

Abstract
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is an inherited disorder associated with arrhythmias and sudden death. A recessive mutation in the gene encoding plakoglobin has been shown to cause Naxos disease, a cardiocutaneous syndrome characterized by ARVC and abnormalities of hair and skin. Here, we report, for the first time, a dominant mutation in the gene encoding plakoglobin in a German family with ARVC but no cutaneous abnormalities. The mutation (S39_K40insS) is predicted to insert an extra serine residue at position 39 in the N-terminus of plakoglobin. Analysis of a biopsy sample of the right ventricle from the proband showed markedly decreased localization of plakoglobin, desmoplakin, and connexin43 at intercalated discs in cardiac myocytes. A yeast-two-hybrid screen revealed that the mutant protein established novel interactions with histidine-rich calcium-binding protein and TGFβ induced apoptosis protein 2. Immunoblotting and confocal microscopy in human embryonic kidney 293 (HEK293) cell lines transfected to stably express either wild-type or mutant plakoglobin protein showed that the mutant protein was apparently ubiquitylated and was preferentially located in the cytoplasm, suggesting that the S39_K40insS mutation may increase plakoglobin turnover via proteasomal degradation. HEK293 cells expressing mutant plakoglobin also showed higher rates of proliferation and lower rates of apoptosis than did cells expressing the wild-type protein. Electron microscopy showed smaller and fewer desmosomes in cells expressing mutant plakoglobin. Taken together, these observations suggest that the S39_K40insS mutation affects the structure and distribution of mechanical and electrical cell junctions and could interfere with regulatory mechanisms mediated by Wnt-signaling pathways. These results implicate novel molecular mechanisms in the pathogenesis of ARVC.
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a heart-muscle disorder characterized by fibrofatty replacement of cardiac myocytes. Right ventricular features predominate, but left ventricular involvement can also arise with disease progression and may be the dominant presentation. The main clinical complications are arrhythmias, heart failure, and sudden cardiac death.1
ARVC exhibits age-related expression. Penetrance is low (∼30%) and often incomplete. There is phenotypic variation in both its presentation and its clinical course, even within affected individuals from the same family. Clinical diagnosis relies on demonstration of structural, functional, and electrophysiological abnormalities, which reflect the electrical and mechanical alterations in the right ventricle. Despite thorough investigation, a definitive diagnosis is often problematic. This diagnostic uncertainty is a barrier to disease-gene mapping and identification.1 ARVC is familial in 30%–50% of cases and is typically inherited in an autosomal dominant fashion, although recessive forms are recognized. One of these, Naxos disease (MIM #601214), a cardiocutaneous syndrome characterized by ARVC, nonepidermolytic palmoplantar keratoderma, and woolly hair, is caused by a 2-bp deletion in the gene encoding plakoglobin (PK2157del2 [MIM *173325]).2
Plakoglobin is a major component of cell-cell adhesion complexes, which are abundant in many tissues. It is also a signaling molecule with roles in desmosome assembly and development and in regulation of gene expression.3 Mutations in other desmosomal genes have also been implicated in the pathogenesis of ARVC, including those encoding desmoplakin (MIM +125647),4 plakophilin-2 (MIM *602861),5 desmoglein-2 (MIM *125671),6 and desmocollin-2 (MIM *125645).7,8 It is now widely believed that ARVC is a disease caused by abnormal cell-cell adhesion due to defects in desmosomes, intercellular adhesive junctions that anchor cytoskeletal filaments at membrane-associated plaques. Desmosomes provide tissues with mechanical strength and are most abundant in tissues exposed to high levels of mechanical stress, such as the epidermis and cardiac muscle.9
Here, we report a novel mutation (S39_K40insS) in the gene encoding plakoglobin that has been identified in an ARVC-affected patient of German origin. We also provide evidence derived from in vitro experiments that points toward its potential mechanism of pathogenesis. To our knowledge, S39_K40insS is the first dominant ARVC-causing mutation in plakoglobin to be reported to date.
Methods and Material
Clinical Screening
Patients suspected of having ARVC are evaluated at a consultant-led ARVC clinic at the University Hospital of Münster. All patients undergo detailed noninvasive evaluation, including two-dimensional and Doppler echocardiography, 12-lead and signal-averaged electrocardiography, 24-h Holter monitoring, and exercise testing. Patients whose noninvasive-test results indicate life-threatening ventricular tachyarrhythmias and/or the presence of ARVC also undergo magnetic resonance imaging, right ventricular contrast angiography, endomyocardial biopsy, and invasive electrophysiological study, for further characterization of disease expression and risk stratification.
Genetic Screening and Mutation Identification
The proband in this study (II:1 in fig. 1) was part of a larger cohort of ARVC-affected patients who were investigated for mutations in desmosomal genes associated with the disorder. DNA was extracted from blood samples and was amplified by PCR. PCR products were analyzed by agarose-gel electrophoresis and were amplified for direct sequencing. All participants gave informed consent for genetic screening.
Figure 1. .
