

Abstract
Phenylketonuria (PKU) is a genetic disease caused by mutations in human phenylalanine hydroxylase (PAH). Most missense mutations result in misfolding of PAH, increased protein turnover, and a loss of enzymatic function. We studied the prediction of the energetic impact on PAH native-state stability of 318 PKU-associated missense mutations, using the protein-design algorithm FoldX. For the 80 mutations for which expression analyses have been performed in eukaryote systems, in most cases we found substantial overall correlations between the mutational energetic impact and both in vitro residual activities and patient metabolic phenotype. This finding confirmed that the decrease in protein stability is the main molecular pathogenic mechanism in PKU and the determinant for phenotypic outcome. Metabolic phenotypes have been shown to be better predicted than in vitro residual activities, probably because of greater stringency in the phenotyping process. Finally, all the remaining 238 PKU missense mutations compiled at the PAH locus knowledgebase (PAHdb) were analyzed, and their phenotypic outcomes were predicted on the basis of the energetic impact provided by FoldX. Residues in exons 7–9 and in interdomain regions within the subunit appear to play an important structural role and constitute hotspots for destabilization. FoldX analysis will be useful for predicting the phenotype associated with rare or new mutations detected in patients with PKU. However, additional factors must be considered that may contribute to the patient phenotype, such as possible effects on catalysis and interindividual differences in physiological and metabolic processes.
Phenylketonuria (PKU [MIM 261600]) is a human metabolic disease caused by mutations in the phenylalanine hydroxylase gene (PAH) and is inherited in an autosomal recessive Mendelian fashion. Phenylalanine hydroxylase (PAH, also known as “phenylalanine 4-monooxygenase” [EC 1.14.16.1]) catalyzes the rate-limiting step in l-phenylalanine (l-Phe) catabolism in liver, using tetrahydrobiopterin (BH4) and dioxygen as additional cosubstrates. PKU mutations are associated with impairment of PAH activity, leading to accumulation of l-Phe in plasma—that is, hyperphenylalaninemia (HPA)—and neurological damage in untreated patients.1,2 Two parameters have traditionally been used to classify patients with PKU into different phenotypic groups: plasma l-Phe levels (under normal feeding conditions) and daily l-Phe tolerance (with l-Phe levels kept within therapeutic ranges). The main determinant of the metabolic phenotype in patients with PKU is the mutant genotype, although the fact that >500 mutations in the PAH gene have been reported makes PKU a highly heterogeneous disease from both genetic and metabolic perspectives.3,4 About two-thirds of PKU mutations are missense, causing single amino acid changes in the PAH sequence. The effects of ∼100 of these mutations on PAH function and stability have been identified by means of in vitro expression analysis (extensively compiled in the PAH locus knowledgebase [PAHdb]).5 The predominant molecular mechanism in PKU appears to be a loss-of-function pathogenesis due to decreased stability and/or folding efficiency in the PAH protein with mutation.4,6–9 Thus, a large proportion of all missense mutations studied, including many of the most common mutations found in patients, display stability and folding defects when they are expressed in vitro.4,8–10 These variant PAH proteins are found to aggregate when expressed in prokaryote systems, where they appear as both soluble and insoluble aggregates.6–9 But the presence of amyloid fibrils or insoluble aggregates has not been reported in the liver of PKU-affected individuals, and PKU has, in fact, been classified as a cytosol-associated protein-misfolding disease.11 The current understanding of PKU is, thus, that the PAH mutant proteins are degraded in vivo more rapidly than wild-type (WT) protein by the proteasome or other protein quality-control proteolytic systems in hepatocytes.4,10,12 A loss-of-function mechanism associated with increased degradation rather than aggregation of the mutants has also been reported as the molecular basis of other important genetic diseases, such as cystic fibrosis, Fabry disease, hypogonadotrophic hypogonadism, and familiar hypercholesterolemia.13,14
For many PKU mutations, a correlation between genotype (mutant alleles) and in vivo metabolic phenotype (plasma l-Phe and l-Phe tolerance) in patients and in vitro–expressed mutant residual activity can be semiquantitatively established (for studies including large sets of mutations, see the work of Gjetting et al.8 and Pey et al.9; for a recent and very comprehensive review, see the work of Güttler3). The absence of a quantitative relationship between in vivo phenotypes and in vitro residual activity seems to be related to the use of different cellular model systems (eukaryotic and prokaryotic), which may exacerbate or attenuate folding and stability defects, because of their different abilities to support folding.10 For some other mutations, the inconsistencies reported are possibly related to PKU misclassification, since there are no generally accepted international guidelines and because phenotypes are sensitive to age and BMI.3 Interindividual differences in other metabolic and physiological processes, such as intestinal absorption or transport of l-Phe, may also contribute to inconsistencies in the genotype-phenotype correlations in PKU mutations.15
Several crystal structures of truncated forms of mammalian PAH16–19 have provided an experimental framework to qualitatively explain the effects of many PKU mutations on PAH activity and stability.20,21 Stability and folding defects observed in in vitro expression analyses have been rationalized in terms of altered interactions in the native structure because of mutation,20–22 possibly leading to reduced kinetic or thermodynamic stability.4,10 However, PAH displays a quite complex protein architecture, organized as an asymmetric dimer of dimers, and contains three functional domains per monomer.22 Moreover, the enzyme does not undergo reversible chemical or thermal denaturation, which is a prerequisite for the quantitative study and comparison of the thermodynamic stability of WT and mutant proteins. For this reason, it has been assumed that the lowered solubility or “operational” thermal stability in vitro (see, e.g., the work of Gámez et al.7 and Pey et al.9) reflects reduced thermodynamic stability.4,10 Nevertheless, to date, no attempt has been made to quantitatively correlate mutational effects on PAH stability with in vitro residual activity or phenotypic outcome in patients.
In this article, we evaluate the energetic impact of all missense PKU mutations (N=318) identified to date on PAH native-state stability, by using the available crystal structures and structural models for the human PAH protein. The energetic penalties were evaluated using the new FoldX energy function.23 Changes in PAH stability as a result of mutation (in terms of ΔΔG values) were then compared with both the residual activities determined in vitro in eukaryotic expression systems and the patient phenotypes associated with these mutations in the available literature. This is the first large-scale study, to our knowledge, to demonstrate relatively good mutation-based prediction of disease outcome. We were able to confirm and quantify changes in protein stability as the main molecular pathogenic mechanisms in PKU, except in a few mutations that are mostly catalytic. Our results help to identify the amino acids and regions with the greatest effects on the stability of the protein and represent a tool potentially capable of assisting individual patients on the basis of the outcome predicted by FoldX.
Data and Methods
PAH Structural Models
Human and rat PAH show 90% sequence identity. The humanized versions of the crystal structures of dimeric rat PAH (residues 19–427) in the phosphorylated (Protein Data Bank [PDB] 1PHZ) and nonphosphorylated (PDB 2PHM) forms were prepared by changing the residues that differ between rat and human PAH and then regularizing the structure with use of the BuildModel option of FoldX in the new version, 2.65. Energy calculations show compatibility of the human variations with the rat structure. Calculations were also performed on the N-terminal truncated tetrameric human PAH, residues 111–452 (PDB 2PAH). Moreover, calculations were attempted on a composite model of the full-length human tetrameric structure.24 However, this model, although apparently correct, contains numerous van der Waals clashes that could not be resolved, even after various runs of minimization and molecular-dynamics simulations. This was not the case for the humanized dimeric rat structures that could be modeled without any problem.
FoldX Force Field
The FoldX energy function includes terms that have been found to be important for protein stability. The free energy from the unfolding (ΔG) of a target protein is calculated using equation (1):
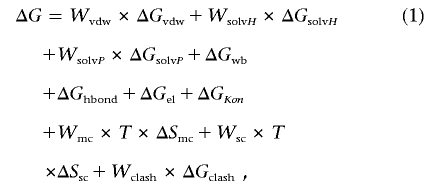 |
where ΔGvdw is the sum of the van der Waals contributions of all atoms with respect to the same interactions with the solvent. ΔGsolvH and ΔGsolvP are the differences in solvation energy for apolar and polar groups, respectively, when these groups change from the unfolded to the folded state. ΔGhbond is the free-energy difference between the formation of an intramolecular hydrogen bond and the formation of an intermolecular hydrogen bond (with solvent). ΔGwb is the extra stabilizing free energy provided by a water molecule that makes more than one hydrogen bond to the protein (water bridges) and that cannot be taken into account with nonexplicit solvent approximations.25 ΔGel is the electrostatic contribution of charged groups, including the helix dipole. ΔSmc is the entropy cost of fixing the backbone in the folded state; this term is dependent on the intrinsic tendency of a particular amino acid to adopt certain dihedral angles.26 Finally, ΔSsc is the entropic cost of fixing a side chain in a particular conformation,27 and the ΔGclash term provides a measure of the steric overlaps between atoms in the structure.
When we are working with oligomeric proteins or protein complexes, two extra terms are involved: ΔGkon, which reflects the effect of electrostatic interactions on the association constant kon (this applies only to the subunit binding energies),28 and ΔStr, which is the loss of translational and rotational entropy that ensues after formation of the complex. The latter term cancels out when we are looking at the effect of point mutations on complexes.
The energy values of ΔGvdw, ΔGsolvH, ΔGsolvP, and ΔGhbond attributed to each atom type have been derived from a set of experimental data, and ΔSmc and ΔSsc have been taken from theoretical estimates. The terms Wvdw, WsolvH, WsolvP, Wmc, and Wsc correspond to the weighting factors applied to the raw energy terms. They are all 1, except for the van der Waals contribution (Wvdw), which is 0.33 (the van der Waals contributions are derived from vapor-to-water energy transfer, whereas, in the protein, we are going from solvent to protein). For a detailed explanation of the FoldX force field, see the work of Schymkowitz et al.23,29 and the FoldX Web server.
FoldX Modeling
To model the mutations on the humanized structures, we used the BuildModel option of FoldX, version 2.65. This command reads the PDB and duplicates it internally. Then, it mutates the selected position in one molecule to itself and, in the other, to the variant selected, while moving the neighboring side chains. We ensure that the moving side chains and the rotamer set for them are the same in both cases. In this way, we prevent artefactual changes in energy due to the release, for example, of a clash in a neighboring side chain in the mutant. The effect of the mutation is then computed by subtracting the energy of the self-mutated WT from that of the mutant. When working with a dimer or tetramer, all mutations and self-mutations are performed at the same time. The ΔΔG values are provided in kilocalories per mole of dimer for all model structures.
Residual Activities of the Mutant Forms: “In Vitro” Phenotype
To the best of our knowledge, 96 PAH mutant alleles have been expressed to date in vitro in at least one expression system (compiled in PAHdb and references therein5,30,31), and all were evaluated for inclusion in this study. Nine of these mutations were not included for further classification and analysis because they are known or expected to affect protein sequence beyond a single amino acid substitution20,21,32: M1V (affecting mRNA translation), F39del and I94del (amino acid deletions), [T63P;H64N] (double mutant), EX6-96A→G (aberrant splicing/deletion), IVS10-11G→A (aberrant splicing/inframe insertion), R243X and G272X (truncations), and IVS12+1G→A (aberrant splicing/truncation). It is worth noting that some additional mutations initially classified as missense (or silent) can mask splicing defects.33,34 These effects are not considered further in our analysis, since they must be assessed experimentally. Of the remaining 87 mutant alleles, 7 were discarded because no expression analyses have been reported in eukaryotic systems (E178G, S231P, R270K, A300S, A313T, I318T, and A373T). We thus used a set of 80 mutations (table 1) that have been expressed at least once in a eukaryotic expression system (mammalian transfected cells or cell-free systems based on reticulocyte extracts [TnT systems]) and whose activity measurements have been reported. These mutations are distributed all over the PAH structure, include 31 of the 36 most frequent missense mutations reported in patients (92.1% of reported alleles), and represent >47.5% of the total mutant alleles reported so far. The mutations were classified into three groups (types I–III) (see table 1) on the basis of the mean residual activity (compared with WT activity in each study).
Table 1. .
Classification of 80 PKU Mutations into Three Types (I–III), Depending on Their Residual In Vitro Activities
| Type and Mutanta |
Residual Activityb (%) |
| Ic (n=19): | |
| R53H | 79 |
| D59Y | 92 |
| R68G | 100 |
| R68S (3) | 57±36 |
| P69S | 69 |
| E76G | 85 |
| T92I | 76 |
| D143G (2) | 68 |
| P211T | 72 |
| G218V (3) | 64±35 |
| V230I | 63 |
| P244L (2) | 51 |
| V245A | 50 |
| R261Q (6) | 51±26 |
| K274E | 100 |
| A322G | 75 |
| E390G | 75 |
| A403V (2) | 66 |
| D415N | 72 |
| IId (n=33): | |
| L41F | 10 |
| K42I | 12 |
| G46S (2) | 16 |
| A47V | 12.5 |
| L48S | 39 |
| L52S | 27 |
| I65T (5) | 41±33 |
| S70P | 20 |
| S87R | 25 |
| G103S | 39 |
| A104D (2) | 27 |
| P122Q | 22 |
| T124I | 42 |
| R158Q (3) | 16±11 |
| G171A | 27 |
| I174T | 12 |
| R176L | 42 |
| E178V | 18 |
| R241C | 25 |
| R241H | 23 |
| R243Q (3) | 13±5 |
| I283F | 10 |
| L293M | 41 |
| I306V | 39 |
| A309V (2) | 40 |
| A342T | 26 |
| L348V (4) | 35±8 |
| V388M (4) | 28±16 |
| A395P | 16 |
| R408Q (3) | 46±43 |
| R413S | 32 |
| R413P | 35±45 |
| Y414C | 42±26 |
| IIIe (n=28): | |
| R157N | 5 |
| F161S | 7 |
| W187C | 1 |
| V245L | 7 |
| V245E | 7 |
| G247V | 4 |
| R252W (2) | 0 |
| R252Q (2) | 2.5 |
| R252G (2) | 3.0 |
| L255V (2) | 7.0 |
| L255S (2) | 2.0 |
| A259T (2) | 5.5 |
| A259V (3) | 2.0±1.7 |
| R270S (3) | 1.7±1.5 |
| Y277D | 0 |
| T278I | 1 |
| E280K (4) | 2.2±2.6 |
| P281L (3) | 2.3±3.2 |
| D282N | 2 |
| F299C | 3 |
| L311P (2) | .5 |
| G332V | 0 |
| L333F | 7 |
| S349P (3) | .5±.4 |
| S349L (2) | 0 |
| S391I | 0 |
| R408W (4) | 1.2±1.1 |
| A447P | 8 |
In parentheses after the mutant, the number of reported measurements (if more than one) used for the mean in vitro residual activity estimation is given (data from PAHdb; references for the individual expression and activity studies are available from PAHdb, and additional data are reported elsewhere30,31).
Given as % of WT activity obtained in each study. SDs are given for some mutations for which more than one report is available.
Type I mutant activity is ⩾50% of WT activity (mean±SD 71.8%±14.9%).
Type II mutant activity is 10%–50% of WT activity (mean±SD 27.2%±11.4%).
Type III mutant activity is <10% of WT activity (mean±SD 2.9%±2.6%).
Results of residual protein and activity obtained from expression of mutants in Escherichia coli have not been included in the study, since, in such systems, PAH is usually expressed and analyzed as a fusion protein with another protein partner, and folding and/or stability defects are accompanied by protein aggregation, an event that has not been demonstrated to be associated with PKU pathogenesis.11,14
Mutation-Associated Disease Outcome: “In Vivo” Phenotype
Assignment of individual PAH mutations to particular metabolic phenotypes (genotype-phenotype correlation) has classically been performed by comparing phenotypes in large genotype-phenotype correlation studies that have included patients with a certain mutation in homozygosity and/or functional hemizygosity.32,35 Homozygosity (homoallelic mutant genotype) seems to be more predictive (∼90% of correlations are consistent) but less frequent than functional heterozygosity (genotype where the mutation under analysis is present in trans with a null mutation), for which ∼70% of correlations are consistent.32 Mutations are considered null when they “are known or predicted to completely abolish PAH activity,”35(p.73) such as frameshift mutations (e.g., F55fsdelT, K363fsdelG, and P407fsdelC), splice-defective mutations (e.g., IVS12nt1g→a or IVS10nt11g→a), and base substitutions that introduce a premature stop codon (e.g., R111X, R243X, and R261X). Mutations that have been repeatedly reported to show null activity (<3%32), such as R252W, R408W, P281L, S349L, and S349P (table 1), are also considered null. Those mutations that may not completely impair correct mRNA splicing35 or lead to low but detectable activity when expressed in vitro (R158Q or R243Q) are not regarded as null mutations. Occasionally, some studies also include phenotypes that are associated with quite rare mutations, using compound heterozygotes (patients carrying two nonnull mutant alleles) because of the lack of information about these mutations in homozygosity or functional hemizygosity.9,36
Two parameters are used to estimate the severity of metabolic phenotypes in patients with PKU: plasma l-Phe concentrations at the time of diagnosis (typically with a normal diet) and l-Phe tolerance (with l-Phe levels kept within a therapeutic range), both depending on age.3 Different phenotypic groups have been proposed on the basis of these two parameters and have been followed throughout the past 2 decades. The cutoff values used as boundaries between groups differ from author to author, and recommendations for classification vary among countries.3,32,35,37–40 Nevertheless, three consensus phenotypic groups emerge, from the mildest to the more severe phenotype: (i) mild, benign, or non-PKU HPA (MHP), (ii) mild or atypical PKU, and (iii) severe or classic PKU. In some reports, an additional group called “moderate PKU” is also found at the boundary between mild and severe PKU (see below and table 2).
Table 2. .
