic attack, and blood glucose levels.
The initial presentation for stroke in patients with DM may also be less severe for those taking sulfonylureas compared to those on insulin or on other oral agents (Table 2). In the Charité study [29], 18/54 patients not on sulfonylurea versus 1/36 on sulfonylurea were excluded from the outcome study because they presented with NIHSS scores >9. (These exclusions were necessary in order to have comparable groups at the start for fair comparison of outcomes.) Thus, the odds of presenting with a severe stroke (NIHSS>9) were higher in patients not on sulfonylurea (33%) than in patients on sulfonylurea (3%) at the time of their event (odds ratio = 17 in favor of sulfonylurea). A similar advantage was deduced from data published by Kahn et al. [30], in which 30/35 (86%) DM patients not on sulfonylurea versus 12/17 (71%) DM patients on glibenclamide presented with “serious stroke events” (odds ratio = 2.5 in favor of sulfonylurea).
Block of SUR1 - Spinal cord injury
Spinal cord injury (SCI) is another condition in which ischemia / hypoxia is believed to play a prominent role [31]. Acute SCI results in progressive hemorrhagic necrosis (PHN), a poorly understood pathological process characterized by hemorrhage and necrosis that leads to devastating loss of spinal cord tissue, cystic cavitation of the cord, and debilitating neurological dysfunction. A rodent model of severe cervical SCI was used to test the hypothesis that SUR1-regulated NCCa-ATP channels are involved in PHN [25]. In controls, SCI caused a progressively expansive lesion with fragmentation of capillaries, hemorrhage that doubled in volume over 12 hr, tissue necrosis and severe neurological dysfunction. Necrotic lesions were surrounded by widespread upregulation of SUR1 in capillaries and neurons. Patch clamp of cultured endothelial cells exposed to hypoxia showed that upregulation of SUR1 was associated with expression of functional SUR1-regulated NCCa-ATP channels. Following SCI, block of SUR1 by glibenclamide or repaglinide, or gene suppression of SUR1 by phosphorothioated antisense oligodeoxynucleotide, essentially eliminated capillary fragmentation (Fig. 3) and progressive accumulation of blood, was associated with significant sparing of white matter tracts and a 3-fold reduction in lesion volume, and resulted in marked neurobehavioral functional improvement compared to controls (Table 2).
Drug doses and specificity
In the studies cited above on rodent models of stroke and SCI, glibenclamide was administered by constant infusion at 75–200 ng/hr. Pharmacokinetic analysis (clearance, CL=1.25 ml/min [32;33]) indicated that these infusion rates would yield steady-state plasma concentration of 2–6 nM. It seems likely that such low concentrations would preferentially affect only the highest affinity receptors – SUR1, and not other potential targets such as SUR2, which is 1–2 orders of magnitude less sensitive to glibenclamide than SUR1 (Table 1). In addition, for the spinal cord study, identical results were obtained with three highly selective, molecularly distinct agents, glibenclamide, repaglinide, and antisense oligodeoxynucleotide, consistent with the target being SUR1. The marked efficacy of glibenclamide and repaglinide, despite the low doses used, is believed to be attributable, in part, to enhanced bioavailability of these weak acids in their un-ionized form in the local region of injury, due to the reduced pH associated with injury. As a general strategy, when treating a condition associated with acidic pH, such as tissue ischemia, and when targeting receptors that can be accessed only by way of the lipid membrane, such as SUR1, use of a weak acid seems ideal, in that regions of low pH will preferentially accumulate drug and normal tissues will be minimally affected.
At higher doses, glibenclamide can also inhibit SUR2, which forms the regulatory subunit of cardiovascular KATP channels (Table 1). In rodents, cerebrovascular SUR2-regulated KATP channels are important for hypoxic / hypercarbic cerebral vasodilation [34;35]. Thus, higher doses of glibenclamide, with potency augmented by low pH [26], could potentially compromise collateral blood flow after stroke. Indeed, unpublished data from our laboratory suggest that the efficacy of glibenclamide in a rodent model of stroke is lost when high doses are used. Fortunately, hypercapnia-induced cerebral vasodilation is not altered by glibenclamide in humans [36], a finding that could account for the positive results observed in the study with humans [29], who were taking 10× or more drug than what was used in the rodent models.