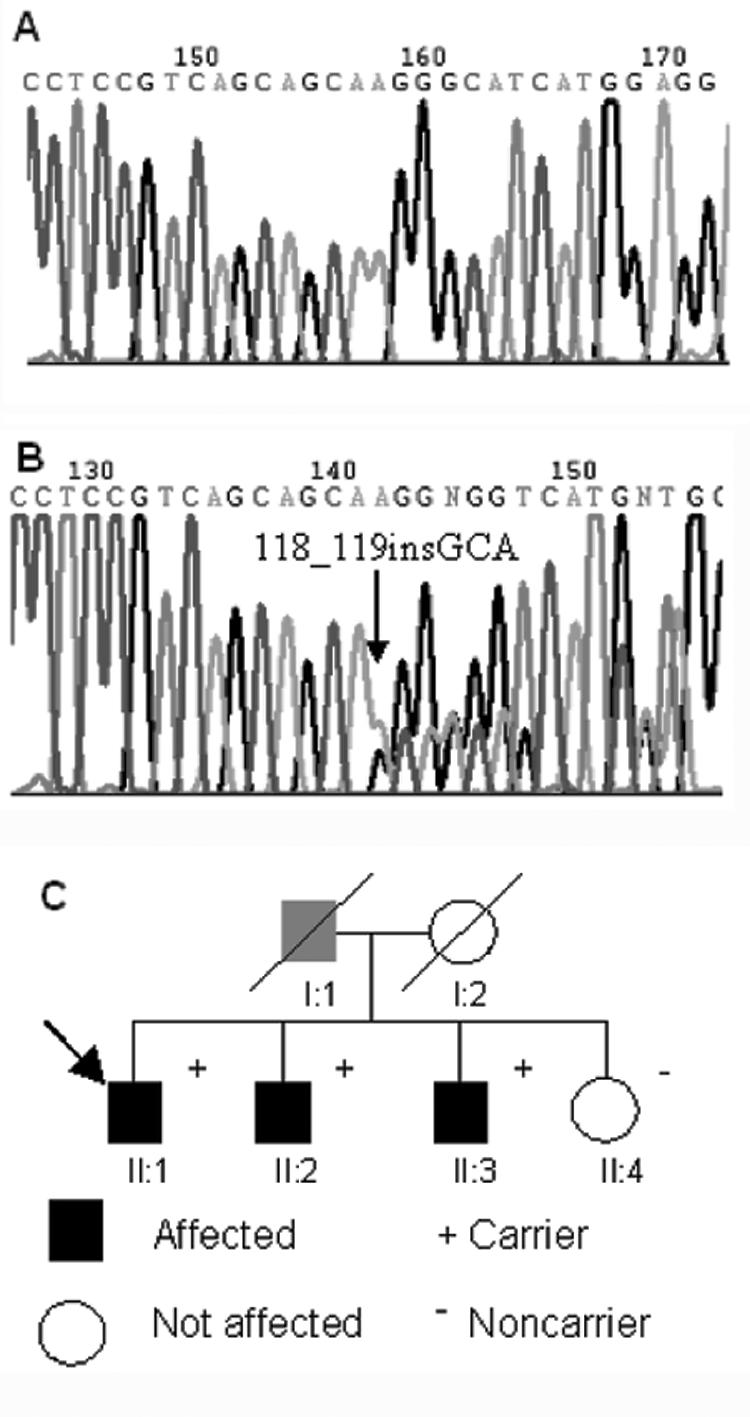
Electropherograms showing wild-type (A) and mutant (B) sequences of exon 2 of the gene encoding plakoglobin. The position of the insertion is marked with an arrow. C, Pedigree of family with ARVC. Squares denote males; circles denote females. The gray symbol denotes a deceased individual who did not undergo clinical evaluation but who was presumed to have been affected. Affected individuals fulfilled the Task Force criteria for ARVC.10 The index patient is marked with an arrow.
Tissue Acquisition and Immunohistochemistry
A right ventricular biopsy sample obtained from the proband (II:1) during a right heart catheterization was immunostained using methods that were validated in a previous study.11 Myocardial specimens obtained at autopsy from three age-matched individuals who had no clinical history or pathological evidence of heart disease were subjected to the same staining protocol and were used as controls. Primary antibodies included polyclonal rabbit anti-connexin43 (Cx43) (Zymed), monoclonal mouse anti-plakoglobin (Sigma), polyclonal rabbit anti-desmoplakin (SeroTec), monoclonal mouse anti-N-cadherin (Sigma), and monoclonal mouse anti-plakophilin-2 (Biodesign) antibodies. Immunostained preparations were analyzed by laser-scanning confocal microscopy (Sarastro Model 2000 [Molecular Dynamics]) as described elsewhere.11
Yeast-Two-Hybrid Screening
This method was used to detect ESTs from a human heart cDNA library that bound wild-type or mutant plakoglobin. The S.c. EasyComp Transformation Kit (Invitrogen) was used to generate competent Saccharomyces cerevisiae cells and to transform them with the plasmid of interest, according to the manufacturer’s instructions. Recombinant pDEST32 (bait, 10 μg) and pEXP-AD502 (library, 10 μg) were introduced into Library-Scale Frozen Competent MaV203 yeast cells (Invitrogen), which were subsequently plated on 10-cm plates with synthetic complete medium lacking leucine and tryptophan to estimate the total number of transformants. Transformants were selected and tested for lacZ with use of a β-galactosidase colony–lift assay. Plasmids were extracted from the yeast cells and were used to transform competent Escherichia coli, which were then plated on antibiotic-selective plates. DNA (500–600 ng) was used to set up a sequencing reaction, and the obtained data were compared against the Human Reference DNA Sequences by a sequence similarity–searching computer program (BLAST).
In Vitro Protein-Binding Assay
BL21 star cells (Invitrogen) were transfected with recombinant plasmids constructed for the expression of glutathione S-transferase (GST) as well as the GST-fused N-terminal, central, and C-terminal domains of the histidine-rich calcium-binding protein (HRC-BP [MIM *142705]). Expression of the recombinant construct was induced by isopropyl-β-d-thiogalactopyranoside (IPTG). Cell pellets obtained after centrifugation were resuspended in Tris buffer (50 mM Tris-HCl [pH 8.0], 0.1% Triton, and 100 mM NaCl, supplemented with protease inhibitors), were sonicated, and were centrifuged, and the supernatant fraction was mixed with G-Sepharose beads. Coomassie-blue staining was used to visualize the purified protein fragments. Pellets of stably transfected human embryonic kidney 293 (HEK293) cells were resuspended in 0.1% Tween in PBS and and were sonicated and centrifuged. Aliquots of the supernatant fractions containing 0.5 mg of protein were added to HRC-fragment– or GST-only–bound Sepharose beads, were rocked, and were centrifuged. The beads were washed in 0.1% Tween in PBS and were loaded on a Tris-glycine gel, and a western blot for plakoglobin expression was performed as described in the “Western Immunoblotting” section.