Classification of 46 PKU Mutations into Phenotypic Groups
| Group, Classification,a and Mutation |
References |
| Group 1, MHP: | |
| MHP (n=13): | |
| A47V | Gjetting et al.,8 Kayaalp et al.32 |
| D59Y | Pey et al.,9 Desviat et al.36 |
| E76G | Pey et al.,9 Desviat et al.36 |
| S87R | Jennings et al.,21 Kayaalp et al.,32 Guldberg et al.35 |
| T92I | Jennings et al.,21 Kayaalp et al.32 |
| R176L | Gjetting et al.,8 Jennings et al.,21 Kayaalp et al.,32 Desviat et al.36 |
| V230I | Güttler,3 Gjetting et al.,8 Jennings et al.,21 Desviat et al.36 |
| R241C | Jennings et al.21 |
| V245A | Güttler,3 Gjetting et al.,8 Jennings et al.,21 Desviat et al.36 |
| I306V | Gjetting et al.,8 Jennings et al.,21 Desviat et al.36 |
| A322G | Desviat et al.36 |
| R413S | Jennings et al.21 |
| D415N | Güttler,3 Kayaalp et al.,32 Desviat et al.36 |
| Group 2, mild PKU: | |
| MHP-mild PKU (n=6): | |
| P122Q | MHP-mild PKU in Pey et al.9; mild PKU in Desviat et al.36 |
| G171A | MHP in Guldberg et al.35; mild PKU in Kayaalp et al.32 |
| E390G | MHP in Güttler,3 Gjetting et al.,8 Desviat et al.36; mild PKU-MHP in Kayaalp et al.32; mild PKU in Aulehla-Scholz et al.41 |
| A403V | MHP in Güttler,3 Desviat et al.36; MHP-mild PKU in Kayaalp et al.32 |
| R408Q | MHP in Kayaalp et al.32; MHP-mild PKU in Pey et al.9; mild PKU in Güttler,3 Gjetting et al.,8 Desviat et al.36 |
| Y414C | MHP in Pey et al.9; mild PKU in Güttler,3 Gjetting et al.,8 Jennings et al.,21 Desviat et al.36 |
| Mild PKU (n=4): | |
| R68S | Güttler,3 Jennings et al.,21 Desviat et al.36 |
| A104D | Güttler,3 Gjetting et al.,8 Kayaalp et al.,32 Desviat et al.36 |
| R241H | Jennings et al.21 |
| P244L | Pey et al.,9 Desviat et al.36 |
| Mild-moderate PKU (n=3): | |
| I65T | Mild PKU in Pey et al.,9 Desviat et al.42; moderate PKU in Güttler,3 Gjetting et al.,8 Jennings et al.21 |
| L348V | Mild PKU in Desviat et al.36; moderate PKU in Güttler,3 Gjetting et al.,8 Jennings et al.,21 Aulehla-Scholz et al.41 |
| V388M | Mild PKU in Desviat et al.36; moderate PKU in Güttler,3 Gjetting et al.,8 Jennings et al.21 |
| Group 3, severe PKU: | |
| Moderate PKU (n=1): | |
| A309V | Pey et al.,9 Desviat et al.42 |
| Severe PKU (n=19): | |
| I174T | Jennings et al.21 |
| P211T | Kayaalp et al.32 |
| V245L | Gjetting et al.8 |
| R252W | Pey et al.,9 Jennings et al.,21 Kayaalp et al.,32 Desviat et al.36 |
| R252Q | Jennings et al.,21 Kayaalp et al.,32 Guldberg et al.35 |
| R252G | Jennings et al.,21 Guldberg et al.35 |
| A259V | Jennings et al.21 |
| Y277D | Pey et al.,9 Desviat et al.36 |
| E280K | Gjetting et al.,8 Kayaalp et al.,32 Desviat et al.36 |
| P281L | Güttler,3 Gjetting et al.,8 Kayaalp et al.,32 Desviat et al.36 |
| D282N | Jennings et al.,21 Guldberg et al.35 |
| I283F | Kayaalp et al.32 |
| F299C | Jennings et al.,21 Kayaalp et al.,32 Aulehla-Scholz et al.41 |
| L311P | Jennings et al.,21 Desviat et al.36 |
| A342T | Gjetting et al.,8 Jennings et al.21 |
| S349P | Gjetting et al.,8 Jennings et al.,21 Kayaalp et al.,32 Desviat et al.36 |
| S349L | De Lucca et al.43 |
| A395P | Gjetting et al.8 |
| R408W | Gjetting et al.,8 Pey et al.,9 Kayaalp et al.,32 Desviat et al.36 |
Classification according to plasma l-Phe levels and/or daily l-Phe tolerance, typically measured after newborn screening or at age 5 years. The phenotypic groups are based on the reports cited, where the cutoff values for plasma l-Phe levels or daily l-Phe tolerance vary among reports and under different national guidelines.21,32,35,36,38,39,42,44,45 MHP, also called “mild,” “benign,” or “non-PKU” HPA, with plasma l-Phe <360–600 μM and very high l-Phe tolerance (>1,000 mg/d). Patients typically display normal development in the absence of l-Phe restriction and need only minor changes in dietary protein intake. Mild PKU, also called “atypical” or “variant” PKU, with plasma l-Phe in the 360–1,200 μM range and daily l-Phe tolerance of 400–1,000 mg/d. If untreated, patients develop abnormally to different degrees, and their intelligence quotient is significantly affected. Moderate PKU, an intermediate group used by some authors to distinguish between mild and severe phenotypes, with plasma l-Phe values of 900–1,800 μM and daily l-Phe tolerance of 350–500 mg/d. Severe PKU, also called “classic” PKU, with plasma l-Phe levels >1,200–1,800 μM and daily l-Phe tolerance <250–400 mg/d. If untreated, the patient's development rapidly and irreversibly deteriorates, leading to severe mental retardation. See the “Mutation-Associated Disease Outcome: “In Vivo Phenotype” subsection for further details about the classification into groups.
Among the mutations in table 1, 16 mutants have not been identified in the literature as being explicitly associated with a particular phenotype in patients, whereas 18 have no clear association with a phenotypic group (table 3). These are inconsistently associated with a broad range of phenotypes, from mild to severe, or are described as “unclassified.” We thus classified the remaining 46 mutations into three patient phenotypic groups (table 2). Group 1 (mean±SD in vitro residual activity 53.0%±25.8%; n=13) corresponds to the very mild phenotype in patients (MHP). In this group, we put the unambiguously MHP mutations found in table 2. Group 2 (mean±SD in vitro residual activity, 41.5%±16%; n=13) corresponds to the mild PKU phenotype, and it was formed by combining mild MHP (reported as either “MHP” or “mild” in different reports), mild PKU (reported as “mild”), and mild-moderate PKU (reported as either “mild” or “moderate” in the literature) mutants. The in vitro residual activities of the mutants in this group are actually quite similar (46%±21%, 40%±17%, and 35%±7% for the mild MHP [n=6], mild PKU [n=4], and mild-moderate PKU [n=3] mutants, respectively). Group 3 (mean in vitro residual activity 6.9%±11.1%; n=20) corresponds to severe PKU and includes the only moderate mutant (A309V), in addition to a large group of mutations unambiguously reported as “severe.”
Table 3. .
PKU Mutations Discarded for Further Analysis because of Lack of Reported or Inconsistent Phenotype Classification
| Mutationa | Reference(s) |
| L41F | Guldberg et al.35 |
| G46S | Gjetting et al.,8 Jennings et al.,21 Kayaalp et al.,32 Guldberg et al.,35 Desviat et al.,36 Eiken et al.46 |
| L48S | Jennings et al.,21 Guldberg et al.,35 Desviat et al.,36 Aulehla-Scholz et al.41 |
| D143G | Jennings et al.21 |
| R158Q | Gjetting et al.,8 Jennings et al.,21 Kayaalp et al.,32 Guldberg et al.,35 Desviat et al.,36 Aulehla-Scholz et al.41 |
| F161S | Jennings et al.21 |
| E178V | Jennings et al.21 |
| G218V | Gjetting et al.,8 Jennings et al.,21 Guldberg et al.35 |
| R243Q | Jennings et al.,21 Guldberg et al.,35 Desviat et al.36 |
| V245E | Gjetting et al.8 |
| G247V | Jennings et al.21 |
| L255V | Jennings et al.21 |
| L255S | Jennings et al.21 |
| A259T | Jennings et al.21 |
| R261Q | Gjetting et al.,8 Jennings et al.,21 Kayaalp et al.,32 Guldberg et al.,35 Aulehla-Scholz et al.,41 Desviat et al.42 |
| R270S | Jennings et al.21 |
| L333F | Jennings et al.21 |
| R413P | Jennings et al.21 |
The 18 mutations associated with an inconsistent phenotype are shown. Mutations lacking a reported phenotype classification (n=16; not shown in the table) are K42I, L52S, R53H, R68G, P69S, S70P, G103S, T124I, R157N, W187C, K274E, T278I, L293M, G332V, S391I, and A447P. Phenotypes regarded as inconsistent are those reported as very disparate for one mutation in the same report or different reports. G46S is reported as “mild,” “severe-mild,” or “severe”; L48S as “mild,” “moderate,” or “undefined”; R158Q (a mutation traditionally defined as “severe”) as “severe,” “mild-severe,” or “mild”; R243Q (another mutation traditionally considered “severe”) as “severe” or “mild”; G218V as “severe” or “unclassified”; R261Q as “moderate-severe,” “moderate,” or “mild-severe.” The remaining 12 mutations have not been associated with any specific phenotype (defined only as “unclassified”).
BH4-Responsive Mutations
BH4-responsive PKU refers to the disease in a subgroup of patients with PKU who have a positive response (showing a reduction in their l-Phe plasma levels) to the administration of pharmacological doses of the natural cofactor BH4 and display a high degree of heterogeneity at the genetic (mutational) level. Seventy-three mutations have been found in patients with BH4-responsive PKU (as compiled in the BIOPKU database). Of these mutations, 39 were not included in the FoldX analysis because of their effects on splicing or truncation of PAH protein or their association with patients with nonresponsive PKU.47 The remaining 34 mutations are compiled in table 4. The main molecular mechanism underlying BH4 responsiveness appears to be a chaperone-like effect, by which BH4 increases the stability of mutant PAH proteins in vitro (thermal stability and protection against proteolytic degradation and oxidative inactivation19,30) and also stabilizes PAH WT and mutant proteins levels in cultured hepatoma cells and mouse liver.48–50 Other mechanisms, such as the correction of a high Km value for BH4, have also been shown to operate for a few BH4-responsive mutants in vitro.19,30,51 About 60% of patients with BH4-responsive PKU show a mild phenotype and present mutations that display significant residual activity in vitro.47
Table 4. .
Predicted Effect on ΔΔG and Phenotypic Prediction based on FoldX Analysis for 34 Missense Mutations Found in Patients with BH4-Responsive PKU[Note]
| Energetic Penalty(kcal/mol) |
Linear Fitting |
|||||
| Mutationa | 20 | 15 | 10 | 5 |
y0 (kcal/mol) |
m |
| BH4 responsive (n=16): | ||||||
| F55L | 4.2 | 3.3 | 3.3 | 3.3 | 2.9 | .05 |
| S110L | 7.4 | 6.2 | 5.1 | 3.5 | 2.4 | .25 |
| P119S | 5.7 | 5.7 | 5.7 | 5.7 | 5.7 | <.01 |
| D129G | 11.6 | 10.9 | 10.9 | 10.9 | 10.6 | .04 |
| A132V | 21.2 | 18.5 | 11.0 | 7.9 | 2.8 | .95 |
| V177M | 2.8 | 2.3 | 2.3 | 2.3 | 2.1 | .03 |
| V190A | 2.3 | 2.3 | 2.3 | 2.3 | 2.3 | <.01 |
| P275L | 23.6 | 20.7 | 17.8 | 14.3 | 11.4 | .62 |
| A300S | .5 | .5 | .5 | .5 | .5 | <.01 |
| S310Y | 157 | 126 | 92.3 | 56.4 | 24.1 | 6.71 |
| A313T | 29 | 22 | 16 | 8.2 | 1.2 | 1.39 |
| P314S | 7.6 | 7.6 | 7.6 | 7.6 | 7.6 | <.01 |
| K320N | 1.8 | 1.8 | 1.8 | 1.8 | 1.8 | <.01 |
| A373T | 19 | 16 | 13 | 9.8 | 6.8 | .61 |
| P407S | 3.7 | 3.7 | 3.7 | 3.7 | 3.7 | <.01 |
| Y417H | 7.3 | 7.1 | 7.1 | 7.2 | 7.2 | <.01 |
| Potentially BH4 responsive (n=18): | ||||||
| F39L | 11 | 11 | 11 | 11 | 11 | <.01 |
| L48S | 5.9 | 5.9 | 5.9 | 5.9 | 5.9 | <.01 |
| I65T | 10 | 9.3 | 8.4 | 7.5 | 6.7 | .17 |
| R68S | 6.3 | 6.3 | 6.3 | 6.3 | 6.3 | <.01 |
| A104D | 7.4 | 6.9 | 6.3 | 5.7 | 5.1 | .11 |
| P211T | 5.1 | 5.1 | 5.1 | 5.1 | 5.1 | <.01 |
| R241C | 4.4 | 4.4 | 4.4 | 4.4 | 4.4 | <.01 |
| R241H | 6.4 | 6.4 | 6.4 | 6.4 | 6.4 | <.01 |
| V245A | 4.4 | 4.4 | 4.4 | 4.4 | 4.4 | <.01 |
| R261Q | 7.4 | 7.4 | 7.4 | 7.4 | 7.4 | <.01 |
| I306V | 2.6 | 2.6 | 2.6 | 2.6 | 2.6 | <.01 |
| V388M | 1.7 | 1.7 | 1.7 | 1.7 | 1.7 | <.01 |
| E390G | 1.1 | 1.1 | 1.1 | 1.1 | 1.1 | <.01 |
| A395P | 48 | 38 | 27 | 14 | 3.5 | 2.26 |
| A403V | 15 | 12 | 9.8 | 7.3 | 4.7 | .51 |
| R408Q | 9.5 | 9.5 | 9.5 | 9.5 | 9.5 | <.01 |
| Y414C | 6.0 | 6.0 | 6.0 | 6.0 | 6.0 | <.01 |
| D415N | 7.5 | 7.5 | 7.5 | 7.4 | 7.4 | <.01 |
Note.— All calculations were performed on the unphosphorylated dimeric structure (PDB 2PHM).
See the work of Blau et al.47 and the BIOPKU database for classification of mutations. In brief, BH4-responsive mutations are found in patients with BH4-responsive PKU who are functional hemizygous; potentially BH4-responsive mutations are found in responsive patients (typically compound heterozygous) and display residual PAH activity in in vitro expression analysis.
Results
Calculations in FoldX
One of the problems of modeling mutations within a rigid backbone is the excessive penalization due to the contributions of torsional and van der Waals forces. Thus, in some cases, stability changes of >500 kcal/mol are predicted, which are >50 times as large as the average stability of a protein. A protein with such a destabilizing mutation either will not fold or will undergo a local reorganization, which is not easy to predict and model. As a result, when correlations with the experimental data are performed, these mutations will bias the analysis. This is because a mutation predicted to destabilize the protein by 500 kcal/mol will be as deleterious in a patient as one predicted to be 20 kcal/mol, in that both of them will produce unfolding or major reorganizations that will impair folding and/or activity. Modeling such reorganizations is not straightforward, and, to minimize this problem, we have used an empirical approximation based on the capping of the van der Waals clash between two atoms (before correction by solvent accessibility), using different ceilings: 5, 10, 15 (original capping value in FoldX), and 20 kcal/mol. The underlying assumption was that, above a certain value, the protein will either unfold or reorganize itself, with a consequent decrease in stability and activity. Although there is no evidence that local reorganization is deleterious in general, especially when the reorganization is small, conformational reorganization due to structural relaxation of large van der Waals clashes is expected to significantly affect protein conformation.52,53 In fact, this assumption is validated when mutational effects on ΔΔG values at the different van der Waals ceilings are compared with in vitro activities and patient phenotypes for PKU mutations (see below and figs. 1–4).
Figure 1. .

Mutation-dependent destabilization and in vitro residual activity. A, Plot of calculated ΔΔG (in kcal/mol dimer, at a penalty of 5 kcal/mol) versus in vitro residual activity calculated for the structure of the dephosphorylated dimeric form for 79 individual mutations (all compiled in table 1 except A447P, which is not included in this structure). Seventy-four of these mutations (unblackened triangles), grouped in types I, II, and III according to the residual activities (table 1), were included in the calculation of the mean±SD ΔΔG values (blackened circles) for types I, II, and III (4.3±2.8 kcal/mol, 5.7±4.8 kcal/mol, and 14.2±11.7 kcal/mol, respectively). Five mutations were defined as outliers (blackened triangles; R68G is also indicated by an arrow; also see main text for details) and were not included in the calculation of the mean ΔΔG values. The line is only to guide the eye and has no formal significance. B and C, Means±SDs of m and y0 values for the different in vitro activity groups calculated using individual fits for each mutation. P values are obtained from one-way ANOVA; P<.05 is considered statistically significant.
Figure 2. .

ΔΔG values (in kcal/mol dimer) predicted by FoldX with the use of different structural models. Data were obtained from table 5 for the unphosphorylated dimer (based on PDB 2PHM; 74 mutations) (blackened circles), phosphorylated dimer (based on PDB 1PHZ; 74 mutations) (unblackened circles), and the N-terminal truncated tetramer (2PAH; 55 mutations) (blackened triangles). A, Plot of ΔΔG versus in vitro residual activity (types I, II, and II as defined in table 1) (with penalty of 5 kcal/mol). B, Plot of ΔΔG (with penalty of 5 kcal/mol) for PKU mutations classified by phenotypic association found in patients with PKU (as defined in table 2). C and D, Dependence of ΔΔG on different energetic penalties for PKU mutations classified by in vitro activity (C) or association with in vivo phenotypes (D).
Figure 3. .

Effect of the energetic penalizations applied (5–20 kcal/mol) on the ΔΔG values calculated for the unphosphorylated dimeric model (based on PDB 2PHM). A, Seventy-four PKU mutations classified into three mutant types according to their in vitro residual activity (as defined in table 1). Lines are linear fits as follows: type I (blackened triangles), y0=3.9±0.1 kcal/mol, m=0.07±0.01; type II (unblackened circles), y0=3.6±0.1 kcal/mol, m=0.44±0.01; type III (blackened circles), y0=8.4±0.4 kcal/mol, m=1.21±0.03. B, Forty-one PKU mutations classified into three phenotypic groups according to their associated patient phenotypes (as defined in table 2). Lines are linear fits as follows: severe PKU (blackened triangles), y0=7.2±0.5 kcal/mol, m=1.19±0.04; mild PKU (unblackened circles), y0=4.8±0.1 kcal/mol, m=0.19±0.01; and MHP (blackened circles), y0=2.6±0.1 kcal/mol, m=0.00±0.01.
Figure 4. .

Mutation-dependent destabilization and in vivo patient phenotype. A, Calculated effect on ΔΔG (in kcal/mol dimer) for 46 PKU mutants classified by phenotypic groups at a 5-kcal/mol penalty. The mean±SD ΔΔG values (blackened circles) for the three phenotypic groups, calculated using 41 mutations (unblackened triangles), were 2.8±2.2 kcal/mol, 5.7±2.7 kcal/mol, and 13.0±9.5 kcal/mol for MHP (group 1), mild (group 2), and severe (group 3) phenotypes, respectively. Five outliers (blackened triangles) have been removed (see text for details) for the calculation of the mean values. B and C, Means±SDs of m and y0 values for the different phenotypic groups, calculated using individual fits for each mutation. P values are obtained from one-way ANOVA; P<.05 is considered statistically significant.
