

Abstract
Mutation screening of the breast and ovarian cancer–predisposition genes BRCA1 and BRCA2 is becoming an increasingly important part of clinical practice. Classification of rare nontruncating sequence variants in these genes is problematic, because it is not known whether these subtle changes alter function sufficiently to predispose cells to cancer development. Using data from the Myriad Genetic Laboratories database of nearly 70,000 full-sequence tests, we assessed the clinical significance of 1,433 sequence variants of unknown significance (VUSs) in the BRCA genes. Three independent measures were employed in the assessment: co-occurrence in trans of a VUS with known deleterious mutations; detailed analysis, by logistic regression, of personal and family history of cancer in VUS-carrying probands; and, in a subset of probands, an analysis of cosegregation with disease in pedigrees. For each of these factors, a likelihood ratio was computed under the hypothesis that the VUSs were equivalent to an “average” deleterious mutation, compared with neutral, with respect to risk. The likelihood ratios derived from each component were combined to provide an overall assessment for each VUS. A total of 133 VUSs had odds of at least 100:1 in favor of neutrality with respect to risk, whereas 43 had odds of at least 20:1 in favor of being deleterious. VUSs with evidence in favor of causality were those that were predicted to affect splicing, fell at positions that are highly conserved among BRCA orthologs, and were more likely to be located in specific domains of the proteins. In addition to their utility for improved genetics counseling of patients and their families, the global assessment reported here will be invaluable for validation of functional assays, structural models, and in silico analyses.
Sequence-based testing for BRCA1 (MIM 113705) and BRCA2(MIM 600185) mutations is now widely available through both commercial laboratories and research studies. Whereas thousands of BRCA1 and BRCA2 truncating mutations have been associated with increased risk of cancer in carriers, the contribution of other BRCA1 and BRCA2 sequence variants to cancer risk remains largely undefined. These “variants of unknown significance” (VUSs) are mainly missense mutations but also include a number of intronic variants and inframe deletions and insertions (IFDIs). The open-access online Breast Cancer Information Core (BIC) Database, which functions as a repository of sequence alterations in BRCA1 and BRCA2, contains >1,500 distinct sequence variants that are currently reported as having unknown clinical significance. To date, classification of the BRCA1 and BRCA2 VUSs as cancer predisposing or neutral has proven problematic, because it is not known whether these subtle changes alter the function of the proteins sufficiently to predispose to cancer. Determination of their disease relevance on the basis of population or family studies has also proved difficult, because most of the alleles encoding these mutations are very rare and, in some cases, population specific. Although identified in a minority (5%–10%) of individuals tested clinically, the number of such tests performed annually worldwide means that a large number of women (and their families) are affected by the VUS issue. In addition, there is evidence to suggest that minority ethnic populations are disproportionately affected. VUSs in the BRCA1 and BRCA2 genes pose significant problems, because patients and physicians do not know whether the VUSs predispose to cancer or are neutral with respect to cancer risk. As a result, carriers of VUSs and their family members cannot take advantage of the risk assessment, prevention, and therapeutic measures that are available to carriers of known deleterious truncating mutations.1 In addition, carriers of VUSs are sometimes counseled to undergo prophylactic oophorectomy or mastectomy because of the presence of the VUS, but in the absence of any knowledge of the cancer relevance of the VUS. For these reasons, the determination of the clinical relevance of VUSs in BRCA1 and BRCA2 has become an important clinical issue. Although the focus of this article is on BRCA1 and BRCA2, similar concerns and issues are found in virtually all complex, genetically heterogeneous diseases for which genetic testing is performed—for example, colon cancer associated with mutations in mismatch repair genes.2
To address the problem of defining which VUSs are deleterious/disease causing and which are neutral, various types of evidence and classification schemes have been proposed.3–5 These include formal assessment of the cosegregation of the variant with disease in pedigrees,6–8 co-occurrence of the variant in question in trans with a known deleterious mutation,9 and a number of in silico approaches that evaluate phylogenetic conservation and severity of the amino acid substitution.10–12 In studies of other genes, evolutionary sequence-conservation analysis involving protein multiple-sequence alignments have been used to show that missense variants (MVs) at highly conserved/invariant residues are more often deleterious, whereas highly variable changes are more likely neutral.13,14 For BRCA1, Abkevich et al.10 developed a predictive algorithm that combines a measure of cross-species conservation, including nonmammalian BRCA1 sequences, with a measure of the degree of chemical change in amino acids,15 to identify 50 putative deleterious BRCA1 missense mutations.
A number of VUSs have been examined, in terms of their effect on protein, with use of functional assays. Efforts in this area have focused on BRCA1 mutations in the two C-terminal BRCT domains,16,17 which assess the transcriptional activation activity of the BRCT domains with use of mammalian and yeast-based models,18–20 and on the E3 ligase activity associated with the N-terminal BARD1-binding domain of BRCA1.21 Examination of BRCA2 function has focused on assays of homology-directed repair and centrosome amplification in response to ectopic expression of full-length, wild-type VUS containing BRCA2 protein.22 Recently, crystal structures of the BRCA1 BRCT and BRCA2 DNA-binding domains have been used to predict that a number of VUSs predispose to cancer,23–25 but genetic evidence suggests that some of these predictions are incorrect. Although none of the approaches described above have successfully been used to classify the clinical relevance of BRCA1 or BRCA2 VUSs, a number have shown promise in this regard. The development of methods based on functional assays, crystal structure, phylogenetic and sequence analyses, and other approaches has been hampered by the absence of a large number of VUSs that have been proven to be either deleterious or neutral on the basis of direct genetic evidence that can be used in validation of these various assays.
In this article, we examine the genetic evidence for or against disease causality for a large number of variants, with use of a large database of tested individuals collected by Myriad Genetic Laboratories. In addition to analysis of co-occurrence of VUSs with established deleterious mutations and the analysis of VUS cosegregation in individual pedigrees, we apply a novel analysis of personal and family cancer history, using data on >70,000 individuals, to assess the clinical significance of 1,433 distinct sequence variants in BRCA1 and BRCA2.
Methods
Source of Data and Variants Analyzed
The data analyzed for this article come from the large database of full-sequence testing for mutations in the BRCA1 and BRCA2 genes, performed at Myriad Genetic Laboratories. Since our primary focus was on the analysis of personal and family history, we began with all 70,071 individuals receiving full-sequence tests as of December 2005. To avoid potential confusion over which part of the personal/family history was due to which variant, this data set was reduced by eliminating all individuals who had more than one reported sequence variant (including both VUS and pathogenic mutations), which resulted in a set of 61,270 individuals with at most a single BRCA1 or BRCA2 sequence variant. We then divided the family history data set into several ethnic groups, since we postulated that probands of different ethnic backgrounds are expected to present with different distributions of mutations and different distributions of personal and family histories of cancer. After examining the data set with use of some preliminary logistic regressions to examine heterogeneity as a function of reported ethnicity, we constructed three separate groups that had similar family-history and mutation-prevalence profiles. These groups were: (a) western European, central/eastern European, other/none specified, Native American, and Asian; (b) Ashkenazi Jewish; and (c) Latin American/Caribbean and African American. In the first group, 72% were of European ancestry, 24% did not specify an ethnicity, and 4% were reported as of Asian, Middle Eastern, or Native American origin. Individual probands who reported mixed ancestry between these broad groups (e.g., Western European and Caribbean) were not included in the final data set, leaving 60,529 individual probands for analyses of family history. Table 1 shows the distribution by ethnicity and testing results of these individuals. A total of 1,433 distinct unclassified variants in 4,623 tested probands were identified through sequencing and are the subject of this article.
Table 1. .
Results of Full BRCA Sequence Analysis of 60,529 Tested Probands
| No. (%) of Subjects with Genotype |
|||||
| Ethnic Group | Wild Type | Neutral | VUS | BRCA1 Deletion | BRCA2 Deletion |
| Europeana | 41,899 (78.6) | 1,993 (3.7) | 3,972 (7.5) | 3,102 (5.8) | 2,334 (4.4) |
| Ashkenazi Jewish | 3,622 (80.6) | 381 (8.5) | 258 (5.7) | 139 (3.1) | 91 (2.0) |
| African American/Caribbean | 1,667 (60.9) |
398 (14.5) |
393 (14.3) |
176 (6.4) |
104 (3.8) |
| Total | 47,188 (78.0) | 2,772 (4.6) | 4,623 (7.6) | 3,417 (5.6) | 2,529 (4.2) |
See the text for the description of ethnicity for this group.