Restriction Enzyme–Based Cloning
A recombination reaction of pDONR vector (Invitrogen) and plakoglobin cDNA bearing Gateway-compatible attB sites was performed using the BP clonase enzyme. The pcDNA4/His-Max vector (Invitrogen) was converted to a Gateway-compatible form with use of a Gateway vector–conversion reagent system kit (Invitrogen). Plakoglobin was subcloned from the recombinant pDONR plasmid into the pcDNA4/His-Max vector and the pDEST32 vector, with use of the LR clonase enzyme. Plakoglobin was subcloned into the pcDNA5/FRT/TO vector (Invitrogen) with use of the BamHI and EcoRV restriction sites and into the pGFP-C3 vector (Clontech) with use of the BamHI and EcoRI restriction sites. TGFβ-induced apoptosis protein 2 (TAIP-2) cDNA (purchased from Biosearch Technologies) was cloned into the PCR blunt TOPO vector with use of the Zero Blunt TOPO PCR Cloning Kit (Invitrogen) and was subcloned into the pcDNA V5 His 3.1 vector (Invitrogen) with use of the EcoRI restriction sites. Ligation was performed with T4 ligase.
Site-Directed Mutagenesis
A plakoglobin construct bearing the S39_K40insS mutation was generated by subjecting recombinant pcDNA5/FRT/TO, pcDNA4/His-Max, pDEST32, and pGFP-C3 vectors to site-directed mutagenesis, with use of Pfu DNA polymerase enzyme, according to the manufacturer’s instructions.
Generation of Stably Transfected Cell Lines
pcDNA5/FRT/TO constructs expressing wild-type and mutant plakoglobin were introduced in HEK293 cells (Invitrogen) with use of a polyfect transfection reagent. Recombinants were selected with blasticidin (15 μg/ml) and hygromycin (100 μg/ml) and were induced to express the exogenous proteins by incubation in tetracycline (100 ng/ml).
Western Immunoblotting
Cell cultures were harvested, were washed with PBS, and were lysed in radioimmunoprecipitation buffer (50 mM Tris-HCl [pH 7.4], 1% NP-40, 0.25% Na deoxycholate, 150 mM NaCl, 1 mM EDTA, 1 mM Na3VO4, 1 mM NaF, and protease-inhibitor cocktail). Cell lysate samples were loaded on Tris-glycine gels, were electrophoresed, were transferred to nitrocellulose membranes, and were incubated, first with a primary antibody (anti-plakoglobin, 1:500 [Santa Cruz Biotech], or anti-ubiquitin, 1:5,000 [AbCam]) and then with a horseradish peroxidase–linked anti-mouse immunoglobulin G. Blots were developed with ECL reagents (Amersham).
Transient Transfection and Coimmunoprecipitation
HEK293 cells were transiently transfected using a polyfect reagent to express either wild-type or mutant plakoglobin constructs that were tagged with green fluorescent protein (GFP) to facilitate identification of the expressed protein by fluorescence microscopy. These cells were also used in immunoprecipitation assays. In preparation for immunoprecipitation studies, cells were grown to confluence and were harvested as described in the “Western Immunoblotting” section. The cell pellets were resuspended in immunoprecipitation buffer (25 mM HEPES-KOH [pH 7.5], 150 mM KCl, 1 mM EDTA, 12.5 mM MgCl2, and 0.1% NP-40), were sonicated, and were centrifuged at 16,110 g at 4°C for 5 min. Aliquots of the supernatant fraction (equivalent to 2 mg of protein) were mixed with an epitope-specific antibody and protein G-Sepharose beads slurry in IP buffer and were rotated overnight at 4°C. After centrifugation, the supernatant was discarded, and the pellet was washed and resuspended in IP buffer. A western blot was performed using the other epitope-specific antibody as described in the “Western Immunoblotting” section.
Cell-Proliferation Assay
Proliferation rates in HEK293 cells expressing either wild-type or mutant plakoglobin were measured using the CellTiter 96 Cell proliferation assay (Promega) according to the manufacturer’s instructions.
Caspase-3 Assay
Cells were grown in 10-cm dishes, were harvested, and were resuspended in cell lysis buffer (10 mM HEPES [pH 7.4], 2 mM EDTA, 0.1% CHAPS, 5 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 10 mg/ml pepstatin A, 10 mg/ml aprotinin, and 20 mg/ml leupeptin). After repeated freeze-thaw cycles, lysates were centrifuged, and aliquots of the supernatant fraction equivalent to 40 μg of protein were obtained. Caspase-3 activity was measured using the CaspACE assay system (Promega) according to the manufacturer’s instructions.
Electron Microscopy of Tissue Culture Cells
Cells were grown to confluence on glass coverslips and were then fixed for 24 h in 2% glutaraldehyde in 0.1 mM cacodylate buffer (pH 7.4). Fixed cells were postfixed in 1% OsO4, were dehydrated in ethanol, and were embedded in Spurr’s low-viscosity epoxy resin. Ultrathin sections were cut in a plane parallel to the plane of the culture dish and were examined with a JEOL 100SX transmission electron microscope. The number and size of individual desmosomes were measured in 10 randomly selected electron micrographs (final magnification, 9,000×) of cells stably expressing wild-type or mutant plakoglobin.