Both small errors in structure determination and crystal contacts could result in prediction errors. To minimize this problem, we used various available crystal structures to model the mutations—that is, two dimeric structural models from human PAH, residues 19–428 (based on the rat structures PDB 1PHZ and 2PHM), which include the regulatory and catalytic domains and the dimerization motif, and the tetrameric human structure, residues 111–452 (PDB 2PAH), which includes the catalytic and oligomerization domains. Eighty mutations were initially modeled (table 1) (i) both in the dimeric and tetrameric structures (60 mutations at the catalytic domain and dimerization motif), (ii) only in the dimeric structures (19 mutations at the N-terminal regulatory domain), and (iii) only in the tetrameric model (one mutation at the tetramerization motif, i.e., A447P). Mutations were divided into three different groups depending on their in vitro residual activity (table 1). The predicted ΔΔG values are shown in table 5, and the relationship between residual activity and ΔΔG (in kcal/mol), calculated on the structure of the dephosphorylated dimeric form for 79 individual mutations (A447P is not included in this structure), is represented in figure 1A. Correlations obtained with the other structural models are shown in figure 2A. The results show that the structural model chosen for the FoldX analysis does not significantly affect the overall outcome for the different groups (notably, when a 5-kcal/mol energy penalty is used) (fig. 2A). Moreover, if higher energetic penalties are used, all three models provide similar mean ΔΔG values and dependencies on the energetic penalty for the different activity groups (fig. 2C), except for a larger slope in type III for the tetrameric structure. This appears to be the result of a large overdestabilizing effect for some mutations at the 20-kcal/mol penalty with this structure, compared with the dimeric structural models. The large SDs observed in these plots (fig. 2C), especially at increasing energetic penalties, are the consequence of differences in the slopes of these dependencies for individual mutations within each group (see table 6). At an energetic penalty of 20 kcal/mol, ΔΔG values (mean±SD) for types I, II, and III were calculated to be 5.4±5.0 kcal/mol, 12.3±15.1 kcal/mol, and 32.4±41.8 kcal/mol, respectively, whereas, at a 5-kcal/mol penalty, the corresponding values were 4.3±2.8 kcal/mol, 5.7±4.8 kcal/mol, and 14.2±11.7 kcal/mol, respectively.
Table 5. .
Effects on ΔΔG Caused by Mutation, Evaluated Using Four Different Energetic Penalizations in Three Different Structural Models
| 2PHM (Unphosphorylated Dimer) |
||||||||||||||
| Energy Penalty(kcal/mol) |
Linear Fitting |
1PHZ (Phosphorylated Dimer) Energy Penalty(kcal/mol) |
2PAH (Tetramer) Energy Penalty(kcal/mol) |
|||||||||||
| Mutation | 20 | 15 | 10 | 5 | m |
y0 (kcal/mol) |
20 | 15 | 10 | 5 | 20 | 15 | 10 | 5 |
| L41Fa | 9.7 | 7.6 | 5.5 | 3.3 | .43 | 1.2 | 33 | 25 | 18 | 11 | … | … | … | … |
| K42I | 1.5 | 1.5 | 1.5 | 1.5 | 0 | 1.5 | 2.7 | 2.7 | 2.7 | 2.7 | … | … | … | … |
| G46S | 6.6 | 5.9 | 5.1 | 4.3 | .15 | 3.6 | 6.5 | 6.0 | 5.5 | 4.7 | … | … | … | … |
| A47V | 14.0 | 9.3 | 4.9 | .7 | .69 | −1.9 | 12 | 8.6 | 5.1 | 1.6 | … | … | … | … |
| L48S | 5.9 | 5.9 | 5.9 | 5.9 | 0 | 5.9 | 6.3 | 6.3 | 6.3 | 6.3 | … | … | … | … |
| L52S | 4.1 | 4.1 | 4.1 | 4.1 | 0 | 4.1 | 3.6 | 3.6 | 3.6 | 3.6 | … | … | … | … |
| R53H | .5 | .5 | .5 | .5 | 0 | .5 | 1.4 | 1.4 | 1.4 | 1.4 | … | … | … | … |
| D59Y | .1 | .1 | .1 | .1 | 0 | .1 | −.3 | −.3 | −.3 | −.3 | … | … | … | … |
| I65Ta | 10 | 9.3 | 8.4 | 7.5 | .17 | 6.7 | 5.5 | 5.5 | 5.5 | 5.4 | … | … | … | … |
| R68G | 8.7 | 8.7 | 8.7 | 8.7 | 0 | 8.7 | 8.5 | 8.5 | 8.5 | 8.5 | … | … | … | … |
| R68S | 6.3 | 6.3 | 6.3 | 6.3 | 0 | 6.3 | 6.9 | 6.9 | 6.9 | 6.9 | … | … | … | … |
| P69S | 5.4 | 5.4 | 5.4 | 5.4 | 0 | 5.4 | 6.1 | 6.1 | 6.1 | 6.1 | … | … | … | … |
| S70P | 59 | 46 | 32 | 17 | 2.8 | 3.5 | 47 | 37 | 28 | 17 | … | … | … | … |
| E76G | 4.1 | 4.1 | 4.1 | 4.1 | 0 | 4.1 | 3.4 | 3.4 | 3.4 | 3.4 | … | … | … | … |
| S87R | −.9 | −.9 | −.9 | −.9 | 0 | −.9 | −.3 | −.3 | −.3 | −.3 | … | … | … | … |
| T92I | 2.6 | 2.6 | 2.6 | 2.6 | 0 | 2.6 | 2.6 | 2.6 | 2.6 | 2.6 | … | … | … | … |
| G103S | 6.8 | 5.4 | 4.0 | 2.6 | .28 | 1.2 | 6.0 | 5.5 | 4.2 | 2.7 | … | … | … | … |
| A104D | 7.4 | 6.9 | 6.3 | 5.7 | .11 | 5.1 | 8.7 | 7.6 | 6.4 | 5.3 | … | … | … | … |
| P122Q | 15 | 15 | 14 | 13 | .14 | 12.5 | 10 | 10 | 10 | 10 | … | … | … | … |
| T124Ia | 2.5 | 2.5 | 2.5 | 2.5 | 0 | 2.5 | −.3 | −.3 | −.3 | −.3 | 5.7 | 5.7 | 4.5 | 3.6 |
| D143G | 3.7 | 3.7 | 3.7 | 3.7 | 0 | 3.7 | 2.4 | 2.4 | 2.4 | 2.4 | .3 | .3 | .3 | .3 |
| R157N | 11 | 11 | 11 | 11 | 0 | 11 | 10 | 10 | 10 | 10 | 8.5 | 8.5 | 8.5 | 8.5 |
| R158Qb | 2.5 | 2.5 | 2.5 | 2.5 | 0 | 2.5 | 3.7 | 3.7 | 3.7 | 3.7 | 1.0 | 1.0 | 1.0 | 1.0 |
| F161S | 8.9 | 8.9 | 8.9 | 8.9 | 0 | 8.9 | 9.1 | 9.1 | 9.1 | 9.1 | 8.8 | 8.8 | 8.8 | 8.8 |
| G171A | 5.8 | 4.5 | 3.3 | 2.0 | .25 | .8 | 10.0 | 7.7 | 5.4 | 3.1 | 9.6 | 9.6 | 4.8 | 2.4 |
| I174T | 6.8 | 6.8 | 6.8 | 6.8 | 0 | 6.8 | 6.0 | 6.0 | 6.0 | 6.0 | 5.8 | 5.8 | 5.8 | 5.8 |
| R176L | 3.2 | 3.2 | 3.2 | 3.2 | 0 | 3.2 | .4 | .4 | .4 | .4 | .8 | .8 | .8 | .8 |
| E178V | .8 | .8 | .8 | .8 | 0 | .8 | 1.4 | 1.4 | 1.4 | 1.4 | 1.3 | 1.3 | 1.3 | 1.3 |
| W187C | 12 | 12 | 12 | 12 | 0 | 12 | 12 | 12 | 12 | 12 | 10.9 | 10.9 | 10.9 | 10.8 |
| P211T | 5.1 | 5.1 | 5.1 | 5.1 | 0 | 5.1 | 7.6 | 7.6 | 7.6 | 7.6 | 7.0 | 7.0 | 7.0 | 7.0 |
| G218Va | 9.1 | 9.1 | 9.1 | 8.3 | .05 | 8.3 | 15 | 13 | 11 | 9.1 | 5.3 | 5.3 | 5.3 | 5.3 |
| V230Ia | 2.0 | 2.0 | 2.0 | 2.0 | 0 | 2.0 | 9.3 | 7.9 | 6.6 | 5.2 | −.3 | −.3 | −.3 | −.3 |
| R241C | 4.4 | 4.4 | 4.4 | 4.4 | 0 | 4.4 | 2.3 | 2.3 | 2.3 | 2.3 | 3.1 | 2.4 | 3.1 | 3.1 |
| R241Ha | 6.4 | 6.4 | 6.4 | 6.4 | 0 | 6.4 | 3.4 | 3.4 | 3.4 | 3.4 | 2.7 | 2.7 | 2.7 | 2.7 |
| R243Q | 3.1 | 3.1 | 3.1 | 3.1 | 0 | 3.1 | .5 | 1.2 | 1.2 | 1.2 | .9 | 1.0 | .9 | .9 |
| P244L | 19 | 15 | 12 | 8 | .72 | 4.5 | 16 | 14 | 11 | 8.8 | 14 | 14 | 10 | 7.9 |
| V245La | 13.0 | 11 | 8.9 | 6.6 | .43 | 4.5 | 2.3 | 2.3 | 2.3 | 1.3 | .8 | .8 | .8 | .8 |
| V245Ea | 7.8 | 7.8 | 7.8 | 7.8 | 0 | 7.8 | 3.8 | 3.8 | 3.8 | 3.8 | 3.2 | 3.2 | 3.2 | 3.2 |
| V245A | 4.4 | 4.4 | 4.4 | 4.4 | 0 | 4.4 | 3.2 | 3.2 | 3.2 | 3.2 | 3.3 | 3.3 | 3.3 | 2.8 |
| G247Va | 20 | 16 | 12 | 7.7 | .82 | 3.7 | 19 | 15 | 12 | 7.3 | 2.0 | 2.0 | 2.0 | 2.0 |
| R252W | 41 | 33 | 25 | 15 | 1.72 | 7 | 49 | 40 | 30 | 19 | 14 | 14 | 10 | 8.9 |
| R252Qa | 6.3 | 6.3 | 6.3 | 6.3 | 0 | 6.3 | 4.7 | 4.7 | 4.6 | 4.6 | 3.0 | 3.0 | 3.0 | 3.0 |
| R252G | 9.1 | 9.1 | 9.1 | 9.1 | 0 | 9.1 | 8.9 | 8.9 | 8.9 | 8.9 | 7.1 | 7.1 | 7.1 | 7.1 |
| L255Va | 8.4 | 8.4 | 8.4 | 8.4 | 0 | 8.4 | 14 | 11 | 8.9 | 6.6 | 22 | 22 | 16 | 11 |
| L255S | 6.5 | 6.5 | 6.5 | 6.5 | 0 | 6.5 | 8.1 | 8.1 | 8.1 | 8.1 | 7.4 | 7.7 | 7.4 | 7.4 |
| A259T | 38.0 | 30 | 22 | 14 | 1.6 | 6 | 40 | 33 | 27 | 19 | 38 | 38 | 23 | 15 |
| A259V | 46.0 | 36 | 27 | 16 | 1.98 | 6.5 | 50 | 41 | 30 | 17 | 41 | 41 | 24 | 14 |
| R261Q | 7.4 | 7.4 | 7.4 | 7.4 | 0 | 7.4 | 6.0 | 6.0 | 6.0 | 6.0 | 6.4 | 6.4 | 6.4 | 6.4 |
| R270Sa,b | 4.2 | 4.2 | 4.2 | 4.2 | 0 | 4.2 | 8.3 | 8.3 | 8.1 | 7.4 | 4.2 | 4.2 | 4.2 | 4.2 |
| K274E | .4 | .4 | .4 | .4 | 0 | .4 | .6 | .6 | .6 | .6 | .6 | .7 | .6 | .6 |
| Y277Da,b | 1.9 | 1.9 | 1.9 | 1.9 | 0 | 1.9 | 7.3 | 6.6 | 5.9 | 5.2 | 2.3 | 2.3 | 2.3 | 2.3 |
| T278Ia | 10.0 | 9.5 | 8.3 | 5.9 | .27 | 5.1 | 4.6 | 4.5 | 4.4 | 4.3 | 20 | 18 | 9.0 | 4.8 |
| E280Kb | −.2 | −.2 | −.3 | −.3 | .01 | −.4 | 3.2 | 3.2 | 3.2 | 3.2 | −3.8 | .0 | .1 | .1 |
| P281L | 6.7 | 6.7 | 6.7 | 6.2 | 0 | 6.7 | 4.9 | 4.9 | 4.9 | 4.9 | 6.3 | 12 | 8.9 | 7.6 |
| D282Na | 15 | 13 | 12 | 11 | .26 | 9.5 | 16 | 16 | 15 | 14 | 11 | 4.0 | 4.0 | 4.0 |
| I283F | 47 | 37 | 28 | 17 | 1.98 | 7.5 | 58 | 45 | 33 | 20 | 73 | 43 | 25 | 16 |
| L293M | .3 | .3 | .3 | .3 | 0 | .3 | 2.8 | 2.8 | 2.8 | 2.8 | 1.0 | .5 | .5 | .5 |
| F299C | 7.8 | 7.8 | 7.8 | 7.8 | 0 | 7.8 | 8.4 | 8.4 | 8.4 | 8.4 | 7.2 | 6.3 | 6.3 | 6.1 |
| I306V | 2.6 | 2.6 | 2.6 | 2.6 | 0 | 2.6 | 2.6 | 2.6 | 2.6 | 2.6 | 2.1 | 2.1 | 2.0 | 2.0 |
| A309V | 31 | 23 | 16 | 7.6 | 1.54 | .1 | 31 | 24 | 16 | 8.9 | 41 | 20 | 9.3 | 3.6 |
| L311P | 69 | 56 | 43 | 27 | 2.78 | 14 | 68 | 56 | 41 | 25 | 98 | 68 | 41 | 28 |
| A322G | 3.8 | 3.8 | 3.8 | 3.8 | 0 | 3.8 | 3.9 | 3.9 | 3.9 | 3.9 | 2.6 | 2.6 | 2.5 | 2.5 |
| G332V | 190 | 140 | 102 | 54 | 8.92 | 10 | 202 | 150 | 110 | 60 | 300 | 200 | 110 | 61 |
| L333F | 43 | 35 | 26 | 17 | 1.74 | 8.5 | 54 | 47 | 39 | 26 | 59 | 45 | 23 | 13 |
| A342T | 23 | 17 | 12 | 6.3 | 1.1 | .8 | 17 | 14 | 11 | 7.0 | 30 | 21 | 16 | 13 |
| L348Va | 14 | 12 | 9.5 | 6.8 | .48 | 4.5 | 16 | 14 | 11 | 8.5 | 31 | 14 | 8.7 | 6.0 |
| S349P | 35 | 27 | 19 | 11 | 1.6 | 3 | 33 | 25 | 17 | 8.5 | 53 | 33 | 17 | 8.7 |
| S349La | 46 | 37 | 28 | 19 | 1.8 | 10 | 42 | 34 | 26 | 17 | 28 | 28 | 27 | 18 |
| V388M | 1.7 | 1.7 | 1.7 | 1.7 | 0 | 1.7 | 2.8 | 2.8 | 2.8 | 2.8 | 1.9 | 3.0 | 3.0 | 3.0 |
| E390G | 1.1 | 1.1 | 1.1 | 1.1 | 0 | 1.1 | 1.5 | 1.5 | 1.5 | 1.5 | −1.4 | 1.8 | 1.8 | 1.8 |
| S391I | 17 | 15 | 12 | 9.7 | .5 | 7.2 | 17 | 16 | 14 | 12 | 18 | 13 | 11 | 9.5 |
| A395Pa | 48 | 38 | 27 | 14 | 2.26 | 3.5 | 35 | 27 | 20 | 12 | 110 | 44 | 26 | 16 |
| A403Va | 15 | 12 | 9.8 | 7.3 | .51 | 4.7 | 12 | 9.6 | 7.0 | 4.3 | 25 | 13 | 7.0 | 4.0 |
| R408Qa | 9.5 | 9.5 | 9.5 | 9.5 | 0 | 9.5 | 9.0 | 9.0 | 9.0 | 9.0 | 3.4 | 2.6 | 2.5 | 2.5 |
| R408Wa | 110 | 93 | 69 | 42 | 4.56 | 21.5 | 11 | 9.1 | 7.6 | 6.0 | 130 | 61 | 35 | 21 |
| R413Sa | 1.8 | 1.8 | 1.8 | 1.8 | 0 | 1.8 | .6 | .6 | .6 | .6 | −.2 | 4.3 | 4.3 | 4.3 |
| R413Pa | 27 | 23 | 19 | 13 | .92 | 9 | 23 | 18 | 14 | 9.2 | 41 | 25 | 18 | 14 |
| Y414C | 6.0 | 6.0 | 6.0 | 6.0 | 0 | 6.0 | 6.1 | 6.1 | 6.1 | 6.1 | 6.1 | 6.3 | 6.6 | 6.6 |
| D415N | 7.5 | 7.5 | 7.5 | 7.4 | .02 | 7.5 | 9.2 | 9.2 | 9.2 | 9.2 | 5.3 | 7.5 | 7.6 | 7.6 |
| A447P | … | … | … | … | … | … | … | … | … | … | 47.6 | 36 | 25.7 | 14.7 |
Mutations for which energetic prediction (ΔΔG values) depends on the structural model.
Catalytic mutations (R158Q, R270S, Y277D, and E280K); see main text.
Table 6. .
Frequency Distribution and Mean Values of the m and y0 Parameters Determined from the Linear Fitting of ΔΔG versus Energetic Penalizations
|
m |
y0 |
|||||||
| Frequency |
Frequency |
|||||||
| PKU Mutation Classification |
m<.1 | .1<m⩽.3 | m>.3 | Mean±SD | y0⩽3 | 3<y0⩽7 | y0>7 | Mean±SD (kcal/mol) |
| Activitya (% of WT): | ||||||||
| Type I: ⩾50% | .889 | 0 | .111 | .07±.20 | .333 | .500 | .167 | 4.00±2.50 |
| Type II: 10%–50% | .516 | .194 | .290 | .44±.75 | .452 | .419 | .129 | 3.64±3.40 |
| Type III: <10% | .417 | .083 | .500 | 1.18±2.05 | .042 | .291 | .667 | 8.28±3.88 |
| Phenotypeb: | ||||||||
| Group 1: MHP | 1 | 0 | 0 | 0 | .556 | .444 | 0 | 2.71±1.44 |
| Group 2: mild PKU | .177 | .411 | .411 | .15±.25 | .25 | .417 | .083 | 4.72±2.71 |
| Group 3: severe PKU | .333 | .056 | .611 | 1.19±1.14 | .167 | .444 | .389 | 7.16±5.04 |
It was interesting to note that, as well as the activity groups, each individual mutation displayed a linear dependence of ΔΔG on the magnitude of the energetic penalty applied (5–20 kcal/mol) (table 5 and fig. 2C; further highlighted in fig. 3A for the unphosphorylated dimeric model). A linear fitting of these data provides m (slope) and y0 (intercept) values as fitting parameters (compiled in table 5). The m values reflect the intrinsic dependence of ΔΔG on energetic penalties (i.e., the extra energetic penalization due to structural rearrangements caused by mutation), whereas the y0 values appear to be associated with the intrinsic penalization in the local environment of the mutated residue. Thus, in spite of the large SD values shown in figure 2A and 2C, calculation of the frequencies of different m values shows that higher in vitro residual activities are associated with lower m values and, to a lesser extent, with lower y0 values (table 6).