As before,3 we assumed that all variants can be classified as deleterious mutations (M) or neutral variants (V). Our aim was to calculate a likelihood ratio (LR) of the form
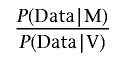 |
for each variant, given the available data. These ratios (or Bayes factors) can be combined with prior probabilities to obtain an absolute probability that each variant is deleterious. In this work, we first focused on the data sources that are most straightforwardly quantifiable from pedigree data: co-occurrence with known deleterious mutations, family history, and cosegregation. These three sources of data are independent, and the LR can therefore be derived by multiplying the LRs from the three components. We then used an admixture model to examine the influence of the type of mutation.
Co-Occurrence with Known Deleterious Mutations
To include information on the co-occurrence of a VUS in trans phase with a deleterious mutation as evidence against the variant being disease causing, we followed the likelihood-ratio calculation described in our previous work.3 We assumed that, for BRCA1, compound heterozygotes for two deleterious mutations are vanishingly rare (occur in the testing population with a frequency of 1 in 10,000), given both biological and genetic evidence that homozygotes or compound heterozygotes for deleterious BRCA1 mutations are embryonically lethal,26,27 whereas, for BRCA2, we assumed that the probability is 1 in 1,000, the increased frequency reflecting the fact that viable compound heterozygotes have been reported as Fanconi anemia, type D1.28,29 For these analyses, we used an updated data set based on ∼100,000 tests with observed frequencies of BRCA1 and BRCA2 deleterious mutations of 6.6% and 5.0%, respectively. The phase of the VUS with an observed deleterious mutation was determined through examination of haplotypes of common polymorphisms known in other examples of each variant or when the VUS and the deleterious mutation were in the same PCR fragment, as determined by direct examination of the sequence traces. In some cases, phase could not be determined, and we assumed that the first deleterious mutation was in cis phase and that subsequent different deleterious mutations were in trans phase. The LR for deleterious versus neutral is expressed as
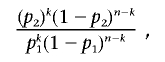 |
where n is the total number of times the variant was observed, k is the number of those that were in trans with a known deleterious mutation, p1 is the overall frequency of pathogenic mutations in the data set, and p2 is the probability of being a compound heterozygote for two pathogenic mutations (e.g., p1=0.066 and p2=1/10,000).
Of the 1,433 VUSs studied here, 476 were observed at least five times, and 248 had at least 10 occurrences. Of the 1,433 variants, 44 were observed to occur with at least one deleterious mutation.
Classification by Personal and Family History of Tested Probands
We developed likelihood models on the basis of family history,3 which, given the availability of the large Myriad Genetic Laboratories data set, is an extremely powerful tool for classification of VUSs. The method is based on the difference in personal and family-history profiles between individuals found to carry a true deleterious mutation and those with a wild-type BRCA sequence. The rationale is that mutation prevalence is known to be strongly dependent on certain key factors (e.g., disease status of the proband, age at diagnosis, and number and age of relatives with breast cancer [BRCA] or ovarian cancer), so these characteristics should also predict the probability of a new disease-causing missense mutation, whereas that of a neutral mutation should be independent of family history.
We suppose that we have a set of probands drawn from a population with a certain distribution of family history. Suppose first that family history is classified into n types. Let pj be the probability that an individual with a mutation has family history type j, whereas qj is the corresponding probability for neutral variants. For a VUS that is deleterious, the predicted distribution of family history should be the same as that for known pathogenic mutations. The LR for a VUS with family history in category j drawn from the same population is therefore
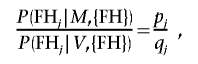 |
where {FH} is the set of all family histories in the study population. In theory, the probabilities pj and qj could be calculated from the proportions of individuals with each category of family history who are found to carry a deleterious mutation. In practice, however, there are large numbers of possible constellations of family history, and the numbers of mutations in some categories would be very small. Instead, we estimated these probabilities by fitting a logistic-regression model. Thus, we fitted a model in which the outcome was the occurrence of a deleterious mutation versus a normal sequence, and summaries of personal and family history were included as covariates.
This model then provides predicted probabilities rk that individual k is a mutation carrier, given his or her FH, is
Let r0 be the corresponding probability under the null hypothesis that the mutation is unrelated to family history or, equivalently, the prior probability of a deleterious mutation in the population. This is estimated by the overall proportion of individuals who have a deleterious mutation (rather than a normal sequence). For example, with use of the data in table 1, for the European group for BRCA1,
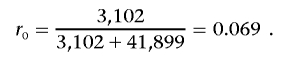 |
Then
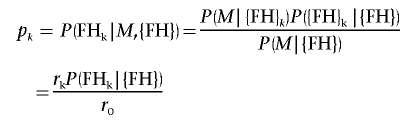 |
and
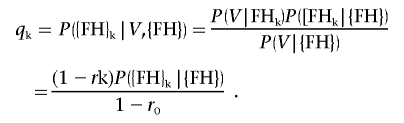 |
Therefore, the required LR is
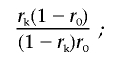 |
that is, the odds ratio (OR) of the predicted probability that the individual with the given family history is a mutation carrier against the corresponding probability under the null hypothesis. Where there are multiple individuals with the same VUS, the LRs can be multiplied. We note that this formulation implicitly assumes that all pathogenic mutations in the same gene confer identical risks—that is, that there is no allelic heterogeneity.
To implement this analysis, the logistic-regression model was fitted separately for each ethnic group and separately for BRCA1 and BRCA2. To construct a predictive model to be applied to the VUS probands, we used a logistic-regression model within each ethnic group, comparing the personal and family histories of individuals with known deleterious sequences versus those with normal sequences. The specific factors used in the prediction were categorized as
Personal history of tested individual
BRCA
-
1.
None,
-
2.
Ductal carcinoma in situ (DCIS) only, or
Diagnosis of BRCA at age
-
3.
⩾60,
-
4.
50–59,
-
5.
40–49,
-
6.
30–39, or
-
7.
<30.
Ovarian cancer
-
1.
None or
Diagnosis of ovarian cancer at age
-
2.
⩾60,
-
3.
50–59,
-
4.
40–49, or
-
5.
<40.
Male BRCA
-
1.
Yes or
-
2.
No.
Bilateral BRCA
-
1.
Yes or
-
2.
No.
and
Reported family history of tested proband (assessed as twice the number of first-degree relatives with given cancer type plus the number of second-degree relatives with given cancer type) (table 2).
Table 2. .
ORs and Associated 95% CIs from Logistic-Regression Analysis of the Myriad Genetic Laboratories Family-History Data of 47,335 White Probands
| OR (95% CI) for Gene versus Wild Type |
||
| History and Cancer Type (Age at Onset, in Years) |
BRCA1 | BRCA2 |
| Personal history of cancer for proband: | ||
| DCIS only | 1.58 (1.2–2.1) | 2.34 (1.9–2.9) |
| BRCA (⩾60) | 1.25 (1.0–1.6) | 1.55 (1.3–1.9) |
| BRCA (50–59) | 1.67 (1.4–2.0) | 2.07 (1.7–2.5) |
| BRCA (40–49) | 3.40 (3.0–3.9) | 2.89 (2.5–3.4) |
| BRCA (30–39) | 9.65 (8.4–11.1) | 4.97 (4.2–5.8) |
| BRCA (<30) | 15.3 (12.4–19.1) | 4.71 (3.5–6.4) |
| Bilateral BRCA | 2.40 (2.1–2.7) | 1.46 (1.3–1.7) |
| Male BRCA | 1.91 (.9–4.1) | 12.0 (8.9–16.3) |
| Ovarian (⩾60) | 4.60 (3.7–5.8) | 4.52 (3.6–5.7) |
| Ovarian (50–59) | 11.8 (9.7–14.4) | 7.92 (6.4–9.8) |
| Ovarian (40–49) | 18.0 (14.8–21.9) | 4.05 (3.0–5.5) |
| Ovarian (<40) | 7.06 (5.1–9.8) | .52 (.18–1.36) |
| Family history of cancera: | ||
| 1 BRCA (<50) | 2.16 (1.9–2.5) | 1.82 (1.6–2.1) |
| 2 BRCA (<50) | 3.33 (3.0–3.7) | 2.20 (2.0–2.5) |
| 3 BRCA (<50) | 5.60 (4.7–6.7) | 3.84 (3.2–4.6) |
| ⩾4 BRCA (<50) | 8.31 (7.2–9.8) | 4.20 (3.5–5.0) |
| 1 BRCA (⩾50) | .92 (.82–1.04) | 1.26 (1.1–1.4) |
| 2 BRCA (⩾50) | .82 (.73–.93) | 1.36 (1.2–1.5) |
| 3 BRCA (⩾50) | .75 (.6–.94) | 1.45 (1.2–1.7) |
| ⩾4 BRCA (⩾50) | .56 (.41–.57) | 1.33 (1.1–1.7) |
| 1 Ovarian | 2.56 (2.2–2.9) | 1.56 (1.3–1.8) |
| 2 Ovarian | 4.75 (4.2–5.3) | 1.87 (1.6–2.2) |
| 3 Ovarian | 9.06 (7.3–11.3) | 3.30 (2.5–4.4) |
| ⩾4 Ovarian | 11.1 (8.5–14.4) | 1.83 (1.2–2.9) |
Assessed as twice the number of first-degree relatives with given cancer type plus the number of second-degree relatives with given cancer type.