CONCLUSIONS
The standard view of neuroprotection, which has long been exclusively neurocentric, is no longer accepted and instead has been replaced by a more integrative approach that recognizes the importance of dynamic interactions between cells that form the “neurovascular unit”, i.e., endothelial cells, astrocytes and neurons [37;38]. In so far as the SUR1-regulated NCCa-ATP channel has now been shown to be newly upregulated in each member of the neurovascular unit following injury, and that this channel appears to be critic1ally involved in pathophysiological processes at both the cellular and organ levels, this channel appears to be a particularly attractive target for neuroprotection. Emerging data on the salutary effects of blocking this channel in rodent models of cerebral ischemia and spinal cord injury are promising, especially in light of the apparent beneficial effect of sulfonylureas in human diabetics with stroke. Much work remains to be done, however, especially in determining the therapeutic index and therapeutic window, before targeting of SUR1 can to go forth as a treatment for human CNS ischemia and trauma.
Acknowledgements
This work was supported by grants to JMS from the National Heart, Lung and Blood Institute (HL051932, HL082517), the National Institute of Neurological Disorders and Stroke (NS048260), the Department of Veterans Affairs (Baltimore, MD), and the Christopher and Dana Reeve Foundation; to VG from the National Institute on Drug Abuse (DA018329) and the American Heart Association (0455634U).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest JMS has applied for a US patent, "A novel non-selective cation channel in neural cells and methods for treating brain swelling" (application number 10/391,561).
Reference List
- 1.Shuaib A, Lees KR, Lyden P, Grotta J, Davalos A, Davis SM, Diener HC, Ashwood T, Wasiewski WW, Emeribe U. NXY-059 for the treatment of acute ischemic stroke. N.Engl.J.Med. 2007;357:562–571. doi: 10.1056/NEJMoa070240. [DOI] [PubMed] [Google Scholar]
- 2.Simard JM, Kent TA, Chen M, Tarasov KV, Gerzanich V. Brain oedema in focal ischaemia: molecular pathophysiology and theoretical implications. Lancet Neurol. 2007;6:258–268. doi: 10.1016/S1474-4422(07)70055-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aguilar-Bryan L, Bryan J. Molecular biology of adenosine triphosphatesensitive potassium channels. Endocr.Rev. 1999;20:101–135. doi: 10.1210/edrv.20.2.0361. [DOI] [PubMed] [Google Scholar]
- 4.Seino S. ATP-sensitive potassium channels: a model of heteromultimeric potassium channel/receptor assemblies. Annu.Rev.Physiol. 1999;61:337–362. doi: 10.1146/annurev.physiol.61.1.337. [DOI] [PubMed] [Google Scholar]
- 5.Campbell JD, Sansom MS, Ashcroft FM. Potassium channel regulation. EMBO Rep. 2003;4:1038–1042. doi: 10.1038/sj.embor.7400003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ashcroft FM. From molecule to malady. Nature. 2006;440:440–447. doi: 10.1038/nature04707. [DOI] [PubMed] [Google Scholar]
- 7.Bryan J, Munoz A, Zhang X, Dufer M, Drews G, Krippeit-Drews P, guilar-Bryan L. ABCC8 and ABCC9: ABC transporters that regulate K+ channels. Pflugers Arch. 2007;453:703–718. doi: 10.1007/s00424-006-0116-z. [DOI] [PubMed] [Google Scholar]
