

Abstract
Mucolipidosis type IV (MLIV) is an autosomal recessive lysosomal storage disorder caused by mutations in the MCOLN1 gene, which encodes the 65-kDa protein mucolipin-1. The most common clinical features of patients with MLIV include severe mental retardation, delayed motor milestones, ophthalmologic abnormalities, constitutive achlorhydria, and elevated plasma gastrin levels. Here, we describe the first murine model for MLIV, which accurately replicates the phenotype of patients with MLIV. The Mcoln1−/− mice present with numerous dense inclusion bodies in all cell types in brain and particularly in neurons, elevated plasma gastrin, vacuolization in parietal cells, and retinal degeneration. Neurobehavioral assessments, including analysis of gait and clasping, confirm the presence of a neurological defect. Gait deficits progress to complete hind-limb paralysis and death at age ∼8 mo. The Mcoln1−/− mice are born in Mendelian ratios, and both male and female Mcoln1−/− mice are fertile and can breed to produce progeny. The creation of the first murine model for human MLIV provides an excellent system for elucidating disease pathogenesis. In addition, this model provides an invaluable resource for testing treatment strategies and potential therapies aimed at preventing or ameliorating the abnormal lysosomal storage in this devastating neurological disorder.
Mucolipidosis type IV (MLIV [MIM 252650]) belongs to a group of inherited metabolic diseases known as the lysosomal storage disorders (LSDs). It was first described in 1974 as a new variant of the mucolipidoses that displayed corneal clouding and abnormal systemic storage bodies.1,2 MLIV is a progressive neurological disease that presents during the 1st year of life with mental retardation, corneal opacities, elevated blood gastrin levels with achlorhydria, and delayed motor milestones.1,3 Biochemical studies demonstrated abnormal storage of sphingolipids, phospholipids, and acid mucopolysaccharides in the lysosomes of patients with MLIV.4 MLIV is a rare disorder, and the majority of patients reported to date are of Ashkenazi Jewish (AJ) descent.5 The MLIV disease gene, MCOLN1 (GenBank accession number NM_020533), maps to chromosome 19p13.2-13.3 and encodes a 580-aa protein with a predicted molecular weight of 65 kDa, named “mucolipin-1” (TRPML1). Structural analysis of the amino acid sequence predicts that the protein has six transmembrane domains, and a comparison of the protein sequence against known protein motifs identified a transient receptor potential (TRP) cation-channel domain and an internal channel pore. This TRP domain spans transmembrane domains 3–6, with the pore-forming loop between the fifth and sixth domains.5–8 The major AJ mutation, present on 72% of the AJ MLIV alleles, is an A→G transition at the 3′ acceptor site of intron 3.9 The minor AJ mutation, found on 23% of the AJ MLIV alleles, is a 6,434-bp genomic deletion that spans exons 1–6 and the first 12 bp of exon 7.10,11 Discovery of the disease gene has led to the successful implementation of genetic screening for MLIV in the AJ population.10,12 To date, >20 independent mutations have been described in MCOLN1.11,13–15
Lipid storage disorders are most often caused by defective lysosomal hydrolases or activator proteins. However, studies performed before the cloning of the MCOLN1 gene suggested that MLIV was more likely the result of a defect in sorting and transport pathways, rather than a degradation defect, since it had been shown that the lysosomal hydrolases involved in the catabolism of the stored material in MLIV were normal.16,17 Lysosomal storage occurs in cells from every tissue and organ of patients with MLIV.4,16–21 The composition of the stored material varies from tissue to tissue,21 is heterogeneous, and includes sphingolipids (mostly gangliosides), phospholipids, and acid mucopolysaccharides. Experiments in cultured fibroblasts suggested that phospholipids and gangliosides accumulate in MLIV because of a defect in the process of endocytosis of membranous components. The defect appears to lead to excessive transport of these macromolecules into the lysosomes, which is consistent with the heterogeneity of the stored materials in MLIV.16 Further studies examined movement of a lipid analogue along the lysosomal pathway and implicated a defect in the late steps of the endocytic pathway.17
Numerous studies of TRPML1 function have been performed since it was implicated in MLIV; however, the precise pathogenic mechanism that leads to disease remains to be elucidated. TRPML1 belongs to the TRPML family, which also includes the closely related proteins mucolipin-2 (TRPML2) and mucolipin-3 (TRPML3).5 The TRP superfamily consists of a diverse group of Ca2+-permeable nonselective cation channels that bear structural similarities to the Drosophila melanogaster TRP gene. To date, >20 TRP-related channels have been identified, and they play roles in extremely diverse cellular processes.22,23 TRP proteins are widely expressed in the nervous system, and, in nonexcitable cells, these channels may be the primary mode of calcium entry. TRPML1 was initially reported as a novel nonselective cation channel that was permeable to Ca2+ and was regulated by changes in Ca2+ concentration.24 Subsequent studies performed in endosomal vesicles from MLIV cells, as well as in liposomes containing the in vitro translated protein, suggested that the channel can be inhibited by pH reduction and Ca2+ transport.25,26 Recently, demonstration of a strong outwardly rectifying current in whole-cell recordings has led to the suggestion that TRPML1 might function to regulate lysosomal pH and to maintain the required acidity for normal lysosomal hydrolase function.27,28 Several studies have shown that TRPML1 is localized to late endosomes and lysosomes.29–31 An N-terminal dileucine motif is sufficient for lysosomal targeting of full-length TRPML1, and this targeting is AP1 dependent.9,31,32 Endogenous TRPML1 is cleaved in its long luminal loop between the first and second transmembrane domains; however, the role of this cleavage in regulating protein function has yet to be determined.31,33 Homo- and heteromultimerization of TRPML1 with TRPML2 and TRPML3 was recently described in cells overexpressing all three proteins.32 Significant mitochondrial fragmentation and decreased mitochondrial Ca2+ buffering efficiency have also been described in MLIV cells. Jennings et al. speculate that a defect in mitochondrial recycling leads to increased sensitivity to apoptosis induced by Ca2+, possibly explaining the degenerative cell death found in MLIV and other LSDs.34 Defective lysosomal exocytosis has also been reported in fibroblasts isolated from patients with MLIV.35
The Caenorhabditis elegans mucolipin-1 gene ortholog, cup-5, was isolated during a screen for endocytosis-defective mutants.36 Mutations in cup-5 result in an enhanced rate of uptake of fluid-phase markers, decreased degradation of endocytosed protein, and accumulation of large vacuoles. Interestingly, human TRPML1 was able to rescue the abnormal lysosomal accumulation in these mutants. Recent studies in C. elegans revealed that a mutation in the ATP-binding cassette transporter MRP-4 compensates the degradation defect caused by the absence of cup-5.37
The phenotype of MLIV is variable, with psychomotor delay and corneal opacities by age 1 year as the most common presentation.21,38 The brain pathology in patients with MLIV is characterized by pigmented cytoplasmic granules in nondistended neurons and microglial cells.21 Electron microscopy (EM) reveals accumulation of lamellated membrane structures and amorphous material in lysosomes in every cell type examined.21,38 Cultured skin fibroblasts derived from patients with MLIV contain autofluorescent lysosomes.39 Storage vacuoles are present in the corneal epithelium, as well as in many other cell types, including pancreatic acinar cells, macrophages, and chondrocytes, and in the renal collecting ducts and hepatic bile duct.38,40 Diagnosis of MLIV is suggested for patients with characteristic neurodevelopmental delay, elevated blood gastrin levels, and corneal clouding. Currently, confirmatory molecular diagnosis can be made via targeted mutation analysis of the two most common mutations, IVS3-2 A→G and 511→6994 del, which together account for 95% of mutations in the AJ population.10
To understand the pathogenic mechanism by which complete loss of TRPML1 causes MLIV, we generated a mouse with a targeted disruption of the Mcoln1 gene (GenBank accession number NM_053177). This Mcoln1 knockout, designated “Mcoln1−/−,” exhibits many clinical and cellular features of the human disease. Murine Mcoln1 is highly similar to human MCOLN1, showing 86% nucleotide identity and 91% amino acid identity. In addition, all six of the transmembrane domains, the putative cation channel, and the dileucine (L-L-X-X) motif at the C-terminus are all highly conserved.41 Our mouse model will be an invaluable resource for understanding the role of TRPML1 in lysosomal function and vesicular trafficking, mitochondrial recycling, and other cellular functions that play a role in the pathophysiology of MLIV. Furthermore, Mcoln1−/− mice will be crucial for testing therapeutic agents aimed at ameliorating the abnormal lysosomal storage in MLIV.