In total, therefore, there were 24 parameters estimated in each of six logistic regressions: BRCA1 versus wild type and BRCA2 versus wild type for each of the three ethnicity groupings. The predicted probabilities pk were obtained using the “predict” option in STATA.
As a simple validation of the process outline above, we examined the family history ORs for the known deleterious variants. Of the 1,106 distinct deleterious variants examined, 123 had odds of >100:1 in favor of being deleterious, 216 had odds of >10:1, and no known variants were excluded at this same threshold. Although we recognize that these summaries may overstate the model’s out-of-sample predictive ability, since the deleterious variants were used to construct the logistic regressions, they do provide reassurance that, in general, the method is performing as we expected. Further confirmation of this is obtained by looking at the similar calculation for 44 variants already classified as neutral and not used in either the logistic regression or in the subsequent analysis of VUSs. Of these 44 variants, 33 had odds against being deleterious of >100:1, and the other 11 all had odds against being deleterious of a lesser magnitude.
Cosegregation of VUSs in Pedigrees
Elsewhere, we developed a general-pedigree likelihood method for evaluating cosegregation of variants with disease in families that can be used to assess disease causality for unclassified sequence variants.6 The pedigree likelihood is evaluated under the hypothesis that the variant under consideration has the same penetrance as the “average” known deleterious mutation, compared with the hypothesis that the variant segregates independent of disease in the pedigree(s) under study.
As part of an effort to assess the clinical significance of VUSs, individuals found, through the full-sequence testing by Myriad Genetic Laboratories, to carry a VUS are offered free testing of additional family members on a research basis. These additional individuals can be unaffected or affected with BRCA or ovarian cancer and can be male or female. Typically, only 10% of individual probands with VUS provide such samples, and, in most cases, only one or two samples per family are obtained. The pedigree information available in the Myriad Genetic Laboratories database is somewhat descriptive in nature, and, for this reason, we used a simplified model of BRCA1/2 penetrance, with seven liability classes corresponding to (1) unaffected and aged <20 years (or male), (2) unaffected and aged 21–49 years, (3) unaffected and aged ⩾50 years, (4) affected with BRCA at age <50 years, (5) affected with BRCA at age ⩾50, (6) affected with ovarian cancer at age <50, and (7) affected with ovarian cancer at age ⩾50. For each liability class, penetrance values were taken from the 2003 study of Antoniou et al.,30 by collapsing the incidence rates across the broader categories used in that study. These pedigrees were then analyzed using a modification of the LINKAGE package,31 as described by Thompson et al.,4 to obtain the required LR.
Because the analysis of cosegregation relies on the distribution of the additional genotype data in the pedigree, conditional on the pedigree (including the proband) phenotypes, it is statistically independent from the family/personal information. Similarly, the co-occurrence data is independent from the other two sources, since individuals who also have known deleterious mutations are not considered in these analyses. Thus, the three individual LRs can be multiplied to arrive at a combined LR.
Evaluation of Heterogeneity by Variant Class
To estimate the proportion of mutations in the data set that are likely to be clinically significant as a function of location, conservation, class, etc., we performed a heterogeneity analysis analogous to that routinely used in linkage analysis. Specifically, the required likelihood is given by
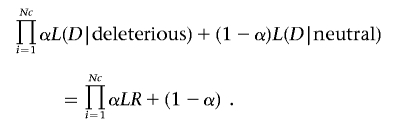 |
This likelihood (in practice, the log likelihood) is then maximized over α, and 95% CIs were constructed by finding those values of 2ln(likelihood) that differed by <3.84 from the maximum value. Hypotheses regarding differences in values of α as a function of partitions of the total variant space are performed using likelihood-ratio tests.
The 1,433 variants were subdivided into the following groups:
-
1.
Variants that changed the resulting protein—for example, MVs and IFDIs;
-
2.
Variants that were extremely likely, on the basis of studies of many genes and experimental data, to affect splicing, defined for our purposes as (a) all IVS± 1/2 or (b) if the last nucleotide of the exon is G and if the first five bases of the subsequent intron are not GTRAG. Note that the latter set includes some missense mutations as well as silent substitutions; mutations that were missense changes as well as splice variants were counted only in the splice variant group.
-
3.
Other variants, consisting primarily of intronic variants not located within 2 bp of the intron/exon splice junction, exonic variants likely to affect splicing but not meeting the strict definition above, and also a small number of variants in the 5′ UTRs and truncating variants at the extreme 3′ end of the genes.
For the missense/IFDI category, we further subdivided the variants by their location within one of two recognized functional domains of the proteins: the C-terminus region containing the BRCA1 BRCT repeats, defined loosely here as “amino acids 1396–1862,” and the BRCA2 DNA-binding domain (amino acids 2500–3098). We also categorized these variants according to whether the wild-type residue involved in the substitution/deletion was evolutionarily conserved through the pufferfish Tetraodon, using multiple-sequence alignments constructed by S.V.T.10 and available on the Align GVGD Web site. In the case of three inframe insertions, these were denoted as “conserved” if the alignment across species was stable in the region surrounding the location of the insertion.
Statistical analyses were performed using STATA version 9.0 (StataCorp). The heterogeneity analysis was performed using the R programming language.
Results
We first built the logistic-regression model comparing probands found with wild-type BRCA sequence with those with known deleterious mutations, using the factors detailed in the “Methods” section. As might be expected with the very large sample sizes used in the analysis, the logistic regressions were highly significant with good predictive power; areas under the ROC curve were ∼0.80 for BRCA1 and ∼0.70 for BRCA2. Not surprisingly, the most highly significant predictive factors associated with a deleterious BRCA1 mutation were associated with personal and family history of ovarian cancer, with high ORs for early-onset BRCA as well. For BRCA2, the most important factors were a personal history of male BRCA and personal/family history of early-onset BRCA. Interestingly a strong family history of BRCA diagnosed after age 50 years was associated with an increased probability of having a BRCA2 mutation but a decreased risk of carrying a BRCA1 deleterious mutation. For both genes, there were strong dose-response effects for decreasing age at onset of BRCA (or ovarian cancer) and also for increasing numbers of affected relatives. The details of the parameter estimates are provided in table 2 for the large European sample set; those for the other two ethnic classifications were broadly similar. The ethnicity- and gene-specific logistic-regression equations were then applied to a total of 3,951 family and personal histories of tested probands found to carry a VUS, to obtain log-ORs as described in the “Methods” section. The average number of such scores per variant was 2.76, with a range of 1–77; over half of the studied variants had only a single family history available for study. One hundred eighty-two distinct variants had 5 or more family-history scores; of these, 72 had at least 10 family-history scores. The associated log-odds of causality per variant ranged from −10.6 to 11.2, with a mean of −0.26. Sixteen variants had total family history log10-likelihood–ratio (LLR) scores >2, whereas 55 VUSs had scores <−2, indicating odds of 100:1 for and against the variant being deleterious, respectively, on the basis of the family-history analysis alone.
