

Abstract
In both multiple sclerosis (MS) patients and experimental autoimmune encephalomyelitis (EAE) animals, axon loss has been demonstrated to correlate with neurological disability. However, it is difficult to accurately determine the location and severity of axonal damage since the lesion in MS or EAE is disseminated and is frequently in a relapsing-remitting mode. The corticospinal system is the only direct pathway from the motorsensory cortex to the spinal cord, and the major neural pathway for control of voluntary movement. Moreover, it is frequently involved in the pathological process of the disease. To evaluate corticospinal tract (CST) axon loss in EAE mice, we developed a direct tracing method with a fluorescent neuronal tracer DiI which was injected into the primary motor cortex and sensorimotor cortex to label the pyramidal neurons. The lesion location in the spinal cord and axon disruption were indicated by dye leakage. Using the EAE induced axon reduction as an index of the extent of axonal damage, our data showed a high correlation between the axonal loss and the behavioral outcome score in the EAE mice. The results were consistent with the axonal Bielschowsky silver staining. Thus, this CST tracing method permits monitoring of the axonal damage in EAE.
Keywords: DiI, corticospinal tract, experimental autoimmune encephalomyelitis, axon loss, mice
1. Introduction
Multiple sclerosis (MS) is a chronic immune-mediated inflammatory disease of the central nervous system (CNS), and is the most common cause of neurological disability in young adults (Hickey, 1999). The disease may initially show a relapsing-remitting course but finally leads to permanent neurological impairment. Experimental autoimmune encephalomyelitis (EAE) is a widely used murine model for investigating the disease since it is characterized by extensive autoimmune-induced inflammatory lesions, demyelination, axonal damage, and neuronal death within the CNS (Gold et al., 2000). Classically, demyelination induced nerve action potential conduction block was considered to be the primary contributor of clinical symptoms and neural deficits in MS. However, recent studies indicate that neuronal and axonal dysfunction at the earliest clinical stages, and axonal disruption and subsequent axonal loss at later phases may be the major cause of progressive and persistent, irreversible disability of the disease (De Stefano et al., 1998; Filippi et al., 2003; Papadopoulos et al., 2006; Trapp et al., 1998).
In many MS patients and EAE animals, the pathological processes are predominantly found in the spinal cord but not in the brain (Bettelli, 2007). Moreover, neurological disability in patients with MS or animals with EAE has been correlated with the magnitude of degeneration of spinal cord axons (Bjartmar et al., 2000; Bjartmar et al., 2003; Medana and Esiri, 2003). Protective treatment for axonal degeneration also significantly reduces neurological deficit in the EAE model (Bechtold et al., 2006; Black et al., 2006; Kaneko et al., 2006; Zhang et al., 2006). The descending axons of the corticospinal tract (CST) are the only direct pathway from the motorsensory cortex to the spinal cord, and the major neural pathway for control of voluntary movement (Heffner and Masterton, 1983), and are often affected in MS (DeLuca et al., 2004). Most CST axons of the rodent decussate in the caudal medulla and descend caudally in the most-ventral part of the dorsal funiculus of the spinal cord (Brown, 1971). Therefore, the loss of CST axons in the spinal cord may be a neuroanatomical parameter to evaluate disease severity, and the effects of treatment in the EAE model. However, it is difficult to accurately detect the location and severity of axonal damage since the disease is disseminated and relapsing. In the present study, we inject a fluorescent neuronal tracer DiI into the primary motor and sensorimotor cortex to label the pyramidal neurons, and thereby establish a direct labeling method to evaluate the CST axon loss in EAE mice.
2. Materials and methods
Adult female SJL/J mice (n=10, 4 for normal control, 6 for EAE; 8-10 weeks old; Jackson Laboratory, Bar Harbor, ME, USA) were used throughout this study.
2.1. Induction and scoring of EAE
Myelin proteolipid protein (PLP; p139-151, HSLGKWLGHPDKF; SynPep Corporation, Dublin, CA, USA) was used for immunization. The purity of the peptide was greater than 95% as measured by high-performance liquid chromatography. EAE was induced following previously published procedures (Youssef et al., 2002; Zhang et al., 2005). Briefly, the mice were immunized by subcutaneous injection with 25 μg PLP dissolved in 50 μl complete Freund's adjuvant (CFA; Difco Laboratories, Livonia, MI, USA). On the day of immunization and 48 hr later, pertussis toxin (PT; List Biological Laboratories, Campbell, CA, USA) at 200 μg in 0.2 ml phosphate-buffered saline (PBS) was injected into the mouse tail vein. Immunized mice were weighed and scored daily for clinical symptoms of EAE, as follows: 0, healthy; 1, loss of tail tone; 2, ataxia and/or paresis of hindlimbs; 3, paralysis of hindlimbs and/or paresis of forelimbs; 4, tetraparalysis; and 5, moribund or dead (Furlan et al., 1999). The first neurological attack was defined as a concomitant increase in clinical score and loss of body weight. The peak severity of acute EAE was defined as the highest score achieved by each mouse during the first 5 days of the first attack. In virtually every case, this score was maintained for at least 2 consecutive days.