Statistical Analysis
Data are expressed as mean ± SD and were analyzed by Student’s t test. Statistical significance was defined as P<.05.
Results
Mutation in Plakoglobin
The proband was initially screened for mutations in plakoglobin, desmoplakin, and plakophilin-2. We identified an insertion of 3 bases in plakoglobin (118_119insGCA) in one family (fig. 1). This mutation has not been reported elsewhere. It does not disrupt the frame of translation but is predicted to result in the insertion of an additional serine residue at amino acid position 39 within the N-terminal domain of plakoglobin (S39_K40insS). It also does not disrupt any restriction sites and, therefore, could not be confirmed by restriction digest. S39_K40insS was absent in 400 ethnically matched control chromosomes. Following the recent implication of two new genes (encoding desmoglein-2 and desmocollin-2) in the pathogenesis of ARVC,6–8 the proband was also investigated for mutations in these two genes and was found to be negative.
Clinical and Pathological Findings
Individuals expressing the S39_K40insS mutation showed an autosomal dominant mode of inheritance of ARVC, and the mutation cosegregated with the disease phenotype in the family (fig. 1). In 1990, the proband (II:1) experienced syncope at age 39 years. After a documented episode of sustained ventricular tachycardia, he was admitted to the hospital for further diagnostic evaluation and management. Electrocardiography demonstrated QRS prolongation, T-wave inversion, and late potentials in right precordial leads. Angiography showed moderate global right ventricular dilatation and regional wall-motion abnormalities without left ventricular involvement. Sustained monomorphic ventricular tachycardia of left bundle branch block morphology was induced at electrophysiological study. Biopsy of an endomyocardial sample showed extensive fibrofatty replacement of right ventricular muscle and patchy mononuclear inflammatory infiltrate (fig. 2). These phenotypes fulfilled criteria for the diagnosis of ARVC from the Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology.10 An implantable cardioverter-defibrillator was implanted in 1997, which has since discharged appropriately. The proband’s skin and hair appeared grossly normal on physical examination. Individuals II:2 and II:3 (fig. 1) received the diagnosis of ARVC after cardiac evaluation at their local medical facilities, on the basis of 12-lead and 24-h electrocardiography and echocardiographic abnormalities. Both subjects II:2 and II:3 have offspring who refused clinical and genetic screening and who were not included in the family pedigree. Individual I:1 died at age 70 years of heart failure and is, therefore, presumed to have been a carrier of S39_K40insS.
Figure 2. .

Microscopic appearance of the cardiac biopsy sample from the proband (by Masson’s trichrome stain).
Immunohistochemical Analysis of Intercalated Disk Proteins
To examine the effect of S39_K40insS on the distribution and expression levels of intercalated disk proteins, immunohistochemistry was performed on the proband’s cardiac biopsy sample (fig. 3). Myocardial specimens from three age-matched autopsy subjects who had no clinical history or pathological evidence of heart disease were subjected to the same staining protocol and were used as controls. The ARVC and control myocardium showed comparable levels of expression of N-cadherin (MIM *114020) and plakophilin-2, but there was a marked decrease in the amount of immunoreactive signal at intercalated discs for plakoglobin, desmoplakin, and the gap-junction protein Cx43 (MIM *121014) (fig. 3).
Figure 3. .

Confocal immunofluorescence microscopy analysis of control and proband left ventricular myocardium, showing the amount of immunoreactive signal for selected junctional proteins at intercalated disks.
Yeast-Two-Hybrid Screening
To investigate the effects of the S39_K40insS mutation on the binding properties of plakoglobin, yeast-two-hybrid screening was performed. Both wild-type and mutant plakoglobin were shown to bind previously identified binding partners, including desmoplakin and cadherins. However, the yeast-two-hybrid screen revealed an interaction between mutant plakoglobin and two clones that have not previously been associated with the protein: TAIP-2 and the sarcoplasmic HRC-BP.
Potential interactions between plakoglobin and TAIP-2 were confirmed by coimmunoprecipitation studies in transiently transfected HEK293 cells. With use of this approach, both wild-type and mutant plakoglobin were found to bind TAIP-2 (fig. 4). Potential interactions between plakoglobin and HRC-BP were verified by in vitro protein-binding assays. Although wild-type plakoglobin did not bind HRC-BP, mutant plakoglobin was able to bind all three fragments of the sarcoplasmic protein in vitro (fig. 4).
Figure 4. .

A, Coomassie-blue staining of GST-fused N-terminal (N), central (mid), and C-terminal (C) domains of HRC-BP and GST. B, In vitro protein-binding assay, which shows that wild-type plakoglobin does not bind any of the three HRC-BP fragments (lanes 1–3), whereas mutant plakoglobin binds all three fragments in vitro (lanes 4–6). C, Lysates of cell lines induced to express wild-type (lane 1) and mutant (lane 2) plakoglobin, shown for control purposes. Coimmunoprecipitation of wild-type PG and TAIP-2 (lane 3), mutant PG and TAIP-2 (lane 4), wild-type PG and V5 vector (lane 5), mutant PG and V5 vector (lane 6), His Max vector and TAIP-2 construct (lane 7), and His Max vector and V5 vector only (lane 8), showing that both wild-type and mutant plakoglobin bind TAIP-2 in vitro.
