

Abstract
Whole-genome association studies present many new statistical and computational challenges due to the large quantity of data obtained. One of these challenges is haplotype inference; methods for haplotype inference designed for small data sets from candidate-gene studies do not scale well to the large number of individuals genotyped in whole-genome association studies. We present a new method and software for inference of haplotype phase and missing data that can accurately phase data from whole-genome association studies, and we present the first comparison of haplotype-inference methods for real and simulated data sets with thousands of genotyped individuals. We find that our method outperforms existing methods in terms of both speed and accuracy for large data sets with thousands of individuals and densely spaced genetic markers, and we use our method to phase a real data set of 3,002 individuals genotyped for 490,032 markers in 3.1 days of computing time, with 99% of masked alleles imputed correctly. Our method is implemented in the Beagle software package, which is freely available.
Multilocus analysis can provide improved power to detect associations between complex traits and densely spaced genetic markers, compared with that of single-marker methods.1 Most methods for multilocus analysis that are suitable for whole-genome association data require phased haplotypes because methods that allow for uncertainty in haplotype phase typically use small sliding windows of markers, which cannot make full use of the correlation structure in the data. Inference of haplotype phase for whole-genome association data can be performed with a high degree of accuracy, as we demonstrate, but is computationally challenging and requires methods that scale well to thousands of individuals, as well as to hundreds of thousands or millions of genetic markers. It has been common to compare haplotype-inference methods with HapMap data2,3; however, because each of the ethnicities in the HapMap study has at most 60 unrelated individuals, results from such comparisons are not necessarily good predictors of haplotype-phasing performance for data sets with thousands of individuals.
We propose a new haplotype-inference method and show that our method outperforms existing methods in terms of both computational speed and measures of accuracy for large whole-genome data sets with thousands of individuals and hundreds of thousands of or even a million genetic markers. The method is one or two orders of magnitude faster than the most accurate competing methods, enabling accurate haplotype phasing of data from whole-genome association studies in a few days of computing time, instead of months or years. An efficient software implementation of our method is freely available in version 2.1 of the Beagle genetic analysis software package,1 which is written in Java and includes software for haplotype and missing-data inference, single-marker and multilocus association analysis, and permutation testing.
Early methods for haplotype inference were based on a multinomial model for haplotype frequencies that used no prior information about the haplotype frequency distribution.4–6 Such methods include those commonly referred to as “expectation-maximization (EM) methods,” reflecting the algorithm that is used to maximize the likelihood. These methods should be referred to as “multinomial model methods,” because EM algorithms are also used in methods employing more-complex statistical models, such as fastPHASE7 and HaploRec.8 The multinomial model methods work fairly well on a small handful of markers but break down with larger numbers of markers. When the number of markers increases, so does the number of observable haplotypes, and the frequencies of these haplotypes become too small to estimate directly. Also, the computational time quickly becomes intractable, since all feasible haplotypes must be considered. Extensions such as partition-ligation (PL)–EM9 have extended the usefulness of the multinomial model to a somewhat larger number of markers, but multinomial methods are still less accurate than are other types of models.8
PHASE10 and later related methods applied a coalescent model to haplotype frequencies. This model implies that haplotypes similar to ones we have already seen are more likely to be seen than are completely different haplotypes, since changes to haplotypes occur through recombination and mutation. Although it produces very accurate results, application of the coalescent model is computationally intensive, limiting its applicability for large data sets.
A variety of other approaches has been proposed, some of which make use of haplotype blocks.11 Although the concept of haplotype blocks is useful, haplotype blocks do not adequately explain all the correlation structure between markers, because linkage disequilibrium (LD) can extend beyond block boundaries and can have complex patterns within blocks.2
Another haplotype-phasing method is needed because all existing methods for haplotype inference either are too slow for routine application to whole-genome association studies or have severely suboptimal accuracy. We address these two issues with the method that we propose here. Our method is a novel application of the recently proposed localized haplotype-cluster model that has been used for association testing.1,12 The localized haplotype-cluster model is an empirical LD model that adapts to the local structure in the data. Relative to other methods, it does particularly well with large sample sizes, where the data are, in a sense, allowed to speak for themselves. The model can be fit to haplotypes by use of an algorithm that is very fast, and we apply an iterative approach to haplotype phasing in which an initial guess of haplotype phase is made, the model is fit, improved estimates of haplotype phase are obtained, the model is refit, and so forth. This is essentially an EM approach, as are most other non-Bayesian haplotype-inference methods. (Bayesian methods also employ iteration, by means of Markov chain–Monte Carlo techniques.)
We compared the speed and accuracy of our proposed method with those of a selection of alternative haplotype-inference methods. We excluded any method that is excessively slow or whose current implementation does not allow it to be applied to at least a few hundred densely spaced genetic markers. We also excluded methods that showed significantly lower accuracy than that of competing methods in the only previous study that considered large numbers of individuals.8 In particular, we excluded Gerbil13 (version 1.0) and PL-EM9 (version 1.5), which had low accuracy relative to that of HaploRec.8 The methods we chose for comparison, on the basis of these criteria, were fastPHASE (version 1.2.3), HaploRec (version 2.1), HAP (version 2.9), and 2SNP (version 1.5.1, for 64-bit architecture). All methods were used with the default settings except as noted.
fastPHASE7 uses a haplotype-clustering model with a fixed number of clusters. For data sets with small numbers of individuals, such as the HapMap data, fastPHASE is almost as accurate as PHASE but is much faster.7 Although an earlier study found that fastPHASE performed poorly compared with HaploRec for large data sets,8 we included it because we knew that we could decrease the running time by turning off the default cross-validation procedure (as recommended for data sets with only several hundred markers) and because we felt that the default choices of 5, 10, and 15 for the number of clusters, K, used in the previously published comparison of fastPHASE version 1.1.3 and HaploRec were too low for accurate phasing of large numbers of individuals. Because the default numbers of clusters that are considered by fastPHASE version 1.2.3 in the cross-validation procedure (which was omitted to reduce running times) are K=10 and K=20, we ran fastPHASE with each of these values.
HaploRec8 uses frequencies of haplotype fragments in a segmentation model (HaploRec-S) or in a variable-order Markov-chain model (HaploRec-VMM). HaploRec was run with increased memory allocation (as was necessary for its operation) and with both the segmentation model and the variable-order Markov model. The segmentation model is generally slower but more accurate than is the variable-order Markov model.8 One limitation of HaploRec is that it does not currently have the capability to impute missing data, which limits its usefulness for genetic association studies.
HAP11 uses haplotype blocks with imperfect phylogeny constraints that limit inference of haplotypes that would imply back-mutations or recombinations within blocks. 2SNP14 uses a very fast algorithm based on consideration of two-marker haplotypes.
Methods
We first describe the localized haplotype-cluster model, which is a special class of directed acyclic graph. We show how the localized haplotype-cluster model defines a hidden Markov model (HMM) that can be used to sample haplotype pairs or to find the most likely haplotype pair for each individual conditional on the individual’s genotypes. We then describe the phasing algorithm, which involves iteratively sampling haplotype pairs and building the localized haplotype-cluster model from the sampled haplotype pairs. We conclude with a description of the real and simulated data sets and the metrics we used to compare haplotype-inference algorithms.
The Localized Haplotype-Cluster Model
Correlation between markers is a localized phenomenon, since LD decays with distance. If this localization is not considered when genotype data are phased over an extended region, noise will be introduced by sampling variation, resulting in apparent correlations observed between distant markers, which reduces the accuracy of the haplotype inference. Existing approaches to this problem include explicitly modeling the recombination in the coalescent model,15 using haplotype blocks,11,13 taking a sliding-window approach with window size varying with LD structure,16 employing an HMM,7 and using frequent haplotype fragments.8 Our approach to making use of the localized LD structure is a localized haplotype-cluster model,1,12 which empirically models haplotype frequencies on a local scale.