2.2. Dye injection
For corticospinal tracing, the dye injection was performed seven days before sacrifice. Under deep anesthesia with an intraperitoneal injection of Ketamine (100 mg/kg) and Xylazine (10 mg/kg), an incision was made through the skin covering the skull. Two elongated craniotomies were made at 1 mm lateral to the midline on both sides from 1 to -1 mm rostral to the bregma on the skull (Paxinos and Franklin, 1997). A high-speed drill (Foredom Electric, Bethel, CT, USA) was carefully used to reduce the skull thickness by about 80%. Residual skull was completely removed by gently scraping with microsurgical forceps.
2.5% solution of 1,1″-Dioleyl-3,3,3″,3″-tetramethylindocarbocyanine methanesulfonate (DiI; AnaSpec, San Jose, CA, USA) in DMSO was injected through a finely drawn glass capillary with an electric micro injector into six points in the primary motor cortex and motorsensory cortex (Paxinos and Franklin, 1997) (40 nl per injection; stereotaxic coordinates: 1, 0, and −1 mm to the bregma, 1 mm bilateral to the midline, 0.5 mm depth from the surface of the cortex). The micropipette remained in place for 4 min after completion of the injection. If cortical vessels were encountered at the intended incision site, the site was moved immediately rostral or caudal to avoid the cortical vessel. After completion of the injections, the scalp was sutured, and mice were allowed to awaken.
2.3. Tissue preparation and DiI measurement
Animals were sacrificed under deep Ketamine anesthesia at 50 days after EAE onset. Mice were perfused transcardialy with saline, followed by 4% paraformaldehyde. The entire spinal cord was extracted from the vertebra and then immersed in 4% paraformaldehyde overnight. The spinal cord segments of C1-2 and L3-4 were embedded in paraffin. C2-3 and L2-3 were cut for transverse vibratome sections (100 μm). The remaining part of the spinal cord was cut into 5 mm segments for longitudinal vibratome sections (100 μm). The fluorescent labeled CST axons in the spinal dorsal funiculus were digitized using a 3-CCD color video camera (DXC-970MD; Sony, Tokyo, Japan) interfaced with an Micro Computer Imaging Device system (MCID, Imaging Research, St. Catharines, Ontario, Canada) or a Bio-Rad MRC 1024 (argon and krypton) laser-scanning confocal imaging system mounted onto a Zeiss microscope (Bio-Rad, Cambridge, MA, USA). Microscopic data were acquired with a 10× objective or a 40× oil-immersion objective lens.
2.4. Bielschowsky silver staining
Bielschowsky silver staining was performed on 6 μm-thick paraffin coronal sections of the cervical and lumbar cord (Litchfield and Nagy, 2001). Briefly, the rehydrated spinal sections were placed in 20% silver nitrate at room temperature in the dark for 15 min. The solution was washed off with distilled water and replaced by ammoniacal silver solution for 10 min in the dark. Then ammonium hydroxide/formaldehyde was added to stain slides until the tissue turned brown with a gold background, and was followed by treatment with gold chloride and sodium thiosulphate. The average axonal number in the ventral-most third part of the dorsal funiculus at the cervical and lumbar levels was counted on 3 slides from each animal, under a 40× objective (BX40; Olympus Optical, Tokyo, Japan) using a 3-CCD color video camera (DXC-970 MD; Sony) interfaced with the MCID computer analysis system (Imaging Research).
2.5. Quantification and statistical analysis
The numbers of DiI-labeled CST axons in the cervical and lumbar dorsal funiculus were determined on the single laser-scanning confocal images of three transverse spinal vibratome sections from each animal by using NIH image software (Image J). All data are presented as mean ± SD. Two-sample t-test was used to test the difference in means of labeled axon numbers between normal and EAE groups. The extent of CST damage for an individual animal in the EAE group was analyzed by the percentage of CST fiber reduction between the lumbar and cervical levels, which was corrected with the mean reduction of CST fibers between the respective levels in normal animals as fibers entered into the gray matter using following expression:
| Eq. 1: |
R = percentage of EAE induced CST fiber reduction, Nel = number of DiI-labeled axons at the lumbar level in the EAE mice, Nec = number of DiI-labeled axons at the cervical level in the EAE mice, Nnl = mean number of DiI-labeled axons at the lumbar level in the normal mice, Nnc = mean number of DiI-labeled axons at the cervical level in the normal mice. Pearson's correlation coefficients were calculated between the axonal damage extent and the behavioral scores.