Subcellular Distribution of Plakoglobin Altered by S39_K40insS
To elucidate potential pathogenic mechanisms of S39_iK40insS, HEK293 cells that stably expressed wild-type or mutant (S39_K40insS) plakoglobin were analyzed by multiple methods. Western blotting revealed differences in electrophoretic mobility in plakoglobin expressed by the transfected cells. In cells expressing wild-type plakoglobin, one band was observed that matched the predicted size of plakoglobin (82 kDa) and corresponded to the endogenous protein expressed by nontransfected HEK293 cells (fig. 5A). In contrast, two bands were observed in cells expressing the S39_K40insS mutation: a lower band (82 kDa) corresponding to native plakoglobin and a higher band (∼90 kDa) that was the mutant plakoglobin (fig. 5A). Because cytoplasmic plakoglobin is degraded by the ubiquitin-proteasomal system,12 we investigated the possibility that the mutant protein migrated with an apparent molecular weight of 90 kDa because it was bound to ubiquitin, which has a molecular weight of 8 kDa. Western blotting with an anti-ubiquitin antibody typically reveals a smearing band pattern reflecting the abundance of ubiquitin and its numerous interactions. However, brief film exposure revealed two major bands, a lower one that matched the size of free ubiquitin (∼8 kDa) and a higher one (∼90 kDa) at the same position seen in the plakoglobin blot prepared from cells expressing the mutant protein (fig. 5B).
Figure 5. .

A, Four noninduced cell lines (lanes 2, 4, 6, and 8) that express the endogenous plakoglobin only (82 kDa), compared with four induced cell lines (lanes 1, 3, 5, and 7) that express both endogenous plakoglobin (82 kDa) and the mutant form of plakoglobin, which migrates at ∼90 kDa. Signal for endogenous plakoglobin is faint in lanes 2, 3, 5, 7, and 8. B, Bands showing that ubiquitin is present in the cell line expressing the highest levels of the exogenous construct (from lane 1 in panel A), both in its free form (8 kDa) and at a form (∼90 kDa) consistent with its being covalently bound to mutant plakoglobin.
If S39_K40insS does enhance the interaction of plakoglobin with ubiquitin, the mutant protein might be expected to show increased cytoplasmic localization. To investigate the cellular distribution of S39_K40insS plakoglobin as distinguished from that of endogenous plakoglobin, HEK293 cells were transiently transfected to express wild-type or mutant plakoglobin tagged with GFP. Transfected cells were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) to mark nuclei before examination by confocal microscopy to visualize the GFP-tagged plakoglobin. In cells expressing wild-type plakoglobin, the GFP signal was localized mainly at cell junctions. In contrast, the signal in cells expressing mutant plakoglobin was markedly shifted to the cytoplasm (fig. 6). These results support the hypothesis that mutant plakoglobin is driven away from cell junctions by its increased association with the cytoplasmic degradation machinery.
Figure 6. .

Confocal microscopy showing the distribution of wild-type and mutant plakoglobin in transiently transfected HEK293 cells. The preparations were counterstained with DAPI to show nuclei.
Effects of S39_K40insS on Cell Proliferation and Apoptosis
Apart from its role in cell junctions, plakoglobin is also known to participate in Wnt-signaling pathways, where it associates with Tcf/Lef transcription factors13 and regulates functions such as cell proliferation14 and apoptosis.15 To investigate the effects of S39_K40insS on cell proliferation, a CellTiter 96 Cell Proliferation Assay was performed. HEK293 cells stably expressing S39_K40insS plakoglobin showed higher proliferation rates than did cells expressing the wild-type protein; the difference was statistically significant on the 4th and 5th d of the assay (fig. 7A).
Figure 7. .

A, CellTiter 96 Cell Proliferation Assay over the course of 5 consecutive d. A single asterisk (*) indicates P=.014; a double asterisk (**) indicates P=.006. B, CaspACE assay. Specific caspase activity was measured in 10 individual wild-type (wt) and mutant (mut) cultures. An asterisk (*) indicates P=.038.
The effect of S39_K40insS on programmed cell death was examined using the CaspACE Assay method. HEK293 cells stably expressing S39_K40insS plakoglobin showed significantly less caspase activity than did cells expressing the wild-type protein (fig. 7B), suggesting that there are lower rates of apoptosis in cells expressing the mutant protein.
Effects of S39_K40insS on Desmosomal Ultrastructure
Reduced amounts of S39_K40insS plakoglobin at junctional sites, potentially related to increased association with cytoplasmic ubiquitin, might be expected to affect the size and shape of desmosomes. To investigate this hypothesis, HEK293 cells stably expressing wild-type or mutant plakoglobin were examined by electron microscopy. Mutant cells formed fewer (3.2±1.2 vs. 6.4±1.9 desmosomes per micrograph; P<.001) and smaller (0.87±0.42 vs. 2.68±0.83 μm per desmosome profile; P<.001) desmosomes than did wild-type cells. Representative micrographs are shown in figure 8.
Figure 8. .

Electron micrographs of HEK293 cells stably expressing wild-type or mutant plakoglobin. The arrows indicate adhesive junctions between adjacent cells.
Discussion
We have identified a novel autosomal dominant plakoglobin mutation (S39_K40insS) in a German family affected with ARVC. Recently, an uncommon polymorphism in plakoglobin (L697M) was identified to cosegregate with a nonsense mutation in desmoplakin (R1267X) in a family with early-onset ARVC.16 It has been suggested that L697M has a negative modifier effect on the cardiac phenotype in the presence of R1267X.16 In the family presented here, the L697M polymorphism was not present, and genotyping of other desmosomal candidate genes—including those encoding desmoplakin, plakophilin2, desmoglein2, and desmocollin2—did not reveal additional mutations. S39_K40insS cosegregated with the disease phenotype and was not found in 400 ethnically matched control chromosomes. It is likely, therefore, that this mutation is pathogenic rather than a modifying polymorphism.
The index patient was part of a larger cohort of ARVC-affected patients who were screened for mutations in candidate desmosomal genes. The cohort included >200 individuals of English origin, 70 individuals of German origin, and 20 individuals of Greek/Cypriot origin. The mutation presented in our study was the only plakoglobin mutation identified in the cohort. This suggests that plakoglobin mutations are uncommon in an unselected population of ARVC-affected patients. In contrast, several mutations in the genes coding for desmoplakin, plakophilin2, desmoglein2, and desmocollin2 were identified in the cohort.