The localized haplotype-cluster model clusters haplotypes at each marker to improve prediction of alleles at markers t+1, t+2, t+3,…, given alleles at markers t, t-1, t-2,… on a haplotype. This is achieved by defining clusters according to a Markov property—given cluster membership at position t, the sequence of alleles at markers t, t-1, t-2,… is irrelevant for predicting the sequence of alleles at markers t+1, t+2, t+3,…. The clustering is localized, so that haplotypes in the same cluster at position t are likely to be in the same cluster at position t+1 but need not be. This model has a number of important advantages. By clustering the haplotypes, a parsimonious model is obtained, which is important for obtaining good estimates of haplotype frequencies. By allowing the number of clusters and the relationships between clusters at different positions to be determined largely by the data rather than by a restrictive model, the model adapts to the data, which is particularly useful when the number of individuals in the sample is large, as it will be for well-powered association studies. Finally, the model can be fit using a computationally efficient algorithm1,12,17 that is extremely fast.
Suppose that we have a sample of haplotypes for M markers and that the haplotypes have no missing alleles. A localized haplotype-cluster model for this sample is a directed acyclic graph with the following four properties:
-
1.
The graph has one root (initial) node with no incoming edges and has one terminal node with no outgoing edges. The root node represents all haplotypes before any markers are processed, whereas the terminal node represents all haplotypes after all markers are processed.
-
2.
The graph is leveled with M+1 levels. Each node A has a level, m. All incoming edges to A have the parent (originating) node at level m-1, and all outgoing edges from A have the child (destination) node at level m+1. The root node has level 0, and the terminal node has level M.
-
3.
For each m=1,2,…,M, each edge with the child node at level m is labeled with an allele for the mth marker. Two edges originating from the same parent node cannot be labeled with the same allele.
-
4.
For each haplotype in the sample, there is a path from the root node to the terminal node, such that the mth allele of the haplotype is the label of the mth edge of the path. Each edge of the graph has at least one haplotype in the sample whose path traverses the edge.
Each edge, e, of the graph represents a cluster of haplotypes consisting of all haplotypes whose path from the initial node to the terminal node of the graph traverses e. Haplotypes are defined over the whole chromosome, but haplotypes within a cluster corresponding to an edge at level m will tend to have similar patterns of alleles at markers immediately to the right of marker m. Thus, each edge defines a localized haplotype cluster that is determined by local LD patterns.
For each edge, e, of a localized haplotype-cluster model, we define the edge count, n(e), to be the number of haplotypes in the sample whose path traverses the edge, and we define the parent node count, np(e), to be the number of haplotypes in the sample whose path traverses the parent node of the edge.
Table 1 and figure 1 illustrate these concepts. The data in table 1 do not correspond to any real data set but are merely for illustration. The bold-line edges from the root node to the terminal node in figure 1 represent the haplotype 2112. Edge eF includes haplotypes 1111, 1112, 2111, and 2112, with total count n(eF)=21+79+25+112=237 (counts taken from table 1). The node marked by an asterisk (*) is the parent node for edge eF. This node includes the four haplotypes whose paths traverse edge eF and, additionally, haplotypes 1122 and 2122, whose paths traverse edge eG, and it has count
Table 1. .
Example Haplotype Counts for Figure 1
| Haplotype | Count |
| 1111 | 21 |
| 1112 | 79 |
| 1122 | 95 |
| 1221 | 116 |
| 2111 | 25 |
| 2112 | 112 |
| 2122 | 152 |
Figure 1. .

Example of a directed acyclic graph representing the localized haplotype-cluster model for four markers, with the haplotype counts given in table 1. For each marker, allele 1 is represented by a solid line, and allele 2 by a dashed line. The bold-line edges from the root node to the terminal node represent the haplotype 2112. The node marked by an asterisk (*) is the parent node for edge eF.
Localized haplotype-cluster models are especially well suited for modeling LD structure. Recombination between haplotypes is modeled as merging edges (i.e., edges with the same child node).12 Unlike haplotype-block–based models, which permit recombination only between haplotype blocks, localized haplotype-cluster models can model the complex recombination patterns found in real data. Unlike models with fixed numbers of clusters at each locus, the localized haplotype-cluster model is flexible and can vary the number of clusters at each locus to model the data.
A computationally efficient algorithm for fitting a localized haplotype model to haplotype data has been described in detail elsewhere.1,12 The algorithm scales linearly in the number of markers and slightly more than linearly (less than quadratically) in the number of individuals,1 so that it can quickly process large-scale data sets. Since the model-fitting algorithm is a published algorithm, we do not repeat the details of the model-fitting algorithm in this work but refer the reader to our earlier work,12 which contains a detailed worked example of fitting a localized haplotype-cluster model to the haplotype data in table 1.
We show below that localized haplotype-cluster models can be interpreted as a special class of HMMs. By viewing localized haplotype-cluster models as HMMs, we are able to extend the model to diplotypes and to use efficient HMM sampling algorithms. In “The Beagle Phasing Algorithm” section, we describe an iterative process for haplotype phasing that involves fitting a localized haplotype model to estimated haplotype data and sampling haplotype estimates conditional on the fitted localized haplotype model and the genotype data.
The Induced HMM
A localized haplotype-cluster model determines an HMM for which the states of the HMM are the edges of the localized haplotype-cluster model, and the emitted symbol for each state is the allele that labels the edge of the localized haplotype-cluster model. To specify the HMM, we must specify the emission probabilities, the initial-state probabilities, and the transmission probabilities.18 Each state (i.e., edge) emits with probability 1 the allele that labels the edge. Thus, the state uniquely determines the observed allele, but the observed allele for a marker does not generally determine the state, because edges with distinct parent nodes at the same level of the graph can be labeled with the same allele.
The initial-state probabilities and the transition probabilities are computed from the edge counts. The initial-state probabilities are P(e)=n(e)/np(e) if the parent node of edge e is the root node and are P(e)=0 otherwise. The transition probabilities are P(e1|e2)=n(e1)/np(e1) if the parent node of edge e1 is the child node of edge e2 and are P(e1|e2)=0 otherwise. Note that, if edges e2 and e3 both have the same child node, then P(e1|e3)=P(e1|e2). For example, by considering the haplotype counts in table 1 and the corresponding graph in figure 1, P(eF|eC)=P(eF|eE)=n(eF)/np(eF)=237/484=0.49, whereas P(eC|eE)=0 because the parent node of eC is not the child node of eE.
We have described a haploid HMM; however, a diploid HMM is needed because we observe diploid data (i.e., genotypes) rather than haploid data. We create a diploid HMM from ordered pairs of edges in each level of the graph. Since the graph is leveled, the states of the haploid HMM (the edges of the graph) can be partitioned into classes Lm, where m=1,2,…,M is the level of the edge’s child node in the graph. For example, in figure 1, L1={eA,eB}, L2={eC,eD,eE}, L3={eF,eG,eH}, and L4={eI,eJ,eK,eL}.
For the diploid HMM, the state space is the union over m of (Lm×Lm), and the emitted symbol for each state is the unordered pair of alleles that label the state’s ordered pair of edges. If (e1,e2) is a state of the diploid HMM, then the unordered genotype determined by the two alleles that label edges e1 and e2 is emitted with probability 1. We assume Hardy-Weinberg equilibrium, so that the diploid initial and transition probabilities are the product of the corresponding haploid probabilities: P(e1,e2)=P(e1)P(e2) and P[(e1,e2)|(e3,e4)]=P(e1|e3)P(e2|e4). Note that the edge pairs are ordered, so that factors of 2 are not needed when the ordered edge pairs represent heterozygote genotypes.