Correlation between In Vitro Residual Activity and Energetic Prediction by FoldX
As figure 1A shows, analysis of the mutation-associated ΔΔG values versus in vitro activity is not straightforward because of the high degree of scatter. However, and as discussed above, if we consider the frequency distribution of the m and y0 values for the individual mutations, as well as the averaged m and y0 values determined for each group (types I, II, and III), we see a negative correlation between the in vitro activities of the mutations and the values of these parameters (table 6 and fig. 1B and 1C). Actually, in some cases, m and y0 values differ significantly between milder groups (types I and II) and the severe group (type III) (fig. 1). Mutations in residues identified as catalytic were considered outliers (blackened triangles in fig. 1A) and were excluded from the calculation of the mean values and the classification into activity groups. These outliers were Y277D9 (affecting hydrogen bonding to substrate), R270S54,55 (affecting hydrogen bonding/electrostatic network at the active site and electrostatic bridge to the substrate), and R158Q and E280K9 (strongly affecting specific activity and reaction coupling).56
An additional outlier is R68G (shown in fig. 1A), since the predicted effect on protein stability (ΔΔG=8.7 kcal/mol) is abnormally high with respect to its very high in vitro residual activity (table 1) and mild folding effect.57 Arg68 is located at the subunit interface in the dimer,58 and a high ΔΔG value for the R68G mutant appears to be partly explained by a relatively high contribution of the subunit binding energy to the change in stability (see the “Effects of Subunit Binding Energies in Oligomer Stability” subsection). Other mutations for which inconsistencies are encountered using different models are marked in table 5. The different energetic values can be explained by local differences in the structure due to crystal packing or by small errors in structure determination arising from ambiguities in the electron density, due to the different resolution of the crystal structures used (i.e., 2.1 Å, 2.6 Å, and 3.1 Å for the phosphorylated and unphosphorylated dimeric forms and the tetrameric form, respectively). There is no objective way of choosing one of the three structural models to individually analyze the mutations, although the resolution of the structures points to the phosphorylated dimeric form as the best structure with which to investigate the energetic effect of the mutations. Moreover, the large number of mutations present at the regulatory domain and which could not be analyzed in the truncated tetrameric domain also supports the selection of this dimeric structure to run the prediction of the phenotype (see below) in all missense mutations, notably after determination that the overall conclusions and the criteria for prediction of these analysis are not dependent on the structural model selected (fig. 2).
Effects of Subunit Binding Energies in Oligomer Stability
The contribution of subunit binding energies within the dimeric structure (monomer-monomer interactions) was also calculated. Only 6 mutations (L48S, 1.25 kcal/mol; R68G, 2.36 kcal/mol; R68S, 3.44 kcal/mol; P69S, 4.48 kcal/mol; Y414C, 5.79 kcal/mol; and D415N, 4.85 kcal/mol) of the 79 studied in this structural model, all located at subunit interfaces, show a contribution >1 kcal/mol (energetic penalty 5 kcal/mol), indicating that the most frequent and largest effects on stability arise from destabilization of the PAH monomer. This is in agreement with a lower abundance of damaging mutations at the dimer or tetramer interfaces, as also shown in previous structural analyses,20,21 suggesting that there is a higher degree of flexibility in these regions than within the monomers. Removal of these subunit binding energies from the FoldX analysis does not significantly modify the overall results—that is, the averaged values varied from 4.3±2.8 kcal/mol to 3.6±2.5 kcal/mol for type I, from 5.7±4.8 kcal/mol to 5.3±3.3 kcal/mol for type II, and from 14.2±11.7 kcal/mol to 14.1±11.2 kcal/mol for type III. However, in some individual cases, the contribution from binding energies is substantial (21% and 27.1% of the total ΔΔG for L48S and R68G, respectively) or predominant (54.6%, 65.5%, 82.9%, and 96.5% of the total ΔΔG for R68S, D415N, P69S, and Y414C, respectively). In those cases, and notably for the outliers R68G and D415N, subtraction of the binding energy results in ΔΔG values that correspond better to in vitro residual activity for the mutant. Thus, in spite of the minor contribution of subunit binding energies to the predicted effects of PKU mutations on PAH stability in general, detailed predictions for individual mutations should include evaluations of these binding energies.
Protein Misfolding and Aggregation
The observed correlation between in vitro residual activity and energetic prediction by FoldX indicates that the destabilization produced by the mutations appears to be the major cause of the reduced in vitro residual activity, as indeed pointed out in reports of previous structural20,21 and expression6,8,9,59 studies. Protein destabilization could lead to unfolding and, in turn, to either degradation or precipitation due to the exposure to the solvent of aggregation-prone regions. It is also possible that some mutations could produce an increase in aggregation tendency, thus exacerbating the destabilizing effect. We therefore checked the effect of the mutations on the aggregation tendency of the protein, using the sequence-based TANGO analysis,60 in spite of the fact that increased degradation, rather than massive aggregation, of the PKU mutants has been suggested as the molecular loss-of-function mechanism in vivo.4,14 It was particularly relevant to investigate the effect for mutations showing ΔΔG values that are highly dependent on the structural model employed (table 5). Only in some cases we did see a significant increase in the aggregation tendency per residue in the mutated sequence (i.e., WT aggregation tendency of 2.9% per residue compared with 3.3% in the case of G247V [data not shown]). Protein destabilization thus appears to be the major determinant of the in vitro residual activity observed.
Correlation between In Vivo Metabolic Phenotypes and Energetic Prediction by FoldX
More interesting than the correlations between predicted ΔΔG values for the mutations and in vitro residual activity is the comparison of the predicted changes in PAH stability with the mutation-associated phenotype in patients. Table 2 includes the mutations from table 1 for which “consistent” mutation-associated phenotypes are found in the literature (according to the criteria described in the “Data and Methods” section). The results of this correlation with use of the dimeric unphosphorylated model are shown in figure 4A, although similar results are obtained if other structural models are used for the correlation (fig. 2B and 2D). Of 46 individual mutations (table 2), 41 were used to calculate the mean ΔΔG for the three in vivo phenotypic groups. Five outliers were not included in the calculation of the average values: two mutations in the MHP group (A47V and D415N) and one in the mild group (P122Q), which display abnormally high energetic penalties, and two in the severe group (Y277D, ΔΔG=1.9 kcal/mol, and E280K, ΔΔG=-0.3 kcal/mol), which are catalytic (see above).
Milder phenotypes have a lower energetic impact on PAH stability (MHP<mild<severe) (fig. 4A). The errors associated with each group in this plot are lower than those observed for the correlations of ΔΔG with the residual activity (fig. 1A). This is most probably because the phenotyping process is more strict (i.e., more care is taken to phenotypically classify a patient-associated mutation) and because the classification is based on a large number of patients, whereas some residual activities have been measured in only one report or have been reported as discrepant residual activities by different reports (therefore, activities intrinsically carry a higher level of noise). It has been documented that genotypes correlate with metabolic phenotype (l-Phe tolerance), whereas in vitro residual activities frequently do not.3 The high frequency of “null” mutations (predicted to abolish activity) makes “functional hemizygosity” a frequent event, allowing us to phenotypically classify many mutations not found in homozygosity.3,32 In vitro expression analysis has, in fact, been a powerful tool for the study of pathogenic mechanisms in PKU and for the rank-ordering of the severity of each mutation. However, different experimental protocols in the transient expression and in activity measurements may contribute to the scatter found in the experimental data.10
The frequency distributions of m and y0 values also differ more among phenotypic groups than among the classifications based on in vitro activities (table 6). Significantly lower m values are associated with milder phenotypes (severe>mild>MHP) (fig. 4B), whereas y0 values of milder groups (mild or MHP) are also significantly lower than those for severe phenotypes (MHP≈mild<severe) (fig. 4C). Interestingly, m and y0 values estimated both from the activity (fig. 1B and 1C) and from the phenotypic classification (fig. 4B and 4C) are relatively similar. These results indicate that FoldX analysis is capable of correlating mutational effects on stability with both high in vitro activities and mild in vivo phenotypes (low m and y0 values), whereas mutations that display low activities are linked to severe phenotypes (high m and/or y0 values).
Phenotypic Prediction
The correlations obtained between energetic penalties and the predicted ΔΔG values (table 6 and fig. 4) suggest that both fitting parameters y0 and m might be simple tools to provide guides for predicting an in vivo phenotype and, to a lesser extent, for in vitro residual activity associated with a certain PKU mutation, as follows: (a) MHP phenotypes, m=0–0.1 and y0⩽3 kcal/mol (associated with in vitro residual activities >50% of WT); (b) mild phenotypes, m=0.1–0.3 and y0=3–7 kcal/mol (associated with in vitro activities ∼10%–50% of WT); and (c) severe phenotypes, m>0.3 and y0>7 kcal/mol (associated with low activity, ⩽10% of WT). As we pointed out above, m values reflect the extra energetic penalization due to structural rearrangements caused by mutation, whereas y0 values are the intrinsic penalization in the local environment of the mutated residue. We therefore treated y0 values as a first prediction of the phenotype associated with a certain mutation, whereas m values are consequently considered when they predict a more severe phenotype (consider, e.g., mutations such as S349P, which is associated with patients with severe phenotypes and which displays y0=3.0 kcal/mol but a very large m value [1.6]; certainly, in cases like this, m values must dominate in the final prediction).
We then calculated the energetic impact (ΔΔG, m, and y0 values) and the predicted phenotype for 238 missense mutations for which in vitro residual activity has not been reported—mostly because these mutations are rare—and the results are summarized in table 7. With use of these criteria, 32.6%, 26.4%, and 41.0% of the 238 missense mutations predicted by FoldX are associated with MHP, mild, and severe phenotypes, respectively.
Table 7. .
Phenotypic Prediction Based on the Calculation of the Energetic Impact (ΔΔG) for 318 PKU Missense Mutations[Note]
| Energetic Penalty(kcal/mol) |
Linear Fitting |
|||||||
| Data Set and Mutation | 20 | 15 | 10 | 5 | m |
y0 (kcal/mol) |
In Vivo Phenotypea | FoldX Predicted Phenotypeb |
| Trainingc: | ||||||||
| F39L | 11.0 | 11 | 11 | 11 | 0 | 11 | … | Severe |
| L41F | 9.7 | 7.6 | 5.5 | 3.3 | .43 | 1.2 | … | Severe |
| K42I | 1.5 | 1.5 | 1.5 | 1.5 | 0 | 1.5 | … | MHP |
| G46S | 6.6 | 5.9 | 5.1 | 4.3 | .15 | 3.6 | … | Mild |
| A47Vd | 14.0 | 9.3 | 4.9 | .7 | .69 | −1.9 | MHP8,32 | Severe |
| L48S | 5.9 | 5.9 | 5.9 | 5.9 | 0 | 5.9 | … | Mild |
| L52S | 4.1 | 4.1 | 4.1 | 4.1 | 0 | 4.1 | … | Mild |
| R53H | .5 | .5 | .5 | .5 | 0 | .5 | … | MHP |
| D59Y | .1 | .1 | .1 | .1 | 0 | .1 | MHP9,36 | MHP |
| I65T | 10 | 9.3 | 8.4 | 7.5 | .17 | 6.7 | Mild3,8,9,21,42 | Mild |
| R68G | 8.7 | 8.7 | 8.7 | 8.7 | 0 | 8.7 | … | Severe |
| R68S | 6.3 | 6.3 | 6.3 | 6.3 | 0 | 6.3 | Mild3,21,36 | Mild |
| P69S | 5.4 | 5.4 | 5.4 | 5.4 | 0 | 5.4 | … | Mild |
| S70P | 59 | 46 | 32 | 17 | 2.8 | 3.5 | … | Severe |
| E76G | 4.1 | 4.1 | 4.1 | 4.1 | 0 | 4.1 | MHP9,36 | Mild |
| S87R | −.9 | −.9 | −.9 | −.9 | 0 | −.9 | MHP21,32,35 | MHP |
| T92I | 2.6 | 2.6 | 2.6 | 2.6 | 0 | 2.6 | MHP21,32 | MHP |
| G103S | 6.8 | 5.4 | 4.0 | 2.6 | .28 | 1.2 | … | Mild |
| A104D | 7.4 | 6.9 | 6.3 | 5.7 | .11 | 5.1 | Mild3,8,32,36 | Mild |
| P122Qd | 15 | 15 | 14 | 13 | .14 | 12.5 | Mild9,36 | Severe |
| T124I | 2.5 | 2.5 | 2.5 | 2.5 | 0 | 2.5 | … | MHP |
| D143G | 3.7 | 3.7 | 3.7 | 3.7 | 0 | 3.7 | … | Mild |
| R157N | 11 | 11 | 11 | 11 | 0 | 11 | … | Severe |
| R158Q | 2.5 | 2.5 | 2.5 | 2.5 | 0 | 2.5 | … | MHP |
| F161S | 8.9 | 8.9 | 8.9 | 8.9 | 0 | 8.9 | … | Severe |
| G171A | 5.8 | 4.5 | 3.3 | 2.0 | .25 | .75 | Mild32,35 | Mild |
| I174T | 6.8 | 6.8 | 6.8 | 6.8 | 0 | 6.8 | Severe9,42 | Mild |
| R176L | 3.2 | 3.2 | 3.2 | 3.2 | 0 | 3.2 | MHP8,21,32,36 | Mild |
| E178V | .8 | .8 | .8 | .8 | 0 | .8 | … | MHP |
| W187C | 12 | 12 | 12 | 12 | 0 | 12 | … | Severe |
| P211T | 5.1 | 5.1 | 5.1 | 5.1 | 0 | 5.1 | Severe32 | Mild |
| G218V | 9.1 | 9.1 | 9.1 | 8.3 | .05 | 8.3 | … | Severe |
| V230I | 2.0 | 2.0 | 2.0 | 2.0 | 0 | 2.0 | MHP3,8,21,36 | MHP |
| R241C | 4.4 | 4.4 | 4.4 | 4.4 | 0 | 4.4 | MHP21 | Mild |
| R241H | 6.4 | 6.4 | 6.4 | 6.4 | 0 | 6.4 | Mild21 | Mild |
| R243Q | 3.1 | 3.1 | 3.1 | 3.1 | 0 | 3.1 | … | Mild |
| P244L | 19 | 15 | 12 | 8.0 | .72 | 4.5 | Mild9,36 | Severe |
| V245L | 13 | 11 | 8.9 | 6.6 | .43 | 4.5 | Severe8 | Severe |
| V245E | 7.8 | 7.8 | 7.8 | 7.8 | 0 | 7.8 | … | Severe |
| V245A | 4.4 | 4.4 | 4.4 | 4.4 | 0 | 4.4 | MHP3,8,21,36 | Mild |
| G247V | 20 | 16 | 12 | 7.7 | .82 | 3.7 | … | Severe |
| R252W | 41 | 33 | 25 | 15 | 1.72 | 7 | Severe9,21,32,36 | Severe |
| R252Q | 6.3 | 6.3 | 6.3 | 6.3 | 0 | 6.3 | Severe21,32,35 | Mild |