In total, 351 pedigrees of probands with 127 (45 BRCA1; 82 BRCA2) distinct variants were analyzed for cosegregation with disease in families. Odds based on the pedigree data alone ranged from 138:1 in favor of causality based on six pedigrees to >250,000:1 against causality based on analysis of 17 pedigrees. It should be noted that the variants targeted for this analysis were selected on the basis of prior evidence that such analysis could contribute to the likelihood calculation. In the co-occurrence analysis, 121 variants were observed at least once in trans with a known deleterious mutation, and 21 were observed two or more times. The LLRs for this source of information ranged from <−8 (for the variant BRCA1*N723D seen 82 times, of which 4 were in trans with a deleterious mutation) to 1.1 (BRCA1*A1708E, seen 76 times, never with a deleterious mutation). The combined evidence from all three sources ranged from odds of >1011:1 in favor of disease causality to 1012:1 against causality.
Figure 1 contains the frequency distribution by gene of the combined log-odds of causality for the 1,433 VUSs. From this figure, we see that, whereas 133 variants could be classified as neutral with odds of >100:1, only 24 variants (18 BRCA1; 6 BRCA2) achieved this level of classification in favor of causality. The vast majority of variants analyzed are relatively uninformative and do not individually provide information (fig. 1). Figure 2 displays the distribution of the log-odds of causality for missense and IFDIs as a function of whether they are located in the BRCT domain of BRCA1 or the DNA-binding domain of BRCA2. Although 27 of the 30 variants with odds in favor of causality >10:1 are in one of these two domains, it should be noted that 29 variants within these domains have odds in favor of neutrality of >100:1.
Figure 1. .

Frequency histogram of combined LLR for 1,433 sequence VUSs, by gene (BRCA1 or BRCA2).
Figure 2. .

Frequency histogram of combined LLR for 1,177 MVs and IFDIs, by location within either DNA-binding domain (DBD) of BRCA2, or BRCT domain of BRCA1. For definitions of domains, see the text. Yes = within DBD/BRCT; no = located outside these domains.
Table 3 displays the list of all 43 variants with odds in favor of the VUS being deleterious of at least 20:1, showing each component of the total score and information on species conservation. Table 4 lists the odds for the 133 variants that have odds of >100:1 against causality and thus are likely to be of no or little clinical significance on the basis of our observations. For those variants with both family history and cosegregation information, there was a modest but significant correlation between the log-odds scores for these two sources (r=0.32; P<.001).
Table 3. .
BRCA Sequence VUSs with Odds of ⩾20:1 in Favor of Causality
| LLR |
|||||
| Gene and Variant | Family-History LLR | Co-Occurrence | Cosegregation | Combined LLR | Odds in Favor of Causality |
| BRCA1: | |||||
| A1708E | 11.22 | 1.10 | .00 | 12.32 | 2,110,000,000,000 |
| IVS20+1G→A | 11.11 | .52 | .00 | 11.63 | 431,000,000,000 |
| IVS13+1G→A | 6.09 | .60 | .00 | 6.68 | 4,818,331 |
| R1495M (4603G→T) | 5.28 | .81 | .00 | 6.10 | 1,249,808 |
| R1699W | 4.34 | .26 | .00 | 4.60 | 39,978 |
| G1788V | 2.37 | .42 | 1.06 | 3.85 | 7,054 |
| IVS5+3A→G | 3.04 | .12 | .00 | 3.15 | 1,417 |
| G1706E | 2.35 | .13 | .29 | 2.77 | 589 |
| IVS19-12G→A | 4.52 | .10 | −2.06 | 2.56 | 363 |
| L1764P | 1.56 | .09 | .89 | 2.54 | 350 |
| V1688del (5181del3) | 1.13 | .07 | 1.23 | 2.43 | 268 |
| IVS6-3C→G | 2.28 | .07 | .00 | 2.35 | 225 |
| IVS15+1G→A | 2.11 | .19 | .00 | 2.30 | 199 |
| IVS17+1G→A | 2.21 | .09 | .00 | 2.29 | 196 |
| T1685I | 2.10 | .04 | .00 | 2.15 | 140 |
| I1766S | 1.11 | .16 | .87 | 2.14 | 139 |
| G1738R | 1.94 | .12 | .00 | 2.06 | 114 |
| T1685A | 2.00 | .03 | .00 | 2.03 | 107 |
| IVS6-1G→C | 1.84 | .01 | .00 | 1.85 | 72 |
| M1689R | 1.49 | .17 | .00 | 1.67 | 46 |
| S1715R | 1.56 | .09 | .00 | 1.65 | 45 |
| IVS12+2del21insA | 1.46 | .10 | .00 | 1.56 | 36 |
| M18T | 1.41 | .09 | .00 | 1.49 | 31 |
| A1623G (4987C→G) | 1.02 | .04 | .43 | 1.49 | 31 |
| IVS11-1G→A | 1.36 | .01 | .00 | 1.38 | 24 |
| IVS18-1G→C | 1.31 | .06 | .00 | 1.37 | 24 |
| BRCA2: | |||||
| R2659K (8204G→A) | 3.19 | .34 | .00 | 3.52 | 3,339 |
| G2748D | 2.41 | .16 | .83 | 3.40 | 2,494 |
| I2627F | 2.61 | .11 | .30 | 3.02 | 1,046 |
| E2663V | .05 | .18 | 2.14 | 2.37 | 233 |
| P3039P (9345G→A) | 1.81 | .35 | .00 | 2.16 | 146 |
| D2723G | 1.88 | .20 | .00 | 2.08 | 121 |
| IVS15-1G→A | 1.51 | .46 | .00 | 1.97 | 94 |
| T2722R | .84 | .10 | 1.03 | 1.97 | 93 |
| IVS24-1G→C | 1.68 | .06 | .00 | 1.74 | 55 |
| R2336H (7235G→A) | 1.31 | .39 | .00 | 1.70 | 50 |
| W2626C | 1.36 | .33 | .00 | 1.68 | 48 |
| IVS21+4A→G | .35 | .13 | 1.08 | 1.56 | 36 |
| L2653P | 1.56 | .06 | −.24 | 1.38 | 24 |
| D3095E | 1.50 | .22 | −.37 | 1.35 | 23 |
| IVS5-2A→G | 1.26 | .07 | .00 | 1.34 | 22 |
| R2502C | .83 | .50 | .00 | 1.32 | 21 |
| IVS19+1G→A | 1.12 | .20 | .00 | 1.32 | 21 |
Table 4. .
