

Abstract
Solid tumors can be highly aneuploid and many display high rates of chromosome missegregation in a phenomenon called chromosomal instability (CIN). In principle, aneuploidy is the consequence of CIN, but the relationship between CIN and aneuploidy has not been clearly defined. In this study, we use live cell imaging and clonal cell analyses to evaluate the fidelity of chromosome segregation in chromosomally stable and unstable human cells. We show that improper microtubule–chromosome attachment (merotely) is a cause of chromosome missegregation in unstable cells and that increasing chromosome missegregation rates by elevating merotely during consecutive mitoses generates CIN in otherwise stable, near-diploid cells. However, chromosome missegregation compromises the proliferation of diploid cells, indicating that phenotypic changes that permit the propagation of nondiploid cells must combine with elevated chromosome missegregation rates to generate aneuploid cells with CIN.
Introduction
Solid tumors can be highly aneuploid, with typical karyotypes ranging from 40 to 60 chromosomes but occasionally exceeding 70 or more chromosomes (http://cgap.nci.nih.gov/Chromosomes/Mitelman). How tumor cells acquire extra chromosomes and maintain them during cell division is currently unknown. In some cases, the unusually high numbers of chromosomes are segregated faithfully, and the aneuploid karyotype displays little variation over time (Storchova and Pellman, 2004). However, in other cases, aneuploid cells are genetically unstable and frequently missegregate whole chromosomes in a phenomenon called chromosomal instability (CIN; Lengauer et al., 1997). The elevated rate of chromosome missegregation in aneuploid tumor cells with CIN causes phenotypic changes that contribute to tumor cell evolution and pose therapeutic challenges (Gao et al., 2007). For example, chromosomal differences could change the growth properties of metastatic cells compared with the primary tumor (Kuukasjarvi et al., 1997). Increased chromosome missegregation rates in aneuploid tumor cells with CIN suggest an underlying defect in mitotic fidelity, and it has been presumed that persistent chromosome missegregation causes the high level of aneuploidy observed in tumor cells (Lengauer et al., 1998). Defects in both bipolar spindle assembly (e.g., multipolar spindles) and the spindle assembly checkpoint have been identified in some CIN tumor cell lines (Cahill et al., 1998; Lingle et al., 2002). However, those mitotic defects explain CIN in only a handful of cells, leaving the underlying cause of CIN in most aneuploid cells undetermined. Moreover, the question of whether CIN drives cells into a highly aneuploid state remains unanswered.
Results and discussion
Chromosomes display characteristic movements during alignment and segregation in mitosis that are directly visible by video microscopy. Defects in mitotic processes such as spindle checkpoint function or multipolar spindle assembly predictably impair chromosome behavior by inducing premature entry into anaphase before chromosome alignment and multipolar anaphase, respectively. Thus, to identify the mechanism of chromosome missegregation in aneuploid human tumor cell lines with CIN, we used live cell video microscopy to examine chromosome behavior in both chromosomally stable and unstable human cells. We selected two chromosomally stable cell lines (HCT116 and RPE-1) and three aneuploid and CIN cell lines (HT29, MCF-7, and Caco2) for in-depth analyses in this study because these cells have been karyotypically defined (Table S1, available at http://www.jcb.org/cgi/content/full/jcb.200712029/DC1), represent different tissues of origin, and have each been established in culture for many years.