3. Results
3.1. Characterization of the experimental model
In most SJL mice, onset of clinical symptoms occurred at days 10-15 after immunization, and the peak score for the clinical symptoms was reached between day 3 and day 5 after the onset of clinical deficits. Mice displayed moderate symptoms rarely exceeding a score of 4 in the period of the first attack (Fig. 1). Then a slow and incomplete recovery process followed until the next attack. There were two courses of disease within 50 days after clinical symptom onset when the animals were sacrificed.
Figure 1. Course of functional deficient in the EAE mice.
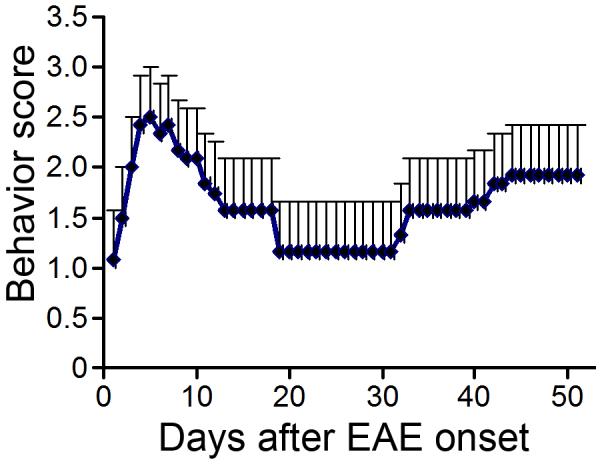
Data represent mean behavior scores measured daily for EAE animals. At 50 days after the first clinical onset, the animals were in the second attack courses.
3.2. CST labeling in normal mice
Using the precise micro injector, a total volume of 240 nl for six injections of DiI was injected stereotactically into the motorsensory cortex layer V to label the pyramidal neurons without any leakage out the cortex (Fig. 2A and B). In the rodent, most of the CST decussates to the opposite side in the medulla oblongata and descends in the most-ventral part of spinal dorsal funiculus. One week after dye injection, the DiI diffused throughout the whole length of the spinal cord (C-E).
Figure 2. Anterograde DiI-labeling of the CST.
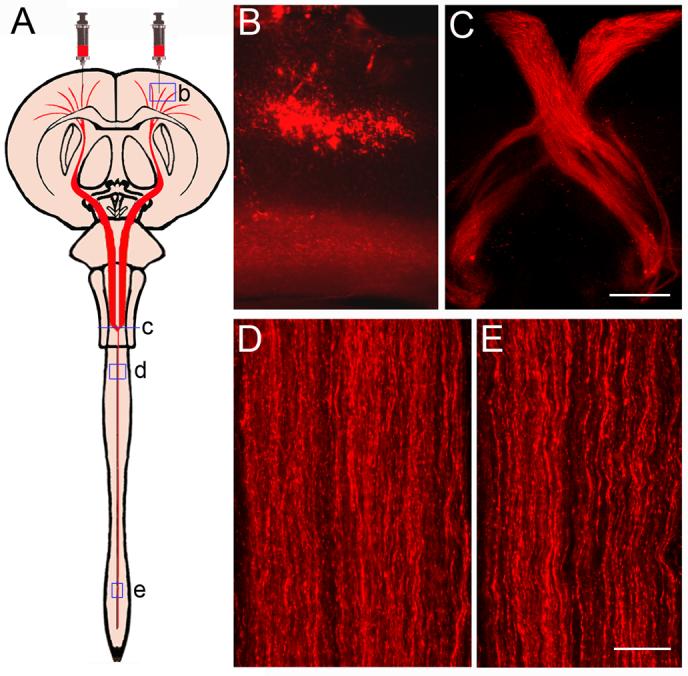
A schematic diagram shows the bilateral cortical injections for CST labeling (A). A small volume of DiI solution was delivered into the layer V of motor cortex through a finely drawn glass capillary with a microinjection system to label the pyramidal neurons (B) in a representative normal mouse. Fluorescent images show that the CST fibers were labeled by DiI one week after injection in a coronal section at the decussation level (C) and in longitudinal sections of cervical (D) and lumbar (E) enlargements. Scale Bars in B and C = 250 μm, in D and E = 25 μm.