- 8.Chen M, Dong Y, Simard JM. Functional coupling between sulfonylurea receptor type 1 and a nonselective cation channel in reactive astrocytes from adult rat brain. J.Neurosci. 2003;23:8568–8577. doi: 10.1523/JNEUROSCI.23-24-08568.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Simard JM, Chen M, Tarasov KV, Bhatta S, Ivanova S, Melnitchenko L, Tsymbalyuk N, West GA, Gerzanich V. Newly expressed SUR1-regulated NC(Ca-ATP) channel mediates cerebral edema after ischemic stroke. Nat.Med. 2006;12:433–440. doi: 10.1038/nm1390.** First description of the role of SUR1-regulated NC(Ca-ATP) in stroke, showing the novel mechanism involved in edema formation
- 10.Tucker SJ, Gribble FM, Zhao C, Trapp S, Ashcroft FM. Truncation of Kir6.2 produces ATP-sensitive K+ channels in the absence of the sulphonylurea receptor. Nature. 1997;387:179–183. doi: 10.1038/387179a0. [DOI] [PubMed] [Google Scholar]
- 11.Liss B, Roeper J. Molecular physiology of neuronal K-ATP channels. Mol.Membr.Biol. 2001;18:117–127. [PubMed] [Google Scholar]
- 12.Ammala C, Moorhouse A, Gribble F, Ashfield R, Proks P, Smith PA, Sakura H, Coles B, Ashcroft SJ, Ashcroft FM. Promiscuous coupling between the sulphonylurea receptor and inwardly rectifying potassium channels. Nature. 1996;379:545–548. doi: 10.1038/379545a0. [DOI] [PubMed] [Google Scholar]
- 13.Simard JM, Tarasov KV, Gerzanich V. Non-selective cation channels, transient receptor potential channels and ischemic stroke. Biochim.Biophys.Acta. 2007;1772:947–957. doi: 10.1016/j.bbadis.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cartier EA, Conti LR, Vandenberg CA, Shyng SL. Defective trafficking and function of KATP channels caused by a sulfonylurea receptor 1 mutation associated with persistent hyperinsulinemic hypoglycemia of infancy. Proc.Natl.Acad.Sci.U.S.A. 2001;98:2882–2887. doi: 10.1073/pnas.051499698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yan FF, Casey J, Shyng SL. Sulfonylureas correct trafficking defects of disease-causing ATP-sensitive potassium channels by binding to the channel complex. J.Biol.Chem. 2006;281:33403–33413. doi: 10.1074/jbc.M605195200. [DOI] [PubMed] [Google Scholar]
- 16.Partridge CJ, Beech DJ, Sivaprasadarao A. Identification and pharmacological correction of a membrane trafficking defect associated with a mutation in the sulfonylurea receptor causing familial hyperinsulinism. J.Biol.Chem. 2001;276:35947–35952. doi: 10.1074/jbc.M104762200. [DOI] [PubMed] [Google Scholar]
- 17.Gullo D, Rabuazzo AM, Vetri M, Gatta C, Vinci C, Buscema M, Vigneri R, Purrello F. Chronic exposure to glibenclamide impairs insulin secretion in isolated rat pancreatic islets. J.Endocrinol.Invest. 1991;14:287–291. doi: 10.1007/BF03346813. [DOI] [PubMed] [Google Scholar]
- 18.Kawaki J, Nagashima K, Tanaka J, Miki T, Miyazaki M, Gonoi T, Mitsuhashi N, Nakajima N, Iwanaga T, Yano H, Seino S. Unresponsiveness to glibenclamide during chronic treatment induced by reduction of ATP-sensitive K+ channel activity. Diabetes. 1999;48:2001–2006. doi: 10.2337/diabetes.48.10.2001. [DOI] [PubMed] [Google Scholar]
- 19.Bruno A, Williams LS, Kent TA. How important is hyperglycemia during acute brain infarction? Neurologist. 2004;10:195–200. doi: 10.1097/01.nrl.0000131800.77824.dd. [DOI] [PubMed] [Google Scholar]