None of the individuals affected by the S39_K40insS mutation showed apparent cutaneous abnormalities, in contrast to abnormalities seen in patients with Naxos disease.2 This might be explained by the dominant mode of inheritance of S39_K40insS, because, to date, only homozygous carriers of ARVC-causing recessive mutations have been documented to manifest hair and skin abnormalities.4 It is likely that the different clinical phenotypes produced by S39_K40insS and 2057del2 may result from differential effects of the mutations on the various plakoglobin-binding partners and the downstream effects on signaling, but this must be analyzed in detail in future studies.
Previous immunohistochemical studies of cardiac tissue from patients affected with Naxos disease or Carvajal syndrome, a cardiocutaneous syndrome caused by a recessive deletion mutation in desmoplakin, demonstrated markedly decreased localization of intercellular junction protein at intercalated disks.17,18 To investigate the effects of S39_K40insS on intercellular junction protein interactions, a myocardial biopsy sample from the proband was analyzed by immunohistochemistry. N-cadherin and plakophilin-2 were expressed at control levels. As a major component of adherens junctions, N-cadherin is not anticipated to be affected by S39_K40insS. Plakophilin-2 interacts with desmosomal cadherins through its N-terminal domain, but this initial interaction is not mediated by plakoglobin,19 which may explain why S39_K40insS does not affect the localization patterns of plakophilin-2 at intercalated discs either. In contrast, localization of plakoglobin, desmoplakin, and Cx43 at intercalated disks was significantly reduced in the ARVC sample. The small size of the biopsy sample and its preservation in paraffin prevented supportive immunoblotting studies. However, the fact that some but not all intercalated disc proteins were affected precludes nonspecific postmortem effects. Furthermore, a similar pattern has been observed in previous studies.17,18 Plakoglobin binds the N-terminal domain of desmoplakin and targets it to junctions,20 which may explain the altered localization pattern of desmoplakin observed in the proband tissue. Diminished accumulation of Cx43 at intercalated discs has also been observed in patients with Naxos disease17 and Carvajal syndrome.18 Indeed, it has been suggested that, when adhesive junctions are disturbed, gap junctions cannot be formed and maintained normally, which may slow electrical conduction, enhance conduction heterogeneity, and act synergistically with the characteristic structural defects in ARVC to promote fatal arrhythmias.17
Plakoglobin consists of 13 imperfect armadillo repeats flanked by N- and C-termini. The first three armadillo repeats mediate interactions with desmosomal cadherins.21 Repeats 6–8 are important for interactions between plakoglobin and classic cadherins,22 whereas part of the N-terminal domain and the first armadillo repeat (residues 109–137) control interactions with α-catenin (MIM *116805).23 The last three armadillo repeats are not known to interact with any junctional components but seem to target the protein to cell borders and stabilize desmosomal interactions.24 S39_K40insS is predicted to lead to the insertion of a serine residue at position 39 of plakoglobin. Despite the numerous bonds that plakoglobin establishes with many adhesion proteins, this part of the protein does not seem to be involved in any of the above-mentioned protein-protein interactions. However, a yeast-two-hybrid screen revealed that plakoglobin bearing the S39_K40insS mutation bound two novel partners: HRC-BP and TAIP-2. HRC-BP localizes at the sarcoplasmic reticulum of cardiac and skeletal muscle cells and is responsible for controlling Ca2+ release.25 One of the first genes known to be associated with ARVC was RYR2 (MIM *180902), the gene encoding the cardiac ryanodine receptor.25 Mutations in RYR2 are thought to permit leakage of Ca2+ into the cytoplasm and thereby trigger arrhythmias.26 It is possible that the interaction of the mutant plakoglobin with a sarcoplasmic reticulum protein may result from the altered subcellular distribution of plakoglobin and have no direct role in the pathogenesis of ARVC. Alternatively, this interaction could affect Ca2+ homeostasis and thereby promote myocyte injury and/or contribute to electrical instability. Although TAIP-2 has not been implicated previously in the pathogenesis of ARVC, there is evidence that links plakoglobin to the mechanism of apoptosis.15 In any event, these novel protein interactions must be confirmed in vivo before their potential role in disease pathogenesis can be established.
The N-terminus plays an important role in determining stability and degradation of the armadillo proteins. This region appears to be a target for N- and O-glycosylation27 and is also involved in the ubiquitin-proteasomal–degradation pathway. In the absence of Wnt signaling, plakoglobin associates with GSK-3β through the scaffolding proteins axin and anaphase promoting complex and undergoes phosphorylation at a number of serine residues on its N-terminus (residues 22–39), which targets the protein for ubiquitylation and degradation by the proteasomal system. In response to Wnt signaling, GSK-3β is prevented from phosphorylating plakoglobin, which, as a result, becomes stabilized, enters the nucleus, and participates in regulating gene expression.12 There is a fine balance among the amount of plakoglobin engaged in cell junctions, the fraction of plakoglobin involved in signaling, and the quantity of plakoglobin that is degraded. A plausible hypothesis is that S39_K40insS disrupts this balance, perhaps because the presence of an extra N-terminal serine increases protein-turnover rate. The resultant decrease in the total amount of plakoglobin may limit its accumulation at cell junctions, leading to impaired intercellular adhesion and development of ARVC. To test this hypothesis, HEK293 cell lines were transfected to express the mutant protein. Cells containing the S39_K40insS mutation expressed plakoglobin with a greater molecular weight than that of wild-type plakoglobin. This could be explained by posttranslational modifications, such as glycosylation. However, confocal microscopy showed that more of the mutant protein was localized within the cytoplasm and that less was localized at cell junctions, which could be related to enhanced ubiquitylation of mutant plakoglobin. The distribution of the mutant protein in the heart or skin of affected patients must be analyzed in future studies.