3.3. CST damage in the EAE mice
In the EAE mice, six to eight damage sites indicated by DiI leakage in the CST were found in each animal at 50 days after EAE onset. As shown in Fig. 3A, the number of DiI-labeled intact axons decreased caudally to each damage site. Axonal disruption on individual CST fibers was detected by the presence of focal concentration of DiI leakage inside the lesion on the single scanned confocal images (B). Most CST axons were disrupted in the lesion sites close to the lumbar cord (C).
Figure 3. Longitudinal Images show the CST damage in the spinal dorsal funiculus.

A typical CST lesion site in the spinal cord was indicated with DiI leakage in a MCID image (A). The DiI-labeled axons were decreased caudal to the lesion site. In a single layer confocal image of the CST lesion site, blobs of leaked DiI show the axonal damage site (Arrows in B). In a more caudal lesion site, only few intact axons were labeled (C). The images are oriented with rostral at the top. Scale Bars in A = 250 μm, B and C = 25 μm.
3.4. Quantification of CST lesion severity
In order to assess the CST axon loss in the EAE mice, the numbers of DiI-labeled axons in the dorsal funiculus (Fig. 4A) were counted on the single layer confocal images of spinal coronal sections at the cervical and lumbar levels. As shown in Fig. 4B-D, there was no difference in axon numbers between the normal and EAE mice at the cervical level. However, the DiI-labeled intact axon number at the lumbar level was significantly reduced in the EAE mice compared to that in the normal animals (p < 0.01; Fig. 4B, E and F). At 50 days after EAE onset, 55.8% of the lumbar CST was disrupted.
Figure 4. Axonal quantification.
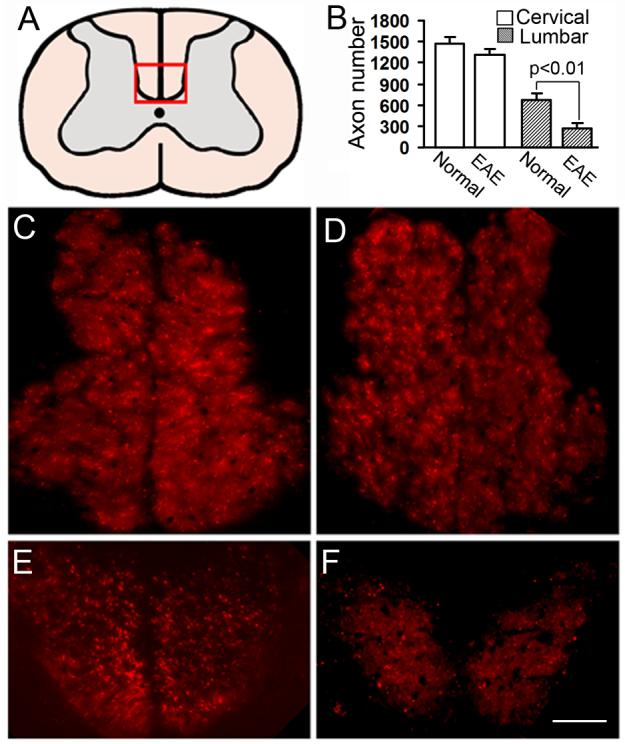
DiI-labeled axon count was performed on the single layer confocal images of high cervical and lumbar cord (A). At the cervical level, the DiI-labeled axon number of EAE mice (B and D) was only slightly decreased compared with normal mice (B and C), while the EAE lesion significantly reduced the DiI-labeled intact axon at the lumbar level (B, E and F). Scale Bars in C-F = 50 μm.
To confirm the CST axonal loss evaluated with DiI tracing method, we further measured the number of CST axons stained with Bielschowsky silver at cervical and lumbar levels in the same animals (Fig. 5). In the ventral-most part of cervical dorsal funiculus, the axon number was 4324 ± 239 and 4140 ± 241 in the normal and EAE mice, respectively. However, the CST number was significantly decreased to 952 ± 241 in EAE mice compared with 1589 ± 226 in normal mice at the lumbar level. Using the same expression (Eq. 1) as for the evaluation of DiI, an average of 49.4% of EAE induced lumbar CST loss was observed at 50 days after onset.
Figure 5. Bielschowsky silver staining.

CST axons in the ventral-most part of the spinal dorsal funiculus were stained on coronal paraffin sections of the cervical and lumbar cord in normal (A and C) and EAE mice (B and D). There was no significant decrease of cervical CST number in the EAE mice, while an average loss of 49.4% of CST number in the lumbar cord after EAE was estimated with Bielschowsky silver staining. Scale Bar = 50 μm.