- 20.Zhu CZ, Auer RN. Optimal blood glucose levels while using insulin to minimize the size of infarction in focal cerebral ischemia. J.Neurosurg. 2004;101:664–668. doi: 10.3171/jns.2004.101.4.0664. [DOI] [PubMed] [Google Scholar]
- 21.Jeremitsky E, Omert LA, Dunham CM, Wilberger J, Rodriguez A. The impact of hyperglycemia on patients with severe brain injury. J.Trauma. 2005;58:47–50. doi: 10.1097/01.ta.0000135158.42242.b1. [DOI] [PubMed] [Google Scholar]
- 22.Yamada K, Inagaki N. Neuroprotection by KATP channels. J.Mol.Cell Cardiol. 2005;38:945–949. doi: 10.1016/j.yjmcc.2004.11.020.**Thoughtful review on the role of K(ATP) channels in neuroprotection
- 23.Ballanyi K. Protective role of neuronal KATP channels in brain hypoxia. J.Exp.Biol. 2004;207:3201–3212. doi: 10.1242/jeb.01106. [DOI] [PubMed] [Google Scholar]
- 24.Chen M, Simard JM. Cell swelling and a nonselective cation channel regulated by internal Ca2+ and ATP in native reactive astrocytes from adult rat brain. J.Neurosci. 2001;21:6512–6521. doi: 10.1523/JNEUROSCI.21-17-06512.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Simard JM, Tsymbalyuk O, Ivanov A, Ivanova S, Bhatta S, Geng Z, Woo SK, Gerzanich V. Endothelial sulfonylurea receptor 1-regulated NC(Ca-ATP) channels mediate progressive hemorrhagic necrosis following spinal cord injury. J.Clin.Invest. 2007;117:2105–2113. doi: 10.1172/JCI32041.** First description of the role of SUR1-regulated NC(Ca-ATP) in spinal cord injury, showing a novel molecular mechanism responsible for the mysterious autodestructive process known as “progressive hemorrhagic necrosis”
- 26.Findlay I. Effects of pH upon the inhibition by sulphonylurea drugs of ATPsensitive K+ channels in cardiac muscle. J.Pharmacol.Exp.Ther. 1992;262:71–79. [PubMed] [Google Scholar]
- 27.Nelson DA, Bryan J, Wechsler S, Clement JP, Aguilar-Bryan L. The high-affinity sulfonylurea receptor: distribution, glycosylation, purification, and immunoprecipitation of two forms from endocrine and neuroendocrine cell lines. Biochemistry. 1996;35:14793–14799. doi: 10.1021/bi960777y. [DOI] [PubMed] [Google Scholar]
- 28.Nedergaard M, Kraig RP, Tanabe J, Pulsinelli WA. Dynamics of interstitial and intracellular pH in evolving brain infarct. Am.J.Physiol. 1991;260:R581–R588. doi: 10.1152/ajpregu.1991.260.3.R581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kunte H, Schmidt S, Eliasziw M, Del Zoppo GJ, Simard JM, Masuhr F, Weih M, Dirnagl U. Sulfonylureas Improve Outcome in Patients With Type 2 Diabetes and Acute Ischemic Stroke. Stroke. 2007 doi: 10.1161/STROKEAHA.107.482216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kahn SE, Haffner SM, Heise MA, Herman WH, Holman RR, Jones NP, Kravitz BG, Lachin JM, O'Neill MC, Zinman B, Viberti G. Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N.Engl.J.Med. 2006;355:2427–2443. doi: 10.1056/NEJMoa066224. [DOI] [PubMed] [Google Scholar]
- 31.Tator CH. Update on the pathophysiology and pathology of acute spinal cord injury. Brain Pathol. 1995;5:407–413. doi: 10.1111/j.1750-3639.1995.tb00619.x. [DOI] [PubMed] [Google Scholar]
- 32.bd Elaziz MA, Al-Dhawailie AA, Tekle A. The effect of stress on the pharmacokinetics and pharmacodynamics of glibenclamide in diabetic rats. Eur.J.Drug Metab Pharmacokinet. 1998;23:371–376. doi: 10.1007/BF03192296. [DOI] [PubMed] [Google Scholar]