Chromosomes align efficiently at metaphase, segregate synchronously at anaphase, and form equivalent daughter cells after cytokinesis in the chromosomally stable colon cancer cell line HCT116 (Fig. 1 and Video 1, available at http://www.jcb.org/cgi/content/full/jcb.200712029/DC). From 35 cells examined by video microscopy, mitotic defects were detected in <10% of cells, and these were lagging chromatids at anaphase and chromatin bridges. Similar frequencies of these mitotic errors are observed in fixed cells that did not express GFP–histone H2B (7.2 ± 1.6% [SEM], n = 400), demonstrating that the video results are representative (Fig. S1 A). Chromosomes in the chromosomally unstable colon carcinoma (HT29 and Caco2) and breast cancer (MCF-7) cell lines also align efficiently on bipolar spindles in metaphase, and no cells enter anaphase before the alignment of all chromosomes (Fig. 1 and Videos 2 and 3), demonstrating checkpoint proficiency as previously suggested (Tighe et al., 2001). Lagging chromatids in anaphase is the most common mitotic defect detected and is observed in 24% of HT29 cells (n = 34), 75% of Caco2 cells (n = 28), and 42% of MCF-7 cells (n = 33) examined by video microscopy (Fig. 1). In some cases, the lagging chromatids form micronuclei after cytokinesis. Similar frequencies of anaphase cells with lagging chromatids are observed in fixed cells that did not express GFP–histone H2B (19.0 ± 2.5% [SEM] in HT29 cells, n = 300; 66.0 ± 1.7% [SEM] in Caco2 cells, n = 300; 50.0 ± 2.6% [SEM] in MCF-7 cells, n = 300), demonstrating that these results are representative (Fig. S1 A). Kinetochores on lagging chromatids are often distorted and display microtubule bundles extending toward both spindle poles, demonstrating that the failure to segregate properly at anaphase onset is caused by merotelic attachment of the kinetochore to spindle microtubules (Fig. S1 C; Cimini et al., 2001, 2004). High percentages of anaphase cells with lagging chromatids is common in CIN cell lines derived from a variety of tumor tissues, including colon, breast, and lung (Fig. S1 A), indicating that this mitotic defect is independent of tissue of origin or oncogenic mutation (e.g., p53 and adenomatous polyposis coli are reported to be wild type in MCF-7 cells but mutated in Caco2 cells; www.sanger.ac.uk/perl/genetics/CGP/cosmic; Liu and Bodmer, 2006). The number of lagging chromatids per anaphase in chromosomally unstable cells is between 3- and 14-fold higher than chromosomally stable cells. The magnitude of this increase cannot be accounted for by the increase in the modal chromosome number in each cell line. Thus, in addition to the few cell lines reported with checkpoint defects (Cahill et al., 1998), frequent and persistent merotelic attachment of kinetochores to spindle microtubules is a common mechanism of chromosome missegregation in aneuploid tumor cells with CIN. Increases in lagging chromatids at anaphase have been reported after perturbation of diverse proteins like BRCA1 (Joukov et al., 2006), Sgo2 (Huang et al., 2007), adenomatous polyposis coli (Fodde et al., 2001; Green and Kaplan, 2003), kinesin-13 proteins (mitotic centromere-associated kinesin and Kif2a; Kline-Smith et al., 2004; Ganem et al., 2005), Mad2 (Michel et al., 2001), aurora kinase (Hauf et al., 2003; Knowlton et al., 2006), cdc4 (Rajagopalan et al., 2004), cyclin E overexpression (Rajagopalan et al., 2004), and the Ndc80 complex (DeLuca et al., 2006), reflecting the complex nature of establishing and maintaining proper kinetochore microtubule attachments (Cimini and Degrassi, 2005; Salmon et al., 2005). Merotelic kinetochore attachments elude spindle assembly checkpoint detection (Cimini et al., 2001), indicating that CIN may arise through reductions in the efficiency of merotelic correction.
Figure 1.
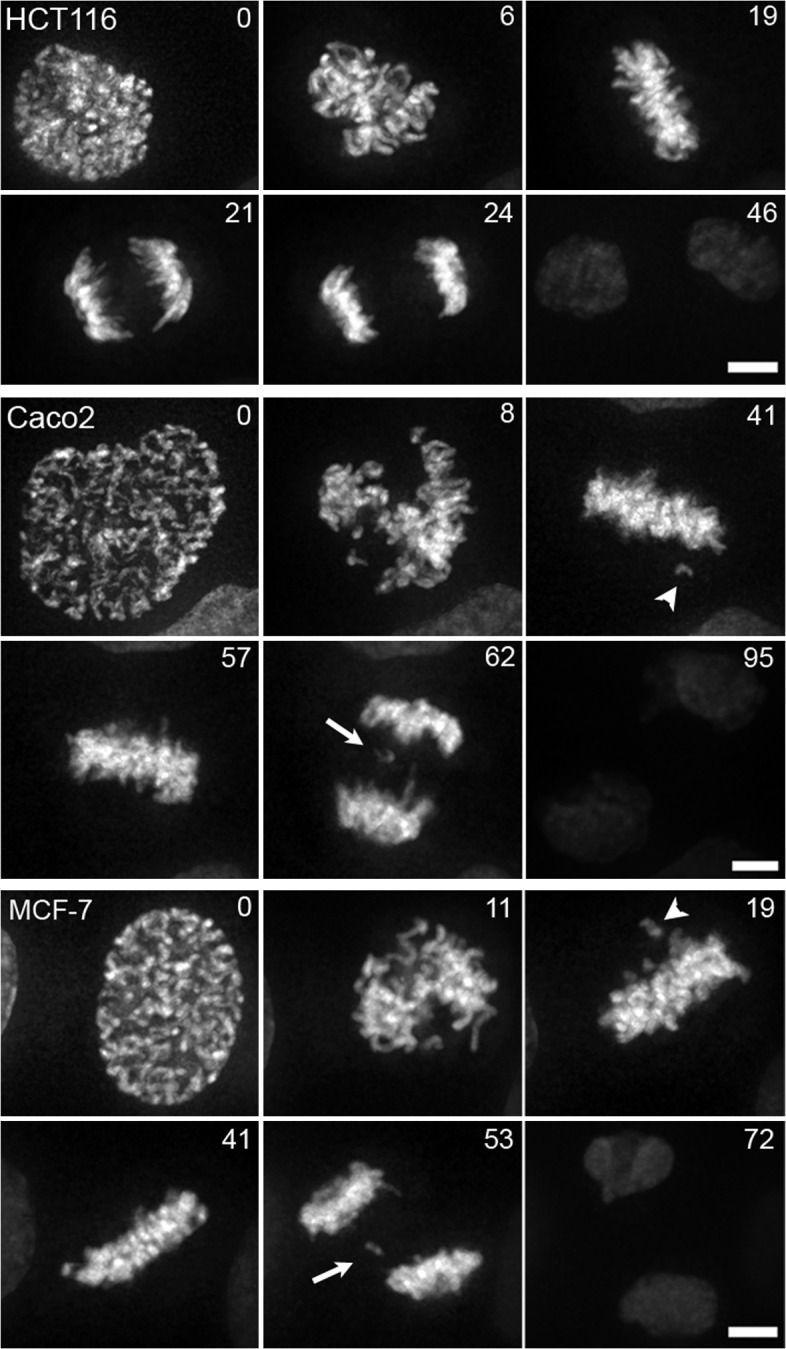
Chromosome segregation in human tumor cell lines.GFP–histone H2B was expressed in colon carcinoma HCT116 and Caco2 cells and in breast cancer MCF-7 cells. Images selected from time-lapse videos are shown with times given in minutes. Arrowheads identify unaligned chromosomes in prometaphase, and arrows identify lagging chromatids at anaphase. Bars, 5 μm.