Using the corrected percentage of EAE induced lumbar CST reduction as an index for the extent of damage severity, the correlation coefficient was calculated between the axon damage and behavior score for each EAE mouse (Fig. 6). Statistical data showed that the CST axon loss was highly correlated to the functional deficit (r = 0.80, p < 0.05 for DiI and r = 0.77, p < 0.05 for Bielschowsky silver).
Figure 6. Correlation between the axonal loss and functional deficient.
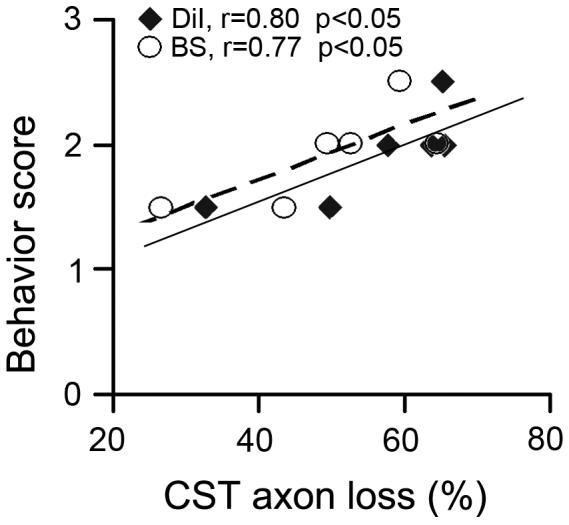
Significant correlations between the axonal damage and behavior score in individual EAE mice at 50 days after the first attack were demonstrated with both DiI tracing method (quadrangle, r=0.80, p<0.05) and Bielschowsky silver staining (circle, r=0.77, p<0.05).
4. Discussion
In order to directly evaluate axonal damage in the EAE model, we successfully established a microinjection method for anterograde labeling of the CST fibers in the spinal cord via cortical delivery of the lipophilic fluorescent tracer, DiI, to the pyramidal neurons. The method is technically simple, both in tracer application and tissue processing. There is an obvious advantage for the use of DiI, in that the labeled axons in the vibratome sections can be measured directly under a fluorescent microscope or a laser-scanning confocal imaging system without additional processing. However, because of slight variations in the location or amount of dye applied, small variations in the number of labeled CST fibers are inevitable. To avoid possible inter-animal variation, the percentage of reduced DiI-labeled axon number in the lumbar spinal cord compared with that at the cervical level in individual EAE mice was used to index the extent of axonal damage severity, which was corrected with the mean CST reduction between the cervical and the lumbar cord in normal animals.
DiI is an excellent anterograde corticospinal tracer due to its high fluorescence intensity, resistance to photo bleaching, and non-cytotoxicity. Although it is possible that DiI can cross “immature synapses” that are present between developing neurons (Vannucchi and Faussone-Pellegrini, 2000), DiI transcellular labeling has been found in the adult rodent spinal cord after DiI applied into the sensorimotor cortex (Tsai et al., 2001). In mouse, as early as five to seven days after DiI is taken up into the pyramidal neurons, selective CST tract labeling was present from the internal capsule to the lumbar cord. Other spinal tracts such as the reticulospinal tract in the brainstem and the propriospinal tracts in the dorsal column, did not show DiI labeling. Moreover, the DiI labeling can not traverse a transection lesion in the spinal cord (Tsai et al., 2001). Therefore, using this approach for the EAE model, only the intact CST fibers and the segments rostral to the disruption site can be labeled. Because of the significance of axonal loss contributed to the neurological functional deficit, it is a crucial substrate of MS therapeutic strategies to protect the axonal integrity. The CST tracing technique may provide a useful quantitative method to evaluate the availability of MS therapeutic approaches.
Currently, neuroanatomical studies for axonal damage and axonal loss in the MS or EAE process are primarily performed using magnetic resonance imaging (MRI) (Matthews et al., 1998; Pike et al., 1999) and immunohistochemical analysis (Ferguson et al., 1997; Kornek et al., 2000; Kuhlmann et al., 2002; Raine and Cross, 1989; Trapp et al., 1998). Axonal damage may occur in areas with acute inflammation and demyelination from an early stage of EAE, and lead to permanent disability with cumulative axonal loss (Ferguson et al., 1997). Thus, there is a high correlation between the axonal loss and the persistent neurological deficit in the chronic-relapsing EAE (De Stefano et al., 1998; van Waesberghe et al., 1999). However, one problem in acute EAE models is that in many cases the structural damage is not reflected in the clinical deficit (Wujek et al., 2002). It is possible that the irreversible axonal damage occurs in the early stage of EAE, but may be limited to a small fraction of axons from a given axonal tract system, which can be compensated by the remaining intact fibers. Thus axon loss must exceed a threshold before progressive un-remitting clinical symptoms appear (Trapp et al., 1999). Only in models with a chronic disease phase is the accumulated structural damage translated into measurable clinical deficits (McGavern et al., 2000). On the other hand, a primary intrinsic limitation of conventional MRI studies as an indirect method to measure the axonal change is the difficulty to identify the particular underlying pathology due to image contrast, since it can be affected by many factors (Arnold, 1999). Immunohistochemical staining with antibodies against the neurofilament proteins or axonal degeneration markers such as nonphosphorylated neurofilament H (SMI-32) and amyloid precursor protein (APP) (Mancardi et al., 2001) is the commonly used technique for axon detection in the EAE model. However, it is difficult to identify the disrupted axonal debris with immunostaining on thin sections of the degenerating distal part of the spinal cord, which may underestimate the extent of axonal loss. In contrast, the DiI can be used to selectively label the intact CST axons by cortical injection after EAE onset, while it may also be used to monitor the axonal degeneration process if the axons were labeled prior to damage (unpublished data).