Material and Methods
The Mcoln1−/− knockout mice were generated and propagated following National Institutes of Health (NIH) guidelines for animal care under an approved protocol of the Animal Care and Use Committee of the National Institutes of Mental Health. All breeding and other procedures performed were reviewed and approved by the Massachusetts General Hospital Subcommittee on Research Animal Care.
Generation of Mcoln1−/− Mice
Mcoln1 genomic DNA clones were isolated by plaque hybridization of a mouse 129/Sv genomic DNA library with full-length mouse Mcoln1 cDNA probes. The targeting vector was constructed as follows (fig. 1A): a neomycin resistance gene (Neor cassette) for positive selection was inserted between exons 2 and 5 of Mcoln1. The herpes simplex virus–thymidine kinase gene (HSV-TK) for negative selection was inserted 3′ of Mcoln1 within exon 13. Methods for generating targeted embryonic stem cells and producing mice from these cells have been described elsewhere.42 Hybrid LC3 (C57BL6×129S6) embryonic stem cells (GlobalStem) were transfected with the Mcoln1 targeting vector by electroporation. After 24 h, cells were fed with medium containing 200 μg/ml of G418. G418-resistant clones were picked after 7 d and were screened for homologous recombination by Southern blotting. Three clones carrying a Mcoln-1 targeted allele were injected into C57BL6 blastocysts, which were then transferred into oviducts or uterus of pseudopregnant outbreed recipient females. Pups were evaluated for chimerism. Chimeras from all three lines were mated to C57BL/6 females. Germline transmission was evidenced by coat color and was confirmed by PCR.
Figure 1. .

Generation of Mcoln1−/− knockout mice. A, Illustration of the Mcoln1−/− targeting strategy. The top line shows the wild-type Mcoln1 allele, with restriction-enzyme sites, the locations of exons (blackened boxes), and the locations of 5′ and 3′ genomic DNA probes (hatched boxes). The second line shows the loxP-flanked PGK-neomycin (PGK-Neo) cassette–targeting vector, with upstream and downstream Mcoln1 genomic DNA. The HSV-TK cassette was inserted at the 3′ end of the construct, for negative selection. ex = exon; KO = knockout. The third line shows the configuration of the properly targeted allele (Mcoln1ex3,4/5neo) created by homologous recombination in embryonic stem cells. Restriction-enzyme sites are EcoRI (E), BamH1 (B), SalI (S), and NotI (N). B, Southern-blot analysis of embryonic stem cell clone DNA with the 5′ probe. Genomic DNA isolated from embryonic stem cell clones was digested with AseI. The 5′ probe detects the wild-type (11.2-kb) and targeted (7.7-kb) bands in Mcoln1+/+ (lanes 2 and 6) and Mcoln1+/+ (lanes 3 to 5) mice. C, Mouse genotyping by analysis of ethidium bromide–stained PCR products from DNA obtained from tail snips. Mcoln1+/+ and Mcoln1−/− mice present only a single band of 400 bp and 187 bp, respectively; Mcoln1+/− mice present both bands. D, Ethidium bromide–stained RT-PCR products were amplified from Mcoln1+/+ and Mcoln1−/− brains by use of Mcoln1 exon 2 forward primer and exon 6 reverse primer. DNA sequencing confirmed the identity of the Mcoln1 band.
Southern-Blot Analysis
For Southern-blot analysis, 5′ and 3′ probes were generated using the following sets of primers: forward 5′-TAAACTTGGTGGAGGCTTGC-3′ and reverse 5′-GTGCTCAGCCACTGGGTAAC-3′ and forward 5′-TAGGCTGATCAACGTCACCA-3′ and reverse 5′-GGGTCTGAGGATGGGAGAGT-3′. Mouse genomic DNA was used as a template for the reactions. DNA extracted from embryonic stem cells was digested with AseI (for 5′-probe hybridization) and with PinA and PspI (for 3′-probe hybridization), was run on a 0.8% agarose gel, was blotted onto GeneScreen Plus membrane (Perkin Elmer), and was hybridized with the respective radiolabeled probes. The 5′ probe detected a 7.7-kb band in the targeted allele and an 11.2-kb band in the normal allele, and the 3′ probe detected a 9.6-kb band in the targeted allele and an 11.2-kb band in the normal allele.
mRNA Expression Analysis
For expression analysis, 1–2 μg of total RNA extracted from brain was used to synthesize cDNA with SuperScript III (Invitrogen Life Technologies) and was used as template for PCR and quantitative real-time PCR (Q-PCR ), as follows. For PCR, exons 2–6 of Mcoln1 were amplified with the primer set forward 5′-CCCCACAGAAGAGGAAGA-3′ and reverse 5′-GTGGATGGTGACATTGACC-3′. PCR products were sequenced to confirm Mcoln1 identity.
For Q-PCR, analysis of Mcoln1 was performed using SYBR Green PCR kit in I-Cycler (Bio-Rad). Standard curves representing five-point 1:3 serial dilution of cDNA of one of the samples in the control group were analyzed in each assay. The r2 values were ⩾0.99 for all standard curves, and the amplification efficiency varied between 90% and 100%. Sample relative quantification was analyzed by the ΔCT method, using CT values from Mcoln1+/+ mice as calibrators, and was corrected by the internal control—hypoxanthine guanine phosphoribosyl transferase (HPRT)—in all experiments. Primers were designed to detect Mcoln1 message within exon 4 forward primer 5′-ATGTGGACCCAGCCAATGATACCT-3′ and exon 6 reverse primer 5′-TGTCTTCAGCTGGAAGTGGATGGT-3′. Mouse HPRT primers were forward 5′-TCGAAGTGTTGGATACAGGCC-3′ and reverse 5′-CAACAGGACTCCTCGTATTTG-3′. All primers were designed using PrimerQuest software (Integrated DNA Technologies).
Genotyping
Genotyping was done by PCR with use of genomic DNA from mouse tail snips, prepared using the method with hot sodium hydroxide and Tris-HCl.43 PCR was performed with HotStart-Red Taq polymerase (Sigma). Three primers were used in the reaction to easily identify Mcoln1+/+ (wild-type), Mcoln1+/− (heterozygote), and Mcoln1−/− (knockout) mice: forward 5′-GACCCAGGAATGACACCTTC-3′, located in intron 3 position 9060 of the Mcoln1 sequence; reverse 5′-CCCCTTGCTGCCATGTAATA-3′, located in the Neor cassette present in the targeted allele; and reverse 5′-GCGCAAACCACATGTGCTTT-3′, located in intron 3 position 9441 of Mcoln1.
Histopathological Analysis and EM
Stomach
Mcoln1+/+ and Mcoln1−/− mice were sacrificed, and gastric tissues were harvested and washed. The tissues were dissected from mice and were fixed in 2.0% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.4) overnight at 4°C. Tissues were stained with hematoxylin and eosin for light microscopy or were postfixed in 1.0% osmium tetroxide (Electron Microscopy Sciences) in cacodylate buffer for EM and infiltrated with Epon resin (Ted Pella). The following day, samples were placed in fresh 100% EPON resin and were embedded in an oven at 60°C overnight. Thin sections were stained with uranyl acetate and lead citrate and were examined at 80 kV in a JEOL 1011 transmission electron microscope (Advanced Microscopy Techniques).
Brain
For studies of brain, mice were deeply anesthetized with sodium pentobarbital and were perfused systemically with 0.9% saline followed by 4.0% paraformaldehyde in 0.1 M phosphate buffer. Tissue blocks for EM were postfixed in 2.5% glutaraldehyde in 0.1 M cacodylate buffer. Before the embedding in EPON, tissues were fixed in 2% osmium tetroxide, were dehydrated in alcohols, and were plastic embedded. Ultrathin sections were cut on an ultramicrotome, were stained with uranyl acetate and lead citrate, and were examined on a Philips CM10 electron microscope. LAMP2 staining of cerebrocortical neurons was done with immunoperoxidase according to the protocol previously described by Zervas et al.44
Eyes
Eyes were enucleated immediately after sacrifice, and the globe was cleaned of all extraneous tissue and then was rinsed with saline. The globe was immediately placed into fixative consisting of 2.5% gluteraldehyde and 2% formaldehyde in 0.1 M cacodylate buffer with 0.08 M CaCl2 at 4°C. After a short 10–15-min fixation, the eye was bisected at the limbus, the anterior segment was separated from the posterior segment, and the parts to be examined were placed back in the fixative. Within 24 h after enucleation, the tissue was washed in 0.1 M cacodylate buffer and was stored at 4°C. The tissue was postfixed for 1.5 h in 2% aqueous osmium tetroxide. Tissue was dehydrated in graded ethanol, was transitioned in propylene oxide, was infiltrated with propylene oxide and EPON mixtures (TAAB 812 resin [Marivac]) embedded in EPON, and was cured for 48 h at 60°C. Sections of 1 μm were cut on a Leica Ultracut UCT and were stained with 1% toluidine blue in 1% borate buffer. For EM, thin sections were cut and were stained with saturated, aqueous uranyl acetate and Sato’s lead stain. Examination was done on a Philips CM10 electron microscope.