| R252G | 9.1 | 9.1 | 9.1 | 9.1 | 0 | 9.1 | Severe21,35 | Severe |
| L255V | 8.4 | 8.4 | 8.4 | 8.4 | 0 | 8.4 | … | Severe |
| L255S | 6.5 | 6.5 | 6.5 | 6.5 | 0 | 6.5 | … | Mild |
| A259T | 38 | 30 | 22 | 14 | 1.6 | 6 | … | Severe |
| A259V | 46 | 36 | 27 | 16 | 1.98 | 6.5 | Severe21 | Severe |
| R261Q | 7.4 | 7.4 | 7.4 | 7.4 | 0 | 7.4 | … | Severe |
| R270S | 4.2 | 4.2 | 4.2 | 4.2 | 0 | 4.2 | … | Mild |
| K274E | .5 | .5 | .5 | .5 | 0 | .5 | … | MHP |
| Y277Dd | 1.9 | 1.9 | 1.9 | 1.9 | 0 | 1.9 | Severe9,36 | MHP |
| T278I | 10 | 9.5 | 8.3 | 5.9 | .27 | 5.1 | … | Mild |
| E280Kd | −.2 | −.2 | −.3 | −.3 | .01 | −.4 | Severe8,32,36 | MHP |
| P281L | 6.7 | 6.7 | 6.7 | 6.2 | 0 | 6.7 | Severe3,8,32,36 | Mild |
| D282N | 15 | 13 | 12 | 11 | .26 | 9.5 | Severe21,35 | Severe |
| I283F | 47 | 37 | 28 | 17 | 1.98 | 7.5 | Severe32 | Severe |
| L293M | .3 | .3 | .3 | .3 | 0 | .3 | … | MHP |
| F299C | 7.8 | 7.8 | 7.8 | 7.8 | 0 | 7.8 | Severe21,32,41 | Severe |
| I306V | 2.6 | 2.6 | 2.6 | 2.6 | 0 | 2.6 | MHP8,21,36 | MHP |
| A309V | 31 | 23 | 16 | 7.6 | 1.54 | .1 | Severe9,42 | Severe |
| L311P | 69 | 56 | 43 | 27 | 2.78 | 14 | Severe21,36 | Severe |
| A322G | 3.8 | 3.8 | 3.8 | 3.8 | 0 | 3.8 | MHP36 | Mild |
| G332V | 190 | 140 | 102 | 54 | 8.92 | 10 | … | Severe |
| L333F | 43 | 35 | 26 | 17 | 1.74 | 8.5 | … | Severe |
| A342T | 23 | 17 | 12 | 6.3 | 1.1 | .8 | Severe8,21 | MHP |
| L348V | 14 | 12 | 9.5 | 6.8 | .48 | 4.6 | Mild3,8,21,36,41 | Severe |
| S349P | 35 | 27 | 19 | 11 | 1.6 | 3 | Severe8,21,32,36 | Severe |
| S349L | 46 | 37 | 28 | 19 | 1.8 | 10 | Severe43 | Severe |
| V388M | 1.7 | 1.7 | 1.7 | 1.7 | 0 | 1.7 | Mild3,8,21,36 | MHP |
| E390G | 1.1 | 1.1 | 1.1 | 1.1 | 0 | 1.1 | Mild3,8,32,36,41 | MHP |
| S391I | 17 | 15 | 12 | 9.7 | .5 | 7.2 | … | Severe |
| A395P | 48 | 38 | 27 | 14 | 2.26 | 3.5 | Severe8 | Severe |
| A403V | 15 | 12 | 9.8 | 7.3 | .51 | 4.7 | Mild3,32,36 | Severe |
| R408Q | 9.5 | 9.5 | 9.5 | 9.5 | 0 | 9.5 | Mild3,8,9,32,36 | Severe |
| R408W | 110 | 93 | 69 | 42 | 4.56 | 21.5 | Severe8,9,32,36 | Severe |
| R413S | 1.8 | 1.8 | 1.8 | 1.8 | 0 | 1.8 | MHP21 | MHP |
| R413P | 27 | 23 | 19 | 13 | .92 | 9 | … | Severe |
| Y414C | 6.0 | 6.0 | 6.0 | 6.0 | 0 | 6.0 | Mild3,8,9,21,36 | Mild |
| D415Nd | 7.5 | 7.5 | 7.5 | 7.4 | .02 | 7.5 | MHP3,32,36 | Severe |
| A447P | 47.6 | 36 | 25.7 | 14.7 | 3.71 | 2.2 | … | Severe |
| Predictede: | ||||||||
| Q20L | 1.9 | 1.9 | 1.9 | 1.9 | <.01 | 1.9 | … | MHP |
| S40L | 3.0 | 3.1 | 3.1 | 3.1 | <.01 | 3.1 | … | MHP |
| L41P | 20.8 | 18.2 | 15.6 | 11.8 | .59 | 9.2 | … | Severe |
| A47E | 3.6 | 3.6 | 3.6 | 3.6 | <.01 | 3.6 | … | Mild |
| R53C | 3.1 | 3.1 | 3.1 | 3.1 | <.01 | 3.1 | … | Mild |
| L54S | 7.4 | 7.4 | 7.4 | 7.4 | <.01 | 7.4 | … | Severe |
| F55L | 4.2 | 3.3 | 3.3 | 3.3 | .05 | 2.9 | … | MHP |
| E56D | 1.3 | 1.3 | 1.3 | 1.3 | <.01 | 1.3 | … | MHP |
| D59V | 3.8 | 3.8 | 3.8 | 3.8 | <.01 | 3.8 | … | Mild |
| N61D | −1.1 | −1.1 | −1.1 | −1.1 | <.01 | −1.1 | … | MHP |
| N61K | −.3 | −.3 | −.3 | −.3 | <.01 | −.3 | MHP36 | MHP |
| T63P | 5.7 | 4.3 | 2.9 | 1.4 | .28 | 0 | … | Mild |
| H64N | −2.3 | −2.3 | −2.3 | −2.3 | <.01 | −2.3 | … | MHP |
| I65N | 7.4 | 8.6 | 8.6 | 8.6 | <.01 | 9.2 | … | Severe |
| I65S | 9.2 | 9.2 | 9.2 | 9.2 | <.01 | 9.2 | … | Severe |
| I65V | .8 | .8 | .8 | .8 | <.01 | .8 | … | MHP |
| S67P | 10 | 8.3 | 6.3 | 4.2 | .39 | 2.3 | … | Severe |
| R71H | 7.4 | 7.4 | 7.4 | 7.4 | <.01 | 7.4 | … | Severe |
| E76A | 2.7 | 2.7 | 2.4 | 2.7 | <.01 | 2.5 | … | MHP |
| E78K | 6.1 | 6.0 | 6.0 | 6.0 | <.01 | 6.0 | … | Mild |
| T81P | 12 | 11.2 | 9.9 | 7.1 | .32 | 6.0 | Severe61 | Severe |
| D84Y | 3.3 | 3.3 | 2.0 | 1.7 | .12 | 1.0 | Severe21,35,61 | MHP |
| P89S | 2.8 | 2.8 | 2.8 | 2.7 | <.01 | 2.8 | … | MHP |
| I94S | 5.8 | 5.8 | 5.8 | 5.7 | <.01 | 5.7 | Severe21,35 | Mild |
| I95F | 29.3 | 23.1 | 18.6 | 13.3 | 1.05 | 8.0 | … | Severe |
| L98S | 6 | 6 | 6 | 6 | <.01 | 6.0 | … | Mild |
| I102T | 6.5 | 6.5 | 6.5 | 6.5 | <.01 | 6.5 | … | Mild |
| S110L | 7.4 | 6.2 | 5.1 | 3.5 | .25 | 2.4 | … | Mild |
| D129G | 11.6 | 10.9 | 10.9 | 10.9 | .04 | 10.6 | Mild36,42 | Severe |
| D129Y | 9.0 | 8.8 | 8.8 | 8.7 | .02 | 8.6 | … | Severe |
| D145Vf | 1.5 | 1.6 | 1.6 | 1.6 | <.01 | 1.5 | Mild61,62 | MHP |
| H146Y | 1.1 | 1.2 | 1.2 | 1.2 | <.01 | 1.3 | … | MHP |
| P147S | 2.9 | 2.9 | 2.9 | 2.9 | <.01 | 2.9 | … | MHP |
| G148S | 35.3 | 30.2 | 23.9 | 16.2 | 1.27 | 10.5 | … | Severe |
| D151E | 6.1 | 6.1 | 6.1 | 6.1 | <.01 | 6.1 | … | Mild |
| D151G | 9.7 | 9.7 | 9.7 | 9.7 | <.01 | 9.7 | Severe36 | Severe |
| D151H | 8.8 | 8.8 | 8.8 | 8.8 | <.01 | 8.8 | … | Severe |
| Y154H | 6.7 | 6.6 | 6.6 | 6.6 | <.01 | 6.6 | … | Mild |
| Y154N | 7.2 | 7.1 | 7.1 | 7.1 | <.01 | 7.1 | … | Severe |
| R155H | 2.0 | 2.0 | 2.0 | 2.0 | <.01 | 2.0 | … | MHP |
| R155P | 33.7 | 30.0 | 22.6 | 13.8 | 1.34 | 8.2 | … | Severe |
| R157K | 8.6 | 8.6 | 8.6 | 8.6 | <.01 | 8.6 | … | Severe |
| R157I | 8.6 | 8.8 | 8.8 | 8.5 | <.01 | 8.7 | … | Severe |
| R158P | 45.6 | 35.8 | 26 | 16.2 | 1.96 | 6.4 | … | Severe |
| R158Wf | 4.5 | 4.5 | 4.5 | 4.5 | <.01 | 4.5 | Severe61 | Mild |
| Q160P | 30.3 | 25.2 | 19.8 | 12.7 | 1.16 | 7.5 | … | Severe |
| I164T | 3.6 | 3.1 | 3.1 | 3.1 | .03 | 2.9 | … | MHP |
| I164V | 2.7 | 2.3 | 2.3 | 2.3 | .03 | 2.0 | … | MHP |
| A165P | 32.0 | 24.5 | 16.7 | 9.0 | 1.54 | 1.3 | … | Severe |
| A165T | −2.2 | −2.7 | −2.7 | −2.7 | .03 | −2.9 | Severe36 | MHP |
| N167I | 4.2 | 3.7 | 3.7 | 3.7 | .03 | 3.5 | Mild36 | Mild |
| N167S | .5 | 0 | 0 | 0 | .03 | −.2 | … | MHP |
| Y168H | 13.5 | 11.3 | 11.3 | 11.3 | .13 | 10.2 | … | Severe |
| R169H | 3.1 | 2.1 | 2.1 | 2.1 | .06 | 1.7 | MHP36 | MHP |
| H170D | 2.6 | 1.6 | 1.6 | 1.6 | .06 | 1.2 | … | MHP |
| H170Q | −1.4 | −1.9 | −1.9 | −1.9 | .03 | −2.09 | … | MHP |
| H170R | 1.5 | 1 | 1 | 1 | .03 | .8 | … | MHP |
| G171R | 9.9 | 8.1 | 5.9 | 3.2 | .45 | 1.2 | … | Severe |
| P173T | −.3 | −.7 | −.8 | −.9 | .03 | −1.1 | … | MHP |
| I174V | 2.6 | 2.2 | 2.1 | 2.1 | .03 | 1.9 | … | MHP |
| P175A | 4.2 | 3.8 | 3.8 | 3.8 | .03 | 3.6 | … | Mild |
| R176P | −.4 | −.8 | −.8 | −.8 | .03 | −1.0 | … | MHP |
| R176Q | 2.0 | 1.6 | 1.6 | 1.6 | .03 | 1.3 | … | MHP |
| V177L | 1.8 | 1.3 | 1.3 | 1.3 | .03 | 1.1 | MHP61 | MHP |
| V177M | 2.8 | 2.3 | 2.3 | 2.3 | .03 | 2.1 | … | MHP |
| E178G | .1 | .1 | .1 | .1 | 0 | .1 | MHP21,35,61 | MHP |
| Y179H | 7.5 | 7.1 | 7.1 | 7.1 | .02 | 6.9 | … | Mild |
| E182G | 3.8 | 3.3 | 3.3 | 3.3 | .03 | 3.1 | … | Mild |
| E183Q | 7.9 | 7.3 | 7.3 | 7.1 | .04 | 6.9 | … | Mild |
| W187R | 11.4 | 11.8 | 11.8 | 11.8 | <.01 | 12 | … | Severe |
| V190A | 2.3 | 2.3 | 2.3 | 2.3 | 0 | 2.3 | … | MHP |
| L194P | 31.5 | 25.6 | 19.2 | 12.9 | 1.24 | 6.7 | Severe21,32,35 | Severe |
| H201R | 1.4 | 1.7 | 1.7 | 1.7 | <.01 | 1.9 | … | MHP |
| H201Y | 2.4 | 2.9 | 2.9 | 3 | <.01 | 3.2 | … | Mild |
| C203Y | 21.3 | 215 | 165 | 113 | 10.2 | 62.3 | … | Severe |
| E205A | 0 | .4 | .4 | .4 | <.01 | .6 | Severe62 | MHP |
| E205K | −1.3 | −1.0 | −1.7 | −2.3 | .07 | −2.4 | … | MHP |
| Y206C | 5.5 | 5.8 | 5.8 | 5.8 | <.01 | 6.0 | … | Mild |
| Y206D | 7.0 | 19.3 | 17.0 | 12.9 | .64 | 10.0 | … | Severe |
| N207S | −2.3 | −1.9 | −1.9 | −1.9 | <.01 | −1.7 | … | MHP |
| N207D | 5.4 | 5.7 | 5.7 | 5.7 | <.01 | 5.9 | … | Mild |
| L212P | 13.2 | 11.6 | 9.8 | 6.7 | .43 | 5.0 | … | Severe |
| L213P | 23.7 | 22.1 | 19.0 | 13.7 | .66 | 11.4 | … | Severe |
| C217G | 3.0 | 3.4 | 3.4 | 3.4 | <.01 | 3.6 | … | Mild |
| C217R | 16.6 | 13.9 | 10.9 | 8.0 | .58 | 5.1 | … | Severe |
| C217Y | 156 | 121 | 85.7 | 50.3 | 7.05 | 15.1 | … | Severe |
| E221G | 3.0 | 3.4 | 3.4 | 3.4 | <.01 | 3.6 | … | Mild |
| D222G | 2.2 | 2.6 | 2.6 | 2.6 | <.01 | 2.8 | … | MHP |
| D222V | 1.0 | 1.4 | 1.4 | 1.4 | <.01 | 1.6 | Severe32 | MHP |
| I224M | 2.8 | 3.2 | 3.2 | 3.2 | <.01 | 3.4 | … | Mild |
| I224T | 6.3 | 6.6 | 6.6 | 6.6 | <.01 | 6.8 | … | Mild |
| P225A | 5.3 | 5.7 | 5.7 | 5.7 | <.01 | 5.9 | … | Mild |
| P225R | 67.4 | 55.0 | 42.0 | 28.4 | 2.6 | 15.8 | … | Severe |
| P225T | 16.6 | 14.4 | 11.7 | 8.7 | .53 | 6.2 | … | Severe |
| Q226H | .3 | 1.5 | 1.4 | 1.5 | <.01 | 2.1 | … | MHP |
| V230G | 7.0 | 8.2 | 8.2 | 8.2 | <.01 | 8.8 | … | Severe |
| S231F | 19.5 | 18.0 | 15.3 | 10.4 | .6 | 8.3 | … | Severe |
| S231P | 56 | 45 | 35 | 23 | 12.5 | 2.2 | Severe21,35 | Severe |
| F233L | 3.3 | 4.5 | 4.5 | 4.5 | <.01 | 5.1 | … | Mild |
| Q235P | 37.6 | 30.5 | 22.6 | 14.1 | 1.57 | 6.6 | … | Severe |
| T238P | 19.4 | 17.9 | 15.2 | 12.5 | .47 | 10.4 | … | Severe |
| G239A | 48.6 | 40.2 | 29.1 | 17.2 | 2.11 | 7.5 | … | Severe |
| G239D | 51.9 | 43.2 | 33.3 | 21.3 | 2.03 | 12.0 | … | Severe |
| G239S | 42.1 | 35.8 | 28.2 | 18.7 | 1.56 | 11.8 | Severe21,32,35 | Severe |
| G239V | 97.9 | 82.2 | 64.3 | 41.2 | 3.76 | 24.4 | … | Severe |
| F240S | 6.8 | 8.1 | 8.1 | 8.1 | <.01 | 8.8 | Severe62 | Severe |
| F240V | 12.6 | 11.1 | 8.4 | 5.8 | .46 | 3.7 | … | Severe |
| R241L | 2.6 | 3.8 | 3.8 | 3.8 | <.01 | 4.4 | … | Mild |
| L242F | 38.7 | 30.4 | 21.0 | 11.6 | 1.8 | 2.8 | … | Severe |
| R243L | −1.2 | .1 | .1 | .1 | <.01 | .8 | … | MHP |
| V245I | 3.1 | 4.8 | 4.8 | 3.4 | <.01 | 4.2 | … | Mild |
| A246D | 9.6 | 9.5 | 7.7 | 5.9 | .26 | 4.9 | … | Mild |
| A246V | 13.9 | 13.5 | 11.4 | 9.2 | .32 | 8.0 | Mild21,35 | Severe |
| G247D | 5.0 | 4.9 | 3.3 | 1.6 | .24 | .7 | … | Mild |
| G247R | 3.9 | 3.6 | 1.7 | −.2 | .28 | −1.3 | … | Mild |
| G247S | 3.9 | 4.0 | 2.6 | 1.1 | .2 | .4 | … | Mild |
| L248P | −3.0 | −1.4 | −1.4 | −1.4 | <.01 | −.6 | … | MHP |
| L248R | −2.5 | −1.0 | −1.0 | −.9 | <.01 | −.21 | … | MHP |
| L249F | 28.9 | 23.4 | 16.2 | 8.7 | 1.36 | 2.3 | Severe61 | Severe |
| L249H | 22.3 | 19.6 | 15.2 | 10.9 | .77 | 7.4 | … | Severe |
| F254I | .2 | 1.9 | 1.9 | 1.9 | <.01 | 2.7 | … | MHP |
| G257C | .9 | 2.7 | 2.7 | 2.7 | <.01 | 2.3 | … | MHP |
| G257D | 21.7 | 18.0 | 12.6 | 7.2 | .98 | 2.6 | … | Severe |
| G257S | −1.6 | .1 | .1 | −.1 | <.01 | .8 | … | MHP |
| G257V | 28.2 | 23.7 | 17.4 | 10.0 | 1.12 | 5.3 | … | Severe |
| R261P | 35.7 | 31.8 | 26.5 | 21.2 | .98 | 16.6 | Severe21,35 | Severe |
| F263L | 7.8 | 8.2 | 7.0 | 5.7 | .15 | 5.3 | … | Mild |
| H264L | 5.1 | 5.0 | 3.2 | 1.4 | .26 | .5 | … | Mild |
| C265G | 3.9 | 5.5 | 5.5 | 5.5 | <.01 | 6.4 | … | Mild |
| C265Y | 138 | 108 | 74.5 | 38.8 | 6.62 | 7.1 | … | Severe |
| T266E | 10.2 | 9.6 | 7.4 | 5.1 | .35 | 3.7 | … | Mild |
| Q267E | 3.9 | 4.7 | 4.7 | 4.7 | <.01 | 5.1 | … | Mild |
| Q267H | 11.7 | 10.0 | 7.5 | 5.5 | .42 | 3.4 | … | Severe |
| Y268H | 6.2 | 7.1 | 7.1 | 7.1 | <.01 | 7.5 | … | Severe |
| I269L | 10.9 | 11.6 | 11.6 | 10.4 | .03 | 10.8 | … | Severe |
| I269Nf | 2.7 | 3.4 | 3.4 | 3.4 | <.01 | 3.7 | Severe21,35 | Mild |
| R270K | 3.5 | 2.8 | 2.4 | 2.4 | 1.85 | .1 | Severe62 | MHP |
| H271Y | −2 | −1.2 | −1.2 | −1.2 | <.01 | −.8 | … | MHP |
| S273F | 1.2 | 1.2 | 1.2 | 1.2 | <.01 | 1.2 | … | MHP |
| P275L | 23.6 | 20.7 | 17.8 | 14.3 | .62 | 11.4 | … | Severe |
| P275Rf | 5.4 | 5.4 | 5.4 | 5.9 | <.01 | 5.9 | Severe62 | Mild |
| P275S | 7.3 | 7.3 | 7.3 | 7.3 | <.01 | 7.3 | … | Severe |
| M276I | 1.2 | 1.2 | 1.2 | 1.2 | <.01 | 1.2 | … | MHP |
| M276K | 1.2 | 1.2 | 1.2 | 1.2 | <.01 | 1.2 | … | MHP |
| M276R | −.3 | −.3 | −.3 | −.3 | <.01 | −.3 | … | MHP |
| M276V | 4.2 | 4.2 | 4.2 | 4.2 | <.01 | 4.2 | … | Mild |
| Y277C | 2.7 | 2.7 | 2.7 | 2.6 | <.01 | 2.6 | … | MHP |
| T278A | 2.8 | 2.7 | 2.7 | 2.7 | <.01 | 2.7 | … | MHP |
| T278N | 2.3 | 2.3 | 2.3 | 2.3 | <.01 | 2.3 | … | MHP |
| E280G | 6.4 | 5.9 | 5.9 | 5.9 | .03 | 5.7 | … | Mild |
| P281A | 4.4 | 4.4 | 4.4 | 4.4 | <.01 | 4.4 | … | Mild |
| P281S | 3.9 | 3.9 | 3.9 | 3.9 | <.01 | 3.9 | … | Mild |
| D282G | 14.3 | 12.6 | 12.6 | 12.6 | .1 | 11.8 | … | Severe |
| I283N | 5.5 | 5.5 | 5.5 | 5.5 | <.01 | 5.5 | … | Mild |
| H285Yf | .5 | .7 | .4 | .2 | .02 | .2 | Severe21,35 | MHP |
| E286K | 10.1 | 10.7 | 10.7 | 10.6 | <.01 | 10.9 | … | Severe |
| L288F | 34.8 | 29.4 | 22.9 | 13.0 | 1.44 | 7.1 | … | Severe |
| H290R | 16.7 | 12.8 | 9.0 | 5.1 | .77 | 1.2 | … | Severe |
| R297C | 9.0 | 9.0 | 9.0 | 9.0 | <.01 | 9.0 | … | Severe |
| R297H | 4.1 | 4.1 | 4.1 | 4.1 | <.01 | 4.1 | … | Mild |
| A300S | −.5 | −.5 | −.5 | −.5 | <.01 | −.5 | MHP21,35,36,61–63 | MHP |
| A300V | 48.3 | 37.5 | 26.7 | 14.1 | 2.27 | 3.3 | … | Severe |
| S303A | .6 | .6 | .5 | .5 | <.01 | .5 | … | MHP |
| S303P | 35.2 | 28.4 | 20.3 | 11.0 | 1.61 | 3.6 | … | Severe |
| Q304R | 7.2 | 7.2 | 7.2 | 7.2 | <.01 | 7.2 | … | Severe |
| L308F | 48.1 | 41.3 | 33.3 | 24.2 | 1.59 | 16.8 | … | Severe |
| L308V | 6.4 | 5.5 | 4.7 | 4.2 | .15 | 3.4 | … | Mild |
| A309D | 24.1 | 21.4 | 18.5 | 13.4 | .7 | 10.6 | … | Severe |
| S310F | 134 | 106 | 77.1 | 43.6 | 6 | 15.2 | … | Severe |
| S310Y | 157 | 126 | 92.3 | 56.4 | 6.71 | 24.1 | … | Severe |
| G312V | 66.6 | 52.3 | 38.0 | 23.2 | 2.89 | 8.9 | … | Severe |
| A313V | 36.9 | 27.6 | 19.5 | 10.8 | 1.73 | 2.1 | … | Severe |
| A313T | 29 | 22 | 16 | 8.2 | 1.7 | 1.4 | … | Severe |
| P314H | 10.2 | 10.2 | 10.2 | 10.2 | <.01 | 10.2 | … | Severe |
| P314S | 7.6 | 7.6 | 7.6 | 7.6 | <.01 | 7.6 | … | Severe |
| P314T | 8.5 | 8.0 | 8.0 | 8.0 | .03 | 7.7 | … | Severe |
| D315Y | .9 | .9 | .9 | .9 | <.01 | .9 | … | MHP |
| I318T | 2.2 | 2.2 | 2.2 | 2.2 | <.01 | 2.2 | Severe62 | MHP |
| K320N | 1.8 | 1.8 | 1.8 | 1.8 | <.01 | 1.8 | … | MHP |
| A322T | 15.2 | 12.7 | 10.1 | 6.5 | .58 | 3.9 | … | Severe |
| Y325C | 9.5 | 9.4 | 9.4 | 9.4 | <.01 | 9.4 | … | Severe |
| F327Lf | 11.4 | 10.2 | 8.9 | 7.7 | .25 | 6.5 | Severe21,35 | Mild |
| T328A | 12.4 | 10.1 | 7.7 | 7.2 | .36 | 4.9 | … | Severe |
| E330D | 8.3 | 8.4 | 8.4 | 8.4 | <.01 | 8.4 | … | Severe |
| F331Cf | 5.8 | 5.8 | 5.8 | 5.8 | <.01 | 5.8 | Severe61 | Mild |
| F331L | 10.5 | 10.5 | 10.5 | 9.8 | .05 | 9.8 | Severe21,35 | Severe |
| G332E | 114 | 89.4 | 65.1 | 38.5 | 5.02 | 14.1 | … | Severe |
| C334S | 12.4 | 23.9 | 21.5 | 17.6 | .63 | 14.7 | … | Severe |
| E336R | 1.8 | 1.7 | 1.7 | 1.7 | <.01 | 1.7 | … | MHP |
| G337V | 6.0 | 5.5 | 5.0 | 4.3 | .11 | 3.9 | … | Mild |
| D338Y | −.4 | −.4 | −.4 | −.4 | <.01 | −.4 | Mild32 | MHP |
| I340T | 3.5 | 3.5 | 3.5 | 3.5 | <.01 | 3.5 | … | Mild |
| K341R | −1.6 | −1.7 | −1.7 | −1.7 | <.01 | −1.8 | … | MHP |
| K341T | 5.5 | 5.5 | 5.5 | 5.5 | <.01 | 5.5 | … | Mild |
| A342P | 18.7 | 15.8 | 11.8 | 7.7 | .74 | 4.3 | … | Severe |
| Y343C | .6 | .6 | .6 | .6 | <.01 | .6 | … | MHP |
| Y343D | 9.6 | 9.6 | 9.6 | 9.6 | <.01 | 9.6 | … | Severe |