In rodents, the dorsal CST is the principal component mediating motor behavior. In the present study, we therefore detected the CST integrity in the spinal cord by direct fluorescent tracing to parallel clinical behavior in the EAE model. At 50 days after EAE onset, most of the CST axon disruptions were in the spinal cord. The intact axon number at the cervical level only slightly decreased. Our data indicate that although the inflammatory onset of EAE is disseminated in the CNS, the lesion in the spinal cord is the principal site of axon loss rather than the brain. Furthermore, there is a strong correlation between the behavior score and the CST axonal damage in the spinal cord of EAE mice. An earlier study demonstrated a 40-50% loss of CST axons at 90 and 180 days post-EAE induction via myelin-oligodendrocyte glycoprotein injection in C57/BL6 or Biozzi mice, and consistent a melioration of CST axon loss, spinal compound action potential amplitudes and areas, and clinical scores in phenytoin-treated animals (Black et al., 2006). In a CST local target EAE model, descending CST axons extend new collaterals above the lesion that establish a “detour” circuit to the lumbar target area, whereas below the lesion, spared CST axons increase their terminal branching (Kerschensteiner et al., 2004). The DiI tracer also is a competent candidate for monitoring such axonal restructuring in EAE model with axonal protective or repair-oriented treatment.
In conclusion, we have developed a tracing method for the evaluation of CST axon loss in the EAE model. This method can be easily applied to monitor the axonal damage in the EAE relapsing-remitting course. The results were sensitive, consistent and reproducible. This quantifiable method may be applied for the evaluation of axonal protective or repair-oriented therapeutic strategies for experimental MS.
Acknowledgments
This work was supported by NINDS grants PO1 NS42345, the Benson Ford Foundation and the Mandell and Madeleine H. Berman Foundation. The authors wish to thank Cindi Roberts and Qing-e Lu for technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arnold DL. Magnetic resonance spectroscopy: imaging axonal damage in MS. J Neuroimmunol. 1999;98:2–6. doi: 10.1016/s0165-5728(99)00074-0. [DOI] [PubMed] [Google Scholar]
- Bechtold DA, Miller SJ, Dawson AC, Sun Y, Kapoor R, Berry D, Smith KJ. Axonal protection achieved in a model of multiple sclerosis using lamotrigine. J Neurol. 2006;253:1542–51. doi: 10.1007/s00415-006-0204-1. [DOI] [PubMed] [Google Scholar]
- Bettelli E. Building different mouse models for human MS. Ann N Y Acad Sci. 2007;1103:11–8. doi: 10.1196/annals.1394.021. [DOI] [PubMed] [Google Scholar]
- Bjartmar C, Kidd G, Mork S, Rudick R, Trapp BD. Neurological disability correlates with spinal cord axonal loss and reduced N-acetyl aspartate in chronic multiple sclerosis patients. Ann Neurol. 2000;48:893–901. [PubMed] [Google Scholar]
- Bjartmar C, Wujek JR, Trapp BD. Axonal loss in the pathology of MS: consequences for understanding the progressive phase of the disease. J Neurol Sci. 2003;206:165–71. doi: 10.1016/s0022-510x(02)00069-2. [DOI] [PubMed] [Google Scholar]
- Black JA, Liu S, Hains BC, Saab CY, Waxman SG. Long-term protection of central axons with phenytoin in monophasic and chronic-relapsing EAE. Brain. 2006;129:3196–208. doi: 10.1093/brain/awl216. [DOI] [PubMed] [Google Scholar]
- Brown LT., Jr Projections and termination of the corticospinal tract in rodents. Exp Brain Res. 1971;13:432–50. doi: 10.1007/BF00234340. [DOI] [PubMed] [Google Scholar]