Radioimmunoassay for Gastrin
Plasma samples from Mcoln1+/+ and Mcoln1−/− mice were obtained from the abdominal aorta before death. Gastrin concentrations for total amidated gastrin concentrations were determined by radioimmunoassay with use of L2 antibody (which reacts with G17 and G34 but not with progastrin or Gly-gastrins) to the COOH terminus of amidated gastrin, as described elsewhere.45 Standard curves were prepared using human G17 as a standard in hormone-free plasma, and samples were diluted to fall in the midrange of the standard curve.
Behavioral Analyses
Studies were performed in Mcoln1−/− and Mcoln1+/+ littermates. Animals were analyzed for clasping, gait, and overall behavior and appearance. Clasping analysis was done by grasping the tail and holding the mouse 12 in from a solid surface for 30 s. The test was considered positive for clasping if limbs were clenched toward the belly for ∼5 s. Gait analysis was done by placing each animal with differentially painted hind paws and forepaws in a paper-lined tunnel apparatus. Measurements of the stride length (rear paw–to–rear paw distance) and base width (rear-paw stance) were determined. Overall behavior assessment was done on the basis of the first phase of the Behavior Primary Screen (SHIRPA protocol), with modifications.46 In brief, each mouse was individually placed in a viewing jar for 5 min, and body position, spontaneous activity, respiration rate, and posture were monitored and recorded without disturbing the animal. All behavior studies were repeated two times for each mouse, on two separate days by two investigators blinded to the phenotype of the animals. Statistical significance was tested with one-way analysis of variance (ANOVA).
Results
Generation of Mcoln1−/− Mice
We have generated a strain of mice with a targeted disruption in the Mcoln1 gene. The knockout construct was generated by replacing exons 3 and 4 and 72 bp of exon 5 of Mcoln1 with a PGK-neomycin cassette (fig. 1A). Because the deletion leads to a frameshift in exon 5, the Mcoln1 knockout allele was expected to be null. Homologous recombination in embryonic stem cells was confirmed by genomic Southern-blot analysis, and analysis revealed the expected restriction fragments, confirming proper targeting (fig. 1B). We identified three positive embryonic stem cell clones from 200 μg/ml of G418. Three correctly targeted embryonic stem cell clones were microinjected into B6 blastocysts to generate chimeras. The chimeras were crossed with B6 mice to establish and maintain the Mcoln1 heterozygous (Mcoln1+/−) mouse line on a mixed 129/B6 hybrid background. Homozygous mice (Mcoln1−/−) were obtained from heterozygote breeding and were genotyped (fig. 1C). No phenotypic differences were ever observed in mice generated from the three targeted ES clones. We confirmed the absence of Mcoln1 transcripts in Mcoln1−/− mice, using RT-PCR of brain cDNA (fig. 1D). cDNA generated from Mcoln1+/+ and Mcoln1−/− brains was analyzed by Q-PCR with specific primers for Mcoln1, which also confirmed the absence of Mcoln1 message in Mcoln1−/− mice (data not shown).
Progressive Hind-Limb Paralysis and Decreased Life Span of Mcoln1−/− Mice
At birth, affected pups were indistinguishable from littermates. Therefore, nine Mcoln1−/− mice were observed as they aged. At age 3 mo, there were no obvious differences in growth, appearance, or behavior. Open field tests showed no difference in activity or distance covered, and growth rate and weight were as-yet indistinguishable between Mcoln1−/− animals and wild-type littermates. However, when strength was assessed in the hanging wire test,47 3-mo-old Mcoln1−/− mice had lost strength, compared with 2-mo-old Mcoln1−/− or 3-mo-old Mcoln1+/+ littermates (data not shown). At age 6 mo, the mice began to show lethargy, compared with their Mcoln1+/− and Mcoln1+/+ littermates. At age ∼6.5 mo, we began to detect noticeable gait changes, which quickly progressed to total hind-limb paralysis and death. The age at onset of the noticeable changes in motor behavior was variable, with the earliest animal showing onset at age ∼6.5 mo and two animals showing onset at age 8.5 mo. Regardless of onset age, the progression was similar, with 3–4 wk from onset to complete hind-limb paralysis and sacrifice. Survival rates were statistically different for Mcoln1−/− mice, with an average age at death of 8 mo (fig. 2).
Figure 2. .

Survival rates in Mcoln1+/+ mice (n=12) versus Mcoln1−/− mice (n=9). Results are expressed as the percentage of live animals plotted against age, ranging from 0 to 10 mo. Mcoln1−/− animals were all severely paralyzed at ages 7–9 mo and were euthanized.
We performed gait and clasping studies in Mcoln1−/− and Mcoln1+/+ animals of matched ages, in the range 6–9 mo. Studies were performed before onset of visible paralysis. Clasping studies done in two separate trials (n=8, each mouse tested twice) revealed clasping in 94% of the Mcoln1−/− tests as compared with clasping in only 19% of the Mcoln1+/+ tests (fig. 3A). Hind-paw and forepaw gait measurements during tunnel walks revealed significantly shortened average stride length (rear paw–to–rear paw distance) for Mcoln1−/− mice (5.96±0.9 cm for Mcoln1+/+ vs. 3.98±0.2 cm for Mcoln1−/−; P<.0001), and no statistical difference was observed in base width (rear-paw stance) (fig. 3B and 3C). Notable differences were observed in total body weight at the time of testing (35.66±6.80 g for Mcoln1+/+ vs. 24.62±3.84 g for Mcoln1−/−; P<.0001) (fig. 3D). This seems to be caused by loss of subcutaneous fat and decreased muscle mass, despite food and sterile water gel being readily available in the bottom of the cages. Other behavioral assessments, including body position, respiration rates, spontaneous activity, and posturing, were compared, and results were not statistically different.
Figure 3. .

Neurologic deficits and decreased body weight in Mcoln1−/− mice. A, Graph showing dramatically increased clasping in Mcoln1−/− mice aged 7–9 mo. B, Gait analysis. Results expressed as average ± SD of stride length in cm (rear paw–rear paw stance) for Mcoln1+/+ mice (n=8; 180 strides) and Mcoln1−/− mice (n=8; 192 strides). Base width (rear-paw stance) was also measured, but no significant difference was detected. C, Gait analysis shown by footprints. Footprints are represented in blue (front paws) and red (hind paws). The Mcoln1+/+ mouse has a stride length of 6 cm at age 7–9 mo, whereas the Mcoln1−/− mouse has stride length of 4.5 cm. D, Total body weight in Mcoln1+/+ versus Mcoln1−/−. Results are expressed as average ± SD of Mcoln1+/+ mice (n=9) and Mcoln1−/− mice (n=15). Statistical significance was assessed by one-way ANOVA.
Clinical manifestations of the gait deficiency progressed until the Mcoln1−/− mice showed dragging of the hind limbs, severely hunched posture, abdominal distension, and scruffy coat (fig. 4A). Three Mcoln1−/− mice showing marked paralysis at ages 7–9 mo were sacrificed, as were three age-matched Mcoln1+/+ controls, for anatomical examination. Brain, heart, and eyes were carefully dissected and weighed. Although there were no statistical differences in these tissues between Mcoln1+/+ and Mcoln1−/− mice, cardiac anatomy suggested a subtle dilated cardiomyopathy, and the Mcoln1−/− brains were slightly smaller. Both of these observations will require further investigation at a variety of ages to determine the cause and extent of the pathology. At the time of sacrifice, the Mcoln1−/− mice exhibited a grossly distended bladder filled with 1–2 ml of turbid urine (fig. 4B). The bony architecture of the severely emaciated Mcoln1−/− mice was demonstrative, and the ribs and spine can be clearly seen after skin removal. The Mcoln1−/− physical examination revealed loss of major muscle bulk, absence of subcutaneous fat stores, and decreased overall weight measurements, despite similar length measurements between the Mcoln1+/+ and Mcoln1−/− groups (fig. 4C). Similar decreased muscle mass and fat storage was noted in hind-limb dissection (fig. 4D).
Figure 4. .