| Y343F | −.2 | 175 | 127 | 80 | 9.47 | 32.7 | … | Severe |
| G344R | 245 | 191 | 137 | 80.7 | 10.9 | 26.7 | … | Severe |
| G344S | 99.7 | 76.7 | 52.7 | 28.0 | 4.78 | 4.5 | Mild21,35 | Severe |
| G344V | 199 | 156 | 112 | 64 | 8.98 | 20.5 | … | Severe |
| A345S | 1.7 | 1.7 | 1.7 | 1.7 | <.01 | 1.7 | … | MHP |
| A345T | 25.6 | 22.9 | 20.0 | 12.6 | .84 | 9.8 | … | Severe |
| G346R | 53.3 | 42.4 | 31.8 | 19.9 | 2.22 | 9.1 | Severe21,35 | Severe |
| L347F | 65.3 | 51.8 | 38.5 | 23.8 | 2.76 | 10.4 | Mild61 | Severe |
| S349A | −.3 | −.4 | −.4 | −.4 | <.01 | −.4 | … | MHP |
| S350T | 2.3 | 2.3 | 2.4 | 2.4 | <.01 | 2.4 | … | MHP |
| S350Y | 2.8 | .8 | −.1 | −.4 | .21 | −1.9 | … | Mild |
| G352C | −.4 | −.4 | −.4 | −.4 | <.01 | −.4 | … | MHP |
| G352Rf | −2.6 | −2.5 | −2.5 | −2.6 | <.01 | −2.5 | Severe21,35 | MHP |
| Y356H | 7.2 | 7.2 | 7.2 | 6.5 | .05 | 6.5 | … | Mild |
| C357G | 2.8 | 2.8 | 2.8 | 2.8 | <.01 | 2.8 | Severe62 | MHP |
| P362T | 18.4 | 18.4 | 18.4 | 17.0 | .08 | 17.0 | … | Severe |
| K363N | 2.4 | 2.4 | 2.4 | 2.4 | <.01 | 2.4 | … | MHP |
| P366H | 2.4 | 2.4 | 2.4 | 2.4 | <.01 | 2.4 | MHP64 | MHP |
| L367P | 16.7 | 162 | 125 | 86.5 | 7.55 | 49.0 | … | Severe |
| K371R | .1 | −.1 | −.1 | −.1 | .01 | −.2 | … | MHP |
| T372S | 3.1 | 3.1 | 3.1 | 3.1 | <.01 | 3.1 | … | Mild |
| A373T | 19 | 16 | 13 | 9.8 | 6.8 | .6 | … | Severe |
| Y377C | 6.3 | 6.3 | 6.3 | 6.3 | <.01 | 6.3 | … | Mild |
| T378S | .4 | 2.8 | 2.8 | 2.8 | <.01 | 2.8 | … | MHP |
| T380M | .6 | .6 | .6 | .6 | <.01 | .6 | MHP32,35,61,63 | MHP |
| Y386C | 8.3 | 8.3 | 8.3 | 8.1 | .01 | 8.1 | … | Severe |
| Y387H | 7.3 | 7.3 | 7.3 | 7.3 | <.01 | 7.3 | … | Severe |
| V388L | 1.8 | 1.8 | 1.8 | 1.8 | <.01 | 1.8 | … | MHP |
| D394A | .2 | .2 | .2 | .1 | .01 | .1 | … | MHP |
| D394H | .3 | .3 | .3 | .3 | <.01 | .3 | … | MHP |
| A395D | 19.5 | 16.0 | 12.6 | 8.9 | .7 | 5.4 | … | Mild |
| A395G | 4.2 | 4.2 | 4.2 | 4.2 | <.01 | 4.2 | … | Mild |
| R400K | −.1 | −.1 | −.2 | −.1 | <.01 | −.1 | … | MHP |
| R400T | 1.4 | 1.4 | 1.4 | 1.4 | <.01 | 1.4 | … | MHP |
| F402L | 4.5 | 4.5 | 4.5 | 4.5 | <.01 | 4.5 | … | Mild |
| I406T | 4.6 | 4.6 | 4.6 | 4.6 | <.01 | 4.6 | … | Mild |
| P407L | 4.1 | 4.1 | 4.1 | 4.1 | <.01 | 4.1 | … | Mild |
| P407S | 3.7 | 3.7 | 3.7 | 3.7 | <.01 | 3.7 | Mild36 | Mild |
| F410C | 7.1 | 7.1 | 7.1 | 7.1 | <.01 | 7.1 | … | Severe |
| F410S | 6.9 | 6.9 | 6.9 | 6.9 | <.01 | 6.9 | … | Mild |
| R413C | 3.0 | 3.1 | 3.1 | 2.8 | .02 | 2.8 | … | MHP |
| Y417H | 7.3 | 7.1 | 7.1 | 7.2 | <.01 | 7.2 | … | Severe |
| Y417N | 4.7 | 4.7 | 4.7 | 4.7 | <.01 | 4.7 | … | Mild |
| T418P | 9.6 | 7.2 | 4.8 | 3.8 | .39 | 1.4 | … | Severe |
| E422K | 1.3 | 1.3 | 1.3 | 1.3 | <.01 | 1.3 | … | MHP |
| L424S | 2.3 | 2.1 | 2.1 | 2.1 | <.01 | 2.0 | … | MHP |
| L430P | 57.1 | 45.9 | 29.0 | 31.9 | 1.85 | 17.8 | … | Severe |
| A434D | 16.9 | 16.4 | 14.8 | 7.0 | .62 | 6.0 | … | Severe |
| A447D | 9.01 | 9.1 | 8.7 | 4.1 | .3 | 3.9 | … | Mild |
Note.— Mutations were compiled from the mutation map (updated August 3, 2006) available at PAHdb. All calculations were performed on the unphosphorylated dimeric structure (PDB 2PHM) except for mutations L430P, A434D, A447D, and A447P, for which calculations were performed on the tetrameric form (PDB 2PAH). For the sake of simplicity, SEs associated with m and y0 values estimated from linear fittings are not shown (typically <5% of the estimated mean values). The first step of the prediction is to provide a preliminary phenotype based on the y0 value (which corresponds to the intrinsic penalization in the local environment of the mutated residue); the second step is either to confirm the phenotype if the m value (which corresponds to the intrinsic dependence of ΔΔG on energetic penalties, related to the extra energetic penalization due to structural rearrangements on mutation) provides a similar or milder phenotype than the y0 value does or to reevaluate the prediction if the m value provides a more severe phenotype than the y0 value does. In the latter case, protein reorganization dominates over local stability effects of the mutation.
Metabolic phenotypes reported in the literature.
Phenotypes predicted on the basis of the following criteria based on y0 and m values: MHP, y0⩽3 kcal/mol, m⩽.1; mild, y0=3–7 kcal/mol, m=.1–.3; severe, y0>7 kcal/mol, m>.3.
Mutations expressed in vitro in a eukaryote expression system.
Mutations withdrawn from the testing data set prior to overall analysis.
Mutations without reported in vitro expression results.
Mutations predicted to be catalytic.
Validation of FoldX-Based Prediction of Metabolic Phenotypes
First, we cross-validated the predictive power of FoldX for the mutation associated with in vivo phenotypes, by testing our m and y0 predictive guidelines on the mutants for which phenotypic classification has been reported (referred to as the “training data set”) (tables 2 and 7) and which were actually used to calculate the cutoff values for m and y0 parameters for the various phenotypic groups' criteria (fig. 4) (see also the “Phenotypic Prediction” subsection). Of the MHP mutations, 54.5% were predicted to be associated with the MHP phenotype, whereas the remaining 45.5% were predicted to be associated with mild PKU phenotypes. In the mild PKU phenotype group, 50.0%, 16.7%, and 33.3% were predicted to be associated with mild, MHP, and severe PKU phenotypes, respectively, whereas 77.8% of the severe mutations were correctly predicted as severe PKU (the remaining 22.2% were predicted as mild PKU). Thus, although there is some degree of uncertainty, we are able to conclude that our FoldX-based analysis is a satisfactory tool that can correctly predict a significant fraction of the phenotypes associated with PKU mutations. Uncertainty may be related to different factors, such as imprecision in the energy calculation by FoldX or errors in the structural models used, interindividual phenotypic variability of some mutations, and differences in potential modifiers of the metabolic outcome.15 Moreover, there is no absolute consensus in the international guidelines for phenotypic classification.3
Further cross-validation was attempted by analyzing the correlation of the predicted PKU phenotype associated with some of the 238 mutations for which in vitro residual activity has not been reported (i.e., the predicted data set) (table 7). For 38 of these mutations, we have found associations with a certain phenotype in the literature (shown in table 7). All the mutations reported as MHP mutations (n=7) were correctly predicted by our FoldX approach. However, the predictive power of FoldX was significantly lower in the mild and severe PKU groups. Structural analysis shows that, for nine mutations (eight in the severe PKU group and one in the mild PKU group) for which severity in metabolic phenotype is underestimated by FoldX analysis, the mutation likely affects PAH catalytic properties. The R158W mutation is expected to affect catalytic efficiency and coupling, as described for the R158Q mutant.56 The H285Y mutation would directly affect iron coordination, as described for the H285S mutant.65 I269N and R270K are expected to distort important ionic interactions toward l-Phe at the substrate binding site.66 F331C and F327L are located at the substrate and cofactor binding sites, respectively, where they likely affect proper l-Phe and BH4 binding. D145V, P275R, and G352R are located at the entrance of the l-Phe binding site, and these mutations might also affect proper substrate binding. If these mutations are withdrawn from the predicted data set, FoldX prediction is improved in the severe group (from 39.1% to 60% correct prediction of severe PKU phenotypes) but not so much in the mild group (from 25.0% to 29.1%). A possible explanation of the higher prediction success for the mutations in the training data set is that they are among the most frequent PKU mutations, and genotype-phenotype correlations have been reported by different groups that used several patients for each mutation. In the predicted data set, often only one report or even one patient is reported (table 7); therefore, genotype-phenotype correlations based on patient phenotypes are not so robust. For the rest of the ∼200 mutations in the predicted data set, little or no information about homozygotic or functional hemizygotic patients is found in the PAHdb or BIOPKU databases; thus, no reliable clinical phenotype can be assigned and compared with the FoldX-predicted phenotypes.
BH4-Responsive Mutants
BH4-responsive HPA/PKU is a PKU subtype that has recently been put forward to designate the disease in a subgroup of patients who respond to supplementation with high doses of the natural cofactor BH4 (>30% l-Phe reduction in plasma within 24 h after BH4 administration).47,67 These patients carry specific mutations in the PAH gene leading to either single amino acid alterations or small inframe insertions or deletions. Table 4 shows ΔΔG values estimated for missense mutations detected in BH4-responsiveness studies in patients with PKU. Mutations have been divided into two groups (BH4-responsive and BH4–potentially responsive mutations) on the basis of previously established criteria.47 Both groups cluster around similar mean y0 values (5.7±5.9 kcal/mol and 5.5±2.5 kcal/mol for BH4-responsive and BH4–potentially responsive groups, respectively; P=.88), whereas m values are not significantly different either (m=0.66±1.67 and 0.17±0.54 for BH4-responsive and BH4–potentially responsive groups, respectively; P=.23). Phenotypic predictions of mutations of both groups revealed that a large fraction of mutations are predicted to be associated with mild phenotypes (31.2% and 16.7% of MHP and 18.7% and 50% of mild for BH4-responsive and BH4–potentially responsive groups, respectively). These predictions are consistent with clinical studies and biochemical analyses of BH4 responsiveness, which have shown that most (∼60%) of the patients47 who responded to BH4 have mild phenotypes and carry at least one mutant allele with relatively high in vitro residual activity (>30% of WT activity19,30,47). Our results thus suggest that one of the main molecular mechanisms underlying BH4 responsiveness is BH4-induced stabilization of PAH mutants with mild stability defects, as we have proposed elsewhere on the basis of in vitro expression analysis19 and in vivo studies.48 BH4 thus can be added to the list of chemical and pharmacological chaperones that emerge as potential therapeutic agents for correction of protein destabilization and misfolding.13,68,69
Hotspots for Destabilization
Previous attempts to predict the frequency of HPA/PKU mutations on the basis of the occurrence of CpG dinucleotides have shown that the predictions do not correlate with the actual occurrence of mutations among the population.20 All the PAH mutations encountered so far are, in fact, spread all over the three-dimensional structure of PAH.20 Figure 5 shows the location of all mutations compiled in table 7, as represented on the composite full-length subunit structure24 according to the final phenotypic prediction (fig. 5A), the y0 parameter (fig. 5B), and the m parameter (fig. 5C). We can see that most of the mutations that impair PAH stability affect residues that are partly or totally buried, as also inferred from previous structural interpretations of the effect of PKU mutations,20,21 whereas surface mutations have little effect on stability, especially when they are in loops (fig. 5). The most destabilizing mutations are those that introduce large van der Waals clashes in a buried position (by increasing the volume of the side chain because of mutation), which would either prevent protein folding or induce large conformational rearrangements (therefore displaying large m values) (fig. 5C). Such mutations will be predicted to be associated with severe phenotypes according to the criteria elaborated in this work (fig. 5A; see above and table 7 for criteria). Our results suggest that PAH structure and stability are highly optimized in terms of atomic packing and are very sensitive to mutations that increase side-chain size (table 7).
Figure 5. .

Mutation-dependent destabilization represented on the dimeric structure (PDB 2PHM). All mutations summarized in table 7 have been represented according to the following criteria. Mutated residues are colored according to (A) the predicted phenotype: MHP (blue), mild (yellow), and severe (red); (B) the y0 parameter: y0⩽3 kcal/mol (blue), 3<y0⩽7 kcal/mol (yellow), and y0>7 kcal/mol (red); and (C) the m parameter: m⩽0.1 (blue), 0.1<m⩽0.3 (yellow), and m>0.3 (red). For the cases with several mutations in the same residue, that position has been colored according to the mutation with the most severe effect.
The frequency distribution of the predicted phenotypes (including all mutations in table 7) along the PAH amino acid sequence is rather similar in segments 1–225 and 331–452 (MHP, 33.8%–35.9%; mild, 22.5%–38.3%; severe, 27.7%–43.7%). However, in the 226–330 region, there is a high predominance of predicted severe phenotypes (MHP, 21.7%; mild, 27.0%; severe, 51.3%) (table 7 and fig. 5). This segment largely comprises exons 7–9 in the PAH protein and covers most of the catalytic site. Expression analyses have demonstrated elsewhere a serious destabilizing effect of recurrent exon 7 (residues 235–281) mutations.6 In fact, however, the highest frequency of mutations predicted as severe (70.4%) is found in exons 8–9 (residues 282–323), which appear to contribute strongly to PAH stability by preserving the arrangement of the iron-coordinating residues (fig. 6). An important role in maintaining PAH native-state stability can thus be assigned to the region of residues 226–330 (highlighted in fig. 6), beyond their possible roles in catalysis (some of these residues also have a proven catalytic function—i.e., R270S, Y277D, and E280K). Other residues that are regarded as playing an important structural role are those involved in interdomain interactions in a monomer, such as Ser70–Arg71, P122, Leu308–L311, and Arg40820 (fig. 6). Mutations at most of these residues are also predicted to be severe and, in many cases, display large m values (table 7).
Figure 6. .