- De Stefano N, Matthews PM, Fu L, Narayanan S, Stanley J, Francis GS, Antel JP, Arnold DL. Axonal damage correlates with disability in patients with relapsing-remitting multiple sclerosis. Results of a longitudinal magnetic resonance spectroscopy study. Brain. 1998;121(Pt 8):1469–77. doi: 10.1093/brain/121.8.1469. [DOI] [PubMed] [Google Scholar]
- DeLuca GC, Ebers GC, Esiri MM. Axonal loss in multiple sclerosis: a pathological survey of the corticospinal and sensory tracts. Brain. 2004;127:1009–18. doi: 10.1093/brain/awh118. [DOI] [PubMed] [Google Scholar]
- Ferguson B, Matyszak MK, Esiri MM, Perry VH. Axonal damage in acute multiple sclerosis lesions. Brain. 1997;120(Pt 3):393–9. doi: 10.1093/brain/120.3.393. [DOI] [PubMed] [Google Scholar]
- Filippi M, Bozzali M, Rovaris M, Gonen O, Kesavadas C, Ghezzi A, Martinelli V, Grossman RI, Scotti G, Comi G, Falini A. Evidence for widespread axonal damage at the earliest clinical stage of multiple sclerosis. Brain. 2003;126:433–7. doi: 10.1093/brain/awg038. [DOI] [PubMed] [Google Scholar]
- Furlan R, Martino G, Galbiati F, Poliani PL, Smiroldo S, Bergami A, Desina G, Comi G, Flavell R, Su MS, Adorini L. Caspase-1 regulates the inflammatory process leading to autoimmune demyelination. J Immunol. 1999;163:2403–9. [PubMed] [Google Scholar]
- Gold R, Hartung HP, Toyka KV. Animal models for autoimmune demyelinating disorders of the nervous system. Mol Med Today. 2000;6:88–91. doi: 10.1016/s1357-4310(99)01639-1. [DOI] [PubMed] [Google Scholar]
- Heffner RS, Masterton RB. The role of the corticospinal tract in the evolution of human digital dexterity. Brain Behav Evol. 1983;23:165–83. doi: 10.1159/000121494. [DOI] [PubMed] [Google Scholar]
- Hickey WF. The pathology of multiple sclerosis: a historical perspective. J Neuroimmunol. 1999;98:37–44. doi: 10.1016/s0165-5728(99)00079-x. [DOI] [PubMed] [Google Scholar]
- Kaneko S, Wang J, Kaneko M, Yiu G, Hurrell JM, Chitnis T, Khoury SJ, He Z. Protecting axonal degeneration by increasing nicotinamide adenine dinucleotide levels in experimental autoimmune encephalomyelitis models. J Neurosci. 2006;26:9794–804. doi: 10.1523/JNEUROSCI.2116-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerschensteiner M, Bareyre FM, Buddeberg BS, Merkler D, Stadelmann C, Bruck W, Misgeld T, Schwab ME. Remodeling of axonal connections contributes to recovery in an animal model of multiple sclerosis. J Exp Med. 2004;200:1027–38. doi: 10.1084/jem.20040452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornek B, Storch MK, Weissert R, Wallstroem E, Stefferl A, Olsson T, Linington C, Schmidbauer M, Lassmann H. Multiple sclerosis and chronic autoimmune encephalomyelitis: a comparative quantitative study of axonal injury in active, inactive, and remyelinated lesions. Am J Pathol. 2000;157:267–76. doi: 10.1016/S0002-9440(10)64537-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlmann T, Lingfeld G, Bitsch A, Schuchardt J, Bruck W. Acute axonal damage in multiple sclerosis is most extensive in early disease stages and decreases over time. Brain. 2002;125:2202–12. doi: 10.1093/brain/awf235. [DOI] [PubMed] [Google Scholar]
- Litchfield S, Nagy Z. New temperature modification makes the Bielschowsky silver stain reproducible. Acta Neuropathol (Berl) 2001;101:17–21. doi: 10.1007/s004010000248. [DOI] [PubMed] [Google Scholar]
- Mancardi G, Hart B, Roccatagliata L, Brok H, Giunti D, Bontrop R, Massacesi L, Capello E, Uccelli A. Demyelination and axonal damage in a non-human primate model of multiple sclerosis. J Neurol Sci. 2001;184:41–9. doi: 10.1016/s0022-510x(00)00490-1. [DOI] [PubMed] [Google Scholar]
- Matthews PM, De Stefano N, Narayanan S, Francis GS, Wolinsky JS, Antel JP, Arnold DL. Putting magnetic resonance spectroscopy studies in context: axonal damage and disability in multiple sclerosis. Semin Neurol. 1998;18:327–36. doi: 10.1055/s-2008-1040884. [DOI] [PubMed] [Google Scholar]