Anatomical differences in Mcoln1−/− animals. A, Comparison of Mcoln1−/− and Mcoln1+/+ littermates, showing complete hind-limb paralysis in Mcoln1−/− mice. B, Dramatically distended bladder, seen in all Mcoln1−/− mice at age 8 mo. C and D, Comparison of Mcoln1+/+ and Mcoln1−/− mice after removal of skin, showing loss of major muscle bulk and absence of subcutaneous fat stores.
CNS Inclusion Formation in Mcoln1−/− Mice
To evaluate inclusion-body formation in Mcoln1−/− mice, we investigated CNS tissues of Mcoln1−/− mice and littermates at age 8 mo. Ultrastructural studies in murine MLIV disease revealed inclusions in neurons, astrocytes, oligodendroglia, microglia, and endothelial cells. Some neurons contained large numbers of inclusions, mostly confined to the basilar cytoplasm, as shown in the large pyramidal neuron (fig. 5A). The inclusions closely resembled what were referred to in an earlier study of MLIV in a 7-year-old child as “compound bodies.”38 That is, they consisted of an unusual intermixture of compact lamellae and a granular matrix, with the latter occasionally presenting as a crystalline array (fig. 5B and 5C). At times, the inclusions formed as a large commingled mass concentrated within localized regions of the neuronal perikaryon. Purkinje neurons in the cerebellum exhibited inclusions that were very similar to cortical pyramidal neurons. Generally, neurons appeared to exhibit more inclusions than did glial cells. Inclusions in oligodendroglia in the cerebral cortex were similar to those in neurons but often possessed a more open, swirling pattern of compact lamellae (fig. 5D). Inclusions in astrocytes also typically resembled those seen in neurons but were observed only occasionally (fig. 5E). Microglial inclusions were often numerous and possessed densely packed sheets of lamellae intermixed with granular matrixes (data not shown). To confirm the lysosomal identity of the storage vesicles, we performed LAMP2 staining with peroxidase, which demonstrated the increased number and size of LAMP2-positive vesicles in Mcoln1−/− neurons (fig. 5F–5I). Overall, the ultrastructure of the inclusions observed in Mcoln1−/− mice match what has been described in human MLIV. The MLIV inclusions are unusual in terms of the combination of dense lamella and granular matrices, and, as such, they do not resemble those typically seen in the glycosphingolipidoses, the mucopolysaccharidoses, or most other types of lysosomal disease.
Figure 5. .

CNS lysosomal storage in 8-mo-old Mcoln1−/− mice. A, Large cortical pyramidal neuron (based on cell size and morphology), exhibiting a large cache of granulomembranous storage bodies located principally beneath the nucleus in the base of the perikaryon (arrow). B and C, Higher magnification view of inclusion bodies in neurons. Note the presence of both membranous lamella and a granular, dense core matrix. D, An oligodendroglial cell (based on nuclear and cell morphology) exhibiting a few membranous inclusions at one end of the cell (arrow). E, Astrocyte (based on presence of longitudinal bundles of filaments) containing several inclusions (arrow) resembling those of neurons. The calibration bar in panel A equals 2.25 μm, whereas it equals 0.65 μm in panel B, 0.30 μm in panel C, and 1.75 μm in panels D and E. F–I, LAMP2 staining of pyramidal neurons in the cortex from 8-mo-old Mcoln1+/+ mice (F and H) and Mcoln1−/− mice (G and I). Immunoperoxidase labeling of neurons in Mcoln1+/+ versus Mcoln1−/− cerebral cortex shows increased number and size of LAMP2-positive vesicles in affected neurons (unblackened arrows). The calibration bar is 10 mm in panels F and G and is 5 mm in panels in H and I. N = nucleus.
Gastric Pathology of Mcoln1−/− Mice
Patients with MLIV are constitutively achlorhydric as a result of a block in gastric acid secretion. Parietal cells from patients with MLIV have very large lysosomes that contain lamellar, concentric, microgranular inclusions.48 Analysis of the gastric epithelium of Mcoln1−/− mice revealed similar dramatic vacuolization (figs. 6 and 7). Examination of the gross morphology shows extremely large vesicles in the gastric mucosa (fig. 6B). Vesicles are visible along the length of the gastric pit (fig. 6D). Overall, the morphology of the gastric mucosa is not dramatically different in the Mcoln1−/− mice, with only minor reductions in the number of granular pit cells on the surface and zymogenic cells in the base of the gastric pit. Ultrastructural examination shows that, in addition to the large vesicles observed in parietal cells (fig. 7B), numerous smaller vesicles are also present in the mucus-producing neck cells (fig. 7C). The vesicles in both cell types contain storage material, which is an unusual mixture of compact lamellae and granular matrix (fig. 7B and 7C). Interestingly, intact vesicles were observed in the lumen adjacent to the epithelial cells in the neck region (fig. 7B and 7C), suggesting that the secretory vesicles fail to fuse with the apical membrane of the gastric epithelium. Although the overall morphology of the gastric mucosa is not different in the Mcoln1−/− mice, minor reductions in the granular pit cells on the surface and zymogenic cells in the base are seen. However, the subepithelial mucosa or the gastric muscle layers are unaffected in the Mcoln1−/− mice.
Figure 6. .

Morphology of stomach mucosa. A, Representative image of the antral region of the mouse stomach, showing normal gross morphology of the gastric epithelium. B, Representative image of the antral region of the mouse stomach, showing the gross morphology of the gastric epithelium of Mcoln1−/− mouse. In panels A and B, magnification is 63×. C and D, Detailed morphology of the gastric glands. Increased frequency of the parietal cells in the neck region of the gastric epithelium was observed in the mutant mice with enlarged and multiple secretary vacuoles. GP = gastric pit; L = lumen. In panels C and D, magnification is 630×. In all panels, staining is toluidine blue.
Figure 7. .
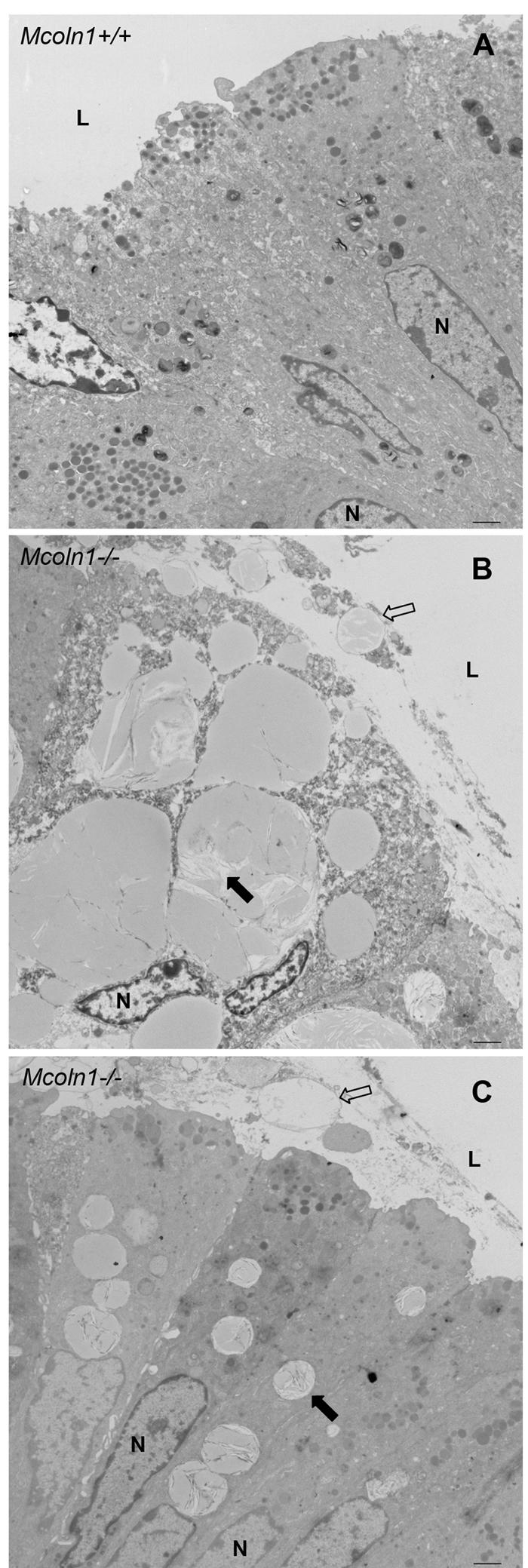
EM of gastric epithelium. Ultrastructural figures of the neck region of the gastric epithelium for Mcoln1+/+ and Mcoln1−/− mice are shown. A, Normal gastric epithelium at the neck region of the Mcoln1+/+ mice, compared with that of Mcoln1−/− mice. B and C, Parietal (B) and neck (C) cells, both showing a pronounced defect with multiple enlarged vesicles. Vesicles are retained in Mcoln1−/− epithelium and fail to fuse with the apical surface (unblackened arrow), and granular storage material is seen within vesicles (blackened arrow). In addition to large intact vacuoles within the cells, vesicles are also observed outside the cells in the lumen of the stomach. L = lumen; N = nucleus. In panels B and C, magnification is 6,000×.