The identification of merotely as a cause of chromosome missegregation in CIN cell lines provided us the opportunity to directly test the relationship between CIN and aneuploidy using human cultured cells. It has been shown that merotelic kinetochore attachments increase in mitotic cells recovering from drug treatments that perturb spindle organization (Cimini et al., 1999, 2001; Knowlton et al., 2006). Those treatments alter the spatiotemporal assembly of bipolar spindles, leading to increases in merotelic kinetochore attachment, but they do not inhibit the cell's ability to eventually undergo anaphase with bipolar spindles. Thus, we used mitotic recovery from nocodazole or monastrol treatment as a strategy to increase the incidence of merotelic kinetochore attachments during mitosis in two chromosomally stable, near-diploid cell lines (HCT116 and RPE-1) to evaluate the relationship between CIN and aneuploidy. As expected, we observe increases in lagging chromatids at anaphase in both cell lines recovering from nocodazole or monastrol treatment in mitosis (Fig. S1 B). Elevated frequencies of lagging chromatids required mitotic recovery from these drugs because lagging chromatid frequencies returned to basal rates if treatments were suspended. Fixed cell analyses verify that kinetochores of lagging chromatids are merotelically attached to spindle microtubules (Fig. S1 C). Time-lapse video microscopy confirms that cells recovering from monastrol treatment do not enter anaphase before bipolar spindle assembly and chromosome alignment but display lagging chromatids at anaphase (Fig. S2 A and Video 4, available at http://www.jcb.org/cgi/content/full/jcb.200712029/DC1). Also, these treatments do not impair cell cycle transit (Fig. S2 C) or induce DNA damage as judged by γH2AX histone isoform expression (Fig. S2 B).
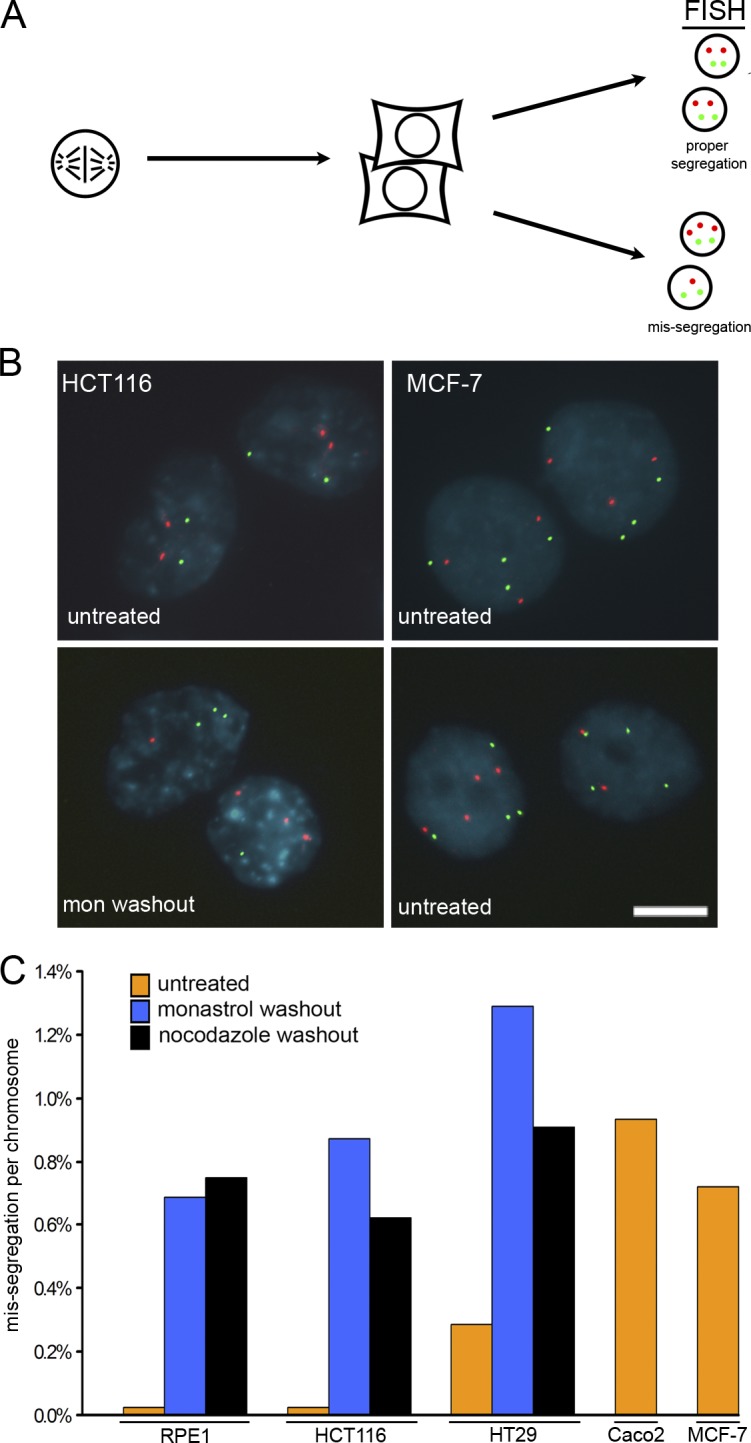
To assess mitotic fidelity, we quantified the modal chromosome number and the percentage of cells deviating from that mode in isolated colonies grown for 20–30 generations using FISH with chromosome-specific centromeric α-satellite DNA probes (Fig. 2 A). Colonies were either untreated or forced to recover from nocodazole- or monastrol-induced mitotic delay for one or more consecutive days during the growth of single-cell colonies (Fig. 2 B). Chromosome counts were performed on at least three independent colonies for each treatment (Tables S2 and S3, available at http://www.jcb.org/cgi/content/full/jcb.200712029/DC1). HCT116 cells did not survive monastrol-induced mitotic recovery for 10 or more consecutive days, so monastrol recovery in these cells was limited to nine or fewer consecutive days or at every other day throughout growth of the cell colony.