Sampling from an HMM
Our phasing algorithm samples from the diploid HMM conditional on the observed data by use of a forwards-backwards algorithm (see the work of Rabiner18 for a review of the forwards-backwards and Viterbi algorithms for HMMs, and see section 7.1 in the work of Thompson19 for an example of conditional HMM sampling). For a given individual, let gm be the observed unordered genotype at marker m, and let the state sm=(e1,e2) be an ordered pair of edges in Lm×Lm. For any sm in the diploid HMM and with the individual’s genotype {g1,g2,…,gM}, define the forward variables as αm(sm)=P(g1,g2,…,gm,sm). The αm(sm) for m=1,2,…,M can be computed inductively by the forward algorithm as follows.
-
1.
Initiation: α1(s1)=P(g1,s1)=P(s1)P(g1|s1).
-
2. Induction:

For our purposes, the αm need be known only up to a constant of proportionality for each m. Appendix A gives a worked example of forward calculation.
The probabilities P(gm+1|sm+1) are always either zero or one, depending on whether the genotype is consistent with the labels on the ordered pair of edges. Our model could be extended to incorporate genotype error by allowing these probabilities to take intermediate values; however, for low error rates, we expect that genotype-error modeling will not add significant value. Our results (see the “Results” section) show that our method outperforms existing methods for real data, despite the undoubted presence of some genotype errors.
In our application, one or both alleles in a genotype may be missing. If both alleles of gm are missing, then P(gm|sm)=1 in the forward algorithm and the sampling algorithm. If one allele of gm is missing, then P(gm|sm)=1 if the nonmissing allele labels one of the ordered edges of sm, and P(gm|sm)=0 otherwise.
Sampling of hidden states conditional on the individual’s genotype proceeds backward by induction as follows.
-
1.
Initiation: randomly choose the state sM with probability proportional to αM(sM).
-
2. Induction: given states sm+1, sm+2,…,sM, choose state sm with probability

The sampled path of hidden states corresponds to an ordered pair of haplotypes that are consistent with the individual’s genotype. A worked example of sampling the hidden state (and thus the haplotype pair) conditional on genotype data is given in appendix A.
We have given a sampling algorithm for the diploid HMM. It is also possible to determine the most likely ordered pair of haplotypes conditional on the genotype data and the diploid model by use of the Viterbi algorithm.18
The Beagle Phasing Algorithm
The Beagle phasing algorithm is conceptually simple: at each iteration of the algorithm, phased input data are used to build a localized haplotype-cluster model as described elsewhere.1,12 After the localized haplotype-cluster model is built, phased haplotypes for each individual are sampled from the induced diploid HMM conditional on the individual’s genotypes. The sampled haplotypes are the input for the next iteration. In the final iteration, instead of sampling haplotypes, we use the Viterbi algorithm to select the most-likely haplotypes for each individual, conditional on the diploid HMM and the individual’s genotype data, and these most-likely haplotypes are the output of the phasing algorithm. We have found that, when starting from randomly phased data, 10 iterations gives good accuracy and that using >10 iterations yields very little improvement in accuracy.
We have made two enhancements to this algorithm that increase accuracy. First, at each iteration of the phasing algorithm, we reverse the marker order, processing the chromosome from left to right in the odd-numbered iterations and from right to left in the even-numbered iterations. Second, we sample multiple haplotype pairs per individual for use in building the model at the next iteration, taking into account the correlation between haplotype pairs from the same individual; this yields significant improvements in accuracy when small numbers of individuals are phased.
Let R⩾1 be the number of samples per individual. In the initialization step, we copy the genotype data for each individual R times, so that, if there are N distinct individuals, we have NR individuals after copying. For each copy of each individual, missing alleles are randomly imputed according to allele frequencies, and the data for each individual are phased by randomly ordering the genotypes. The randomly phased data are the input for the first iteration of the phasing algorithm.
The model-building algorithm uses scale and shift parameters that control the complexity of the model.1 When building the localized haplotype-cluster model, we use a scale parameter of 1.0 if there are R=1 samples per individual. If there are more than one sample per individual (R>1), the haplotypes are not independent, and, to account for the decreased effective sample size, we increase the scale parameter. If the haplotypes were determined without error, each haplotype pair from the same individual would be identical, and a scale factor of 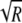 would be appropriate. However, since the haplotype pairs are not perfectly correlated, we gain accuracy by using a scale factor of
would be appropriate. However, since the haplotype pairs are not perfectly correlated, we gain accuracy by using a scale factor of 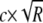 , where c is a positive constant <1. We use c=0.75 because, in real and simulated data sets, we found that this value works well for a variety of sample sizes and marker densities (authors' unpublished data), although the optimal choice of c will differ slightly from one data set to another. The shift parameter is always set to 0.0 and does not depend on the number of sampled haplotypes, R, for each individual.
, where c is a positive constant <1. We use c=0.75 because, in real and simulated data sets, we found that this value works well for a variety of sample sizes and marker densities (authors' unpublished data), although the optimal choice of c will differ slightly from one data set to another. The shift parameter is always set to 0.0 and does not depend on the number of sampled haplotypes, R, for each individual.
After the localized haplotype-cluster model is built, R phased haplotype pairs are sampled for each individual, conditional on the genotypes for the individual and the diploid HMM model. The NR sampled haplotype pairs are used as input in the next iteration. The output phased haplotype pair for each individual is the most likely haplotype pair conditional on the individual’s genotype and the diploid HMM in the last iteration of the phasing algorithm.
Our haplotype-phasing software, Beagle, will sample R=4 haplotype pairs per individual with the default settings. Increasing the number of sampled haplotype pairs per individual improves the accuracy but also increases the computing time. The results of our experiments below show that, for data sets with small numbers of individuals when the computing time is not an issue, it is worthwhile to increase the number of samples (e.g., to R=25, as we do in this study), whereas, for very large data sets with thousands of individuals, the use of one sample per individual (R=1) results in significant savings in computational time with an insignificant loss of accuracy.
The computational time required by our algorithm scales linearly with the number of markers for a given marker density. We must estimate the scaling of our algorithm with respect to sample size empirically because the running time depends on the rate at which the number of nodes and edges at each level grows as the sample size increases.
The algorithm that we have described is similar to a stochastic EM algorithm20,21; however, our model does not include a likelihood. In place of maximizing a likelihood, we fit a localized haplotype-cluster model. An advantage of using a stochastic EM–type algorithm is that such algorithms are less likely to get stuck in local maxima than are regular EM algorithms. Sampling multiple haplotype pairs per individual, as described above, also helps to stop the algorithm from getting stuck in local maxima, particularly for data sets with small numbers of individuals. The localized haplotype-cluster model also helps because “merging edges” in the model cause the model to assign nonzero probability to many haplotypes not seen in the input data.
Simulated Data
We compared the accuracy of our new method with that of other phasing algorithms, using realistic simulated data generated by Cosi,22 with parameters calibrated to empirical human data. Three sample sizes were simulated: small (200 haplotypes), medium (2,000 haplotypes), and large (10,000 haplotypes). For each sample size, multiple data sets with 1 Mb of marker data were simulated using the “best-fit” parameters obtained from fitting a coalescent model to real data.22 Samples were taken from a “European” population, and each simulated data set has a recombination rate sampled from a distribution matching the deCODE map,22,23 with recombination clustered into hotspots.
