

Abstract
The gene of mouse kappa opioid receptor (KOR) utilizes two promoters, P1 and P2. P1 is active in various brain areas and constitutively in P19 mouse embryonal carcinoma cells. P2 is active in limited brain stem areas of adult animals and only in late differentiated cells of P19 induced for neuronal differentiation in the presence of nerve growth factor (NGF). NGF response of P2 was found to be mediated by a specific binding site for transcription factor activation protein 2 (AP2) located in P2. Electrophoretic gel shift assay showed specific binding of this AP2 site by AP2β, but not AP2α. Knockdown of endogenous AP2β with siRNA abolished the stimulating effect of NGF on the expression of transcripts driven by P2. Binding of endogenous AP2β on the endogenous KOR P2 chromatin region was also confirmed by chromatin immunoprecipitation. The effect of NGF was inhibited by LY2942002 (phosphatidylinositol 3-kinase, PI3K inhibitor), suggesting that PI3K was involved in signaling pathway mediating the effect of NGF stimulation on KOR P2. The chromatin of P2 in P19 was found to be specifically modified following NGF stimulation, which included demethylation at Lys9 and dimethylation at Lys4 of histone H3 and was consistent with the increased recruitment of RNA polymerase II to this promoter. This study presents the first evidence for epigenetic changes occurred on a specific KOR promoter triggered by NGF in cells undergoing neuronal differentiation. This epigenetic change is mediated by recruited AP2β to this promoter and involves PI3K system.
Keywords: Kappa opioid receptor, epigenetic, NGF, retinoic acid, P19 neuronal differentiation, AP2
Introduction
It is known that the effects of opioid drugs are mediated through, primarily, three opioid receptors μ-, δ- and κ (KOR)-opioid receptor. Opioid receptors belong to the superfamily of G protein-coupled seven transmembrane receptors. Activated opioid receptors convey opioid signals via inhibition of adenylyl cyclase activity (Prather et al., 1993; Smart et al., 1997; Ozawa et al., 1999), increased phospholipase C activity and transient surge in intracellular Ca2+ levels (Johnson et al., 1994; Spencer et al., 1997), activation of inward-rectifying K+ channels (Henry et al., 1995), inhibition of Ca2+ channels (Tallent et al., 1994; Piros et al., 1995), and activation of the mitogen-activated protein kinases Erk1/2 (Fukuda et al., 1996; Li and Chang, 1996). The three opioid receptors are encoded by three different genes. Each gene produces multiple mRNA variants through alternative promoter usage, splicing and/or polyadenylation (Zimprich et al., 1995; Pan et al., 2001; Pan, 2003; Wei et al., 2004).
The activity of these receptors is regulated at different levels, including the protein level such as receptor phosphorylation and desensitization (Pei et al., 1995; Arden et al., 1995; Appleyard et al., 1997), the transcriptional level (Wei and Loh, 2002; Law et al., 2004), and the post-transcriptional level like alternative splicing and polyadenylation (Lu et al., 1997; Wei et al., 2000; Pan et al., 2001; Hu et al., 2002), mRNA stability (Wei et al., 2000), mRNA transport (Bi et al., 2003, 2006, 2007) and translation (Tsai et al., 2006). Distribution of opioid receptors and their mRNAs has been extensively examined (Fowler and Fraser, 1994; Hu et al., 2002). These studies demonstrate that these receptors are expressed in different, but slightly overlapping, patterns, and that each gene contains various specific regulatory elements upstream of the somewhat similar promoter region. However, it remains unclear as to how these genes can be specifically activated in neurons where the activities and functions of these receptors are mostly expected.