Figure 2.

CIN analyses.(A) Examples of FISH data from HCT116 colonies that were either untreated or subjected to recovery from monastrol-induced mitotic delay (monastrol washout) every third division during colony growth as indicated. Cells were fixed and stained with DAPI (blue) to visualize nuclei and with probes specific for centromeric α-satellite DNA of chromosomes 7 (green) and 8 (red). Arrowheads identify nuclei in which one or both chromosomes deviate from the modal number of two. Bar, 20 μm. (B) Cells were isolated by mitotic shake-off, plated at low density, and grown for several generations to form individual colonies with (bottom) or without (top) recovery from monastrol or nocodazole treatment sequentially for various numbers of cell divisions during colony growth. After ∼30 generations, cells were harvested, and interphase nuclei were labeled with centromere-specific FISH probes. (C) Percentage of nuclei that display deviation from the modal chromosome number of two for four different chromosomes in representative single colonies of HCT116 cell clones that were untreated or subjected to recovery from monastrol- or nocodazole-induced mitotic delay for various days during colony growth (data for all clones can be found in Table S2, available at http://www.jcb.org/cgi/content/full/jcb.200712029/DC1). *, P < 0.05, χ2 test. (D) Percentage of nuclei that display deviation from the modal chromosome number (chromosomal modes for each cell line are provided in Table S1) for four different chromosomes in representative single colonies in the untreated CIN cell lines HT29, Caco2, and MCF-7 (data for all clones can be found in Table S4); RPE-1 cells subjected to recovery from monastrol- or nocodazole-induced mitotic delay for 25 consecutive days (data for all clones can be found in Table S3); or HCT116 cells subjected to recovery from monastrol-induced mitotic delay for nine consecutive days or nocodazole-induced mitotic delay for 20 consecutive days (data for all clones can be found in Table S2).
Untreated HCT116 and RPE-1 cells are chromosomally stable and maintain a near-diploid karyotype (Lengauer et al., 1997) as judged by small deviations from a modal number of two for each chromosome (Fig. 2 C and Tables S2 and S3). Rare treated HCT116 colonies show increases in the modal number of one or all chromosomes examined. Trisomy for one chromosome must occur by missegregation at the earliest stages of colony growth, and tetraploidy most likely arises when cells escape mitotic arrest in the presence of monastrol or nocodazole. However, cells in these colonies show no significant increase in deviation from the modal chromosome number, confirming that in this cell line, alterations in ploidy itself do not increase the rate of chromosome missegregation (Lengauer et al., 1997). Other colonies of treated HCT116 and RPE-1 cells display significant deviation from a modal chromosome number of two for one or more chromosomes when the incidence of merotely is increased during consecutive mitoses (Tables S2 and S3). The extent of chromosome deviations was higher in colonies of HCT116 cells compared with RPE-1 cells and may reflect differences between transformed (HCT116) and immortalized but not transformed (RPE-1) cells. Fig. 2 C presents the percentage of cells with chromosome deviations from representative single colonies of HCT116 cells and illustrates that chromosome missegregation is more prevalent when mitotic cells recover from monastrol compared with nocodazole and that maximal deviation occurs after approximately five to seven consecutive days of mitotic recovery (Fig. 2 C). Importantly, both chromosome gains and losses contribute to deviation from diploidy (Fig. 2 A), and individual colonies differ in which chromosomes deviate from the mode (Tables S2 and S3). Also, chromosome numbers among cells within individual colonies vary (Fig. 2 A), demonstrating that multiple, independent chromosome missegregation events occur during colony growth. These chromosomal changes are characteristic of cells with CIN and demonstrate that chromosome missegregation does not obligatorily impair cytokinesis as was recently proposed (Shi and King, 2005). Thus, elevating the incidence of merotely in otherwise stable, near-diploid cells induces chromosome missegregation with the characteristics of CIN.