Variants with Odds of ⩾100:1 in Favor of Neutrality
| LLR |
|||||
| Gene and Variant | Family History | Co-Occurrence | Cosegregation | Combined LLR | Odds in Favor of Neutrality |
| BRCA1: | |||||
| R866C | −7.7 | −1.76 | −2.63 | −12.1 | 1.25×1012 |
| P142H | −8.92 | −2.16 | 0 | −11.08 | 1.20×1011 |
| T1720A | −6.23 | .49 | −5.22 | −1.95 | 9.00×1010 |
| N810Y | −2.42 | −7.28 | −1.24 | −1.94 | 8.77×1010 |
| S186Y | −3.57 | −3.54 | −2.3 | −9.42 | 2.61×109 |
| P1614S | −.51 | −7.12 | −1.02 | −8.64 | 4.40×108 |
| V1534M | −4.63 | −3.99 | 0 | −8.62 | 4.18×108 |
| E597K | −6.55 | −1.92 | 0 | −8.47 | 2.95×108 |
| N723D | 1.13 | −8.94 | 0 | −7.81 | 6.44×107 |
| E1214K | −2.41 | −2.13 | −2.93 | −7.47 | 2.96×107 |
| S1101N | −7.06 | .55 | 0 | −6.51 | 3.23×106 |
| IVS16-20A→G | −2.57 | −3.69 | 0 | −6.25 | 1.78×106 |
| IVS8-17G→T | −4.42 | −1.62 | 0 | −6.04 | 1.10×106 |
| P1238L | −5.25 | −1.31 | .54 | −6.02 | 1.05×106 |
| Y105C | −.83 | −4.57 | .12 | −5.29 | 1.94×105 |
| V191I | −1.86 | −2.23 | −1.02 | −5.11 | 1.30×105 |
| P334L | .95 | −4.85 | −.97 | −4.88 | 7.52×104 |
| I1275V | −5.28 | .49 | 0 | −4.79 | 6.16×104 |
| IVS18-6C→A | −2.14 | −2.45 | 0 | −4.59 | 3.90×104 |
| P1859R | −2.74 | −2.26 | .52 | −4.47 | 2.98×104 |
| R504H | −3.19 | .57 | −1.66 | −4.28 | 1.92×104 |
| S1266T | −1.87 | −2.39 | 0 | −4.26 | 1.80×104 |
| D67Y | −3.23 | .28 | −1.08 | −4.03 | 1.06×104 |
| I1044V | −1.54 | −2.4 | 0 | −3.94 | 8,723 |
| V1247I | −1.58 | .17 | −2.5 | −3.91 | 8,123 |
| IVS2-11delT | −1.49 | −2.39 | 0 | −3.88 | 7,598 |
| IVS18-13A→G | −.39 | −2.42 | −1.02 | −3.83 | 6,799 |
| I925L | −1.41 | −2.39 | 0 | −3.8 | 6,251 |
| N473S | −1.29 | −2.49 | 0 | −3.78 | 6,049 |
| IVS6+7G→A | −1.43 | −2.16 | −.17 | −3.75 | 5,601 |
| R1203Q | −1.71 | .15 | −2.18 | −3.75 | 5,589 |
| G890V | −3.86 | .16 | 0 | −3.7 | 5,006 |
| E842G | −1.22 | −2.46 | 0 | −3.68 | 4,741 |
| M1652T | −1.74 | .09 | −1.92 | −3.57 | 3,728 |
| D369N | −3.77 | .22 | 0 | −3.56 | 3,593 |
| D369del (1225del3) | −1.31 | −2.21 | 0 | −3.52 | 3,328 |
| V1736A | −1.5 | −2.1 | .25 | −3.35 | 2,219 |
| K862E | −2.93 | .19 | −.57 | −3.31 | 2,059 |
| R1028H | −1.23 | −2.08 | 0 | −3.31 | 2,045 |
| E143K | −2.3 | .12 | −1.09 | −3.27 | 1,871 |
| D642H | −.75 | .07 | −2.49 | −3.16 | 1,455 |
| IVS2-13C→G | −1.16 | −1.91 | 0 | −3.07 | 1,166 |
| D1546Y | −.56 | −2.47 | 0 | −3.04 | 1,088 |
| A622V | −3.43 | .45 | 0 | −2.98 | 963 |
| V1804D | −2.61 | .57 | −.88 | −2.92 | 828 |
| IVS12+10G→C | −.47 | −2.43 | 0 | −2.9 | 803 |
| I1405V | −.42 | −2.46 | 0 | −2.88 | 752 |
| Q804H | −1.25 | .33 | −1.95 | −2.86 | 729 |
| K1109N | −.51 | −2.32 | 0 | −2.82 | 664 |
| IVS11-11T→C | −.37 | −2.45 | 0 | −2.81 | 648 |
| Q155E | −.27 | −2.45 | 0 | −2.72 | 524 |
| P1637L | −1.83 | −.4 | −.46 | −2.69 | 493 |
| T1349M | −2.71 | .04 | 0 | −2.66 | 462 |
| I1858L | −.14 | −2.5 | 0 | −2.65 | 444 |
| N1468H | −.04 | −2.49 | 0 | −2.53 | 337 |
| M297I | 0 | −2.5 | 0 | −2.51 | 322 |
| E1682K | 0 | −2.5 | 0 | −2.5 | 317 |
| IVS21-8C→T | −1.89 | .13 | −.74 | −2.5 | 313 |
| A280G | .06 | −2.49 | 0 | −2.43 | 266 |
| IVS17-9A→T | .09 | −2.43 | 0 | −2.34 | 218 |
| E1419Q | −2.37 | .06 | 0 | −2.31 | 203 |
| F1662S | .16 | −2.45 | 0 | −2.28 | 192 |
| H1402Y | −.07 | −2.45 | .29 | −2.22 | 165 |
| R1751Q | −2.66 | .19 | .3 | −2.17 | 150 |
| I124V | −2.21 | .04 | 0 | −2.17 | 147 |
| IVS15-7C→T | −.24 | .12 | −1.97 | −2.1 | 125 |
| N132K | −2.32 | .23 | 0 | −2.09 | 123 |
| D420Y | −2.16 | .07 | 0 | −2.09 | 123 |
| L668F | −2.85 | .73 | .1 | −2.02 | 105 |
| M1361L | −2.09 | .07 | 0 | −2.02 | 104 |
| BRCA2: | |||||
| K2950N | −1.6 | −.25 | 0 | −1.85 | 7.15×1010 |
| R2108H | −2.72 | −2.5 | −5.52 | −1.74 | 5.48×1010 |
| S1733F | −8.88 | .76 | 0 | −8.12 | 1.33×108 |
| G1529R | −4.12 | −3.97 | 0 | −8.09 | 1.23×108 |
| I2285V | −5.49 | −2.41 | 0 | −7.9 | 7.86×107 |
| L1019V | −3.81 | .54 | −3.18 | −6.46 | 2.86×106 |
| A75P | −3.52 | −.31 | −2.51 | −6.34 | 2.20×106 |
| T3349A | −.71 | −1.23 | −3.4 | −5.35 | 2.22×105 |
| R1190W | −1.99 | −1.17 | −2.12 | −5.27 | 1.88×105 |
| P1819S | −4.47 | .77 | −1.53 | −5.23 | 1.70×105 |
| T630I | −4.64 | −.58 | 0 | −5.23 | 1.68×105 |
| G1771D | −4.97 | −.15 | 0 | −5.12 | 1.33×105 |
| K1690N | −4.49 | −.53 | 0 | −5.02 | 1.05×105 |
| S1172L | −3.36 | −.56 | −.89 | −4.81 | 6.48×104 |
| Q2384K | −1.7 | −3.35 | .26 | −4.79 | 6.11×104 |
| C554W | −1.31 | .3 | −3.7 | −4.71 | 5.15×104 |
| D2312V | .06 | −2.73 | −1.91 | −4.57 | 3.76×104 |
| IVS26-20C→T | −1.3 | −3.71 | .53 | −4.48 | 3.04×104 |
| G602R | −.89 | .17 | −3.59 | −4.3 | 2.02×104 |
| E462G | −1.39 | −.64 | −1.82 | −3.84 | 6,960 |
| N56T | −.97 | .11 | −2.96 | −3.82 | 6,666 |
| H2074N | −3.5 | −.31 | 0 | −3.81 | 6,513 |
| R2973C | −1.39 | .26 | −2.63 | −3.75 | 5,685 |
| C3198R | −1.65 | −1.22 | −.87 | −3.74 | 5,541 |
| I1929V | −.37 | −1.93 | −1.3 | −3.59 | 3,914 |
| N1228D | −.35 | −1.18 | −2.04 | −3.57 | 3,726 |
| H1918Y | −2.3 | −1.25 | 0 | −3.55 | 3,552 |
| V1306I | −1.89 | −1.21 | −.37 | −3.47 | 2,979 |
| Y3098H | −3.45 | −.71 | .7 | −3.46 | 2,892 |
| V2969M | −2.06 | −.82 | −.57 | −3.45 | 2,806 |
| V894I | −.9 | −1.01 | −1.48 | −3.39 | 2,440 |
| I1349T | −.64 | −2.72 | 0 | −3.37 | 2,320 |
| Q1396R | −.02 | −1.74 | −1.6 | −3.36 | 2,280 |
| N2113S | −1.03 | −.99 | −1.23 | −3.24 | 1,749 |
| R2842H | −.85 | −1.23 | −1.12 | −3.2 | 1,574 |
| IVS25+9A→C | −.48 | −1.01 | −1.7 | −3.19 | 1,553 |
| V3079I | −.8 | −2.38 | 0 | −3.19 | 1,545 |
| IVS11-20T→A | −.83 | .23 | −2.52 | −3.11 | 1,288 |
| R2888C | −1.11 | −1.35 | −.64 | −3.1 | 1,246 |
| T2250A | −1.3 | .29 | −1.98 | −3 | 1,005 |
| F1524V | −1.99 | −1.18 | .17 | −2.99 | 982 |
| L1904V | −.33 | −1.24 | −1.42 | −2.99 | 976 |
| T582P | −.32 | −2.64 | 0 | −2.96 | 910 |
| D3170G | −1.54 | .07 | −1.42 | −2.89 | 772 |
| D1280V | −2.57 | −1.01 | .72 | −2.85 | 714 |
| P168T | −1.72 | −1.09 | 0 | −2.81 | 646 |
| N2048I | −.75 | −1.28 | −.78 | −2.81 | 639 |
| K2729N | −2.21 | .8 | −1.38 | −2.79 | 614 |
| D2665G | −.61 | −.89 | −1.1 | −2.61 | 406 |
| R3052Q | −1.24 | −1.31 | 0 | −2.55 | 354 |
| G1194D | −1.41 | .15 | −1.24 | −2.5 | 316 |
| P375S | −1.26 | −1.24 | 0 | −2.5 | 315 |
| V2908G | −1.17 | −1.24 | 0 | −2.41 | 256 |
| D806H | −.57 | −1.18 | −.66 | −2.41 | 255 |
| T1354M | −1.12 | .27 | −1.56 | −2.4 | 254 |
| N1102Y | −2.53 | .14 | 0 | −2.39 | 246 |
| Y3092C | −2.34 | .11 | .01 | −2.22 | 166 |
| IVS20-16C→G | −1.71 | .27 | −.78 | −2.22 | 165 |
| C1365Y | −.57 | −1.23 | −.38 | −2.18 | 150 |
| N2436I | −.39 | −1.32 | −.41 | −2.12 | 132 |
| K2411T | −.84 | −1.24 | 0 | −2.08 | 119 |
| L2396F | −.67 | −1.38 | 0 | −2.04 | 111 |
| K513R | −.74 | −1.29 | 0 | −2.03 | 108 |
As described in the “Methods” section, we estimated the proportion of the variants included here that are predicted to be deleterious in terms of cancer risk, both overall and in classes of VUSs defined by characteristics on the basis of mutation type and conservation. These results are shown in table 5.