For each simulated data set, we selected a set of tagging markers, using a greedy pairwise selection algorithm.24 The parameters for the marker-selection algorithm were set to produce either a low-density set with ∼100 SNPs (1 SNP per 10 kb) or a high-density set with ∼333 SNPs (1 SNP per 3 kb). First, a screening set of markers was randomly selected from among those markers with minor-allele frequency >0.05. For the low density, the screening set contained 1 SNP per 4 kb (∼250 markers), and, for the high density, the screening set contained 1 SNP per 0.7 kb (∼1,428 markers). The tag SNP–selection algorithm was applied to 120 randomly selected haplotypes, to identify a set of genotyped markers such that every marker in the screening set with sample minor-allele frequency >0.05 (on the basis of the 120 haplotypes) either was a genotyped marker or had pairwise squared correlation coefficient r2>0.7 (low density) or r2>0.9 (high density) with at least one genotyped marker. The median number of markers selected for the low-density tagging set was 108, with a range of 42–179 markers, and the median number of markers selected for the high-density tagging set was 344, with a range of 51–688 markers.
For the small and medium sample sizes, 100 low-density and 100 high-density data sets were generated. For the large sample sizes, 40 low-density and 10 high-density data sets were generated.
Real Data
We applied our method to data from the Wellcome Trust Case Control Consortium (WTCCC) control group,25 which consists of 1,502 individuals from the 1958 British Birth Cohort and 1,500 individuals from the U.K. Blood Service Control Group (WTCCC Web site). These individuals were genotyped on the Affymetrix GeneChip Human Mapping 500K Array Set.26 We applied our method to the complete set of autosomes (490,032 SNPs) and also analyzed subsets of the data with our method and with the other haplotyping methods considered in the simulation study. The genotypes were called using the Bayesian robust linear model with Mahalanobis distance classifier (BRLMM) algorithm (an extension of RLMM).27 Genotypes with a BRLMM score <0.50 were set to missing, which resulted in 0.8% missing genotypes for the autosomal chromsosomes.
Three sample sizes of real autosomal data were considered: small (100 individuals), medium (1,000 individuals), and large (3,002 individuals). For each sample size and for each chromosome, sets of 200 markers were selected by generating a random positive integer, n⩽4,300, and by taking sets of 200 consecutive markers on that chromosome, beginning at marker (n+4,500×m) for m=0,1,2,…. This resulted in ∼100 sets of 200 markers for each sample size. The marker sets for each of the sample sizes were generated independently.
We used the male X-chromosome data (10,536 SNPs) to create artificial pairs of X chromosomes for which the phasing is known. Male genotypes appear homozygous, except where genotype errors have occurred. Rather than use the reported sex, we used the proportion of heterozygous X-chromosome markers to determine which individuals are male. The individuals clearly clustered into two groups on this basis: 1,535 females had at least 2,115 of 10,536 markers that were heterozygous, whereas 1,465 males had up to 95 markers (0.9%) that were heterozygous. Two individuals had 537 and 1,141 markers that were heterozygous; these are presumably males with high genotype error rates and were removed from all subsequent analyses. To have an even number of male chromosomes to create pairs, the chromosome with 95 heterozygous markers (i.e., the chromosome with the highest apparent error rate for the remaining male X chromosomes) was removed. Thus, 732 pairs of X chromosomes were created. Before the male chromosomes were paired, heterozygous genotypes were turned into missing data, and 193 markers with >10% missing data (including the previously heterozygous markers) in males were removed from all subsequent analyses, leaving 10,343 SNPs. After pairing, when one allele of an artificial genotype was missing at a marker, the second allele was also set to missing, to mimic typical diploid data in which it is always whole genotypes, rather than single alleles, that are missing. The final paired data had 0.4% missing data. For comparison of phasing algorithms, we created 50 sets of 200 nonoverlapping consecutive markers. These were obtained by taking markers 201–400, 401–600,…, and 10,001–10,200.
Measurement of Phasing Accuracy
We consider two measures of accuracy for haplotype inference. Because the regions considered are large, it is unlikely that any algorithm will be able to infer the haplotypes perfectly; however, the inferred haplotypes should be as correct as possible. For most applications, it is most important that the haplotypes are locally correct and is less important for the haplotypes to be entirely correct.
The switch error rate28,29 measures the proportion of successive pairs of heterozygote markers in an individual that are phased incorrectly with respect to each other. For example, suppose the actual haplotypes over seven markers for one individual are ACATGCA and TCACCCT, and the inferred haplotypes are ACATCCT and TCACGCA. The first, fourth, fifth, and seventh markers are heterozygous for this individual. The first and fourth markers are correctly phased with respect to each other in the inferred haplotypes. The fourth and fifth markers are incorrectly phased (a switch error), and the fifth and seventh markers are correctly phased. Thus, for this individual for these markers, the switch error rate is 1/3. To determine the switch error rate, the true haplotypes must be known, which is possible for simulated data but not for real autosomal genotype data from individuals with no genotyped relatives. As the marker density increases, the number of heterozygote markers will increase, and the switch error rate will tend to drop, even if the haplotype phasing does not improve. For that reason, the average number of observed switches per individual is more useful than is the switch error rate as an absolute measure of phasing accuracy. However, for the purposes of comparing methods, the switch error rate is adequate.
The allelic-imputation error rate is the proportion of missing alleles incorrectly imputed and measures the ability to infer missing-genotype data as part of the haplotype-inference procedure. We can determine this error rate from real (or simulated) data by randomly selecting and masking a small percentage of the genotypes and determining the proportion of alleles that are correctly inferred. For the results presented here, we masked 1% of the genotypes. The allelic-imputation error rate is available only for methods that infer missing data, which includes all the methods studied except HaploRec. We note that, although imputation error rate is of interest in its own right, it is also a measure of phasing accuracy, since good haplotype-phase estimates will lead to high rates of imputation accuracy.
Comparison of Running Times
All timing results (unless otherwise noted) were obtained from a Linux server with eight Dual-Core AMD Opteron 8220 SE processors (running at 2.8 GHz, with 1 MB cache, and using a 64-bit architecture) and a total of 32 GB RAM. Times were obtained by adding the user and system times from the Linux “time” command. These times are sums over all processes; thus, multithreaded programs would not receive a reported time advantage from the multiple dual cores on this system (i.e., times are those that would be seen on an equivalent single processor with a single core).
One data set was selected from each class of data (defined by marker density and sample size) for the timing study. In each case, the masked version of the data (with 1% of genotypes set to missing) was used. This makes the timing results more applicable to real data, which generally include some missing data. For the simulated data, a data set was chosen that had approximately the median number of markers for that class of data set (to reflect an “average” data set). For the WTCCC autosomal data sets, the 50th data set (in chromosomal order) was chosen, whereas, for the WTCCC X-chromosome–paired male haplotype data, the 25th data set was used. Thus, timing results are only indicative, but consistent patterns over the various classes of data size allow us to draw conclusions about the relative time performance of the methods.
Results
Tables 2 and 3 give allelic-imputation and switch error rates for each haplotype-inference method, and figure 2 gives the error rates with error bars (mean ±2 SEs) for the best-performing methods. The error bars are larger for the estimated average absolute error than for the estimated average difference in error (i.e., relative error) between two methods. This is because the methods are all applied to the same data sets, so the variability due to differences in data sets partially cancels out when the differences in error rates are viewed. Thus, the relative-error plots are useful for determining whether apparent differences in accuracy between methods are statistically significant. Although we are able to identify statistically significant differences between the best methods, the actual differences are quite small (typically <1%) and may have a negligible effect in a subsequent analysis that uses the inferred haplotypes. We show below that these methods have significant differences in computational speed. Thus, for differentiating between the most accurate methods, differences in computation burden will be important.