A striking outcome of these analyses is that colonies of both RPE-1 and HCT116 cells emerging after 20–30 generations with elevated merotely at each consecutive mitosis show no significant deviation from two for all chromosomes analyzed and maintain a diploid karyotype (Fig. 2, C and D; and Tables S2 and S3). In contrast, aneuploid tumor cells with CIN show significant increases in deviation from the chromosomal mode for multiple chromosomes after a similar number of generations in culture (Fig. 2 D). This raises the possibility that induction of merotely in RPE-1 and HCT116 cells may not significantly increase the rate of chromosome missegregation despite the fact that the percentage of anaphase cells with lagging chromatids are equivalent to aneuploid tumor cells with CIN (Fig. S1, compare A with B). To address this possibility, we used FISH to measure chromosome missegregation rates in RPE-1 cells, HCT116 cells, and aneuploid tumor cells with CIN (Fig. 3). For this assay, we collected mitotic cells by shake-off, spread the cells on slides at very low density, and incubated briefly to permit the completion of mitosis (Fig. 3 A). At low density, daughter cells remain adjacent, and chromosome missegregation rates can be calculated by counting centromeric fluorescent spots in daughter cell nuclei (Fig. 3 B). Chromosome missegregation rates determined by this method probably underestimate the total chromosome missegregation rate because single chromosomes contained in micronuclei are missed in the FISH analyses. Nevertheless, the rate of chromosome missegregation in untreated RPE-1 and HCT116 cells is ∼0.025% per chromosome and increases to 0.6–0.8% per chromosome upon the induction of merotely through mitotic recovery from either monastrol or nocodazole treatment (Fig. 3 C). These basal and induced rates of chromosome missegregation are similar to those previously measured in primary human fibroblasts (Cimini et al., 1999). Assuming all chromosomes behave equivalently, RPE-1 and HCT116 cells missegregate a chromosome every 100 cell divisions unless merotely is experimentally elevated, whereupon they missegregate a chromosome every third cell division. Chromosome missegregation rates in three aneuploid tumor cell lines with CIN range from ∼0.3 to ∼1.0% per chromosome (Fig. 3 C). Depending on the modal chromosome number in each cell line, these cells missegregate a chromosome every cell division (Caco2), every other cell division (MCF-7), or every fifth cell division (HT29). Thus, induction of merotely in otherwise near-diploid cells increases the chromosome missegregation rate to levels equivalent to those of aneuploid tumor cells with CIN. These rates of chromosome missegregation are sufficient to induce significant deviation in modal chromosome numbers in colonies of HT29, MCF-7, and Caco2 cell lines after 20–30 generations in culture but fail to do so in RPE-1 and HCT116 cells (Fig. 2 D and Tables S2–S4, available at http://www.jcb.org/cgi/content/full/jcb.200712029/DC1). Of note, HT29 cells display lower rates of chromosome missegregation than either RPE-1 or HCT116 cells with elevated merotely (Fig. 3 C), yet HT29 cells show significant increases in deviation from the chromosomal mode after 20–30 generations in culture (Fig. 2 D).
Figure 3.
Chromosome missegregation analysis. (A) Mitotic cells were harvested by mitotic shake-off, plated at low density on slides, and allowed to complete mitosis. Daughter cells were fixed and stained with DAPI (blue) to visualize nuclei and with probes specific to centromeric α-satellite DNA (FISH). (B) FISH for chromosomes 7 and 8 are shown for HCT116 and MCF-7 cells and show normal segregation (top) and missegregation (bottom). Bar, 10 μm. (C) Mean missegregation rate per chromosome for no fewer than two chromosomes in RPE-1 cells (untreated, n = 4,300; monastrol recovery, n = 2,620; nocodazole recovery, n = 2,602), HCT116 cells (untreated, n = 4,000; monastrol recovery, n = 2,640; nocodazole recovery, n = 2,660), HT29 cells (untreated, n = 2,680; monastrol recovery, n = 2,640; nocodazole recovery, n = 2,640), Caco2 cells (n = 1,023), and MCF-7 cells (n = 2,002) as indicated. Percentages have been corrected for modal chromosome number in each cell line (chromosomal modes for each cell line are provided in Table S1, available at http://www.jcb.org/cgi/content/full/jcb.200712029/DC1).
These data raise the possibility that chromosome missegregation diminishes the capacity of RPE-1 and HCT116 cells to propagate in culture. To test this idea, we used FISH to determine the percentage of cells with deviant chromosome numbers in populations at different times after monastrol- or nocodazole-induced mitotic recovery (Fig. 4). Populations of untreated RPE-1 and HCT116 cells show, on average, 2.9% and 4.0% of cells, respectively, that deviate from a modal chromosome number of two for the chromosomes examined. Percentages of cells with deviant chromosome numbers increase significantly immediately after recovery from monastrol- or nocodazole-induced mitotic arrest but decline to basal levels upon growth in culture without further treatment to increase merotely (Fig. 4, A and B). Mitotic recovery from nocodazole or monastrol also increases the frequencies of lagging chromatids (Fig. S1 B) and chromosome missegregation rates (Fig. 3 C) in aneuploid HT29 cells. Percentages of HT29 cells that deviate from the chromosomal mode in a population are relatively low but increase significantly immediately after recovery from monastrol- or nocodazole-induced mitotic arrest. However, unlike RPE-1 and HCT116 cells, percentages of HT29 cells with deviant chromosome numbers persist as these cells grow in culture (Fig. 4 C). Thus, a diminished capacity to propagate in culture selects against nondiploid RPE-1 and HCT116 cells, and this continually converges these populations toward an overall diploid karyotype. HT29 (and, by analogy, other aneuploid cells) cells appear to have acquired an ability to grow efficiently with nondiploid karyotypes. Cell viability would be expected to decrease in response to complete chromosome loss (i.e., nullisomy), but the percentage of RPE-1 and HCT116 cells that gained a chromosome also declines in the population, indicating that chromosome imbalance itself imposes a growth disadvantage. The rare colonies of HCT116 cells that we identified to be trisomic for one chromosome or tetraploid for all chromosomes probably reflect an increased capacity of transformed cells to propagate with these changes that is not shared by immortalized RPE-1 cells. These data also demonstrate that the missegregation of one or a few chromosomes does not convert these cells to the CIN phenotype, and the continuous elevation of chromosome missegregation rates, either induced by recovery from monastrol or nocodazole treatment during mitosis or through acquisition of a specific mutation, is necessary for the CIN phenotype.
Figure 4.