Hotspots for destabilization. The modeled composite subunit structure of human PAH is shown in ribbon representation, with regions 235–281 corresponding to exon 7 (green) and regions 282–330 including exons 8 and 9 (orange). Some residues important for interdomain interactions in the monomer (Ser70–Arg71, P122, Leu308–L311, and Arg408) are shown in dark red. The nonheme iron at the active site is shown as a yellow sphere, and the coordinating residues His285, His290, and Glu330 are shown as orange sticks. See the main text for details.
Discussion
The current view of protein folding describes folding reactions as complex processes in which a polypeptide reaches its native structure by searching the conformational space through a multidimensional energy surface, or “energy landscape.”70 During a folding reaction, the polypeptide chain may populate intermediate states to assist the protein to find the native folded state, even though these intermediates might also act as “kinetic traps” and be more prone to misfolding.70 Missense mutations often result in an increased tendency of the newly synthesized protein to misfold, leading to reduced in vivo function.71 In addition, misfolded proteins may be deleterious to cells per se, since they may interact with other misfolded proteins, resulting in toxic intracellular aggregation that can interfere with normal cellular function.70,72 The cell limits the accumulation of misfolded conformations by using a protein quality control (mainly in the cytosol and endoplasmic reticulum) that involves folding assistance by chaperones.73 The protective mechanisms also include elimination of the defective proteins via degradation through the polyubiquitin-dependent proteasome pathways or other protein quality-control proteolytic systems.10,13,74,75 Protein misfolding seems to be an intrinsic property of the folding process of large and complex proteins, and as many as 30% of the newly synthesized polypeptides in eukaryotes may be rapidly degraded after synthesis.74 Despite stringent quality-control mechanisms, mutations that affect protein folding cause a wide range of diseases, both rare and common. Among mutation-induced misfolding diseases, PKU (caused by mutations in PAH) is considered a paradigm of misfolding metabolic diseases.11,76 Several reports have substantiated PAH misfolding due to mutation, as shown by increased proteolytic turnover with expression in reticulocyte cell-free systems and mammalian cells and by modulation of PAH mutant residual activity and protein levels with chaperonin coexpression in E. coli.7,9,19,30,59
Prediction of the misfolding properties of PAH mutants is not straightforward, since it would depend on knowledge of the kinetic partitioning of the protein between native, partially folded, and misfolded states.73 Some approaches based, for instance, on molecular-dynamics simulations of the kinetic competition between folding and misfolding have been shown to be useful for the study of the basic principles of misfolding, but they are limited to simulations of small and simple proteins or model homopolymers.77,78
Alternatively, the effect of misfolding mutations can be considered from the effect of the stability on the folded protein, which would be related to reductions in the half-life or steady-state protein levels in vivo. It has been shown that mutation-dependent destabilization may decrease mutant protein levels by reducing thermodynamic and/or kinetic stability of proteins associated with different misfolding disorders, such as amyotrophic lateral sclerosis, cancer, and familial and systemic amyloidosis.79–85 The destabilizing mutational effect on the native-state stability may reduce protein thermodynamic, as well as kinetic, stabilities, which can be translated into increased aggregation or degradation rates in vivo.86 Thus, an approach based on the prediction of PKU phenotype from mutational effects on native-state stability seems plausible, and it is supported by the fact that thermal denaturation experiments have shown lower stability in most of the PKU mutants studied in vitro so far,7,9 indicating mutational effects on the PAH native-state stability. Different computational approaches have indeed been successfully applied (mainly in small model proteins) to predict the effects on thermodynamic stability caused by mutation. These predictive achievements are in fact remarkable, since development of these force fields to predict mutation-dependent destabilization is a difficult task because ΔG values are small numbers resulting from the almost complete cancellation of large different (enthalpic and entropic) contributions.87 Several of these computational approaches have provided good correlations between protein destabilization, as measured in vitro and as predicted in silico in studies involving large sets of mutants.88–92 These and other similar approaches have been applied, to test whether disease-related missense destabilizing mutations can actually be discriminated from neutral polymorphisms.93–96 However, to our knowledge, no theoretical approaches so far have provided strong evidence of correlation between stability changes and phenotypic outcome in large-scale studies for misfolding diseases related to loss-of-function mechanisms.
During the past several years, major advances have been achieved for the structural-functional characterization of PAH and the elucidation of genotype-phenotype relationships in PKU. In vitro expression analyses have provided strong evidence, for most of the PKU mutations, of misfolding effects on PAH protein, which may lead to increased degradation rates in vivo.8–10,97 Elucidation of the crystal structures of different truncated forms of human and rat PAH16,18,55,98–101 have provided the structural framework to qualitatively correlate mutational effects on structure and function, and, indeed, these structural analyses agree well, in many cases, with results from in vitro expression studies and in vivo metabolic phenotypes.20–22,31 However, many PKU mutations are rare; therefore, genotype-phenotype correlations cannot be assessed from patients’ data. Moreover, expressing all the missense mutant proteins would be a very time-consuming task. Our approach based on energetic calculation of mutation-dependent destabilization of PAH protein, which is based on the PAH crystal structures available and the semiempirical energy function FoldX, aims to help fill this gap in the genotype-phenotype correlations in PKU, especially in the case of rare and new mutations.
Conclusions
PKU is a paradigm of protein-misfolding diseases that are caused by loss-of-function pathogenic mechanisms,4,10,11,14 for which the screening and genotyping of neonatal infants during the past few decades have enabled us to identify patients and to prevent the major problems caused by disease. In this work, we have embarked on a structure-based energetic prediction of the impact of PKU mutations on PAH native-state stability. This approach has made it possible to capture and confirm the main pathogenic mechanism underlying PKU: the larger the ΔΔG or structural effect on PAH native state, the lower the activity in vitro and the more severe the phenotype. In vivo metabolic phenotypes have been shown to be better predicted by FoldX analysis than are in vitro residual activities, probably because of a higher stringency in the phenotyping process and because of the large number of patients who carry these frequent mutations. Additional analyses performed using the TANGO algorithm60 for PKU mutations have shown negligible contributions to misfolding from a greater propensity to aggregation, compared with the destabilization predicted by FoldX, which agrees well with the absence of reports of amyloidosis in patients with PKU. Moreover, PKU has been classified as a “complex trait” genetic disease, in which the phenotypic outcome in individual patients can be significantly modulated by other genetic and nongenetic modifying factors that can contribute to the metabolic and cognitive phenotypes and that would make a genotype-phenotype correlation difficult.15,102 A degree of variability in PAH residual activity associated with a given mutation (or mutations) has occasionally been invoked to explain inconsistencies in genotype-phenotype correlations; these variations are justified by, among other mechanisms, individual differences in potential modifiers, like intestinal absorption and transport of l-Phe, folding machinery, or intracellular levels of BH4 and l-Phe, which affect the residual activity and stability of the mutant proteins.4,9,15,19 The predictive power of FoldX, however, recognizes the classical “monogenic” autosomal recessive factor (protein destabilization caused by mutation of the PAH protein) as the major determinant for this outcome in most cases. Residues in exons 7–9 and in the interdomain regions within the subunit appear to play a remarkable structural role, constituting hotspots for the destabilization of PAH (fig. 6).
Our results show a positive prediction for a testing data set that is representative of a large fraction of mutant phenotypes in vivo, and we consider that this procedure will be useful to predict the phenotype associated with other rare or new mutations detected in patients with PKU (as performed for the 238 missense mutations for which no in vitro expression analysis is available) (table 7). However, additional factors must be considered that may contribute to the patients' phenotypes, such as possible effects on catalysis and the fact that the PAH mutant genotype is the main, but not the only, determinant of the metabolic phenotype. Our approach, which is based on correlations between mutational effects on protein unfolding energetics and metabolic phenotypes (with use of the m and y0 parameters as guides), thus appears as an alternative, or at least as an aid, to current methods of performing or predicting genotype-phenotype correlations based on the determination of in vitro residual activities associated with these mutations (table 1). As a consequence, a predictive guideline associating the actual PKU mutation with in vitro residual activities and metabolic phenotypes, on the basis of the application of different energy penalties in the FoldX calculations, emerges as a result of this work. The FoldX Web server will be modified to automatically predict the phenotypic effects of single amino acid mutations.
The use of protein-design algorithms to model any mutation and to predict the phenotypic effect (except for mutations involving the active site, which affect catalysis with or without affecting protein stability) can be extended to other misfolding diseases for which a loss-of-function mechanism has been described, as long as an adequate three-dimensional structure is available. This predictive tool contributes to the translational concept of from protein energetics to clinical phenotype.
Acknowledgments
This study was supported by grants from the Research Council of Norway and Helse-Vest. We are very grateful to Joost Schymkowitz at Free University of Brussels, Belgium, for his contribution to the development of FoldX.
Web Resources
The URLs for data presented herein are as follows:
- FoldX Web server, http://foldx.embl.de
- International Database of BH4-responsive HPA/PKU (BIOPKU), http://www.bh4.org/biopku.html
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for PKU) [PubMed]
- PAHdb, http://www.pahdb.mcgill.ca/
- PDB, http://www.rcsb.org/pdb/ (for 1PHZ, 2PHM, and 2PAH)
References
- 1.Scriver CR, Eisensmith RC, Woo SLC, Kaufman S (1994) The hyperphenylalaninemias in man and mouse. Annu Rev Genet 28:141–165 10.1146/annurev.ge.28.120194.001041 [DOI] [PubMed] [Google Scholar]
- 2.Scriver CR, Kaufman S (2001) Hyperphenylalaninemia: phenylalanine hydroxylase deficiency. In: Scriver CR, Beaudet AL, Valle D, Sly WS (eds) The metabolic and molecular bases of inherited disease. McGraw-Hill, New York, pp 1667–1724 [Google Scholar]
- 3.Güttler F, Guldberg P (2006) Genotype/phenotype correlations in phenylalanine hydroxylase deficiency. In: Blau N (ed) PKU and BH4: advances in phenylketonuria and tetrahydrobiopterin. SPS Verlagsgesellschaft, Heilbronn, pp 311–320 [Google Scholar]
- 4.Waters PJ (2003) How PAH gene mutations cause hyper-phenylalaninemia and why mechanism matters: insights from in vitro expression. Hum Mutat 21:357–369 10.1002/humu.10197 [DOI] [PubMed] [Google Scholar]
- 5.Scriver CR, Hurtubise M, Konecki D, Phommarinh M, Prevost L, Erlandsen H, Stevens R, Waters PJ, Ryan S, McDonald D, et al (2003) PAHdb 2003: what a locus-specific knowledgebase can do. Hum Mutat 21:333–344 10.1002/humu.10200 [DOI] [PubMed] [Google Scholar]
- 6.Bjørgo E, Knappskog PM, Martínez A, Stevens RC, Flatmark T (1998) Partial characterization and three-dimensional-structural localization of eight mutations in exon 7 of the human phenylalanine hydroxylase gene associated with phenylketonuria. Eur J Biochem 257:1–10 10.1046/j.1432-1327.1998.2570001.x [DOI] [PubMed] [Google Scholar]
- 7.Gámez A, Pérez B, Ugarte M, Desviat LR (2000) Expression analysis of phenylketonuria mutations: effect on folding and stability of the phenylalanine hydroxylase protein. J Biol Chem 275:29737–29742 10.1074/jbc.M003231200 [DOI] [PubMed] [Google Scholar]
- 8.Gjetting T, Petersen M, Guldberg P, Guttler F (2001) In vitro expression of 34 naturally occurring mutant variants of phenylalanine hydroxylase: correlation with metabolic phenotypes and susceptibility toward protein aggregation. Mol Genet Metab 72:132–143 10.1006/mgme.2000.3118 [DOI] [PubMed] [Google Scholar]
- 9.Pey AL, Desviat LR, Gamez A, Ugarte M, Perez B (2003) Phenylketonuria: genotype-phenotype correlations based on expression analysis of structural and functional mutations in PAH. Hum Mutat 21:370–378 10.1002/humu.10198 [DOI] [PubMed] [Google Scholar]
- 10.Waters PJ (2006) Molecular bases of phenylketonuria: insights from functional studies in vitro. In: Blau N (ed) PKU and BH4: advances in phenylketonuria and tetrahydrobiopterin. SPS Verlagsgesellschaft, Heilbronn, pp 277–310 [Google Scholar]
- 11.Gregersen N, Bross P, Vang S, Christensen JH (2006) Protein misfolding and human disease. Annu Rev Genomics Hum Genet 7:103–124 10.1146/annurev.genom.7.080505.115737 [DOI] [PubMed] [Google Scholar]
- 12.Døskeland AP, Flatmark T (1996) Recombinant human phenylalanine hydroxylase is a substrate for the ubiquitin-conjugating enzyme system. Biochem J 319:941–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ulloa-Aguirre A, Janovick JA, Brothers SP, Conn PM (2004) Pharmacologic rescue of conformationally-defective proteins: implications for the treatment of human disease. Traffic 5:821–837 10.1111/j.1600-0854.2004.00232.x [DOI] [PubMed] [Google Scholar]
- 14.Gregersen N (2006) Protein misfolding disorders: pathogenesis and intervention. J Inherit Metab Dis 29:456–470 10.1007/s10545-006-0301-4 [DOI] [PubMed] [Google Scholar]
- 15.Scriver CR, Waters PJ (1999) Monogenic traits are not simple: lessons from phenylketonuria. Trends Genet 15:267–272 10.1016/S0168-9525(99)01761-8 [DOI] [PubMed] [Google Scholar]
- 16.Erlandsen H, Fusetti F, Martínez A, Hough E, Flatmark T, Stevens RC (1997) Crystal structure of the catalytic domain of human phenylalanine hydroxylase reveals the structural basis for phenylketonuria. Nat Struct Biol 4:995–1000 10.1038/nsb1297-995 [DOI] [PubMed] [Google Scholar]
- 17.Fusetti F, Erlandsen H, Flatmark T, Stevens RC (1998) Structure of tetrameric human phenylalanine hydroxylase and its implications for phenylketonuria. J Biol Chem 273:16962–16967 10.1074/jbc.273.27.16962 [DOI] [PubMed] [Google Scholar]
- 18.Kobe B, Jennings IG, House CM, Michell BJ, Goodwill KE, Santarsiero BD, Stevens RC, Cotton RG, Kemp BE (1999) Structural basis of autoregulation of phenylalanine hydroxylase. Nat Struct Biol 6:442–448 10.1038/8247 [DOI] [PubMed] [Google Scholar]
- 19.Erlandsen H, Pey AL, Gamez A, Perez B, Desviat LR, Aguado C, Koch R, Surendran S, Tyring S, Matalon R, et al (2004) Correction of kinetic and stability defects by tetrahydrobiopterin in phenylketonuria patients with certain phenylalanine hydroxylase mutations. Proc Natl Acad Sci USA 101:16903–16908 10.1073/pnas.0407256101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Erlandsen H, Stevens RC (1999) The structural basis of phenylketonuria. Mol Genet Metab 68:103–125 10.1006/mgme.1999.2922 [DOI] [PubMed] [Google Scholar]
- 21.Jennings IG, Cotton RG, Kobe B (2000) Structural interpretation of mutations in phenylalanine hydroxylase protein aids in identifying genotype-phenotype correlations in phenylketonuria. Eur J Hum Genet 8:683–696 10.1038/sj.ejhg.5200518 [DOI] [PubMed] [Google Scholar]
- 22.Flatmark T, Stevens RC (1999) Structural insight into the aromatic amino acid hydroxylases and their disease-related mutant forms. Chem Rev 99:2137–2160 10.1021/cr980450y [DOI] [PubMed] [Google Scholar]
- 23.Schymkowitz JW, Rousseau F, Martins IC, Ferkinghoff-Borg J, Stricher F, Serrano L (2005) Prediction of water and metal binding sites and their affinities by using the Fold-X force field. Proc Natl Acad Sci USA 102:10147–10152 10.1073/pnas.0501980102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thórólfsson M, Ibarra-Molero B, Fojan P, Petersen SB, Sanchez-Ruiz JM, Martínez A (2002) L-phenylalanine binding and domain organization in human phenylalanine hydroxylase: a differential scanning calorimetry study. Biochemistry 41:7573–7585 10.1021/bi0160720 [DOI] [PubMed] [Google Scholar]
- 25.Petukhov M, Cregut D, Soares CM, Serrano L (1999) Local water bridges and protein conformational stability. Protein Sci 8:1982–1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Munoz V, Serrano L (1994) Intrinsic secondary structure propensities of the amino acids, using statistical phi-psi matrices: comparison with experimental scales. Proteins 20:301–311 10.1002/prot.340200403 [DOI] [PubMed] [Google Scholar]
- 27.Abagyan R, Totrov M (1994) Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J Mol Biol 235:983–1002 10.1006/jmbi.1994.1052 [DOI] [PubMed] [Google Scholar]