A hallmark of human MLIV is elevated plasma gastrin,48 which can be increased up to 13 times that of normal. The serum gastrin in Mcoln1−/− mice was significantly elevated compared with that in Mcoln1+/+ mice (fig. 8). The average gastrin level in Mcoln1−/− mice was 158.6 pM compared with 76.25 pM in age- and sex-matched Mcoln1+/+ controls (P=.0006). Iron studies, including assays of free iron and total iron-binding capacity, did not reveal any statistical differences between Mcoln1+/+ mice and Mcoln1−/− mice (data not shown).
Figure 8. .

Significant increase in plasma gastrin in Mcoln1−/− mice (P=.0006). The concentration of plasma gastrin was measured by radioimmunoassay. Mcoln1+/+ mice (n=11) or Mcoln1−/− mice (n=13) were sacrificed for the collection of plasma and tissue samples. The total number of mice (n=24), the mean age, and the numbers of male and female mice were approximately equal for the Mcoln1+/+ and Mcoln1−/− groups.
Severe Retinal Degeneration in Mcoln1−/− Mice
Patients affected with MLIV have defined corneal clouding, retinal degeneration with varying degrees of vascular attenuation, and mild optic-nerve pallor.49 The retinas of Mcoln1−/− mice show significant degeneration, and optic-nerve pallor was evident by gross histology (fig. 9A and 9B). Mcoln1−/− mice have a dystrophic outer nuclear layer with reduced receptor nuclei and a significantly reduced outer plexiform and inner nuclear layer, compared with those of their Mcoln1+/+ controls. Interestingly, there is no evidence of corneal clouding by slit-lamp examination by age ∼7 mo. This finding is corroborated by histological studies, since there is no discernible difference between the Mcoln1−/− and Mcoln1+/+ corneal stroma at age 5 mo in architecture, thickness, or cell morphology (fig. 9C and 9D).
Figure 9. .

Mcoln1−/− ocular pathology. A, Toluidine blue–stained cross section of Mcoln1+/+ mouse retina. B, Mcoln1−/− mouse retina. Note the severe thinning and irregularity of the entire receptor layer (OS/IS) and the dramatic loss of outer nuclear layer (ONL) nuclei. The structure of the retinal pigment epithelium (RPE) appears normal. INL = inner nuclear layer; OS/IS = outer segment/inner segment of the receptor layer. Magnification 40×. C and D, Toluidine blue–stained cross section of cornea from Mcoln1+/+ mice (C) and Mcoln1−/− mice (D) showing five distinct layers: epithelium (I), Bowman’s layer (II), stroma (III), Descemet’s layer (IV), and endothelium (V). No storage bodies or architectural differences in stromal keratinocytes between Mcoln1+/+ and Mcoln1−/− mice were noted. Magnification 40×.
Discussion
In this article, we report the generation and characterization of the first murine model of the human disease MLIV. Our initial studies show that this model accurately replicates many aspects of the human disease. The Mcoln1−/− mouse therefore will be invaluable for future studies aimed at understanding the role of TRPML1 in normal lysosomal function, as well as for the development and testing of potential therapies for MLIV.
The primary genetic defects of most LSDs have been identified and are known to include not only lysosomal enzymes but also a host of nonlysosomal enzymes in the endoplasmic reticulum and Golgi, as well as nonenzyme proteins of endosomes.50 In spite of this remarkable progress, numerous uncertainties remain concerning the precise mechanisms by which many of these protein defects cause cell injury and disease progression. This is particularly true of newly discovered nonenzyme proteins like NPC1, NPC2, CLN3, and, as reported here, TRPML1. In this study, we show that mice lacking TRPML1 exhibit the presence of dense granulomembranous storage bodies in neurons and many other cell types that closely resemble the so-called compound bodies described in human MLIV disease, a type of inclusion that is not generally seen in any other type of LSD.38 Although, in some respects, the granulomembranous storage bodies of MLIV disease resembled the membranous cytoplasmic bodies seen in the gangliosidoses and the so-called zebra bodies of the mucopolysaccharidoses, they also had conspicuous granular components that were more similar to inclusions described in the neuronal ceroid lipofuscinoses (CLN [MIM 204200]).51,52 This finding is compatible with the view that MLIV disease is characterized by multiple substrate storage, possibly related to a generalized compromise in lysosomal function secondary to alterations in lysosomal pH. Indeed, it has been suggested recently that MLIV may, in fact, be caused by chronically acidic lysosomal pH that results in inactivation of lysosomal lipases.28 It is interesting to note, however, that there is no reported neuronal storage in a mouse model of Wolman disease (MIM 278000), which lacks functional lysosomal acid lipase,53 suggesting that a lipase defect alone may not account for the heterogenous storage seen in MLIV. Complicating things further, the literature also contains reports of normal30 and elevated lysosomal pH in MLIV.2 The biochemical mechanism behind the storage-body accumulation—and the resulting effect on cell function—remains to be determined. We have shown that the storage bodies in the Mcoln1−/− mouse model are identical to those observed in patients with MLIV; therefore, this model will be helpful in elucidating the pathways leading to abnormal storage.
The original finding of iron deficiency in patients with MLIV led to the discovery of elevated blood gastrin coupled with achlorhydria. Subsequent gastroscopy studies identified distended parietal cells containing large vacuoles with lamellar, concentric inclusions.48 The gastric phenotype observed in the Mcoln1−/− mice is remarkably similar to what has been described in patients with MLIV and includes both elevated gastrin and cellular vacuolization.48 Four major types of secretory epithelial cells cover the surface of the stomach and extend down into the gastric pits and glands. The neck cells secrete thick mucus that serves as protection against the action of gastric juices. Parietal cells are located within the gastric pits and secrete both HCl− and intrinsic factor. Gastrin is secreted by enteroendocrine G cells in the pyloric glands, and chief cells secrete pepsin, a proteolytic enzyme.54,55 For acid secretion to occur, calcium signaling is required for fusion of the tubulovesicles to the canalicular membranes. Channel proteins are known to play critical roles in membrane-trafficking events, including transport of vesicles and docking and fusion with cellular membranes, suggesting a direct role for TRPML1 in acid secretion. Interestingly, the initial human studies suggested that the large vacuoles are limited to the parietal cells; however, ultrastructural examination of the Mcoln1−/− mucosa clearly shows vacuoles with granular storage material in the mucus-secreting neck cells of the gastric pit. Effective mucus secretion is required for protection of the stomach mucosa, and increasing mucosal atrophy with age has been described in patients with MLIV.48 Given that the Mcoln1−/− mice show decreased subcutaneous fat stores and slightly decreased muscle mass at age 7–8 mo, it will be interesting to determine whether this is due to mucosal atrophy in older animals. The Mcoln1−/− model provides an excellent system in which to elucidate the role of TRPML1 in acid and mucus secretion, as well as to study the long-term physiological effects of mucosal atrophy.
Corneal clouding is a hallmark of human MLIV, and it is frequently the first defect evident in most patients. In fact, diagnosis of MLIV is frequently made after ophthalmologic examination and conjunctival biopsy. Detailed corneal examination of several Mcoln1−/− mice via slit-lamp analysis by trained observers did not reveal any corneal clouding by age 7 mo. Further, EM studies failed to detect any defects in the corneal stroma. This phenotypic discordance has occurred in several other LSD murine models, notably those for mucopolysaccharidosis (MPS) I (MIM 607014),56 MPS IV (MIM 253200),57 and GM1 gangliosidosis (MIM 230500).58,59 Genetic causes of corneal clouding are variable, and it is often the result of abnormal function of the corneal endothelium, which disrupts fluid homeostasis. The mechanism of corneal clouding in LSDs is also variable and is not completely understood. In many LSD animal models, direct storage in stromal keratocytes appears to be the etiology of the clouding, even with a functional endothelium.60–62 In some LSD mouse models that are discordant for corneal clouding, histopathological corneal-architecture differences were noted between affected and wild-type mice, but there was no evidence of clouding on physical examination.63 The etiology of this corneal preservation is unclear, but increased genetic functional redundancy in the mouse may be present. In addition, the composition of proteoglycans in the mouse corneal stroma is different than that in other mammalian species, with keratin sulfate predominantly undersulfated and an overall different immunostaining pattern of chondroitin and dermatin sulfates.64 These differences may contribute to a lower susceptibility to corneal clouding that depends on the precise molecular mechanism leading to storage or endothelium dysfunction.