Chromosome deviations in cell populations. (A–C) RPE1 (A), HCT116 (B), and HT29 (C) cells were grown in flasks and were untreated or treated with monastrol or nocodazole. Mitotic cells were harvested by shake-off, washed to remove the mitotic inhibitor, and plated in growth medium to allow the completion of mitosis. Cells were then harvested for FISH analysis at the indicated times. Mean deviation from the mode for two different chromosomes in each of two independent experiments, with 600 nuclei counted per chromosome per time point and condition for each experiment. Error bars are SEM. *, P < 0.05, χ2 test. (D) Diploid cells showing two pairs of chromosomes (one red and one blue) that segregate faithfully during most mitoses. A chromosome missegregation event occurs, generating two nondiploid daughter cells that do not propagate efficiently, so the overall karyotype of the culture remains diploid (top). A phenotypic change (gray cells) arises either independently of chromosome missegregation (as shown) or coordinated with the missegregation that permits nondiploid cells to propagate (bottom). Chromosome missegregation subsequently generates aneuploid cells in this population as those cells continue to accrue abnormal chromosome numbers.
Collectively, these data show that elevating chromosome missegregation rates alone in human cultured cells is not sufficient to convert stable, near-diploid cells into highly aneuploid cells with karyotypes that resemble those of tumor cells. Extended monastrol or nocodazole treatments might compromise cell growth, permitting only those cells that experienced the shortest duration of treatment, and presumably the most likely to remain diploid, to survive. However, colonies of HCT116 cells progressed through multiple divisions despite extended monastrol treatment at each consecutive division (Fig. S2 C), and other methods for inducing chromosome missegregation that do not use drug washout strategies, such as mutation of cdc4 or haploinsufficiency for Mad2, also generate CIN without generating highly aneuploid cells after many generations in culture (Michel et al., 2001; Rajagopalan et al., 2004). These results suggest that proliferation in culture selects against the accumulation of aneuploid cells in the population. This explains why RPE-1 and HCT116 cells maintain diploid karyotypes during propagation in the laboratory for thousands of population doublings and is reminiscent of the convergence of haploid and tetraploid strains of budding yeast to diploidy upon extended growth in culture (Gerstein et al., 2006). Thus, CIN (elevated rate of chromosome missegregation) and aneuploidy (a state with abnormal numbers of chromosomes) are distinct phenotypes, and a change that permits the efficient propagation of aneuploid cells appears to be required for CIN to convert diploid cells into highly aneuploid cells (Fig. 4 D). Metabolic imbalance, as recently demonstrated for budding yeast (Torres et al., 2007), might decrease aneuploid cell fitness in culture, providing a competitive advantage to their diploid counterparts. Alternatively, chromosome missegregation in otherwise diploid cells might induce senescence, apoptosis, or cell cycle delay through as yet uncharacterized pathways. This multistep view for the generation of highly aneuploid cells is a departure from other views purporting that single mutagenic events responsible for CIN are sufficient to generate aneuploidy (Lengauer et al., 1998; Shi and King, 2005) and implies that overcoming limitations on the propagation of nondiploid cells is the rate-limiting step. Such a growth limitation on nondiploid cells might provide a tumor suppression function in somatic tissues because the targeted mutation of genes in mice that should elevate chromosome missegregation rates suppresses tumor formation in some tissues (Weaver et al., 2007) and only induces tumor formation very late in life in other tissues (Michel et al., 2001). In contrast, the viability of trisomic individuals (Down syndrome) suggests that specific adaptations to nondiploid karyotypes can be acquired by germ or stem cells that can be subsequently maintained in somatic tissues.
Materials and methods
Fixed cell imaging
Cells were fixed in 3.5% PFA and stained with DAPI, tubulin-specific mAb (Sigma-Aldrich), and centromere-specific human serum (provided by K. Sullivan, Scripps Research Institute, La Jolla, CA) detected with FITC and/or Texas red. Images were acquired with a cooled CCD camera (Orca ER; Hamamatsu) mounted on a microscope (Eclipse TE 2000-E; Nikon). 0.25-μm optical sections in the z axis were collected with a planApo 60× 1.4 NA oil immersion objective at room temperature. Iterative restoration was performed using Phylum Live software (Improvision), and images represent four to seven merged planes in the z axis. Autocontrast using Photoshop CS2 (Adobe) was applied to images. Anaphase chromatids were counted as lagging if they contained kinetochore staining and were located in the spindle midzone, separated from kinetochores at the poles.
Live cell imaging
Cells expressing H2B-GFP (BD Biosciences) were mounted in modified rose chambers and maintained at 37°C in an incubator surrounding the stage of the Nikon microscope with an Orca ER cooled CCD camera. 27 0.5-μm optical sections were collected in the z axis at 1-min intervals with a planApo 60× 1.4 NA oil immersion lens. Iterative restoration and noise reduction for z stacks at each time interval were performed using Phylum Live software. Still frames in Fig. 1, Fig. S2 A, and Videos 1–4 are full volume projections. Photoshop CS2 was used to enhance contrast using autocontrast for still frames in Figs. 1 and S2 A. Anaphase chromatids were counted as lagging if they failed to segregate poleward with the chromatid mass.