Table 5. .
Heterogeneity Analysis of VUSs by Selected Characteristics
| No. of VUSs with LLR |
||||
| Variant Class | n | α (95% CI) | >1.3 | <−2 |
| All: | 1,433 | .20 (.16–.25) | 43 | 133 |
| BRCA1 | 510 | .22 (.16–.28) | 26 | 70 |
| BRCA2 | 923 | .18 (.13–.25) | 17 | 63 |
| MV/IFDI: | 1,177 | .12 (.08–.17) | 22 | 117 |
| Domain: | ||||
| In BRCT/DBD | 323 | .35 (.26–.45) | 21 | 29 |
| Other MV/IFDI | 854 | .0 (.0–.04) | 1 | 88 |
| Sequence Conservation: | ||||
| Invarianta | 218 | .46 (.34–.58) | 17 | 14 |
| Variable | 959 | .03 (.0–.07) | 5 | 103 |
| Splice | 79 | 1.0 (.91–1.0) | 14 | 0 |
| Other | 177 | .26 (.15–.39) | 7 | 16 |
In our alignment of vertebrate BRCA1 and BRCA2 sequences, available at the Align GVGD Web site.
Overall, there was no significant difference between BRCA1 and BRCA2 in the estimated proportion of variants that were deleterious (likelihood-ratio test, χ2 1 df=0.69; not significant). However, there was significant heterogeneity among the three classes of sequence variant (missense/IFDI, splice, and other) (χ2 2 df=109.7; P<.0001); 100% of splice-site mutations were estimated to be deleterious, compared with 12% of missense/IFDI mutations and 26% of other unclassified sequence variants. As in the overall set, no significant differences in the fraction of MVs estimated to be deleterious were observed between BRCA1 and BRCA2 variants. A strong association with the position of the mutation was observed with 35% (95% CI 26%–45%) of mutations within BRCT or DBD domains estimated to be deleterious, compared with 0% of mutations outside these domains (χ2 1 df=56.3; P<.0001). There was no significant difference in the estimated proportion of deleterious mutations in the BRCA2 DNA-binding domain (33%) and the BRCA1 BRCT domain (37%). When mutations were classified by conservation among species, 46% (95% CI 32%–60%) of the conserved variants were classified as deleterious, compared with 3% of the nonconserved substitutions (χ2 1 df=59.7; P<.0001). However, here, we did observe marginally statistically significant evidence that a higher proportion of BRCA1 missense substitutions occurring at strongly conserved positions were deleterious compared with those of BRCA2 (64% vs. 35%; P=.013).
Discussion
Using an approach based on three conditionally independent sources of information contained in the Myriad Genetic Laboratories large BRCA testing data set, we evaluated 1,433 sequence variants of unknown clinical significance in the BRCA susceptibility genes BRCA1 and BRCA2. Through this analysis, we were able to classify 93 variants with a threshold of odds of 1,000:1, 157 at 100:1, and 222 with odds of at least 20:1. In each case, the majority (e.g., 133 of 157 for 100:1 threshold) of VUSs classified were found to be of no clinical significance with respect to cancer risk. Although additional BRCA testing data produced by Myriad Genetic Laboratories, as well as similar data potentially available from other testing laboratories, primarily in Europe, could classify additional variants with use of this approach, it is clear that the majority of variants are unlikely to be classified in this manner, which highlights the need for use of other data. However, it should be noted that, because the variants most likely to be classified using our approach are generally the ones with many observations, the number of families who could benefit from our results is relatively large. On the basis of the Myriad database alone and with the assumption of a threshold for causality of 1,000:1 and for neutrality of 100:1, as proposed in Goldgar et al.,3 1,569 probands and their families may now have a more informative test result. Although only 140 of these are associated with a variant classified as a deleterious mutation that can be used in genetics counseling of their families, there is some benefit for the rest in knowing that they are unlikely to carry a clinically significant mutation and that their risk is that conferred by their family history alone.
We emphasize that all the methods used in this article are based on the assumption that these variants are either neutral with respect to cancer risks or that they have the same age- and site-specific risks as the average BRCA mutation as estimated by Antoniou et al.30 If a particular variant were associated with lower penetrance (but still elevated over noncarrier rates), it might provide inconclusive evidence, even given a large amount of available data, and/or there would be conflicts between the different sources of evidence (as well as other such as functional or in silico studies).
Although most of the information for classification in this study is derived from the family-history analysis, in several instances, the three sources were needed to achieve a clinically useful threshold. For example, the BRCA1 variant I1766S (see table 3) shows odds in favor of causality of 13:1 from analysis of five family histories, 7.4:1 from cosegregation analysis of a single pedigree, and 1.44:1 on the basis of its observation seven times, never with a deleterious mutation, yielding overall odds of 139:1 in favor of this variant being deleterious compared with neutral. In contrast, the intronic variant BRCA1*IVS19-12G→A shows convincing odds based on six family histories (>30,000:1), but this conflicts with the pedigree cosegregation data (odds of 115:1 against, on the basis of three pedigrees); the overall combined evidence for this variant is 363:1, similar to that for I1766S. The reasons for this anomaly are unclear. It might reflect an unusual structure of families from the population from which this variant derives, or it might reflect some unusual pattern of risks associated with the mutation that are not well handled by these methods. To resolve this discrepancy, it would be helpful to verify the pedigree data and cancer diagnoses from the original provider/genetics counselor. Additionally, although there are no data from sequence homology to help in this analysis, in vitro tests of splicing might also provide data on the pathogenicity of this variant. Of the different approaches, we regard the cosegregation analysis as the most robust, since it relates directly to the disease risk and requires few assumptions. Unfortunately, relatively few variants can be classified by this approach alone. Our results demonstrate that the family-history classification can be very powerful, but it is more susceptible to bias due to population-specific ascertainment,8 although this bias may be minimized by appropriate stratification into subgroups with different mutation prevalences. This highlights the need for some caution in the use of these predictions, especially when there are discrepancies between the methods.