Despite this, we do see dramatic retinal degeneration in the MLIV mouse model that is similar to that described in patients. The mice show defects in the outer nuclear layer, the outer plexiform layer, and the inner nuclear layer. These specific defects, in addition to optic-nerve pallor, are clinically present in patients with MLIV.49,65,66 It is likely that defective cell signaling leads to increased apoptosis in the Mcoln1−/− retina, but further studies to determine whether the observed defects are the result of progressive degeneration, instead of developmental dysplasia, will be necessary.
The Mcoln1−/− mouse demonstrates a dramatic physical decline toward the later months of disease that is more severe than is the human correlate. This may be, in part, because of the palliation of the disease course in patients affected with MLIV who receive wheelchair assistance and other direct interventions. The availability of murine models is crucial for the development of potential therapies. Proven treatments for LSDs include bone marrow transplantation, somatic gene therapy, and direct enzyme replacement.67–70 Recent developments in potential treatment modalities by use of mouse models include herpes simplex virus type 1 vectors expressing β-glucuronidase for MPS VII (MIM 253220),71 AAV2/8-mediated gene delivery for MPS II (MIM 309900),72 and injection of recombinant human sulfamidase into the cerebrospinal fluid of MPS IIIA mice.73 Small-molecule therapy has also been pioneered for lysosomal diseases—for example, Niemann-Pick disease, type C (NPC [MIM 257220])—by use of mouse models.44 Recent studies have shown that treatment of MLIV fibroblasts with chloroquine or nigericin can reverse the lysosomal storage phenotype,28 a potentially exciting finding that can now be easily tested for in vivo efficacy in the MLIV mouse model.
In conclusion, we present the first murine model for MLIV. The Mcoln1−/− mice show dense granulomembranous storage bodies in neurons that are similar to those described in patients with MLIV yet are unique from other LSDs. Other remarkable findings include neurological deficits, dramatically elevated plasma gastrin, vacuolization in the gastric mucosa, and retinal degeneration. Although the mice appear grossly normal at birth, they show weakness by age 3 mo, and the neurological deficits progress to complete hind-limb paralysis and death by age ∼8 mo. This model will provide access to otherwise unattainable tissues and cell types for future studies of TRPML1 function, which may help clarify its crucial role in lysosomal function. Lastly, as has already been proven for other LSDs, the Mcoln1−/− mice will be crucial for the development and testing of potential therapeutic agents aimed at improving patient outcomes in the future.
Acknowledgments
This work was supported by National Institute of Neurological Disorders and Stroke grant NS39995 (to S.A.S.), National Institute of Diabetes and Digestive and Kidney Diseases grants DK12437 and DK70260 (to N.N.), Child Health and Development grant HD045561 (to S.U.W.), Harvard Medical School NIH Training Grant T32-GM077481-25 (to B.V.), and NIH Individual Ruth L. Kirschstein National Research Service Award (NRSA) 1F32NS052072-01A1 (to M.F.B.).
We thank Harvard Partners Center for Genetics and Genomics (Gene Modification Lab) for engineering the targeting construct, Gloria Stephney for the EM work done in the brain, Matthew Micsenyi for the LAMP2-staining experiments in neurons, and Mary McKee for the EM work in the stomach. Mary McKee is at the Massachusetts General Hospital EM core, which is supported by Inflammatory Bowel Disease Center grant DK43351, Boston Area Diabetes and Endocrinology Research Center grant DK57521, and Harvard Clinical Nutrition Research Center grant DK40561.
Web Resources
Accession numbers and URLs for data presented herein are as follows:
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for MCOLN1 [accession number NM_020533] and Mcoln1 [accession number NM_053177])
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for MLIV, CLN, Wolman disease, MPS I, MPS IV, GM1 gangliosidosis, MPS VII, MPS II, and NPC)
References
- 1.Berman ER, Livni N, Shapira E, Merin S, Levij IS (1974) Congenital corneal clouding with abnormal systemic storage bodies: a new variant of mucolipidosis. J Pediatr 84:519–526 10.1016/S0022-3476(74)80671-2 [DOI] [PubMed] [Google Scholar]
- 2.Zeevi DA, Frumkin A, Bach G (2007) TRPML and lysosomal function. Biochim Biophys Acta 1772:851–858 [DOI] [PubMed] [Google Scholar]
- 3.Altarescu G, Sun M, Moore DF, Smith JA, Wiggs EA, Solomon BI, Patronas NJ, Frei KP, Gupta S, Kaneski CR, et al (2002) The neurogenetics of mucolipidosis type IV. Neurology 59:306–313 10.1001/archneur.59.2.306 [DOI] [PubMed] [Google Scholar]
- 4.Bargal R, Bach G (1988) Phospholipids accumulation in mucolipidosis IV cultured fibroblasts. J Inherit Metab Dis 11:144–150 10.1007/BF01799863 [DOI] [PubMed] [Google Scholar]
- 5.Bargal R, Avidan N, Ben-Asher E, Olender Z, Zeigler M, Frumkin A, Raas-Rothschild A, Glusman G, Lancet D, Bach G (2000) Identification of the gene causing mucolipidosis type IV. Nat Genet 26:118–123 10.1038/79095 [DOI] [PubMed] [Google Scholar]
- 6.Slaugenhaupt SA, Acierno JS Jr, Helbling LA, Bove C, Goldin E, Bach G, Schiffmann R, Gusella JF (1999) Mapping of the mucolipidosis type IV gene to chromosome 19p and definition of founder haplotypes. Am J Hum Genet 65:773–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun M, Goldin E, Stahl S, Falardeau JL, Kennedy JC, Acierno JS Jr, Bove C, Kaneski CR, Nagle J, Bromley MC, et al (2000) Mucolipidosis type IV is caused by mutations in a gene encoding a novel transient receptor potential channel. Hum Mol Genet 9:2471–2478 10.1093/hmg/9.17.2471 [DOI] [PubMed] [Google Scholar]
- 8.Bassi MT, Manzoni M, Monti E, Pizzo MT, Ballabio A, Borsani G (2000) Cloning of the gene encoding a novel integral membrane protein, mucolipidin—and identification of the two major founder mutations causing mucolipidosis type IV. Am J Hum Genet 67:1110–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vergarajauregui S, Puertollano R (2006) Two di-leucine motifs regulate trafficking of mucolipin-1 to lysosomes. Traffic 7:337–353 10.1111/j.1600-0854.2006.00387.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang ZH, Zeng B, Pastores GM, Raksadawan N, Ong E, Kolodny EH (2001) Rapid detection of the two common mutations in Ashkenazi Jewish patients with mucolipidosis type IV. Genet Test 5:87–92 10.1089/109065701753145529 [DOI] [PubMed] [Google Scholar]
- 11.Goldin E, Stahl S, Cooney AM, Kaneski CR, Gupta S, Brady RO, Ellis JR, Schiffmann R (2004) Transfer of a mitochondrial DNA fragment to MCOLN1 causes an inherited case of mucolipidosis IV. Hum Mutat 24:460–465 10.1002/humu.20094 [DOI] [PubMed] [Google Scholar]
- 12.Edelmann L, Dong J, Desnick RJ, Kornreich R (2002) Carrier screening for mucolipidosis type IV in the American Ashkenazi Jewish population. Am J Hum Genet 70:1023–1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Slaugenhaupt SA (2002) The molecular basis of mucolipidosis type IV. Curr Mol Med 2:445–450 10.2174/1566524023362276 [DOI] [PubMed] [Google Scholar]