Monastrol and nocodazole time courses
Cells were treated with 100 μM monastrol or 100 ng/ml nocodazole for 8 h. Mitotic cells were isolated by shake-off and washed with PBS twice before being plated at very low density in fresh medium. Cell clones were sequentially treated with monastrol or nocodazole followed by washout for 1, 3, 5, 7, 9, 10, 15, 20, or 25 cycles as specified in Fig. 2 and in Tables S2 and S3. HCT116 clones designated every third division had the time between treatment extended to 42 h to allow two full cell cycles without the presence of the drug. Colonies were isolated after ∼15–20 divisions using cloning rings. Isolated clones were maintained until they reached ∼30 generations. Experiments with HT29 cells used 400 μM monastrol or 100 ng/ml nocodazole.
FISH
Cells were washed with PBS and treated with 75 mM potassium chloride for 30 min. Cells were fixed in and washed twice with methanol–acetic acid (3:1). FISH was performed using α-satellite probes specific for chromosomes 2, 7, 8, and 15 (MP Biomedicals and Cytocell) according to the manufacturer's protocol, and chromosome signals in at least 300 nuclei were scored according to the criteria of Cimini et al. (1999). FISH images were acquired as 0.25-μm optical sections with the 60× 1.4 NA objective and are projections of four to five merged planes in the z axis.
Phase-contrast/GFP imaging of clones
Mitotic HCT116 cells expressing GFP–histone H2B grown in flasks were collected by shake-off with or without monastrol treatment for 8 h as described in the Monastrol and nocodazole time courses section and were plated at low density in six-well plates. Cell clones were identified 15–16 h later and treated sequentially with or without monastrol for four cycles, and images were acquired using phase-contrast and fluorescence microscopy (GFP signal) with a planFluor 40× 0.6 NA objective 16 h after each washout.
Immunoblotting
RPE-1 cells were treated with culture medium alone (control) or with 100 μM monastrol, 100 ng/ml nocodazole, or 1 μM doxorubicin and incubated at 37°C for 8 h. Total cell protein was separated by size using SDS-PAGE and transferred to a polyvinylidene difluoride membrane. The membrane was incubated with antibodies for γH2AX (Novus Biologicals) and tubulin (Sigma-Aldrich) in 1% milk TBS for 4 h at room temperature. Primary antibodies were detected by incubation with HRP-conjugated secondary antibodies (Bio-Rad Laboratories) in TBS for 1 h at room temperature. Signal was detected by chemiluminescence.
Online supplemental material
Tables S1–S4 display chromosome content in cell lines with and without monastrol or nocodazole washout for various consecutive divisions. Fig. S1 displays lagging chromatid rates in various cell lines with and without monastrol or nocodazole washout in addition to fluorescence images of examples of lagging chromatids in various cell lines. Fig. S2 displays still frames from HCT116 cells expressing GFP–histone 2B after monastrol washout, levels of γH2AX in treated and untreated cells, and colonies of HCT116 cells growing with and without monastrol washout at consecutive divisions. Videos 1–4 display chromosome segregation in HCT116 (Video 1), Caco2 (Video 2), and MCF-7 (Video 3) cells as well as HCT116 cells recovering from monastrol treatment (Video 4). Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200712029/DC1.
Supplementary Material
Acknowledgments
We thank Amity Manning and Tarun Kapoor for comments on the manuscript and Dr. McPeek, Dr. Whitfield, Dr. Sporn, Dr. Pellman, Dr. Sluder, Dr. Sullivan, and Dr. Chang for technical assistance or reagents.
This work was supported by National Institutes of Health grants GM51542 (to D.A. Compton) and GM008704 (to S.L. Thompson).
Abbreviation used in this paper: CIN, chromosomal instability.
References
- Cahill, D.P., C. Lengauer, J. Yu, G.J. Riggins, J.K. Wilson, S.D. Markowitz, K.W. Kinzler, and B. Vogelstein. 1998. Mutations of mitotic checkpoint genes in human cancers. Nature. 392:300–303. [DOI] [PubMed] [Google Scholar]
- Cimini, D., and F. Degrassi. 2005. Aneuploidy: a matter of bad connections. Trends Cell Biol. 15:442–451. [DOI] [PubMed] [Google Scholar]
- Cimini, D., C. Tanzarella, and F. Degrassi. 1999. Differences in malsegregation rates obtained by scoring ana-telophases or binucleate cells. Mutagenesis. 14:563–568. [DOI] [PubMed] [Google Scholar]
- Cimini, D., B. Howell, P. Maddox, A. Khodjakov, F. Degrassi, and E.D. Salmon. 2001. Merotelic kinetochore orientation is a major mechanism of aneuploidy in mitotic mammalian tissue cells. J. Cell Biol. 153:517–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimini, D., L.A. Cameron, and E.D. Salmon. 2004. Anaphase spindle mechanics prevent mis-segregation of merotelically oriented chromosomes. Curr. Biol. 14:2149–2155. [DOI] [PubMed] [Google Scholar]
- DeLuca, J.G., W.E. Gall, C. Ciferri, D. Cimini, A. Musacchio, and E.D. Salmon. 2006. Kinetochore microtubule dynamics and attachment stability are regulated by Hec1. Cell. 127:969–982. [DOI] [PubMed] [Google Scholar]