Of the 1,433 variants examined, the overall estimate of the proportion of those that are deleterious was 20%, indicating that perhaps as many as 286 of these variants could be deleterious mutations. As expected, there was good evidence that variants predicted to disrupt consensus splice donors were, as a group, deleterious; in fact, the estimated proportion of deleterious mutations was 1.0, so that there was no evidence of neutral variants under the selection criteria we used. This group accounted for an estimated 30% of all deleterious variants examined in the series. Among variants that resulted in a predicted altered BRCA protein, it was estimated that 12% were deleterious, and our analyses indicated that most (if not all) deleterious variants in this group fall at strongly conserved residues and/or in the recognized domains encompassing the BRCT repeats in BRCA1 and the DNA-binding domain in BRCA2. Note that mutations in the ring-finger domain of BRCA1 have been classified elsewhere as deleterious and thus are not included in this analysis. As a group, almost half of all missense substitutions (and IFDIs) falling at positions that were invariant in our alignments of vertebrate BRCA1 and BRCA2 sequences were estimated to be deleterious; these variants, representing 218 of the 1,433 variants studied, would require odds of causality of 117:1 to have a posterior probability of 0.99 of being deleterious. Although we cannot exclude the possibility that VUSs in nonconserved residues or variants in other parts of the proteins are deleterious, the upper confidence limits for these groups indicate that it is unlikely that there are very many of these, and substantial evidence would be required to achieve a posterior probability of 0.99 for these variants. Although there was some slight evidence that the group of missense substitutions falling at variable residues includes deleterious substitutions, these were typically cases where the cross-species sequence variation was conservative and the substitution falls well outside the evolutionarily tolerated range of variation. Further, if only the 129 variants falling at invariant positions within the two important domains are considered, the proportion of variants that are estimated to be deleterious rises to 0.73 (0.58–0.86). Among the 177 variants that were located in introns but not at consensus splice sites, in potential exonic splice sites, in the 5′ UTR, or at the extreme 3′ ends of the coding sequence, there was some evidence that this group may harbor deleterious variants (estimated proportion 26%), with 9 variants having odds in favor of causality of >10:1.
For the variants in this analysis, the estimated proportions in table 5 could be used as prior probabilities to estimate posterior probabilities that each variant is deleterious. On the assumption that a posterior probability of 90% would be required to classify a mutation as “probably deleterious,” missense substitutions (prior probability 0.12) require odds of 66:1 in favor of causality to be classified. Ten variants in BRCA1 and five in BRCA2 could be classified on this basis. The data on species conservation provide a more precise classification, however. On the basis of that data, mutations at invariant residues would require only odds of ∼10:1 to be classified as deleterious—12 variants in BRCA1 and 11 in BRCA2 reach this level. In contrast, mutations at variable residues would require odds of ∼300:1, none of which do so.
Although relatively few variants can currently be classified, these data can be combined with data from other sources to provide more-powerful discrimination. These data could include further pedigree cosegregation data, but also data on tumor histopathology, which is highly predictive of BRCA1 mutation status,32 and data on loss of heterozygosity at the BRCA1 and BRCA2 loci.33,34 This approach has recently been used for classification of a small number of BRCA1/2 variants.35–37
The association with sequence conservation and mutation position that we have observed will also be useful for classification of new variants not present in this data set. Thus, these data indicate that a novel mutation identified in a high-risk family will have a substantial probability of being deleterious only if it occurs in a conserved domain. Note, however, that the “prior” probabilities given in table 5 refer strictly to the families and variants ascertained in this database, and their application outside this context should be undertaken with a degree of caution.
Although many investigators have developed in silico approaches for classification and have used evaluation of sequence changes and some have applied functional data, all of these methods require external validation, and all have suffered from a lack of such validation. The data we generated here could serve as a useful source of validation of such approaches, through repeated sampling of each variant as neutral or deleterious on the basis of its posterior probability of causality and then use of the data to estimate parameters of sensitivity and specificity of other assays.
What is the immediate clinical utility of the results from the present analyses? Clearly, variants with odds against causality of >1,000:1 (or even 100:1) could be considered neutral, rare polymorphisms for the purpose of genetics counseling, and the at-risk unaffected family members who carry such variants would be reassured that they are not at high risk of BRCA or ovarian cancer due to BRCA1 or BRCA2, although they could still be at increased risk on the basis of their family history alone. Those who carry 1 of the 10 variants for which the evidence achieves the threshold of 1,000:1, or perhaps the 48 (36 of which are at splice sites) with posterior probabilities >0.99, should logically be offered the same options as carriers of BRCA-truncating mutations. Apart from those variants that meet these thresholds, will it be useful to report intermediate categories of risk, such as “likely deleterious” or “probably neutral,” or should the current odds in favor of or against causality be reported to the provider? These questions will require consideration by specialists in risk communication and other disciplines and, as such, are beyond the scope of this study. Our goal in this effort is to eventually classify, with such information, a large number of variants as either “deleterious” or “neutral” and to incorporate the evidence behind the classification as part of the BIC database, as a centralized resource for clinicians and researchers alike. We feel that, with a systematic approach such as this, the problems of clinical interpretation and patient recommendations caused by these heretofore “uncertain” variants can be minimized.
Acknowledgments
We gratefully acknowledge the assistance of Diane Bateman, at Myriad Genetic Laboratories, for data generation and helpful comments; Linda Wadum, Jennier Mentlick, and Kiley Johnson, at the Mayo Clinic, for assistance with pedigree-data entry; and the support of Mayo Clinic breast cancer SPORE grant P50 CA116201 (to F.J.C.), American Cancer Society award RSG-04-220-01-CCE (to F.J.C.), National Institutes of Health award CA92309 (to A.N.A.M.), and a Florida Breast Cancer Coalition grant (to A.N.A.M.). D.F.E. is a principal research fellow of Cancer Research UK. We also thank the entire BIC Database steering committee for their support of our efforts in BRCA variant classification.
Web Resources
The URLs for data presented herein are as follows:
- Align GVGD, http://agvgd.iarc.fr/
- BIC, http://research.nhgri.nih.gov/bic/
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for BRCA1 and BRCA2)
References
- 1.Burke W, Daly M, Garber J, Botkin J, Kahn MJ, Lynch P, McTiernan A, Offit K, Perlman J, Petersen G, et al (1997) Recommendations for follow-up care of individuals with an inherited predisposition to cancer: II. BRCA1 and BRCA2—Cancer Genetics Studies Consortium. JAMA 277:997–1003 10.1001/jama.277.12.997 [DOI] [PubMed] [Google Scholar]
- 2.Lucci-Cordisco E, Boccuto L, Neri G, Genuardi M (2006) The use of microsatellite instability, immunohistochemistry and other variables in determining the clinical significance of MLH1 and MSH2 unclassified variants in Lynch syndrome. Cancer Biomark 2:11–27 [DOI] [PubMed] [Google Scholar]
- 3.Goldgar DE, Easton DF, Deffenbaugh AM, Monteiro AN, Tavtigian SV, Couch FJ, Breast Cancer Information Core (BIC) Steering Committee (2004) Integrated evaluation of DNA sequence variants of unknown clinical significance: application to BRCA1 and BRCA2. Am J Hum Genet 75:535–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Billack B, Monteiro AN (2004) Methods to classify BRCA1 variants of uncertain clinical significance: the more the merrier. Cancer Biol Ther 3:458–459 [DOI] [PubMed] [Google Scholar]