Table 2. .
Allelic-Imputation Error Rates
| Error Rate(%) |
|||||||||
| Low-Density Simulateda |
High-Density Simulateda |
Affymetrix 500K WTCCCb |
|||||||
| Method | Small | Medium | Large | Small | Medium | Large | Small | Medium | Large |
| Beaglec: | |||||||||
| R=25 | 3.45 | 1.91 | 1.57 | 1.17 | .53 | .13 | 1.21 | .87 | .77 |
| R=4 | 3.65 | 2.01 | 1.57 | 1.36 | .59 | .13 | 1.32 | .88 | .78 |
| R=1 | 4.10 | 2.21 | 1.80 | 1.82 | .70 | .16 | 1.44 | .94 | .84 |
| fastPHASEd: | |||||||||
| K=20 | 3.37 | 2.54 | 2.80 | 1.19 | .90 | .57 | 1.12 | .95 | .93 |
| K=10 | 3.91 | 3.19 | 3.43 | 1.72 | 1.38 | 1.04 | 1.21 | 1.05 | 1.04 |
| HAP | 5.26 | 3.66 | 3.89 | 3.20 | 2.55 | 2.31 | 1.68 | 1.38 | 1.33 |
| 2SNP | 8.52 | 5.76 | 5.14 | 3.61 | 2.33 | 1.26 | 2.50 | 1.84 | 1.71 |
Parameters for selection of tag SNPs for 1 Mb of simulated data were chosen to obtain approximate densities of 1 SNP per 10 kb (low density) or 1 SNP per 3 kb (high density). For each density, three sample sizes were considered: 100 (small), 1,000 (medium), and 5,000 (large) individuals.
For the WTCCC data, sets of 200 consecutive markers were used. Three sample sizes were considered: 100 (small), 1,000 (medium), and 3,002 (large) individuals.
Beagle results are given for R=1, R=4, and R=25 samples per individual.
fastPHASE results are given for K=10 and K=20 clusters per individual.
Table 3. .
Switch Error Rates
| Error Rate(%) |
|||||||
| Low-Density Simulateda |
High-Density Simulateda |
||||||
| Method | Small | Medium | Large | Small | Medium | Large | WTCCC Xb |
| Beaglec: | |||||||
| R=25 | 5.72 | 2.97 | 2.39 | 1.69 | .84 | .05 | 5.75 |
| R=4 | 5.95 | 3.08 | 2.45 | 1.96 | .93 | .07 | 5.79 |
| R=1 | 7.49 | 3.72 | 3.03 | 2.94 | 1.21 | .10 | 6.41 |
| fastPHASEd: | |||||||
| K=20 | 5.29 | 4.11 | 4.59 | 1.66 | 1.55 | .49 | 6.34 |
| K=10 | 5.83 | 4.98 | 5.49 | 1.96 | 1.95 | .75 | 6.83 |
| HaploRec-S | 4.79 | 2.79 | 2.26 | 1.57 | 1.11 | .34 | 6.05 |
| HaploRec-VMM | 6.07 | 3.37 | 2.46 | 6.07 | 3.37 | .37 | 6.52 |
| HAP | 8.51 | 5.22 | 5.40 | 3.77 | 2.80 | 1.75 | 7.44 |
| 2SNP | 9.28 | 8.57 | 8.96 | 3.81 | 3.75 | 2.40 | 7.44 |
Parameters for selection of tag SNPs for 1 Mb of simulated data were chosen to obtain approximate densities of 1 SNP per 10 kb (low density) or 1 SNP per 3 kb (high density). For each density, three sample sizes were considered: 100 (small), 1,000 (medium), and 5,000 (large) individuals.
“WTCCC X” represents 50 nonoverlapping sets of 200 markers on 732 X-chromosome pairs. The chromosome pairs were created from WTCCC control male X chromosomes genotyped on the Affymetrix 500K platform.
Beagle results are given for R=1, R=4, and R=25 samples per individual.
fastPHASE results are given for K=10 and K=20 clusters per individual.
Figure 2. .

Error rates for selected haplotype-phasing methods. Three classes of data were considered: low-density data with ∼1 SNP per 10 kb (left column), high-density data with ∼1 SNP per 3 kb (middle column), and Affymetrix 500K data for the WTCCC controls (right column). Within each plot, three sample sizes (n) are shown. Each row of graphs gives a different measure of accuracy (Y-axis). The relative error graphs show differences in error rate between each method and a reference method, which is Beagle with R=25 samples per individual. All estimates are averaged across the data sets, with error bars showing ±2 SEs.
We examined two measures of accuracy: switch error and allelic-imputation error. Depending on one’s purpose, one might want to minimize a specific error measure. For example, if the data have a high proportion of missing genotypes, one might be particularly interested in obtaining a low allelic-imputation error rate, whereas, if the data have very little missing data, the switch error rate might be of greater interest.
We compared haplotype-phasing methods by using both allelic-imputation error rates and switch error rates for simulated data with six combinations of marker density (1 SNP per 10 kb or 1 SNP per 3 kb) and sample size (100, 1,000, or 5,000 individuals). For the simulated data, we found that the relative rankings by switch error rates were largely consistent with the rankings by allelic-imputation error rates.
We also compared haplotype-phasing methods by using real data. We assessed allelic-imputation error rates for three different sample sizes (100, 1,000, or 3,002 individuals) of phase-unknown, real autosomal data from the WTCCC control panel, and we assessed switch error rates for 732 phase-known pairings of X-chromosome data from the WTCCC control panel. Below we give the four best-performing method and parameter combinations for each class of data set, first ranked by allelic-imputation error rate and then ranked by switch error rate.
Allelic-Imputation Error
For the low-density data sets with 100 individuals, the best-performing methods with regard to allelic-imputation error rate (in order, best first) were fastPHASE with K=20, Beagle with R=25, Beagle with R=4, and fastPHASE with K=10. (Note that HaploRec does not infer missing data and thus was not included in the comparisons of allelic-imputation error rates.) For the high-density data sets with 100 individuals, the best-performing methods (in order, best first) were Beagle with R=25, fastPHASE with K=20, Beagle with R=4, and fastPHASE with K=10. For the Affymetrix 500K data with 100 individuals, the best-performing methods (in order, best first) were fastPHASE with K=20, Beagle with R=25, fastPHASE with K=10, and Beagle with R=4.
For the low- and high-density simulated data with 1,000 and 5,000 individuals and for the Affymetrix 500K autosomal data with 1,000 and 3,002 individuals, the best-performing methods with regard to allelic-imputation error rate (in order, best first) were Beagle with R=25, Beagle with R=4, Beagle with R=1, and fastPHASE with K=20, in each case.
Switch Error Rates
For the low-density simulated data with 100 individuals, the best-performing methods with regard to switch error rate (in order, best first) were Haplorec-S, fastPHASE with K=20, Beagle with R=25, and fastPHASE with K=10. For the low-density simulated data with 1,000 and 5,000 individuals, the best-performing methods (in order, best first) were Haplorec-S, Beagle with R=25, Beagle with R=4, and Haplorec-VMM, in each case.
For the high-density simulated data with 100 individuals, the best-performing methods with regard to switch error rate (in order, best first) were HaploRec-S, fastPHASE with K=20, Beagle with R=25, and Beagle with R=4. For 1,000 individuals, the best-performing methods were Beagle with R=25, Beagle with R=4, HaploRec-S, and Beagle with R=1, and, for 5,000 individuals, the best-performing methods were Beagle with R=25, Beagle with R=4, Beagle with R=1, and HaploRec-S.