The gene of mouse KOR utilizes two promoters: P1 and P2 (Hu et al., 2002; Wei and Loh, 2002). Transcripts driven by P1 are widely detected in the brain of animals at different developmental stages whereas transcripts from P2 are detected only in very limited brain stem areas of adult animals. To study the regulation of these promoters, this lab has utilized a mouse embryonal carcinoma cell culture, P19, which can be induced for neuronal differentiation by retinoic acid (RA) (Bi et al., 2001; Li et al., 2002; Park et al., 2005). In the P19 neuronal differentiation model, P1 is constitutively active whereas P2 is activated only in later stages of RA-induced differentiation in the presence of nerve growth factor (NGF). P1 is constitutively active in P19 because its chromatin is in an open conformation (Park et al., 2005). P2, located within intron 1, is usually silenced in P19 stem cells (Bi et al. 2001) and in early differentiating cells because its chromatin is organized into a nucleosomal array, which is caused by histone deacetylation triggered by transcription factor Ikaros that recruits histone deacetylases to the Ikaros binding site in P2 (Hu et al., 2001). The search for a trigger that can activate P2 in neural tissues in adult animals has been a challenging task. But, detection of its transcripts in later stages of differentiated P19 cells in the presence of NGF suggests that P2 activation probably involves epigenetic changes triggered by neurotrophins.
NGF is one member of the neurotrophin family that also includes brain-derived neurotrophic factor, and neurotophins 3 and 4. These neurotrophins regulate cell survival, proliferation, axon and dendrite growth and patterning, the determination of neural precursors, and regulation of gene expression and protein activity (Huang and Reichardt, 2003). NGF is known to activate TrkA receptor or the pan-neurotrophin receptor p75, resulting in activation of small G protein-mediated pathway and the pathways of phospholipase Cγ (PLCγ), phosphatidylinositol 3-kinase (PI3K), or mitogenic activation protein (MAP) kinase (Huang and Reichardt, 2003; Rong et al., 2003; Wu and Wong, 2005; Santos et al., 2007; Wehrman et al., 2007). This study examines the effects of NGF on P2, and uncovers NGF-triggered epigenetic changes on P2 that involves recruitment of a specific transcriptional activator AP2β to its cognate binding site on P2 and is mediated by the pathway of PI3K, but not PLCγ.
Experimental procedures
Plasmid construction
Reporter constructs were made with luciferase coding sequence inserted following the initiation site of KOR P2 ([Hu et al., 2001] and [Park et al., 2002]) within the backbone of pGL3B (Promega, Madison, WI). K18 contains the full length P2, including sequences of the whole intron 1 (P2) and exon 1. K18m was generated by site-directed mutagenesis kit (Stratagene, La Jolla, CA) that converted the AP2 consensus sequence CCCGAGGG to TTTTAGGG, thus was mutated specifically on the AP2 site. Kd50 was constructed from K18 to retain exon 1 and only AP2 binding site (-404 to -210).
Cell culture, transfection and reporter assays
P19 cells were cultured in α-minimum essential medium supplemented with 7.5% defined calf serum and 2.5% defined fetal bovine serum and treated with 1.0 × 10-6 M all-trans RA. For neuronal differentiation initiated with aggregation, the procedure was performed as described previously (Bi et al., 2001; Park et al., 2005). Briefly, P19 cells were cultured on bacteriological Petri dishes in α-minimum essential medium containing 5% fetal bovine serum and 1.0 × 10-6 MRA. After 4 days, cell aggregates were subcultured onto gelatinized tissue culture dishes and treated with 5 μg/ml cytosine arabinoside in the absence of RA. NGF (1 ng/ml) was then added to differentiated cells without further RA treatment. PC12 cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% donor horse serum and 5% fetal bovine serum. P19 or PC12 cells were transfected with reporter constructs using lipofectamine 2000® (Invitrogen) followed by treatment of indicated reagents.
RNA interference
P19 cells were transfected with 10 nM siRNAs using Hiperfect reagent for 2-3 days (Qiagen). The siRNA was purchased from Qiagen; AP2β (Mm_Tcfap2b_1 HP siRNA, SI00183883). Silencing effect was assessed by RT-PCR and Southern blot.