- 14.Bach G, Webb MB, Bargal R, Zeigler M, Ekstein J (2005) The frequency of mucolipidosis type IV in the Ashkenazi Jewish population and the identification of 3 novel MCOLN1 mutations. Hum Mutat 26:591 10.1002/humu.9385 [DOI] [PubMed] [Google Scholar]
- 15.Dobrovolny R, Liskova P, Ledvinova J, Poupetova H, Asfaw B, Filipec M, Jirsova K, Kraus J, Elleder M (2007) Mucolipidosis IV: report of a case with ocular restricted phenotype caused by leaky splice mutation. Am J Ophthalmol 143:663–671 10.1016/j.ajo.2006.11.049 [DOI] [PubMed] [Google Scholar]
- 16.Bargal R, Bach G (1997) Mucolipidosis type IV: abnormal transport of lipids to lysosomes. J Inherit Metab Dis 20:625–632 10.1023/A:1005362123443 [DOI] [PubMed] [Google Scholar]
- 17.Chen CS, Bach G, Pagano RE (1998) Abnormal transport along the lysosomal pathway in mucolipidosis, type IV disease. Proc Natl Acad Sci USA 95:6373–6378 10.1073/pnas.95.11.6373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crandall BF, Philippart M, Brown WJ, Bluestone DA (1982) Mucolipidosis IV. Am J Med Genet 12:301–308 10.1002/ajmg.1320120308 [DOI] [PubMed] [Google Scholar]
- 19.Bach G, Cohen MM, Kohn G (1975) Abnormal ganglioside accumulation in cultured fibroblasts from patients with mucolipidosis IV. Biochem Biophys Res Commun 66:1483–1490 10.1016/0006-291X(75)90526-4 [DOI] [PubMed] [Google Scholar]
- 20.Caimi L, Tettamanti G, Berra B, Sale FO, Borrone C, Gatti R, Durand P, Martin JJ (1982) (1982) Mucolipidosis IV, a sialolipidosis due to ganglioside sialidase deficiency. J Inherit Metab Dis 5:218–224 10.1007/BF02179146 [DOI] [PubMed] [Google Scholar]
- 21.Folkerth RD, Alroy J, Lomakina I, Skutelsky E, Raghavan SS, Kolodny EH (1995) Mucolipidosis IV: morphology and histochemistry of an autopsy case. J Neuropathol Exp Neurol 54:154–164 [PubMed] [Google Scholar]
- 22.Pedersen SF, Owsianik G, Nilius B (2005) TRP channels: an overview. Cell Calcium 38:233–252 10.1016/j.ceca.2005.06.028 [DOI] [PubMed] [Google Scholar]
- 23.Nilius B, Owsianik G, Voets T, Peters JA (2007) Transient receptor potential cation channels in disease. Physiol Rev 87:165–217 10.1152/physrev.00021.2006 [DOI] [PubMed] [Google Scholar]
- 24.LaPlante JM, Falardeau J, Sun M, Kanazirska M, Brown EM, Slaugenhaupt SA, Vassilev PM (2002) Identification and characterization of the single channel function of human mucolipin-1 implicated in mucolipidosis type IV, a disorder affecting the lysosomal pathway. FEBS Lett 532:183–187 10.1016/S0014-5793(02)03670-0 [DOI] [PubMed] [Google Scholar]
- 25.Raychowdhury MK, Gonzalez-Perrett S, Montalbetti N, Timpanaro GA, Chasan B, Goldmann WH, Stahl S, Cooney A, Goldin E, Cantiello HF (2004) Molecular pathophysiology of mucolipidosis type IV: pH dysregulation of the mucolipin-1 cation channel. Hum Mol Genet 13:617–627 10.1093/hmg/ddh067 [DOI] [PubMed] [Google Scholar]
- 26.Cantiello HF, Montalbetti N, Goldmann WH, Raychowdhury MK, Gonzalez-Perrett S, Timpanaro GA, Chasan B (2005) Cation channel activity of mucolipin-1: the effect of calcium. Pflugers Arch 451:304–312 10.1007/s00424-005-1448-9 [DOI] [PubMed] [Google Scholar]
- 27.Kiselyov K, Chen J, Rbaibi Y, Oberdick D, Tjon-Kon-Sang S, Shcheynikov N, Muallem S, Soyombo A (2005) TRP-ML1 is a lysosomal monovalent cation channel that undergoes proteolytic cleavage. J Biol Chem 280:43218–43223 10.1074/jbc.M508210200 [DOI] [PubMed] [Google Scholar]
- 28.Soyombo AA, Tjon-Kon-Sang S, Rbaibi Y, Bashllari E, Bisceglia J, Muallem S, Kiselyov K (2006) TRP-ML1 regulates lysosomal pH and acidic lysosomal lipid hydrolytic activity. J Biol Chem 281:7294–7301 10.1074/jbc.M508211200 [DOI] [PubMed] [Google Scholar]
- 29.Manzoni M, Monti E, Bresciani R, Bozzato A, Barlati S, Bassi MT, Borsani G (2004) Overexpression of wild-type and mutant mucolipin proteins in mammalian cells: effects on the late endocytic compartment organization. FEBS Lett 567:219–224 10.1016/j.febslet.2004.04.080 [DOI] [PubMed] [Google Scholar]
- 30.Pryor PR, Reimann F, Gribble FM, Luzio JP (2006) Mucolipin-1 is a lysosomal membrane protein required for intracellular lactosylceramide traffic. Traffic 7:1388–1398 10.1111/j.1600-0854.2006.00475.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miedel MT, Weixel KM, Bruns JR, Traub LM, Weisz OA (2006) Posttranslational cleavage and adaptor protein complex-dependent trafficking of mucolipin-1. J Biol Chem 281:12751–12759 10.1074/jbc.M511104200 [DOI] [PubMed] [Google Scholar]
- 32.Venkatachalam K, Hofmann T, Montell C (2006) Lysosomal localization of TRPML3 depends on TRPML2 and the mucolipidosis-associated protein TRPML1. J Biol Chem 281:17517–17527 10.1074/jbc.M600807200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kiselyov K, Soyombo A, Muallem S (2007) TRPpathies. J Physiol 578:641–653 10.1113/jphysiol.2006.119024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jennings JJ Jr, Zhu JH, Rbaibi Y, Luo X, Chu CT, Kiselyov K (2006) Mitochondrial aberrations in mucolipidosis type IV. J Biol Chem 281:39041–39050 10.1074/jbc.M607982200 [DOI] [PubMed] [Google Scholar]
- 35.LaPlante JM, Sun M, Falardeau J, Dai D, Brown EM, Slaugenhaupt SA, Vassilev PM (2006) Lysosomal exocytosis is impaired in mucolipidosis type IV. Mol Genet Metab 89:339–348 10.1016/j.ymgme.2006.05.016 [DOI] [PubMed] [Google Scholar]
- 36.Fares H, Greenwald I (2001) Regulation of endocytosis by CUP-5, the Caenorhabditis elegans mucolipin-1 homolog. Nat Genet 28:64–68 10.1038/88281 [DOI] [PubMed] [Google Scholar]
- 37.Schaheen L, Dang H, Fares H (2006) Basis of lethality in C. elegans lacking CUP-5, the mucolipidosis type IV orthologue. Dev Biol 293:382–391 10.1016/j.ydbio.2006.02.008 [DOI] [PubMed] [Google Scholar]
- 38.Tellez-Nagel I, Rapin I, Iwamoto T, Johnson AB, Norton WT, Nitowsky H (1976) Mucolipidosis IV: clinical, ultrastructural, histochemical, and chemical studies of a case, including a brain biopsy. Arch Neurol 33:828–835 [DOI] [PubMed] [Google Scholar]
- 39.Goldin E, Blanchette-Mackie EJ, Dwyer NK, Pentchev PG, Brady RO (1995) Cultured skin fibroblasts derived from patients with mucolipidosis 4 are auto-fluorescent. Pediatr Res 37:687–692 10.1203/00006450-199506000-00003 [DOI] [PubMed] [Google Scholar]
- 40.Livni N, Merin S (1978) Mucolipidosis IV: ultrastructural diagnosis of a recently defined genetic disorder. Arch Pathol Lab Med 102:600–604 [PubMed] [Google Scholar]
- 41.Falardeau JL, Kennedy JC, Acierno JS Jr, Sun M, Stahl S, Goldin E, Slaugenhaupt SA (2002) Cloning and characterization of the mouse Mcoln1 gene reveals an alternatively spliced transcript not seen in humans. BMC Genomics 3:3 10.1186/1471-2164-3-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nagy A, Gertsenstein M, Vintersten K, Behringer R (2003) Manipulating the mouse embryo. Cold Spring Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 43.Truett GE, Heeger P, Mynatt RL, Truett AA, Walker JA, Warman ML (2000) Preparation of PCR-quality mouse genomic DNA with hot sodium hydroxide and Tris (HotSHOT). Biotechniques 29:52, 54 [DOI] [PubMed] [Google Scholar]