For the 732 X-chromosome pairs from the WTCCC control data, the best-performing methods with regard to switch error rate (in order, best first) were Beagle with R=25, Beagle with R=4, HaploRec-S, and fastPHASE with K=20.
Summary of Allelic-Imputation and Switch Error Results
Beagle gave consistently the best or close to the best accuracy. It does particularly well with high-density genotype data and large sample sizes. Increasing R, the number of samples per individual per iteration, within the range considered (R=1, 4, or 25) always increased the accuracy for the data sizes we considered, with the largest improvements seen when increasing from R=1 to R=4 samples per individual.
HaploRec-S also gave the best or close to the best switch accuracy for the simulated data sets and X-chromosome data. HaploRec-VMM had inconsistent performance, doing poorly with some high-density data sets. HaploRec could not be compared with the other methods for the real autosomal data or in terms of imputation error, because it does not impute missing data.
For the data sets considered here, fastPHASE with K=20 clusters always gave more-accurate results than fastPHASE with K=10. We have found, in general, that, for data sets with fairly large numbers of individuals, increasing the number of clusters, K, is helpful, at least up to values of K=30, so increasing the number of clusters beyond 20 could improve the accuracy of fastPHASE. On the other hand, computing time is quadratic in the number of clusters, so, with K=20 clusters already taking a very long time to compute, it may not be feasible to increase K further for large data sets. For the small data sets, fastPHASE with K=20 gave slightly higher accuracy than did Beagle, but, for the medium and large data sets, it gave slightly lower accuracy than that of Beagle.
HAP and 2SNP had consistently poor accuracy compared with that of the other phasing methods. Their error rates were always at least 40% higher than those of the best methods and often were much higher than that. The gap between these two methods and the others did narrow somewhat for the real data, but a significant difference remained.
Timing Results
On the basis of the computation times given in table 4, we see the following patterns: 2SNP is by far the fastest program. Beagle (with R=1, 4, or 25 samples per individual) is the next fastest, followed in order by HaploRec-VMM, HaploRec-S, fastPHASE with K=10, and fastPHASE with K=20. HAP is much slower than fastPHASE with K=20 for the large data sets but is slightly faster for the small and medium-sized data sets. It is possible that, with a different compiling option or with a little re-engineering, HAP could be faster than fastPHASE with K=20, although it would probably still be slower than fastPHASE with K=10. The differences in times are large enough to have practical significance. Between Beagle with R=1 and fastPHASE with K=20, there is typically a 100–300-fold difference in timing results. Depending on the data set, HaploRec-S was 5–172 times slower than Beagle with R=1, with the largest relative differences in running time resulting from the real data sets genotyped with the Affymetrix 500K SNP array.
Table 4. .
Timing Results
| Time(s per Marker) |
||||||||||
| Low-Density Simulateda |
High-Density Simulateda |
Affymetrix 500K WTCCCb |
||||||||
| Method | Small | Medium | Large | Small | Medium | Large | Small | Medium | Large | Xc |
| Beagled: | ||||||||||
| R=25 | .07 | .58 | 12.95 | .07 | 1.05 | 16.42 | .07 | .88 | 6.50 | .34 |
| R=4 | .04 | .14 | 4.13 | .03 | .31 | 9.62 | .03 | .19 | 1.14 | .11 |
| R=1 | .02 | .10 | 1.80 | .01 | .13 | 4.55 | .02 | .09 | .34 | .07 |
| fastPHASEe: | ||||||||||
| K=20 | 3.03 | 32.33 | 150.69 | 3.49 | 30.25 | 150.60 | 3.02 | 29.74 | 89.12 | 22.00 |
| K=10 | .89 | 9.63 | 44.67 | 1.14 | 11.74 | 44.59 | .89 | 8.92 | 26.67 | 6.43 |
| HaploRec-S | .60 | 6.26 | 27.93 | 1.23 | 8.02 | 22.63 | .48 | 13.78 | 57.76 | 5.02 |
| HaploRec-VMM | .28 | 3.50 | 33.98 | .25 | 6.07 | 17.72 | .28 | 7.63 | 34.04 | 1.15 |
| HAP | 1.75 | 23.31 | 733.02 | .45 | 41.49 | 613.13 | .38 | 15.79 | 95.36 | 4.83 |
| 2SNP | .01 | .07 | 1.02 | .00 | .09 | 1.49 | .01 | .17 | 1.20 | .04 |
Parameters for selection of tag SNPs for 1 Mb of simulated data were chosen to obtain approximate densities of 1 SNP per 10 kb (low density) or 1 SNP per 3 kb (high density). For each density, three sample sizes were considered: 100 (small), 1,000 (medium), and 5,000 (large) individuals.
For the WTCCC data, sets of 200 consecutive markers were used. Three sample sizes were considered: 100 (small), 1,000 (medium), and 3,002 (large) individuals.
“X” represents 732 X-chromosome pairs constructed from male X-chromosome data.
Beagle results are given for R=1, R=4, and R=25 samples per individual.
fastPHASE results are given for K=10 and K=20 clusters per individual.
Regression analysis of timing results for the medium and large sample sizes for the low-density, high-density, and WTCCC autosomal data indicates that the running time for the Beagle phasing algorithm scales slightly less than quadratically in the number of samples (n1.9). We did not place constraints on our model, but improved scaling with sample size would be possible by constraining the size of the model as the sample size increases. For example, if we limited the maximum number of nodes per level, the scaling would be much closer to linear in the number of samples, at the cost of some loss in accuracy.
Although we used a computer with 32 GB of memory when we compared phasing algorithms, the implementation of our method in the Beagle software package has an extremely low memory footprint. We phased the WTCCC chromosome 1 control data (3,002 individuals and 40,220 markers) by using a 1.8-GHz laptop computer with 1 GB RAM running Windows XP. The running time for the laptop to phase chromosome 1 data with R=1 samples per individual was 11 h and 37 min (compared with 4 h and 40 min for the Linux server), and the maximum memory usage was 420 Mb. Phasing the WTCCC autosomal control data (3,002 individuals and 490,032 markers) with R=1 samples per individual took 3.1 d of computing time on the Linux server.
Discussion
Because of the incomplete information contained in a given data set, there is an upper limit to the accuracy of phasing that can be achieved. The upper bound on accuracy increases with the number of individuals and the marker density. It is likely that the most-accurate methods considered in this article—HaploRec-S, fastPHASE with K=20, and Beagle—approach these limits. As these limits are approached, computational speed and feasibility for large data sets become paramount. To the best of our knowledge, Beagle is the first highly accurate phasing algorithm capable of phasing whole-genome data in as little as a few days of computing time. Timing results presented here suggest that the best competing methods would require months or years of computing time to complete this task.
We have shown that the autosomal chromosomes from a whole-genome scan genotyped with the Affymetrix 500K SNP set can be phased using Beagle with R=1 samples per individual in 3.1 d for 3,002 individuals. On the basis of the results for the masked subsets from the WTCCC data (table 2), we would expect allelic-imputation error rates for imputation of missing genotypes to be ∼0.8%. For 1,000 individuals with use of R=4 samples per individual, we expect the phasing of Affymetrix 500K SNP data to take ∼2 d (extrapolating from table 4), with ∼1% allelic-imputation error.
Extrapolating from the results for the simulated low-density data (high-density data), we expect that phasing of the autosomal chromosomes from a whole-genome scan with 300,000 (1 million) tag SNPs for 5,000 individuals would take ∼6 d (48 d) by use of Beagle with R=1. Thus, by parallelizing by chromosome and with use of multiple processors, the phasing of 1 million SNPs genotyped for 5,000 individuals could be achieved within 1 wk. For 1,000 individuals, phasing would take 13 h (4 d) for 300,000 (1 million) tag SNPs phased using Beagle with R=4.