Reverse transcriptase-PCR (RT-PCR) and Southern blot hybridization
Total RNA was isolated from P19 cells using a Trizol® solution (Invitrogen) and endogenous KOR mRNA isoforms were detected with an established RT-PCR protocol (Li et al., 2002). PCR primers specific to each gene are: AP2β Tcfap2b (forward, 5′-CCC AAG CCA TAG CTC GAG ACT C-3′ and reverse, 5′-TGG CGG AGA CAG CAT TGC TGT TG-3′), p75 Ngfr (forward, 5′- ATG AGG AGG GCA GGT GCT GC- 3′, and reverse, 5′-TCG TCT GCC TCC ACA CAG GG-3′), BM88 Cend1 (forward, 5′-GAG GAA AGT CAG CCA GCA GC-3′, and reverse, 5′-TGT TGG ACT CGT CCT CCT CTG-3′), KOR a (forward, 5′-ATC AGC GAT CTG GAG CT-3′), KOR b (forward, 5′-TCA GCG ATC TGG AGC CCC-3′), KOR c (forward, 5′-ACA GGC AAA GTT TGT-3′), and a common reverse primer for KOR (5′-GCA AGG AGC ATT CAA TGA C-3′). Actin-specific primers were included for internal control in each RT-PCR. Amplified DNAs were transferred to nylon membranes and subjected to Southern blot using probes labeled with [α-32P]dCTP, which was able to detect three isotypes of KOR transcripts (Park et al., 2005).
Electrophoretic mobility shift assay
AP2 binding site was found in KOR P2 by computer alignment. Electrophoretic mobility shift assays (EMSA) using three corresponding fragments from P2 were conducted as described previously (Park et al., 2002). Nuclear extracts (10 μg) prepared from P19 cells treated with RA and NGF were incubated in a 16 μl final reaction volume, which contains binding buffer (in mM: HEPES 10, pH 7.3; EDTA 1; dithiothreitol 1; KCl 25; 10% glycerol; 0.1 mg/ml poly[dI-dC] and 0.5% bovine serum albumin) and 2 ng of labeled DNA, at 4 °C for 30 min. For Supershift assay, antibodies against AP2α (sc-8975), AP2β (sc-8976) from Santa Cruz Biotechnology Inc. (Santa Cruz, CA), and hemagglutinin (HA; H9653, Sigma-Aldrich) were incubated for 30 min at room temperature before addition of labeled DNA. Probe sequence used is 5′-CAACGCCCGAGGGTGAA-3′ (underlined is the putative AP2 binding site).
Western blot and chromatin immunoprecipitation (ChIP) assays
Nuclear proteins were extracted from P19 cells treated with 10-6 M RA and 1 ng/ml NGF and resolved on an SDS-acrylamide gel followed by Western blot using the indicated antibodies as described previously (Park et al., 2005). Cells treated with RA only or RA then NGF were cross-linked with 1% formaldehyde (Park et al., 2005). Sonicated cell extracts, which were adjusted to contain the same amount of proteins, were precipitated with 2 μg of the following antibodies at 4 °C overnight, followed by the addition of protein G beads for 1 h. The antibodies against acetylated histone H4 (AcH4; 06-866), Lys4 methylated histone H3 (H3-K4-me2; 07-030), Lys9 methylated histone H3 (H3- K9-me2; 07-422), heterochromatin protein 1α (HP1α, 07-346) and RNA polymerase II (RNA polII, 05-623) were purchased from Millipore (Lake Placid, NY). Ten percent of cell extracts were used as input. Protein G beads were washed extensively, and the captured DNAs were eluted twice with 250 μl of elution buffer (1% SDS and 1 M NaHCO3), which were subjected to reverse cross-link at 65 °C for at least 4 h. Precipitated DNA was amplified using P2-specific primers: forward, 5′-CAG CCA CAG GAG TGG ACA GCA CAA C-3′ and reverse, 5′-GAC TCC ATG GTG AGC GCT GCA GCT GG-3′.