- 5.Monteiro AN, Couch FJ (2006) Cancer risk assessment at the atomic level. Cancer Res 66:1897–1899 10.1158/0008-5472.CAN-05-3034 [DOI] [PubMed] [Google Scholar]
- 6.Thompson D, Easton DF, Goldgar DE (2003) A full-likelihood method for the evaluation of causality of sequence variants from family data. Am J Hum Genet 73:652–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petersen GM, Parmigiani G, Thomas D (1998) Missense mutations in disease genes: a Bayesian approach to evaluate causality. Am J Hum Genet 62:1516–1524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou XI, Iversen ES Jr, Parmigiani G (2005) Classification of missense mutations of disease genes. J Am Stat Assn 100:51–60 10.1198/016214504000001817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Judkins T, Hendrickson BC, Deffenbaugh AM, Eliason K, Leclair B, Norton MJ, Ward BE, Pruss D, Scholl T (2005) Application of embryonic lethal or other obvious phenotypes to characterize the clinical significance of genetic variants found in trans with known deleterious mutations. Cancer Res 65:10096–10103 10.1158/0008-5472.CAN-05-1241 [DOI] [PubMed] [Google Scholar]
- 10.Abkevich V, Zharkikh A, Deffenbaugh AM, Frank D, Chen Y, Shattuck D, Skolnick MH, Gutin A, Tavtigian SV (2004) Analysis of missense variation in human BRCA1 in the context of interspecific sequence variation. J Med Genet 41:492–507 10.1136/jmg.2003.015867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tavtigian SV, Samollow PB, de Silva D, Thomas A (2006) An analysis of unclassified missense substitutions in human BRCA1. Fam Cancer 5:77–88 10.1007/s10689-005-2578-0 [DOI] [PubMed] [Google Scholar]
- 12.Tavtigian SV, Deffenbaugh AM, Yin L, Judkins T, Scholl T, Samollow PB, de Silva D, Zharkikh A, Thomas A (2006) Comprehensive statistical study of 452 BRCA1 missense substitutions with classification of eight recurrent substitutions as neutral. J Med Genet 43:295–305 10.1136/jmg.2005.033878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miller MP, Kumar S (2001) Understanding human disease mutations through the use of interspecific genetic variation. Hum Mol Genet 10:2319–2328 10.1093/hmg/10.21.2319 [DOI] [PubMed] [Google Scholar]
- 14.Sunyaev S, Ramensky V, Koch I, Lathe W III, Kondrashov AS, Bork P (2001) Prediction of deleterious human alleles. Hum Mol Genet 10:591–597 10.1093/hmg/10.6.591 [DOI] [PubMed] [Google Scholar]
- 15.Grantham R (1974) Amino acid difference formula to help explain protein evolution. Science 185:862–864 10.1126/science.185.4154.862 [DOI] [PubMed] [Google Scholar]
- 16.Chapman MS, Verma IM (1996) Transcriptional activation by BRCA1. Nature 382:678–679 10.1038/382678a0 [DOI] [PubMed] [Google Scholar]
- 17.Monteiro AN, August A, Hanafusa H (1996) Evidence for a transcriptional activation function of BRCA1 C-terminal region. Proc Natl Acad Sci USA 93:13595–13599 10.1073/pnas.93.24.13595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ostrow KL, McGuire V, Whittemore AS, DiCioccio RA (2004) The effects of BRCA1 missense variants V1804D and M1628T on transcriptional activity. Cancer Genet Cytogenet 153:177–180 10.1016/j.cancergencyto.2004.01.020 [DOI] [PubMed] [Google Scholar]
- 19.Phelan CM, Dapic V, Tice B, Favis R, Kwan E, Barany F, Manoukian S, Radice P, van der Luijt RB, van Nesselrooij BP, et al (2005) Classification of BRCA1 missense variants of unknown clinical significance. J Med Genet 42:138–146 10.1136/jmg.2004.024711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carvalho MA, Marsillac SM, Karchin R, Manoukian S, Grist S, Swaby RF, Urmenyi TP, Rondinelli E, Silva R, Gayol L, et al (2007) Determination of cancer risk associated with germ line BRCA1 missense variants by functional analysis. Cancer Res 67:1494–1501 10.1158/0008-5472.CAN-06-3297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morris JR, Pangon L, Boutell C, Katagiri T, Keep NH, Solomon E (2006) Genetic analysis of BRCA1 ubiquitin ligase activity and its relationship to breast cancer susceptibility. Hum Mol Genet 15:599–606 10.1093/hmg/ddi476 [DOI] [PubMed] [Google Scholar]
- 22.Wu K, Hinson SR, Ohashi A, Farrugia DJ, Wendt P, Tavtigian SV, Deffenbaugh A, Goldgar D, Couch FJ (2005) Functional evaluation and cancer risk assessment of BRCA2 unclassified variants. Cancer Res 65:417–426 [PubMed] [Google Scholar]
- 23.Mirkovic N, Marti-Renom MA, Weber BL, Sali A, Monteiro AN (2004) Structure-based assessment of missense mutations in human BRCA1: implications for breast and ovarian cancer predisposition. Cancer Res 64:3790–3797 10.1158/0008-5472.CAN-03-3009 [DOI] [PubMed] [Google Scholar]
- 24.Karchin R, Monteiro AN, Tavtigian SV, Carvalho MA, Sali A (2007) Functional impact of missense variants in BRCA1 predicted by supervised learning. PLoS Comput Biol 3:e26 10.1371/journal.pcbi.0030026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang H, Jeffrey PD, Miller J, Kinnucan E, Sun Y, Thoma NH, Zheng N, Chen PL, Lee WH, Pavletich NP (2002) BRCA2 function in DNA binding and recombination from a BRCA2-DSS1-ssDNA structure. Science 297:1837–1848 10.1126/science.297.5588.1837 [DOI] [PubMed] [Google Scholar]
- 26.Hohenstein P, Kielman MF, Breukel C, Bennett LM, Wiseman R, Krimpenfort P, Cornelisse C, van Ommen GJ, Devilee P, Fodde R (2001) A targeted mouse BRCA1 mutation removing the last BRCT repeat results in apoptosis and embryonic lethality at the headfold stage. Oncogene 20:2544–2550 10.1038/sj.onc.1204363 [DOI] [PubMed] [Google Scholar]
- 27.Gowen LC, Johnson BL, Latour AM, Sulik KK, Koller BH (1996) Brca1 deficiency results in early embryonic lethality characterized by neuroepithelial abnormalities. Nat Genet 12:191–194 10.1038/ng0296-191 [DOI] [PubMed] [Google Scholar]
- 28.Howlett NG, Taniguchi T, Olson S, Cox B, Waisfisz Q, Die-Smulders C, Persky N, Grompe M, Joenje H, Pals G, et al (2002) Biallelic inactivation of BRCA2 in Fanconi anemia. Science 297:606–609 10.1126/science.1073834 [DOI] [PubMed] [Google Scholar]
- 29.Alter BP, Rosenberg PS, Brody LC (2007) Clinical and molecular features associated with biallelic mutations in FANCD1/BRCA2. J Med Genet 44:1–9 10.1136/jmg.2006.043257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Antoniou A, Pharoah PD, Narod S, Risch HA, Eyfjord JE, Hopper JL, Loman N, Olsson H, Johannsson O, Borg A, et al (2003) Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet 72:1117–1130 (erratum 73:209) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lathrop GM, Lalouel JM, Julier C, Ott J (1984) Strategies for multilocus linkage analysis in humans. Proc Natl Acad Sci USA 81:3443–3446 10.1073/pnas.81.11.3443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lakhani SR, van de Vijver MJ, Jacquemier J, Anderson TJ, Osin PP, McGuffog L, Easton DF (2002) The pathology of familial breast cancer: predictive value of immunohistochemical markers estrogen receptor, progesterone receptor, HER-2, and p53 in patients with mutations in BRCA1 and BRCA2. J Clin Oncol 20:2310–2318 10.1200/JCO.2002.09.023 [DOI] [PubMed] [Google Scholar]
- 33.Smith SA, Easton DF, Evans DG, Ponder BA (1992) Allele losses in the region 17q12-21 in familial breast and ovarian cancer involve the wild-type chromosome. Nat Genet 2:128–131 10.1038/ng1092-128 [DOI] [PubMed] [Google Scholar]
- 34.Collins N, McManus R, Wooster R, Mangion J, Seal S, Lakhani SR, Ormiston W, Daly PA, Ford D, Easton DF, et al (1995) Consistent loss of the wild-type allele in breast cancers from a family linked to the Brca2 gene on chromosome 13q12-13. Oncogene 10:1673–1675 [PubMed] [Google Scholar]
- 35.Osorio A, Milne RL, Honrado E, Barroso A, Diez O, Salazar R, de la Hoya M, Vega A, Benitez J (2007) Classification of missense variants of unknown significance in BRCA1 based on clinical and tumor information. Hum Mutat 28:477–485 10.1002/humu.20470 [DOI] [PubMed] [Google Scholar]
- 36.Lovelock PK, Healey S, Au W, Sum EY, Tesoriero A, Wong EM, Hinson S, Brinkworth R, Bekessy A, Diez O, et al (2006) Genetic, functional, and histopathological evaluation of two C-terminal BRCA1 missense variants. J Med Genet 43:74–83 10.1136/jmg.2005.033258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chenevix-Trench G, Healey S, Lakhani S, Waring P, Cummings M, Brinkworth R, Deffenbaugh AM, Burbidge LA, Pruss D, Judkins T, et al (2006) Genetic and histopathologic evaluation of BRCA1 and BRCA2 DNA sequence variants of unknown clinical significance. Cancer Res 66:2019–2027 10.1158/0008-5472.CAN-05-3546 [DOI] [PubMed] [Google Scholar]