In comparing the haplotyping methods, we found lower differences in the accuracy of the different phasing algorithms for the WTCCC control data than for the simulated data. Although the simulated data were tuned to real data, it is expected that some differences will exist. Also, the differences in relative performance may be explained partially by differences in marker ascertainment between the simulated data and the real data. The simulated data used tag SNPs, which intentionally eliminate redundant highly correlated SNPs, whereas the SNPs on the Affymetrix 500K array were selected according to other criteria (e.g., technical quality). Consequently, there is substantial redundancy in the Affymetrix 500K array30 that is not present in the simulated marker sets. Also, ∼12% of the Affymetrix 500K SNPs are monomorphic in the British population.31
Like Beagle, fastPHASE uses an HMM approach but with several important differences that have implications for speed and accuracy. The model underlying fastPHASE has parameters that are fit as part of the EM cycle, whereas Beagle’s localized haplotype-cluster model is empirical and is fit using a one-step algorithm based on current haplotype estimates. This means that the fastPHASE model structure has to be kept simple (e.g., with a fixed number of clusters) to enable estimation of parameters at each cycle in a reasonable time, and it also means that more EM iterations are needed (35 iterations per random start and 15 random starts, with the default settings). Because Beagle just updates haplotype estimates at each iteration, a very small number of iterations is needed (we found that 10 iterations suffice). The fixed number of clusters in the model used by fastPHASE means that it cannot adapt locally to the structure of the data to the extent that Beagle can. The performance of fastPHASE is quadratic in the number of clusters, so that, although its accuracy might be increased by increasing the number of clusters above the values considered here, this may not be practical for large data sets.
HaploRec uses haplotype-fragment frequencies to obtain empirically based probabilities for longer haplotypes. The current implementation of HaploRec-S has high memory requirements—for example, >3 GB were needed to analyze 200 SNPs for the 3,002 individuals in the WTCCC control data. (By comparison, Beagle used <0.45 GB of memory to phase all 40,220 SNPs on chromosome 1 for the 3,002 individuals in the WTCCC control data.) To apply HaploRec to larger numbers of markers, it is necessary to analyze overlapping windows of markers,8 which further increases the computing time, whereas Beagle can analyze seamlessly an entire chromosome. We were not able to compare HaploRec for data with missing genotypes for the accuracy metrics that we considered, and we note that previous work indicates that the accuracy of HaploRec-S decreases more than that of other methods as the rate of missing data increases.8
2SNP is fast, but its accuracy is significantly worse than that of fastPHASE, HaploRec-S, and Beagle. Although we did observe a reduction in the difference in performance between 2SNP and the other methods for the real data compared with for the simulated data, the difference remained consequential.
Similarly, HAP’s relative performance was better for the real data than for the simulated data; however, the computational requirements of the version that we used were very high, so that it would not be possible to apply it to a full whole-genome association data set with thousands of genotyped individuals. The version of HAP that we used seemed to have been compiled with a 1-GB memory allocation. We suspect that the computational time for the larger data sets would be significantly reduced if this limitation were removed. On the basis of the timing results for the smaller simulated data sets, we would expect its computing times to be of the same order of magnitude as those of fastPHASE or HaploRec-S. However, HAP was consistently less accurate than were fastPHASE, HaploRec-S, and Beagle for simulated and real data sets.
Thus, Beagle is the best of the algorithms we considered for phasing large-scale data sets. Its efficient heuristic model fitting and its use of HMM methods for sampling mean that Beagle is significantly faster than the other methods, with comparable accuracy. Since the model used by Beagle is not constrained in size, the algorithm is able to exploit the increased information contained in large high-density marker sets to achieve significantly better accuracy than that of fastPHASE or HaploRec-S at the default parameter settings. These other algorithms presumably could be improved for large high-density data sets by adjustment of default parameters to allow growth of the underlying models; however, such improvements would come at the cost of even longer computing times.
Beagle will output the most-likely haplotype pairs by default but also includes an option for sampling haplotype pairs conditional on an individual’s genotypes at the end of the phasing algorithm. Sampled or most-likely inferred haplotype pairs may be used in association testing for case-control status or other traits of interest. Although it is not optimal to first infer phase and then perform multilocus association testing,32 this is a practical approach that can be applied to multilocus analysis of large-scale data sets from whole-genome association studies. For large data sets, the accuracy of inferred haplotypes is very high, and, provided that all individuals are phased together (regardless of trait status) and that trait status is independent of genotype quality (i.e., genotype error rates and missing-data rates), the type I error rate of the downstream association test will not be affected. For large numbers of markers, it is not necessary to have haplotypes that are completely correct, because the association tests will be based on localized haplotypes, rather than on whole haplotypes, so the inferred haplotypes need to be only locally correct. For multilocus association testing based on localized haplotype clustering, switch error rates of 5% resulted in reduced power of a multilocus test, but the multilocus test remained more powerful than single-marker tests for low-frequency susceptibility variants.1
Acknowledgments
This study makes use of data generated by the WTCCC. A full list of the investigators who contributed to the generation of the data is available from the WTCCC Web site. Funding for the WTCCC project was provided by Wellcome Trust award 076113. We thank Eleazar Eskin for providing a stand-alone version of HAP, Dumitru Brinza for increasing the maximum number of individuals in 2SNP, two anonymous reviewers for helpful comments, and the WTCCC for making their whole-genome control data available. This work was supported by a grant from the University of Auckland Research Committee (to S.R.B.) and by National Institutes of Health grant 3R01GM075091-02S1 (to S.R.B. and B.L.B.).
Appendix A
We give a worked example of calculation of forward probabilities and backward sampling, using the model of table 1 and figure 1. A worked example of model building with use of the data in table 1 is given in our previously published work.12 For this example, consider an individual with genotypes g1=?/?, g2=1/1, g3=1/2, and g4=1/2, where ?/? denotes a missing genotype. Using the haplotype counts in table 1, we have n(eA)=311, n(eB)=289, n(eC)=195, n(eD)=116, n(eE)=289, n(eF)=237, n(eG)=247, n(eH)=116, n(eI)=46, n(eJ)=191, n(eK)=247, and n(eL)=116. The calculations that follow use the equations in the section “Sampling from an HMM.”
The initiation step of the forward probability calculation gives
 |
Similarly, α1(eA,eB)=P(eA)P(eB)(1)=(311/600)(289/600), whereas α1(eB,eA)=(289/600)(311/600) and α1(eB,eB)=P(eB)2=(289/600)2.
In the induction step to calculate the values for α2, we use the values for α1 above and note, for example, that P[s2=(eC,eE)|s1=(eA,eB)]=P(eC|eA)P(eE|eB)=(195/311)(289/289), whereas P[s2=(eC,eE)|s1≠(eA,eB)]=0. Then, for example,
 |
Continuing, we find that α2(eE,eC)=(195/600)(289/600), α2(eC,eC)=(195/600)2, and α2(eE,eE)=(289/600)2, whereas the remaining α2 values are zero. Continuing to α3 and omitting zero terms, we have
 |
The only other nonzero α3 is α3(eG,eF), which takes the same value as α3(eF,eG). The nonzero α4 values are α4(eI,eK)=α4(eK,eI)=(46/600)(247/600).
The probability of sampling the path s1=(eA,eB), s2=(eC,eE), s3=(eF,eG), and s4=(eI,eK) (with ordered haplotype pair 1111 and 2122) is obtained as follows.