Results
NGF activates KOR P2 activity in RA-induced differentiating P19 cells
To examine how the silenced P2 of KOR could be activated in the P19 neuronal differentiation model, we used various neurotrophins to stimulate RA-induced differentiating P19 cultures and monitored the expression of P2-specific KOR transcript named isoform c (Fig. 1A) (Bi et al., 2001). As shown in Fig. 1B, isoform c expression could be elevated in NGF-treated P19 cells that had been induced by RA for differentiation. Neuronal differentiation was monitored by the increased expression of the neuronal marker BM88 (Koutmani et al., 2004; Georgopoulou et al., 2006). Two other KOR mRNA isoforms, a and b, that were initiated from P1, remained relatively constant along the course of NGF treatment. The effects of NGF stimulation of P2 transcripts was found to depend upon pre-treating P19 cells with RA for neuronal differentiation. We therefore monitored the expression of NGF receptors in P19. It appeared that the NGF receptor p75 was not expressed in undifferentiated P19 stem cells, but was dramatically induced after the initiation of differentiation (compare lanes 1 and 2) and slightly enhanced by NGF. This result reveals that P2 is activated by NGF only in RA-induced differentiating P19 cells because these cells express NGF receptors.
Fig. 1.
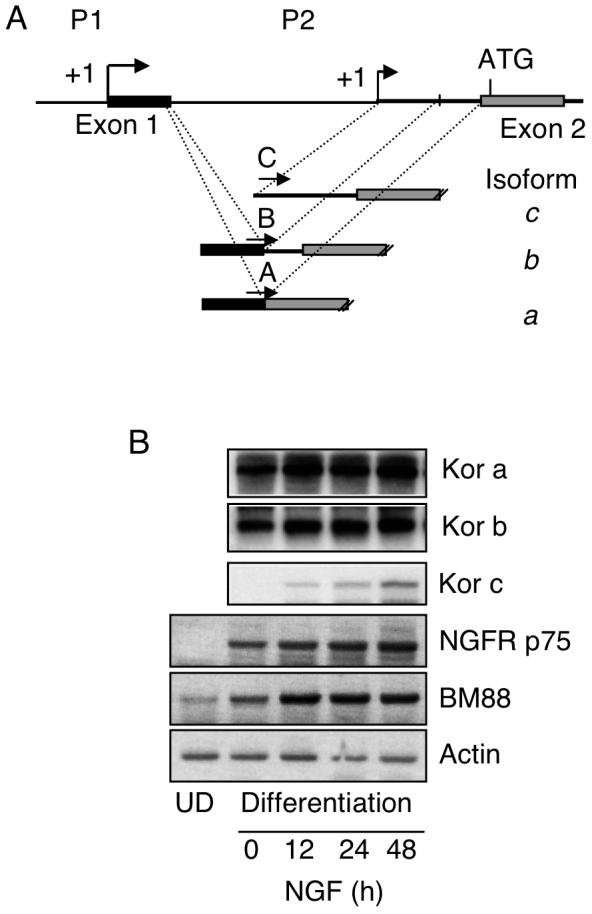
NGF induces expression of KOR c through P2 in RA-induced differentiating P19 cells. (A) The map of KOR gene promoter regions, including P1, P2, exons 1 & 2, and a transcription initiation site of P2 located in intron 1. Splicing variants of P1, isoforms a and b, and P2 transcript isoform c are shown under the map and specific 5′ primers for amplifying each isoform are labeled with A, B and C. The common reverse primer is specific to exon 3. (B) P19 cells were subjected to RA-induced differentiation. After dissociating embryoid bodies and plating onto tissue culture plates, NGF treatment was initiated on day 1 in the absence of RA and cells were harvested on day 3. Isolated RNAs were analyzed by RT-PCR followed by a semi-quantitative Southern blotting procedure using [32P]-probe specific to KOR cDNA (upper left). NGF receptor p75 and neuronal differentiation marker BM88 were also examined on these samples as shown at the bottom. The images shown are the representative of two experiments. UD: Undifferentiated.