- 28.Vijayakumar M, Wong KY, Schreiber G, Fersht AR, Szabo A, Zhou HX (1998) Electrostatic enhancement of diffusion-controlled protein-protein association: comparison of theory and experiment on barnase and barstar. J Mol Biol 278:1015–1024 10.1006/jmbi.1998.1747 [DOI] [PubMed] [Google Scholar]
- 29.Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L (2005) The FoldX web server: an online force field. Nucleic Acids Res 33:W382–W388 10.1093/nar/gki387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pey AL, Perez B, Desviat LR, Martinez MA, Aguado C, Erlandsen H, Gamez A, Stevens RC, Thorolfsson M, Ugarte M, et al (2004) Mechanisms underlying responsiveness to tetrahydrobiopterin in mild phenylketonuria mutations. Hum Mutat 24:388–399 10.1002/humu.20097 [DOI] [PubMed] [Google Scholar]
- 31.Kim SW, Jung J, Oh HJ, Kim J, Lee KS, Lee DH, Park C, Kimm K, Koo SK, Jung SC (2006) Structural and functional analyses of mutations of the human phenylalanine hydroxylase gene. Clin Chim Acta 365:279–287 10.1016/j.cca.2005.09.019 [DOI] [PubMed] [Google Scholar]
- 32.Kayaalp E, Treacy E, Waters PJ, Byck S, Nowacki P, Scriver CR (1997) Human phenylalanine hydroxylase mutations and hyperphenylalaninemia phenotypes: a metanalysis of genotype-phenotype correlations. Am J Hum Genet 61:1309–1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ellingsen S, Knappskog PM, Eiken HG (1997) Phenylketonuria splice mutation (EXON6nt-96A→g) masquerading as missense mutation (Y204C). Hum Mutat 9:88–90 10.1002/(SICI)1098-1004(1997)9:1<88::AID-HUMU21>3.0.CO;2-K [DOI] [PubMed] [Google Scholar]
- 34.Chao HK, Hsiao KJ, Su TS (2001) A silent mutation induces exon skipping in the phenylalanine hydroxylase gene in phenylketonuria. Hum Genet 108:14–19 10.1007/s004390000435 [DOI] [PubMed] [Google Scholar]
- 35.Guldberg P, Rey F, Zschocke J, Romano V, Francois B, Michiels L, Ullrich K, Hoffmann GF, Burgard P, Schmidt H, et al (1998) A European multicenter study of phenylalanine hydroxylase deficiency: classification of 105 mutations and a general system for genotype-based prediction of metabolic phenotype. Am J Hum Genet 63:71–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Desviat LR, Perez B, Gamez A, Sanchez A, Garcia MJ, Martinez-Pardo M, Marchante C, Boveda D, Baldellou A, Arena J, et al (1999) Genetic and phenotypic aspects of phenylalanine hydroxylase deficiency in Spain: molecular survey by regions. Eur J Hum Genet 7:386–392 10.1038/sj.ejhg.5200312 [DOI] [PubMed] [Google Scholar]
- 37.Desviat LR, Perez B, Belanger-Quintana A, Castro M, Aguado C, Sanchez A, Garcia MJ, Martinez-Pardo M, Ugarte M (2004) Tetrahydrobiopterin responsiveness: results of the BH4 loading test in 31 Spanish PKU patients and correlation with their genotype. Mol Genet Metab 83:157–162 10.1016/j.ymgme.2004.06.007 [DOI] [PubMed] [Google Scholar]
- 38.Lindner M (2006) Treatment of phenylketonuria variants: European recommendations. In: Blau N (ed) PKU and BH4: advances in phenylketonuria and tetrahydrobiopterin. SPS Verlagsgesellschaft, Heilbronn, pp 180–187 [Google Scholar]
- 39.Matalon R, Michals-Matalon K (2006) Treatment of phenylketonuria variants: US recommendations. In: Blau N (ed) PKU and BH4: advances in phenylketonuria and tetrahydrobiopterin. SPS Verlagsgesellschaft, Heilbronn, pp 201–219 [Google Scholar]
- 40.Marsden D, Levy H (2006) Classification of PKU. In: Blau N (ed) PKU and BH4: advances in phenylketonuria and tetrahydrobiopterin. SPS Verlagsgesellschaft, Heilbronn, pp 92–103 [Google Scholar]
- 41.Aulehla-Scholz C, Heilbronner H (2003) Mutational spectrum in German patients with phenylalanine hydroxylase deficiency. Hum Mutat 21:399–400 10.1002/humu.9116 [DOI] [PubMed] [Google Scholar]
- 42.Desviat LR, Perez B, Garcia MJ, Martinez-Pardo M, Baldellou A, Arena J, Sanjurjo P, Campistol J, Couce ML, Fernandez A, et al (1997) Relationship between mutation genotype and biochemical phenotype in a heterogeneous Spanish phenylketonuria population. Eur J Hum Genet 5:196–202 [PubMed] [Google Scholar]
- 43.De Lucca M, Perez B, Desviat LR, Ugarte M (1998) Molecular basis of phenylketonuria in Venezuela: presence of two novel null mutations. Hum Mutat 11:354–359 10.1002/(SICI)1098-1004(1998)11:5<354::AID-HUMU2>3.0.CO;2-W [DOI] [PubMed] [Google Scholar]
- 44.Burgard P (2006) Outcome of phenylketonuria variant. In: Blau N (ed) PKU and BH4: advances in phenylketonuria and tetrahydrobiopterin. SPS Verlagsgesellschaft, Heilbronn, pp 251–260 [Google Scholar]
- 45.Michals-Matalon K (2006) Dietary recommendations in the USA. In: Blau N (ed) PKU and BH4: advances in phenylketonuria and tetrahydrobiopterin. SPS Verlagsgesellschaft, Heilbronn, pp 220–231 [Google Scholar]
- 46.Eiken HG, Knappskog PM, Apold J, Flatmark T (1996) PKU mutation G46S is associated with increased aggregation and degradation of the phenylalanine hydroxylase enzyme. Hum Mutat 7:228–238 10.1002/(SICI)1098-1004(1996)7:3<228::AID-HUMU7>3.0.CO;2-6 [DOI] [PubMed] [Google Scholar]
- 47.Blau N, Erlandsen H (2004) The metabolic and molecular bases of tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency. Mol Genet Metab 82:101–111 10.1016/j.ymgme.2004.03.006 [DOI] [PubMed] [Google Scholar]
- 48.Thony B, Ding Z, Martinez A (2004) Tetrahydrobiopterin protects phenylalanine hydroxylase activity in vivo: implications for tetrahydrobiopterin-responsive hyperphenylalaninemia. FEBS Lett 577:507–511 10.1016/j.febslet.2004.10.056 [DOI] [PubMed] [Google Scholar]
- 49.Scavelli R, Ding Z, Blau N, Haavik J, Martinez A, Thony B (2005) Stimulation of hepatic phenylalanine hydroxylase activity but not Pah-mRNA expression upon oral loading of tetrahydrobiopterin in normal mice. Mol Genet Metab Suppl 1 86:S153–S155 [DOI] [PubMed] [Google Scholar]
- 50.Aguado C, Perez B, Ugarte M, Desviat LR (2006) Analysis of the effect of tetrahydrobiopterin on PAH gene expression in hepatoma cells. FEBS Lett 580:1697–1701 10.1016/j.febslet.2006.02.005 [DOI] [PubMed] [Google Scholar]
- 51.Pey AL, Martinez A (2005) The activity of wild-type and mutant phenylalanine hydroxylase and its regulation by phenylalanine and tetrahydrobiopterin at physiological and pathological concentrations: an isothermal titration calorimetry study. Mol Genet Metab Suppl 1 86:S43–S53 [DOI] [PubMed] [Google Scholar]
- 52.Liu R, Baase WA, Matthews BW (2000) The introduction of strain and its effects on the structure and stability of T4 lysozyme. J Mol Biol 295:127–145 10.1006/jmbi.1999.3300 [DOI] [PubMed] [Google Scholar]
- 53.Ventura S, Vega MC, Lacroix E, Angrand I, Spagnolo L, Serrano L (2002) Conformational strain in the hydrophobic core and its implications for protein folding and design. Nat Struct Biol 9:485–493 10.1038/nsb799 [DOI] [PubMed] [Google Scholar]
- 54.Teigen K, Frøystein NÅ, Martínez A (1999) The structural basis of the recognition of phenylalanine and pterin cofactors by phenylalanine hydroxylase: implications for the catalytic mechanism. J Mol Biol 294:807–823 10.1006/jmbi.1999.3288 [DOI] [PubMed] [Google Scholar]
- 55.Andersen OA, Stokka AJ, Flatmark T, Hough E (2003) 2.0 Å resolution crystal structures of the ternary complexes of human phenylalanine hydroxylase catalytic domain with tetrahydrobiopterin and 3-(2-thienyl)-l-alanine or l-norleucine: substrate specificity and molecular motions related to substrate binding. J Mol Biol 333:747–757 10.1016/j.jmb.2003.09.004 [DOI] [PubMed] [Google Scholar]
- 56.Kemsley JN, Wasinger EC, Datta S, Mitic N, Acharya T, Hedman B, Caradonna JP, Hodgson KO, Solomon EI (2003) Spectroscopic and kinetic studies of PKU-inducing mutants of phenylalanine hydroxylase: Arg158Gln and Glu280Lys. J Am Chem Soc 125:5677–5686 10.1021/ja029106f [DOI] [PubMed] [Google Scholar]
- 57.Zekanowski C, Perez B, Desviat LR, Wiszniewski W, Ugarte M (2000) In vitro expression analysis of R68G and R68S mutations in phenylalanine hydroxylase gene. Acta Biochim Pol 47:365–369 [PubMed] [Google Scholar]
- 58.Thórólfsson M, Teigen K, Martínez A (2003) Activation of phenylalanine hydroxylase: effect of substitutions at Arg68 and Cys237. Biochemistry 42:3419–3428 10.1021/bi034021s [DOI] [PubMed] [Google Scholar]
- 59.Waters PJ, Parniak MA, Akerman BR, Scriver CR (2000) Characterization of phenylketonuria missense substitutions, distant from the phenylalanine hydroxylase active site, illustrates a paradigm for mechanism and potential modulation of phenotype. Mol Genet Metab 69:101–110 10.1006/mgme.2000.2965 [DOI] [PubMed] [Google Scholar]
- 60.Fernandez-Escamilla AM, Rousseau F, Schymkowitz J, Serrano L (2004) Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat Biotechnol 22:1302–1306 10.1038/nbt1012 [DOI] [PubMed] [Google Scholar]
- 61.Guttler F, Azen C, Guldberg P, Romstad A, Hanley WB, Levy HL, Matalon R, Rouse BM, Trefz F, de la Cruz F, et al (1999) Relationship among genotype, biochemical phenotype, and cognitive performance in females with phenylalanine hydroxylase deficiency: report from the Maternal Phenylketonuria Collaborative Study. Pediatrics 104:258–262 10.1542/peds.104.2.258 [DOI] [PubMed] [Google Scholar]
- 62.Acosta A, Silva W Jr, Carvalho T, Gomes M, Zago M (2001) Mutations of the phenylalanine hydroxylase (PAH) gene in Brazilian patients with phenylketonuria. Hum Mutat 17:122–130 10.1002/1098-1004(200102)17:2<122::AID-HUMU4>3.0.CO;2-C [DOI] [PubMed] [Google Scholar]
- 63.Zschocke J (2003) Phenylketonuria mutations in Europe. Hum Mutat 21:345–356 10.1002/humu.10192 [DOI] [PubMed] [Google Scholar]
- 64.Daniele A, Cardillo G, Pennino C, Carbone MT, Scognamiglio D, Correra A, Pignero A, Castaldo G, Salvatore F (2007) Molecular epidemiology of phenylalanine hydroxylase deficiency in southern Italy: a 96% detection rate with ten novel mutations. Ann Hum Genet 71:185–193 10.1111/j.1469-1809.2006.00328.x [DOI] [PubMed] [Google Scholar]
- 65.Gibbs BS, Wojchowski D, Benkovic SJ (1993) Expression of rat liver phenylalanine hydroxylase in insect cells and site-directed mutagenesis of putative non-heme iron-binding sites. J Biol Chem 268:8046–8052 [PubMed] [Google Scholar]
- 66.Daubner SC, Fitzpatrick PF (1999) Site-directed mutants of charged residues in the active site of tyrosine hydroxylase. Biochemistry 38:4448–4454 10.1021/bi983012u [DOI] [PubMed] [Google Scholar]
- 67.Kure S, Hou DC, Ohura T, Iwamoto H, Suzuki S, Sugiyama N, Sakamoto O, Fujii K, Matsubara Y, Narisawa K (1999) Tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency. J Pediatr 135:375–378 10.1016/S0022-3476(99)70138-1 [DOI] [PubMed] [Google Scholar]
- 68.Cohen FE, Kelly JW (2003) Therapeutic approaches to protein-misfolding diseases. Nature 426:905–909 10.1038/nature02265 [DOI] [PubMed] [Google Scholar]
- 69.Sawkar AR, D’Haeze W, Kelly JW (2006) Therapeutic strategies to ameliorate lysosomal storage disorders: a focus on Gaucher disease. Cell Mol Life Sci 63:1179–1192 10.1007/s00018-005-5437-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dobson CM (2003) Protein folding and misfolding. Nature 426:884–890 10.1038/nature02261 [DOI] [PubMed] [Google Scholar]
- 71.Welch WJ (2004) Role of quality control pathways in human diseases involving protein misfolding. Semin Cell Dev Biol 15:31–38 10.1016/j.semcdb.2003.12.011 [DOI] [PubMed] [Google Scholar]
- 72.Kopito RR (2000) Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol 10:524–530 10.1016/S0962-8924(00)01852-3 [DOI] [PubMed] [Google Scholar]
- 73.Hartl FU, Hayer-Hartl M (2002) Molecular chaperones in the cytosol: from nascent chain to folded protein. Science 295:1852–1858 10.1126/science.1068408 [DOI] [PubMed] [Google Scholar]
- 74.Goldberg AL (2003) Protein degradation and protection against misfolded or damaged proteins. Nature 426:895–899 10.1038/nature02263 [DOI] [PubMed] [Google Scholar]
- 75.Sitia R, Braakman I (2003) Quality control in the endoplasmic reticulum protein factory. Nature 426:891–894 10.1038/nature02262 [DOI] [PubMed] [Google Scholar]
- 76.Dobson CM (2004) Principles of protein folding, misfolding and aggregation. Semin Cell Dev Biol 15:3–16 10.1016/j.semcdb.2003.12.008 [DOI] [PubMed] [Google Scholar]
- 77.Urbanc B, Borreguero JM, Cruz L, Stanley HE (2006) Ab initio discrete molecular dynamics approach to protein folding and aggregation. Methods Enzymol 412:314–338 [DOI] [PubMed] [Google Scholar]
- 78.Cellmer T, Bratko D, Prausnitz JM, Blanch HW (2007) Protein aggregation in silico. Trends Biotechnol 25:254–261 10.1016/j.tibtech.2007.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bullock AN, Fersht AR (2001) Rescuing the function of mutant p53. Nat Rev Cancer 1:68–76 10.1038/35094077 [DOI] [PubMed] [Google Scholar]
- 80.Lindberg MJ, Tibell L, Oliveberg M (2002) Common denominator of Cu/Zn superoxide dismutase mutants associated with amyotrophic lateral sclerosis: decreased stability of the apo state. Proc Natl Acad Sci USA 99:16607–16612 10.1073/pnas.262527099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hammarstrom P, Jiang X, Hurshman AR, Powers ET, Kelly JW (2002) Sequence-dependent denaturation energetics: a major determinant in amyloid disease diversity. Proc Natl Acad Sci USA Suppl 4 99:16427–16432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hammarstrom P, Wiseman RL, Powers ET, Kelly JW (2003) Prevention of transthyretin amyloid disease by changing protein misfolding energetics. Science 299:713–716 10.1126/science.1079589 [DOI] [PubMed] [Google Scholar]
- 83.Friedler A, Veprintsev DB, Hansson LO, Fersht AR (2003) Kinetic instability of p53 core domain mutants: implications for rescue by small molecules. J Biol Chem 278:24108–24112 10.1074/jbc.M302458200 [DOI] [PubMed] [Google Scholar]
- 84.Lindberg MJ, Bystrom R, Boknas N, Andersen PM, Oliveberg M (2005) Systematically perturbed folding patterns of amyotrophic lateral sclerosis (ALS)-associated SOD1 mutants. Proc Natl Acad Sci USA 102:9754–9759 10.1073/pnas.0501957102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dumoulin M, Kumita JR, Dobson CM (2006) Normal and aberrant biological self-assembly: insights from studies of human lysozyme and its amyloidogenic variants. Acc Chem Res 39:603–610 10.1021/ar050070g [DOI] [PubMed] [Google Scholar]
- 86.Plaza del Pino IM, Ibarra-Molero B, Sanchez-Ruiz JM (2000) Lower kinetic limit to protein thermal stability: a proposal regarding protein stability in vivo and its relation with misfolding diseases. Proteins 40:58–70 10.1002/(SICI)1097-0134(20000701)40:1<58::AID-PROT80>3.0.CO;2-M [DOI] [PubMed] [Google Scholar]
- 87.Cooper A (1999) Thermodynamic analysis of biomolecular interactions. Curr Opin Chem Biol 3:557–563 10.1016/S1367-5931(99)00008-3 [DOI] [PubMed] [Google Scholar]
- 88.Guerois R, Nielsen JE, Serrano L (2002) Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. J Mol Biol 320:369–387 10.1016/S0022-2836(02)00442-4 [DOI] [PubMed] [Google Scholar]
- 89.Capriotti E, Fariselli P, Casadio R (2004) A neural-network-based method for predicting protein stability changes upon single point mutations. Bioinformatics Suppl 1 20:i63–i68 [DOI] [PubMed] [Google Scholar]
- 90.Capriotti E, Fariselli P, Casadio R (2005) I-Mutant2.0: predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res 33:W306–W310 10.1093/nar/gki375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cheng J, Randall A, Baldi P (2006) Prediction of protein stability changes for single-site mutations using support vector machines. Proteins 62:1125–1132 10.1002/prot.20810 [DOI] [PubMed] [Google Scholar]
- 92.Parthiban V, Gromiha MM, Schomburg D (2006) CUPSAT: prediction of protein stability upon point mutations. Nucleic Acids Res 34:W239–W242 10.1093/nar/gkl190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yue P, Li Z, Moult J (2005) Loss of protein structure stability as a major causative factor in monogenic disease. J Mol Biol 353:459–473 10.1016/j.jmb.2005.08.020 [DOI] [PubMed] [Google Scholar]
- 94.Yip YL, Zoete V, Scheib H, Michielin O (2006) Structural assessment of single amino acid mutations: application to TP53 function. Hum Mutat 27:926–937 10.1002/humu.20379 [DOI] [PubMed] [Google Scholar]
- 95.Thusberg J, Vihinen M (2006) Bioinformatic analysis of protein structure-function relationships: case study of leukocyte elastase (ELA2) missense mutations. Hum Mutat 27:1230–1243 10.1002/humu.20407 [DOI] [PubMed] [Google Scholar]
- 96.van der Zee J, Le Ber I, Maurer-Stroh S, Engelborghs S, Gijselinck I, Camuzat A, Brouwers N, Vandenberghe R, Sleegers K, Hannequin D, et al (2007) Mutations other than null mutations producing a pathogenic loss of progranulin in frontotemporal dementia. Hum Mutat 28:416 10.1002/humu.9484 [DOI] [PubMed] [Google Scholar]
- 97.Waters PJ, Parniak MA, Akerman BR, Jones AO, Scriver CR (1999) Missense mutations in the phenylalanine hydroxylase gene (PAH) can cause accelerated proteolytic turnover of PAH enzyme: a mechanism underlying phenylketonuria. J Inherit Metab Dis 22:208–212 10.1023/A:1005533825980 [DOI] [PubMed] [Google Scholar]
- 98.Andersen OA, Flatmark T, Hough E (2001) High resolution crystal structures of the catalytic domain of human phenylalanine hydroxylase in its catalytically active Fe(II) form and binary complex with tetrahydrobiopterin. J Mol Biol 314:279–291 10.1006/jmbi.2001.5061 [DOI] [PubMed] [Google Scholar]
- 99.Andersen OA, Flatmark T, Hough E (2002) Crystal structure of the ternary complex of the catalytic domain of human phenylalanine hydroxylase with tetrahydrobiopterin and 3-(2-thienyl)-l-alanine, and its implications for the mechanism of catalysis and substrate activation. J Mol Biol 320:1095–1108 10.1016/S0022-2836(02)00560-0 [DOI] [PubMed] [Google Scholar]
- 100.Erlandsen H, Bjørgo E, Flatmark T, Stevens RC (2000) Crystal structure and site-specific mutagenesis of pterin-bound human phenylalanine hydroxylase. Biochemistry 39:2208–2217 10.1021/bi992531+ [DOI] [PubMed] [Google Scholar]
- 101.Erlandsen H, Flatmark T, Stevens RC, Hough E (1998) Crystallographic analysis of the human phenylalanine hydroxylase catalytic domain with bound catechol inhibitors at 2.0 A resolution. Biochemistry 37:15638–15646 10.1021/bi9815290 [DOI] [PubMed] [Google Scholar]
- 102.Scriver CR (2002) Why mutation analysis does not always predict clinical consequences: explanations in the era of genomics. J Pediatr 140:502–506 10.1067/mpd.2002.124316 [DOI] [PubMed] [Google Scholar]