We observed that cells expressing mutant plakoglobin exhibited greater proliferation rates than did cells expressing the wild-type protein, suggesting that the S39_K40insS mutation alters plakoglobin-related signaling pathways. Charpentier et al.14 reported that plakoglobin suppresses epithelial proliferation and hair growth in vivo. It is possible, therefore, that, when normal signaling mediated by plakoglobin is disrupted, cell proliferation would be expected to increase. This mechanism might also explain the increased proliferation of keratinocytes that lead to palmoplantar keratoderma in patients with Naxos disease.
Recently, it was shown that suppression of desmoplakin expression with use of small interfering RNA in atrial myocyte cell lines or in heterozygous desmoplakin-deficient mice leads to nuclear localization of plakoglobin, reduction in canonical Wnt signaling through Tcf/Lef transcription factors, and increased myocyte apoptosis.28 Increased myocyte apoptosis has also been reported in patients with ARVC.29 In the present study, we observed that caspase activity is reduced in HEK293 cells expressing mutant plakoglobin, which is consistent with suppressed programmed cell death, but the significance of this observation regarding the pathogenesis of ARVC is unclear. It is possible that the effects of mutant plakoglobin on apoptosis differ in HEK293 cells and cardiac myocytes. In any event, insights about disease mechanisms gained from investigation of noncardiac myocytes cannot necessarily be applied to cardiac myocytes. Further studies characterizing the effects of S39_K40insS on multiple aspects of cardiac myocyte structure and function are needed.
In conclusion, this study presents, for the first time, an autosomal dominant mutation in plakoglobin that appears to be responsible for causing ARVC. Unlike the recessive deletion mutation in plakoglobin known to cause Naxos disease, S39_K40insS is not associated with cutaneous abnormalities and, therefore, may act through a different mechanism, perhaps related to altered turnover kinetics of plakoglobin. Further genetic and molecular studies should improve our understanding of the disease and perhaps provide a more accurate diagnostic algorithm and improved genetics counseling and management strategies.
Acknowledgments
We thank the patients and family members for taking part in this study. We also acknowledge Andreas Langousis for valuable help with the statistics. This work was supported by the British Heart Foundation, the European Commission 5th Framework Program (ARVC/D project QLG1-CT-2000-01091), and a grant from the March of Dimes.
Web Resource
The URL for data presented herein is as follows:
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for Naxos disease, PK2157del2, desmoplakin, plakophilin-2, desmoglein-2, desmocollin-2, HRC-BP, N-cadherin, Cx43, α-catenin, and RYR2)
References
- 1.Thiene G, Nava A, Corrado D, Rossi L, Pennelli N (1988) Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med 318:129–133 [DOI] [PubMed] [Google Scholar]
- 2.McKoy G, Protonotarios N, Crosby A, Tsatsopoulou A, Anastasakis A, Coonar A, Norman M, Baboonian C, Jeffery S, McKenna WJ (2000) Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet 355:2119–2124 10.1016/S0140-6736(00)02379-5 [DOI] [PubMed] [Google Scholar]
- 3.Rubenstein A, Merriam J, Klymkowsky MW (1997) Localizing the adhesive and signaling functions of plakoglobin. Dev Genet 20:91–102 10.1002/(SICI)1520-6408(1997)20:2<91::AID-DVG2>3.0.CO;2-3 [DOI] [PubMed] [Google Scholar]
- 4.Rampazzo A, Nava A, Malacrida S, Beffagna G, Bauce B, Rossi V, Zimbello R, Simionati B, Basso C, Thiene G, et al (2002) Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet 71:1200–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gerull B, Heuser A, Wichter T, Paul M, Basson CT, McDermott DA, Lerman BB, Markowitz SM, Ellinor PT, MacRae CA, et al (2004) Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat Genet 36:1162–1164 10.1038/ng1461 [DOI] [PubMed] [Google Scholar]
- 6.Pilichou K, Nava A, Basso C, Beffagna G, Bauce B, Lorenzon A, Frigo G, Vettori A, Valente M, Towbin J, et al (2006) Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation 113:1171–1179 10.1161/CIRCULATIONAHA.105.583674 [DOI] [PubMed] [Google Scholar]
- 7.Syrris P, Ward D, Evans A, Asimaki A, Gandjbakhch E, Sen-Chowdhry S, McKenna WJ (2006) Arrhythmogenic right ventricular dysplasia/cardiomyopathy associated with mutations in the desmosomal gene desmocollin-2. Am J Hum Genet 79:978–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heuser A, Plovie ER, Ellinor PT, Grossmann KS, Shin JT, Wichter T, Basson CT, Lerman BB, Sasse-Klaassen S, Thierfelder L, et al (2006) Mutant desmocollin-2 causes arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet 79:1081–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Green KJ, Gaudry CA (2000) Are desmosomes more than tethers for intermediate filaments? Nat Rev Mol Cell Biol 1:208–216 10.1038/35043032 [DOI] [PubMed] [Google Scholar]