- 33.Rupp W, Christ O, Fulberth W. Studies on the bioavailability of glibenclamide. Arzneimittelforschung. 1972;22:471–473. [PubMed] [Google Scholar]
- 34.Nakahata K, Kinoshita H, Hirano Y, Kimoto Y, Iranami H, Hatano Y. Mild hypercapnia induces vasodilation via adenosine triphosphate-sensitive K+ channels in parenchymal microvessels of the rat cerebral cortex. Anesthesiology. 2003;99:1333–1339. doi: 10.1097/00000542-200312000-00014. [DOI] [PubMed] [Google Scholar]
- 35.Reid JM, Davies AG, Ashcroft FM, Paterson DJ. Effect of L-NMMA, cromakalim, and glibenclamide on cerebral blood flow in hypercapnia and hypoxia. Am.J.Physiol. 1995;269:H916–H922. doi: 10.1152/ajpheart.1995.269.3.H916. [DOI] [PubMed] [Google Scholar]
- 36.Bayerle-Eder M, Wolzt M, Polska E, Langenberger H, Pleiner J, Teherani D, Rainer G, Polak K, Eichler HG, Schmetterer L. Hypercapnia-induced cerebral and ocular vasodilation is not altered by glibenclamide in humans. Am.J.Physiol Regul.Integr.Comp Physiol. 2000;278:R1667–R1673. doi: 10.1152/ajpregu.2000.278.6.R1667. [DOI] [PubMed] [Google Scholar]
- 37.Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol.Rev. 2005;57:173–185. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- 38.Iadecola C, Goldman SS, Harder DR, Heistad DD, Katusic ZS, Moskowitz MA, Simard JM, Sloan MA, Traystman RJ, Velletri PA. Recommendations of the National Heart, Lung, and Blood Institute working group on cerebrovascular biology and disease. Stroke. 2006;37:1578–1581. doi: 10.1161/01.STR.0000221297.57305.8a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gribble FM, Reimann F. Sulphonylurea action revisited: the post-cloning era. Diabetologia. 2003;46:875–891. doi: 10.1007/s00125-003-1143-3. [DOI] [PubMed] [Google Scholar]
- 40.Karschin C, Ecke C, Ashcroft FM, Karschin A. Overlapping distribution of K(ATP) channel-forming Kir6.2 subunit and the sulfonylurea receptor SUR1 in rodent brain. FEBS Lett. 1997;401:59–64. doi: 10.1016/s0014-5793(96)01438-x. [DOI] [PubMed] [Google Scholar]
- 41.Shi NQ, Ye B, Makielski JC. Function and distribution of the SUR isoforms and splice variants. J.Mol.Cell Cardiol. 2005;39:51–60. doi: 10.1016/j.yjmcc.2004.11.024. [DOI] [PubMed] [Google Scholar]
- 42.Yokoshiki H, Sunagawa M, Seki T, Sperelakis N. ATP-sensitive K+ channels in pancreatic, cardiac, and vascular smooth muscle cells. Am.J.Physiol. 1998;274:C25–C37. doi: 10.1152/ajpcell.1998.274.1.C25. [DOI] [PubMed] [Google Scholar]
- 43.Aguilar-Bryan L, Nelson DA, Vu QA, Humphrey MB, Boyd AE., III Photoaffinity labeling and partial purification of the beta cell sulfonylurea receptor using a novel, biologically active glyburide analog. J.Biol.Chem. 1990;265:8218–8224. [PubMed] [Google Scholar]
- 44.Hansen AM, Christensen IT, Hansen JB, Carr RD, Ashcroft FM, Wahl P. Differential interactions of nateglinide and repaglinide on the human betacell sulphonylurea receptor 1. Diabetes. 2002;51:2789–2795. doi: 10.2337/diabetes.51.9.2789. [DOI] [PubMed] [Google Scholar]
- 45.Proks P, Reimann F, Green N, Gribble F, Ashcroft F. Sulfonylurea stimulation of insulin secretion. Diabetes. 2002;51 Suppl 3:S368–S376. doi: 10.2337/diabetes.51.2007.s368. [DOI] [PubMed] [Google Scholar]