- McGavern DB, Murray PD, Rivera-Quinones C, Schmelzer JD, Low PA, Rodriguez M. Axonal loss results in spinal cord atrophy, electrophysiological abnormalities and neurological deficits following demyelination in a chronic inflammatory model of multiple sclerosis. Brain. 2000;123(Pt 3):519–31. doi: 10.1093/brain/123.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medana IM, Esiri MM. Axonal damage: a key predictor of outcome in human CNS diseases. Brain. 2003;126:515–30. doi: 10.1093/brain/awg061. [DOI] [PubMed] [Google Scholar]
- Papadopoulos D, Pham-Dinh D, Reynolds R. Axon loss is responsible for chronic neurological deficit following inflammatory demyelination in the rat. Exp Neurol. 2006;197:373–85. doi: 10.1016/j.expneurol.2005.10.033. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin K. The Mouse Brain in Stereotaxic Coordinates. Academic Press; San Diego: 1997. [Google Scholar]
- Pike GB, de Stefano N, Narayanan S, Francis GS, Antel JP, Arnold DL. Combined magnetization transfer and proton spectroscopic imaging in the assessment of pathologic brain lesions in multiple sclerosis. AJNR Am J Neuroradiol. 1999;20:829–37. [PMC free article] [PubMed] [Google Scholar]
- Raine CS, Cross AH. Axonal dystrophy as a consequence of long-term demyelination. Lab Invest. 1989;60:714–25. [PubMed] [Google Scholar]
- Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338:278–85. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- Trapp BD, Ransohoff R, Rudick R. Axonal pathology in multiple sclerosis: relationship to neurologic disability. Curr Opin Neurol. 1999;12:295–302. doi: 10.1097/00019052-199906000-00008. [DOI] [PubMed] [Google Scholar]
- Tsai EC, van Bendegem RL, Hwang SW, Tator CH. A novel method for simultaneous anterograde and retrograde labeling of spinal cord motor tracts in the same animal. J Histochem Cytochem. 2001;49:1111–22. doi: 10.1177/002215540104900905. [DOI] [PubMed] [Google Scholar]
- van Waesberghe JH, Kamphorst W, De Groot CJ, van Walderveen MA, Castelijns JA, Ravid R, Lycklama a Nijeholt GJ, van der Valk P, Polman CH, Thompson AJ, Barkhof F. Axonal loss in multiple sclerosis lesions: magnetic resonance imaging insights into substrates of disability. Ann Neurol. 1999;46:747–54. doi: 10.1002/1531-8249(199911)46:5<747::aid-ana10>3.3.co;2-w. [DOI] [PubMed] [Google Scholar]
- Vannucchi MG, Faussone-Pellegrini MS. Synapse formation during neuron differentiation: an in situ study of the myenteric plexus during murine embryonic life. J Comp Neurol. 2000;425:369–81. doi: 10.1002/1096-9861(20000925)425:3<369::aid-cne3>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- Wujek JR, Bjartmar C, Richer E, Ransohoff RM, Yu M, Tuohy VK, Trapp BD. Axon loss in the spinal cord determines permanent neurological disability in an animal model of multiple sclerosis. J Neuropathol Exp Neurol. 2002;61:23–32. doi: 10.1093/jnen/61.1.23. [DOI] [PubMed] [Google Scholar]
- Youssef S, Stuve O, Patarroyo JC, Ruiz PJ, Radosevich JL, Hur EM, Bravo M, Mitchell DJ, Sobel RA, Steinman L, Zamvil SS. The HMG-CoA reductase inhibitor, atorvastatin, promotes a Th2 bias and reverses paralysis in central nervous system autoimmune disease. Nature. 2002;420:78–84. doi: 10.1038/nature01158. [DOI] [PubMed] [Google Scholar]
- Zhang J, Li Y, Cui Y, Chen J, Lu M, Elias SB, Chopp M. Erythropoietin treatment improves neurological functional recovery in EAE mice. Brain Res. 2005;1034:34–9. doi: 10.1016/j.brainres.2004.11.036. [DOI] [PubMed] [Google Scholar]
- Zhang J, Li Y, Lu M, Cui Y, Chen J, Noffsinger L, Elias SB, Chopp M. Bone marrow stromal cells reduce axonal loss in experimental autoimmune encephalomyelitis mice. J Neurosci Res. 2006;84:587–95. doi: 10.1002/jnr.20962. [DOI] [PubMed] [Google Scholar]