- 10.McKenna WJ, Thiene G, Nava A, Fontaliran F, Blomstrom-Lundqvist C, Fontaine G, Camerini F (1994) Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy: Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology. Br Heart J 71:215–218 10.1136/hrt.71.3.215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saffitz JE, Green KG, Kraft WJ, Schechtman KB, Yamada KA (2000) Effects of diminished expression of connexin43 on gap junction number and size in ventricular myocardium. Am J Physiol Heart Circ Physiol 278:H1662–H1670 [DOI] [PubMed] [Google Scholar]
- 12.Sadot E, Simcha I, Iwai K, Ciechanover A, Geiger B, Ben-Ze’ev A (2000) Differential interaction of plakoglobin and β-catenin with the ubiquitin-proteasome system. Oncogene 19:1992–2000 10.1038/sj.onc.1203519 [DOI] [PubMed] [Google Scholar]
- 13.Zhurinsky J, Shtutman M, Ben-Ze’ev A (2000) Plakoglobin and β-catenin: protein interactions, regulation and biological roles. J Cell Sci 113:3127–3139 [DOI] [PubMed] [Google Scholar]
- 14.Charpentier E, Lavker RM, Acquista E, Cowin P (2000) Plakoglobin suppresses epithelial proliferation and hair growth in vivo. J Cell Biol 149:503–520 10.1083/jcb.149.2.503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hakimelahi S, Parker HR, Gilchrist AJ, Barry M, Li Z, Bleackley RC, Pasdar M (2000) Plakoglobin regulates the expression of the anti-apoptotic protein BCL-2. J Biol Chem 275:10905–10911 10.1074/jbc.275.15.10905 [DOI] [PubMed] [Google Scholar]
- 16.Uzumcu A, Norgett EE, Dindar A, Uyguner O, Nisli K, Kayserili H, Sahin SE, Dupont E, Severs NJ, Leigh IM, et al (2006) Loss of desmoplakin isoform I causes early onset cardiomyopathy and heart failure in a Naxos-like syndrome. J Med Genet 43:e5 10.1136/jmg.2005.032904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaplan SR, Gard JJ, Protonotarios N, Tsatsopoulou A, Spiliopoulou C, Anastasakis A, Squarcioni CP, McKenna WJ, Thiene G, Basso C, et al (2004) Remodeling of myocyte gap junctions in arrhythmogenic right ventricular cardiomyopathy due to a deletion in plakoglobin (Naxos disease). Heart Rhythm 1:3–11 10.1016/j.hrthm.2004.01.001 [DOI] [PubMed] [Google Scholar]
- 18.Kaplan SR, Gard JJ, Carvajal-Huerta L, Ruiz-Cabezas JC, Thiene G, Saffitz JE (2004) Structural and molecular pathology of the heart in Carvajal syndrome. Cardiovasc Pathol 13:26–32 10.1016/S1054-8807(03)00107-8 [DOI] [PubMed] [Google Scholar]
- 19.Chen X, Bonne S, Hatzfeld M, van Roy F, Green KJ (2002) Protein binding and functional characterization of plakophilin 2: evidence for its diverse roles in desmosomes and β-catenin signaling. J Biol Chem 277:10512–10522 10.1074/jbc.M108765200 [DOI] [PubMed] [Google Scholar]
- 20.Kowalczyk AP, Bornslaeger EA, Borgwardt JE, Palka HL, Dhaliwal AS, Corcoran CM, Denning MF, Green KJ (1997) The amino-terminal domain of desmoplakin binds to plakoglobin and clusters desmosomal cadherin-plakoglobin complexes. J Cell Biol 139:773–784 10.1083/jcb.139.3.773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Troyanovsky RB, Chitaev NA, Troyanovsky SM (1996) Cadherin binding sites of plakoglobin: localization, specificity and role in targeting to adhering junctions. J Cell Sci 109:3069–3078 [DOI] [PubMed] [Google Scholar]
- 22.Sacco PA, McGranahan TM, Wheelock MJ, Johnson KR (1995) Identification of plakoglobin domains required for association with N-cadherin and α-catenin. J Biol Chem 270:20201–20206 10.1074/jbc.270.34.20201 [DOI] [PubMed] [Google Scholar]
- 23.Witcher LL, Collins R, Puttagunta S, Mechanic SE, Munson M, Gumbiner B, Cowin P (1996) Desmosomal cadherin binding domains of plakoglobin. J Biol Chem 271:10904–10909 10.1074/jbc.271.18.10904 [DOI] [PubMed] [Google Scholar]
- 24.Chitaev NA, Leube RE, Troyanovsky RB, Eshkind LG, Franke WW, Troyanovsky SM (1996) The binding of plakoglobin to desmosomal cadherins: patterns of binding sites and topogenic potential. J Cell Biol 133:359–369 10.1083/jcb.133.2.359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim E, Shin DW, Hong CS, Jeong D, Kim DH, Park WJ (2003) Increased Ca2+ storage capacity in the sarcoplasmic reticulum by overexpression of HRC (histidine-rich Ca2+ binding protein). Biochem Biophys Res Commun 300:192–196 10.1016/S0006-291X(02)02829-2 [DOI] [PubMed] [Google Scholar]
- 26.Tiso N, Stephan DA, Nava A, Bagattin A, Devaney JM, Stanchi F, Larderet G, Brahmbhatt B, Brown K, Bauce B, et al (2001) Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Hum Mol Genet 10:189–194 10.1093/hmg/10.3.189 [DOI] [PubMed] [Google Scholar]
- 27.Hatsell S, Medina L, Merola J, Haltiwanger R, Cowin P (2003) Plakoglobin is O-glycosylated close to the N-terminal destruction box. J Biol Chem 278:37745–37752 10.1074/jbc.M301346200 [DOI] [PubMed] [Google Scholar]
- 28.Garcia-Gras E, Lombardi R, Giocondo MJ, Willerson JT, Schneider MD, Khoury DS, Marian AJ (2006) Suppression of canonical Wnt/β-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J Clin Invest 116:2012–2021 10.1172/JCI27751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamaji K, Fujimoto S, Ikeda Y, Masuda K, Nakamura S, Saito Y, Yutani C (2005) Apoptotic myocardial cell death in the setting of arrhythmogenic right ventricular cardiomyopathy. Acta Cardiol 60:465–470 10.2143/AC.60.5.2004965 [DOI] [PubMed] [Google Scholar]