- Fodde, R., J. Kuipers, C. Rosenberg, R. Smits, M. Kielman, C. Gaspar, J.H. van Es, C. Breukel, J. Wiegant, R.H. Giles, and H. Clevers. 2001. Mutations in the APC tumor suppressor gene cause chromosomal instability. Nat. Cell Biol. 3:433–438. [DOI] [PubMed] [Google Scholar]
- Ganem, N.J., K. Upton, and D.A. Compton. 2005. Efficient mitosis in human cells lacking poleward microtubule flux. Curr. Biol. 15:1827–1832. [DOI] [PubMed] [Google Scholar]
- Gao, C., K. Furge, J. Koeman, K. Dykema, Y. Su, M.L. Cutler, A. Werts, P. Haak, and G.F. Vande Woude. 2007. Chromosome instability, chromosome transcriptome, and clonal evolution of tumor cell populations. Proc. Natl. Acad. Sci. USA. 104:8995–9000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein, A.C., H.-J.E. Chun, A. Grant, and S.P. Otto. 2006. Genomic convergence toward diploidy in Saccharomyces cerevisiae. PLOS Genet. 2:e145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green, R.A., and K.B. Kaplan. 2003. Chromosome instability in colorectal tumor cells is associated with defects in microtubule plus-end attachments caused by dominant mutation in APC. J. Cell Biol. 163:949–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauf, S., R.W. Cole, S. LaTerra, C. Zimmer, G. Schnapp, R. Walter, A. Heckel, J. van Meel, C.L. Rieder, and J.-M. Peters. 2003. The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore-microtubule attachment and in maintaining the spindle assembly checkpoint. J. Cell Biol. 161:281–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, H., J. Feng, J. Famulski, J.B. Rattner, S.T. Liu, G.D. Kao, R. Muschel, G.K.T. Chan, and T.J. Yen. 2007. Tripin/hSgo2 recruits MCAK to the inner centromere to correct defective kinetochore attachments. J. Cell Biol. 177:413–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joukov, V., A.C. Groen, T. Prokhorova, R. Gerson, E. White, A. Rodriguez, J.C. Walter, and D.M. Livingston. 2006. The BRCA1/BARD1 heterodimer modulates Ran-dependent mitotic spindle assembly. Cell. 127:539–552. [DOI] [PubMed] [Google Scholar]
- Kline-Smith, S.L., A. Khodjakov, P. Hergert, and C.E. Walczak. 2004. Depletion of centromeric MCAK leads to chromosome congression and segregation defects due to improper kinetochore attachments. Mol. Biol. Cell. 15:1146–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowlton, A.L., W. Lan, and P.T. Stukenberg. 2006. Aurora B is enriched at merotelic attachment sites, where it regulates MCAK. Curr. Biol. 16:1705–1710. [DOI] [PubMed] [Google Scholar]
- Kuukasjarvi, T., R. Karhu, M. Tanner, M. Kahkonen, A. Schaffer, N. Nupponen, S. Pennanen, A. Kallioniemi, O.-P. Kallioniemi, and J. Isola. 1997. Genetic heterogeneity and clonal evolution underlying development of asynchronous metastasis in human breast cancer. Cancer Res. 57:1597–1604. [PubMed] [Google Scholar]
- Lengauer, C., K.W. Kinzler, and B. Vogelstein. 1997. Genetic instability in colorectal cancers. Nature. 386:623–627. [DOI] [PubMed] [Google Scholar]
- Lengauer, C., K.W. Kinzler, and B. Vogelstein. 1998. Genetic instabilities in human cancers. Nature. 396:643–649. [DOI] [PubMed] [Google Scholar]
- Lingle, W.L., S.L. Barrett, V.C. Negron, A.B. D'Assoro, K. Boeneman, W. Liu, C.M. Whitehead, C. Reynolds, and J.L. Salisbury. 2002. Centrosome amplification drives chromosomal instability in breast tumor development. Proc. Natl. Acad. Sci. USA. 99:1978–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y., and W.F. Bodmer. 2006. Analysis of p53 mutations and their expression in 56 colorectal cancer cell lines. Proc. Natl. Acad. Sci. USA. 103:976–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel, L.S., V. Liberal, A. Chatterjee, R. Kirchwegger, B. Pasche, W. Gerald, M. Dobles, P.K. Sorger, V.V.V.S. Murty, and R. Benezra. 2001. MAD2 haplo-insufficiency causes premature anaphase and chromosome instability in mammalian cells. Nature. 409:355–359. [DOI] [PubMed] [Google Scholar]
- Rajagopalan, H., P.V. Jallepalli, C. Rago, V.E. Velculescu, K.W. Kinzler, B. Vogelstein, and C. Lengauer. 2004. Inactivation of hCDC4 can cause chromosomal instability. Nature. 428:77–81. [DOI] [PubMed] [Google Scholar]
- Salmon, E.D., D. Cimini, L.A. Cameron, and J.G. DeLuca. 2005. Merotelic kinetochores in mammalian tissue cells. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 360:553–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, Q., and R.W. King. 2005. Chromosomal nondisjunction yields tetraploid rather than aneuploid cells in human cell lines. Nature. 437:1038–1042. [DOI] [PubMed] [Google Scholar]
- Storchova, Z., and D. Pellman. 2004. From polyploidy to aneuploidy, genome instability and cancer. Nat. Rev. Mol. Cell Biol. 5:45–54. [DOI] [PubMed] [Google Scholar]
- Tighe, A., V.L. Johnson, M. Albertella, and S.S. Taylor. 2001. Aneuploid colon cancer cells have a robust spindle checkpoint. EMBO Rep. 2:609–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres, E.M., T. Sokolosky, C.M. Tucker, L.Y. Chan, M. Boselli, M.J. Dunham, and A. Amon. 2007. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science. 317:916–924. [DOI] [PubMed] [Google Scholar]
- Weaver, B.A.A., A.D. Silk, C. Montagna, P. Verdier-Pinard, and D.W. Cleveland. 2007. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell. 11:25–36. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}