- 44.Zervas M, Somers KL, Thrall MA, Walkley SU (2001) Critical role for glycosphingolipids in Niemann-Pick disease type C. Curr Biol 11:1283–1287 10.1016/S0960-9822(01)00396-7 [DOI] [PubMed] [Google Scholar]
- 45.Dockray GJ, Hamer C, Evans D, Varro A, Dimaline R (1991) The secretory kinetics of the G cell in omeprazole-treated rats. Gastroenterology 100:1187–1194 [PubMed] [Google Scholar]
- 46.Rogers DC, Fisher EM, Brown SD, Peters J, Hunter AJ, Martin JE (1997) Behavioral and functional analysis of mouse phenotype: SHIRPA, a proposed protocol for comprehensive phenotype assessment. Mamm Genome 8:711–713 10.1007/s003359900551 [DOI] [PubMed] [Google Scholar]
- 47.Sango K, McDonald MP, Crawley JN, Mack ML, Tifft CJ, Skop E, Starr CM, Hoffmann A, Sandhoff K, Suzuki K, et al (1996) Mice lacking both subunits of lysosomal beta-hexosaminidase display gangliosidosis and mucopolysaccharidosis. Nat Genet 14:348–352 10.1038/ng1196-348 [DOI] [PubMed] [Google Scholar]
- 48.Schiffman R, Dwyer NK, Lubensky IA, Tsokos M, Sutliff VE, Latimer JS, Frei KP, Brady RO, Barton NW, Blanchette-Mackie EJ, et al (1998) Constitutive achlorhydria in mucolipidosis type IV. Proc Nat Acad Sci USA 95:1207–1212 10.1073/pnas.95.3.1207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smith JA, Chan CC, Goldin E, Schiffmann R (2002) Noninvasive diagnosis and ophthalmic features of mucolipidosis type IV. Ophthalmology 109:588–594 10.1016/S0161-6420(01)00968-X [DOI] [PubMed] [Google Scholar]
- 50.Walkley SU (2007) Pathogenic mechanisms in lysosomal disease: a reappraisal of the role of the lysosome. Acta Paediatr Suppl 96:26–32 [DOI] [PubMed] [Google Scholar]
- 51.Walkley SU, March PA, Schroeder CE, Wurzelmann S, Jolly RD (1995) Pathogenesis of brain dysfunction in Batten disease. Am J Med Genet 57:196–203 10.1002/ajmg.1320570218 [DOI] [PubMed] [Google Scholar]
- 52.Wisniewski KE, Rapin I, Heaney-Kieras J (1988) Clinico-pathological variability in the childhood neuronal ceroid-lipofuscinoses and new observations on glycoprotein abnormalities. Am J Med Genet Suppl 5:27–46 10.1002/ajmg.1320310607 [DOI] [PubMed] [Google Scholar]
- 53.Du H, Duanmu M, Witte D, Grabowski GA (1998) Targeted disruption of the mouse lysosomal acid lipase gene: long-term survival with massive cholesteryl ester and triglyceride storage. Hum Mol Genet 7:1347–1354 10.1093/hmg/7.9.1347 [DOI] [PubMed] [Google Scholar]
- 54.Samuelson LC, Hinkle KL (2003) Insights into the regulation of gastric acid secretion through analysis of genetically engineered mice. Annu Rev Physiol 65:383–400 10.1146/annurev.physiol.65.092101.142213 [DOI] [PubMed] [Google Scholar]
- 55.Yao X, Forte JG (2003) Cell biology of acid secretion by the parietal cell. Annu Rev Physiol 65:103–131 10.1146/annurev.physiol.65.072302.114200 [DOI] [PubMed] [Google Scholar]
- 56.Clarke LA, Russell CS, Pownall S, Warrington CL, Borowski A, Dimmick JE, Toone J, Jirik FR (1997) Murine mucopolysaccharidosis type I: targeted disruption of the murine alpha-L-iduronidase gene. Hum Mol Genet 6:503–511 10.1093/hmg/6.4.503 [DOI] [PubMed] [Google Scholar]
- 57.Evers M, Saftig P, Schmidt P, Hafner A, McLoghlin DB, Schmahl W, Hess B, von Figura K, Peters C (1996) Targeted disruption of the arylsulfatase B gene results in mice resembling the phenotype of mucopolysaccharidosis VI. Proc Natl Acad Sci USA 93:8214–8219 10.1073/pnas.93.16.8214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hahn CN, del Pilar Martin M, Schroder M, Vanier MT, Hara Y, Suzuki K, Suzuki K, d’Azzo A (1997) Generalized CNS disease and massive GM1-ganglioside accumulation in mice defective in lysosomal acid beta-galactosidase. Hum Mol Genet 6:205–211 10.1093/hmg/6.2.205 [DOI] [PubMed] [Google Scholar]
- 59.Itoh M, Matsuda J, Suzuki O, Ogura A, Oshima A, Tai T, Suzuki Y, Takashima S (2001) Development of lysosomal storage in mice with targeted disruption of the β-galactosidase gene: a model of human GM1-gangliosidosis. Brain Dev 23:379–384 10.1016/S0387-7604(01)00244-3 [DOI] [PubMed] [Google Scholar]
- 60.Kenyon KR (1976) Ocular manifestations and pathology of systemic mucopolysaccharidoses. Birth Defects Orig Artic Ser 12:133–153 [PubMed] [Google Scholar]
- 61.Schwartz MF, Werblin TP, Green WR (1985) Occurrence of mucopolysaccharide in corneal grafts in the Maroteaux-Lamy syndrome. Cornea 4:58–66 [PubMed] [Google Scholar]
- 62.Mollard RJ, Telegan P, Haskins M, Aguirre G (1996) Corneal endothelium in mucopolysaccharide storage disorders: morphologic studies in animal models. Cornea 15:25–34 10.1097/00003226-199601000-00006 [DOI] [PubMed] [Google Scholar]
- 63.Strauch OF, Stypmann J, Reinheckel T, Martinez E, Haverkamp W, Peters C (2003) Cardiac and ocular pathologies in a mouse model of mucopolysaccharidosis type VI. Pediatr Res 54:701–708 10.1203/01.PDR.0000084085.65972.3F [DOI] [PubMed] [Google Scholar]
- 64.Young RD, Tudor D, Hayes AJ, Kerr B, Hayashida Y, Nishida K, Meek KM, Caterson B, Quantock AJ (2005) Atypical composition and ultrastructure of proteoglycans in the mouse corneal stroma. Invest Ophthalmol Vis Sci 46:1973–1978 10.1167/iovs.04-1309 [DOI] [PubMed] [Google Scholar]
- 65.Newell FW, Matalon R, Meyer S (1975) A new mucolipidosis with psychomotor retardation, corneal clouding, and retinal degeneration. Am J Ophthalmol 80:440–449 [DOI] [PubMed] [Google Scholar]
- 66.Riedel KG, Zwaan J, Kenyon KR, Kolodny EH, Hanninen L, Albert DM (1985) Ocular abnormalities in mucolipidosis IV. Am J Ophthalmol 99:125–136 [DOI] [PubMed] [Google Scholar]
- 67.Kaye EM (1995) Therapeutic approaches to lysosomal storage diseases. Curr Opin Pediatr 7:650–654 [DOI] [PubMed] [Google Scholar]
- 68.Malatack JJ, Consolini DM, Bayever E (2003) The status of hematopoietic stem cell transplantation in lysosomal storage disease. Pediatr Neurol 29:391–403 10.1016/j.pediatrneurol.2003.09.003 [DOI] [PubMed] [Google Scholar]
- 69.Pastores GM, Barnett NL (2005) Current and emerging therapies for the lysosomal storage disorders. Expert Opin Emerg Drugs 10:891–902 10.1517/14728214.10.4.891 [DOI] [PubMed] [Google Scholar]
- 70.Grabowski GA (2006) Delivery of lysosomal enzymes for therapeutic use: glucocerebrosidase as an example. Expert Opin Drug Deliv 3:771–782 10.1517/17425247.3.6.771 [DOI] [PubMed] [Google Scholar]
- 71.Berges BK, Yellayi S, Karolewski BA, Miselis RR, Wolfe JH, Fraser NW (2006) Widespread correction of lysosomal storage in the mucopolysaccharidosis type VII mouse brain with a herpes simplex virus type 1 vector expressing β-glucuronidase. Mol Ther 13:859–869 10.1016/j.ymthe.2005.12.017 [DOI] [PubMed] [Google Scholar]
- 72.Cardone M, Polito VA, Pepe S, Mann L, D’Azzo A, Auricchio A, Ballabio A, Cosma MP (2006) Correction of Hunter syndrome in the MPSII mouse model by AAV2/8-mediated gene delivery. Hum Mol Genet 15:1225–1236 10.1093/hmg/ddl038 [DOI] [PubMed] [Google Scholar]
- 73.Hemsley KM, King B, Hopwood JJ (2007) Injection of recombinant human sulfamidase into the CSF via the cerebellomedullary cistern in MPS IIIA mice. Mol Genet Metab 90:313–328 10.1016/j.ymgme.2006.10.005 [DOI] [PubMed] [Google Scholar]