NGF stimulates P2 activity through an AP2 binding site
P2 of KOR gene spans approximately 300 base pairs and contains three major transcription factor-binding sites for regulation, i.e. sites for AP2, c-Myc and Ikaros. The Ikaros binding site has been previously shown as a repressive element that recruits histone deacetylases (Hu et al., 2001). The adjacent E box has been identified to be a binding site for c-Myc that mediates the repressive effect of nitric oxide on this promoter (Park et al., 2002). The remaining transcription factor-binding site is AP2, which is a likely candidate responsible for the positive regulatory effect on P2. In order to first determine if this AP2 site was a functional regulatory element, a full-length P2 reporter (K18) was first generated, and a specific AP2-mutated reporter (K18m) was constructed from K18 by site-specific mutagenesis. These reporters and a negative control were tested in P19 cells as shown in Fig. 2A. We predicted that mutation at this positive regulatory element would drastically reduce P2 activity. Indeed the AP2-mutated P2 activity (K18m) was reduced to a basal level of the minimal promoter activity. To determine if NGF indeed stimulated P2 through this AP2 site, we constructed a deleted reporter by deleting all other regulatory elements from the full length P2 reporter, generating an AP2-specific reporter Kd50. Standard reporter analyses were conducted in P19 cells. Fig. 2B shows that, as predicted, the full-length P2 reporter could be activated by NGF. Importantly, the minimal promoter containing merely the AP2 site (Kd50) could also be effectively activated by NGF. Expression of NGF receptor has been verified in Western blot analysis as shown in Fig. 1B (NGFR p75). These results confirm that the AP2 site on P2 is a functional regulatory element and indeed can mediate the stimulating effect of NGF on P2. Very similar result was obtained when these reporters were analyzed using PC12 cells known to express abundant endogenous NGF receptors (data not shown).
Fig. 2.
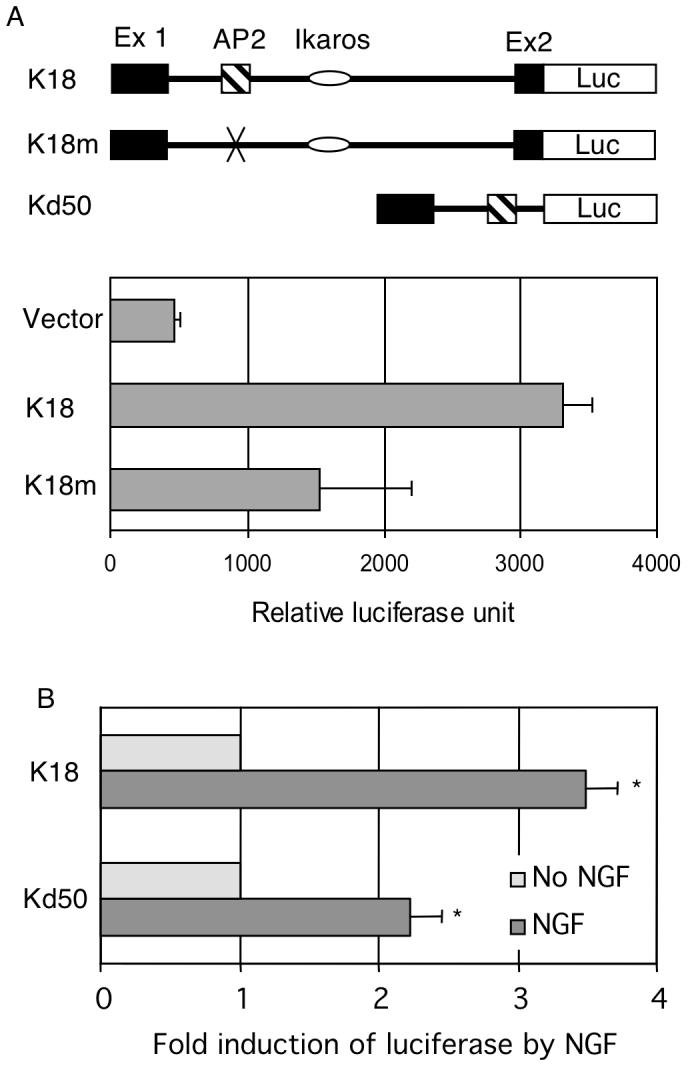
NGF activates P2 through an AP2 site. (A) AP2 site is a functional positive element. The maps of reporter constructs are shown on top: K18 for the full-length P2, K18m for the specific AP2 site mutant of K18, and Kd50 for deletion mutant carrying only the AP2 site. Reporter genes were introduced into P19 cells and luciferase activity as well as internal controls was measured. (B) AP2 site mediates the stimulating effects of NGF. Reporter genes were introduced into P19 cells and luciferase activity as well as internal controls was measured. NGF effect was determined by normalizing the luciferase reading over the internal control for each reporter construct. The data of averages and SDs were collected from three independent experiments. Column NGF *p<0.01, versus No NGF for both constructs.