-
1. For the initiation step, sample s4=(eI,eK) with probability

-
2. Inductively sample s3=(eF,eG) with probability
(It makes sense that the probability of sampling s3=(eF,eG) conditional on sampling s4=(eI,eK) is 1, because (eF,eG) is the only ordered pair of edges leading into the ordered pair (eI,eK).)
-
3. For the next step, sample s2=(eC,eE) with probability

-
4. For the final step, sample s1=(eA,eB) with probability

Thus, this path is sampled with overall probability (0.5)(1)(0.2406)(1)=0.1203. The alternate ordering s1=(eB,eA), s2=(eE,eC), s3=(eG,eF), and s4=(eK,eI), which gives the same pair of haplotypes but in the other order, has the same probability, and, together, this pair of paths has sampling probability 0.2406.
Web Resources
The URLs for data presented herein are as follows:
- Beagle genetic analysis software package, http://www.stat.auckland.ac.nz/~browning/beagle/beagle.html
- WTCCC, http://www.wtccc.org.uk/
References
- 1.Browning BL, Browning SR (2007) Efficient multilocus association mapping for whole genome association studies using localized haplotype clustering. Genet Epidemiol 31:365–375 10.1002/gepi.20216 [DOI] [PubMed] [Google Scholar]
- 2.The International HapMap Consortium (2005) A haplotype map of the human genome. Nature 437:1299–1320 10.1038/nature04226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marchini J, Cutler D, Patterson N, Stephens M, Eskin E, Halperin E, Lin S, Qin ZS, Munro HM, Abecasis GR, et al (2006) A comparison of phasing algorithms for trios and unrelated individuals. Am J Hum Genet 78:437–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Excoffier L, Slatkin M (1995) Maximum-likelihood estimation of molecular haplotype frequencies in a diploid population. Mol Biol Evol 12:921–927 [DOI] [PubMed] [Google Scholar]
- 5.Long JC, Williams RC, Urbanek M (1995) An E-M algorithm and testing strategy for multiple-locus haplotypes. Am J Hum Genet 56:799–810 [PMC free article] [PubMed] [Google Scholar]
- 6.Hawley ME, Kidd KK (1995) HAPLO: a program using the EM algorithm to estimate the frequencies of multi-site haplotypes. J Hered 86:409–411 [DOI] [PubMed] [Google Scholar]
- 7.Scheet P, Stephens M (2006) A fast and flexible statistical model for large-scale population genotype data: applications to inferring missing genotypes and haplotypic phase. Am J Hum Genet 78:629–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eronen L, Geerts F, Toivonen H (2006) HaploRec: efficient and accurate large-scale reconstruction of haplotypes. BMC Bioinformatics 7:542 10.1186/1471-2105-7-542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qin ZS, Niu T, Liu JS (2002) Partition-ligation–expectation-maximization algorithm for haplotype inference with single-nucleotide polymorphisms. Am J Hum Genet 71:1242–1247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stephens M, Smith NJ, Donnelly P (2001) A new statistical method for haplotype reconstruction from population data. Am J Hum Genet 68:978–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Halperin E, Eskin E (2004) Haplotype reconstruction from genotype data using imperfect phylogeny. Bioinformatics 20:1842–1849 10.1093/bioinformatics/bth149 [DOI] [PubMed] [Google Scholar]
- 12.Browning SR (2006) Multilocus association mapping using variable-length Markov chains. Am J Hum Genet 78:903–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kimmel G, Shamir R (2005) GERBIL: genotype resolution and block identification using likelihood. Proc Natl Acad Sci USA 102:158–162 10.1073/pnas.0404730102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brinza D, Zelikovsky A (2006) 2SNP: scalable phasing based on 2-SNP haplotypes. Bioinformatics 22:371–373 10.1093/bioinformatics/bti785 [DOI] [PubMed] [Google Scholar]
- 15.Stephens M, Scheet P (2005) Accounting for decay of linkage disequilibrium in haplotype inference and missing-data imputation. Am J Hum Genet 76:449–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Excoffier L, Laval G, Balding D (2003) Gametic phase estimation over large genomic regions using an adaptive window approach. Hum Genomics 1:7–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ron D, Singer Y, Tishby N (1998) On the learnability and usage of acyclic probabilistic finite automata. J Comput Systems Sci 56:133–152 10.1006/jcss.1997.1555 [DOI] [Google Scholar]
- 18.Rabiner LR (1989) A tutorial on hidden Markov models and selected applications in speech recognition. Proc IEEE 77:257–286 10.1109/5.18626 [DOI] [Google Scholar]
- 19.Thompson EA (2000) Statistical inference from genetic data on pedigrees. Vol 6. Institute of Mathematical Statistics, Beachwood, OH [Google Scholar]
- 20.Celeux G, Diebolt J (1985) The SEM algorithm: a probabilistic teacher algorithm derived from the EM algorithm for the mixture problem. Comput Stat Q 2:73–82 [Google Scholar]
- 21.Tregouet DA, Escolano S, Tiret L, Mallet A, Golmard JL (2004) A new algorithm for haplotype-based association analysis: the stochastic-EM algorithm. Ann Hum Genet 68:165–177 10.1046/j.1529-8817.2003.00085.x [DOI] [PubMed] [Google Scholar]
- 22.Schaffner SF, Foo C, Gabriel S, Reich D, Daly MJ, Altshuler D (2005) Calibrating a coalescent simulation of human genome sequence variation. Genome Res 15:1576–1583 10.1101/gr.3709305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kong A, Gudbjartsson DF, Sainz J, Jonsdottir GM, Gudjonsson SA, Richardsson B, Sigurdardottir S, Barnard J, Hallbeck B, Masson G, et al (2002) A high-resolution recombination map of the human genome. Nat Genet 31:241–247 [DOI] [PubMed] [Google Scholar]
- 24.Carlson CS, Eberle MA, Rieder MJ, Yi Q, Kruglyak L, Nickerson DA (2004) Selecting a maximally informative set of single-nucleotide polymorphisms for association analyses using linkage disequilibrium. Am J Hum Genet 74:106–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.The Wellcome Trust Case Control Consortium (2007) Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447:661–678 10.1038/nature05911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matsuzaki H, Dong S, Loi H, Di X, Liu G, Hubbell E, Law J, Berntsen T, Chadha M, Hui H, et al (2004) Genotyping over 100,000 SNPs on a pair of oligonucleotide arrays. Nat Methods 1:109–111 10.1038/nmeth718 [DOI] [PubMed] [Google Scholar]
- 27.Rabbee N, Speed TP (2006) A genotype calling algorithm for Affymetrix SNP arrays. Bioinformatics 22:7–12 10.1093/bioinformatics/bti741 [DOI] [PubMed] [Google Scholar]
- 28.Lin S, Cutler DJ, Zwick ME, Chakravarti A (2002) Haplotype inference in random population samples. Am J Hum Genet 71:1129–1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stephens M, Donnelly P (2003) A comparison of Bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet 73:1162–1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barrett JC, Cardon LR (2006) Evaluating coverage of genome-wide association studies. Nat Genet 38:659–662 10.1038/ng1801 [DOI] [PubMed] [Google Scholar]
- 31.Plagnol V, Cooper JD, Todd JA, Clayton DG (2007) A method to address differential bias in genotyping in large-scale association studies. PLoS Genet 3:e74 10.1371/journal.pgen.0030074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin DY, Huang BE (2007) The use of inferred haplotypes in downstream analyses. Am J Hum Genet 80:577–579 [DOI] [PMC free article] [PubMed] [Google Scholar]