NGF stimulates specific binding of AP2β to the AP2 site of P2
Transcription factor AP2 is expressed in P19 cells, yet P2 is silenced in these cells. Further, the effect of NGF requires pretreatment of cells with RA to induce neuronal differentiation. A possibility was that NGF stimulated the differentiating P19 nuclear environment to provide activated AP2 for binding to their target sites. This was first assessed by in vitro binding assay, EMSA, with nuclear extracts prepared from NGF-stimulated, differentiating P19 cells treated with RA that, in theory, should contain properly activated AP2. As shown in Fig. 3, the AP2 site (top sequence) was very weakly bound by the control nuclear extract (Fig. 3A, lane 2, arrowhead). The binding by RA-treated P19 extract was slightly enhanced (lane 3) whereas binding by RA-treated and NGF stimulated P19 extract was very robustly increased (lane 4). Specific binding was validated by efficient competition with unlabeled probe (lane 7). Supershift with specific antibodies further confirmed that it was AP2β (lane 9) but not AP2α (lane 8) or a negative control using HA (lane 10) that specifically bound to this site (asterisk). The faster migrating band (arrow) could be the complex of AP2 monomer since its pattern was consistent with the slower migrating band (arrowhead) and could be effectively competed with the cold probe.
Fig. 3.
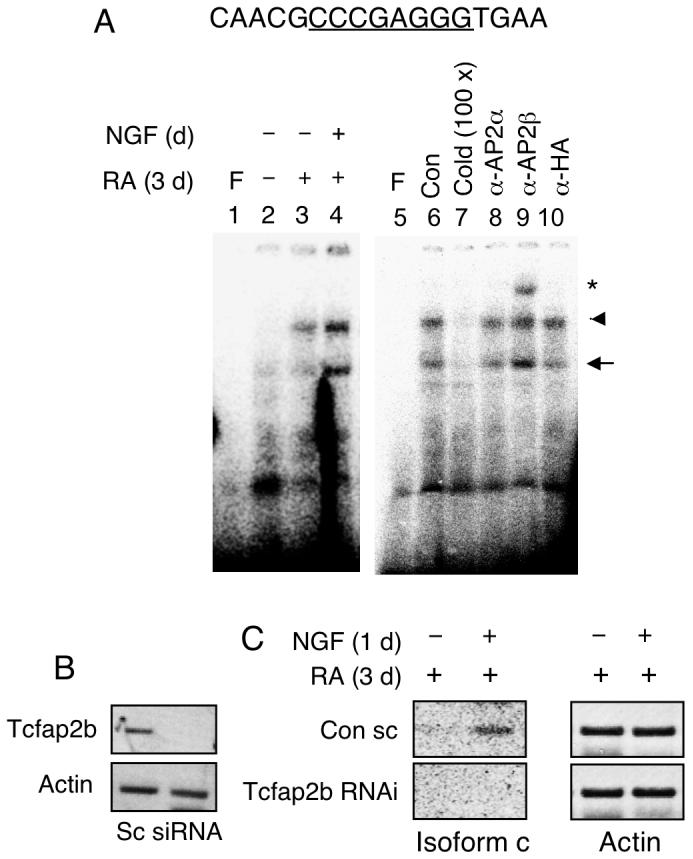
NGF enhanced P19 nuclear extract binding to the AP2β site on P2. (A) P19 cells were induced with RA for 3 days and followed by NGF treatment for 1 day. Nuclear extracts were subjected to EMSA using probes prepared from the P2 DNA segment carrying the AP2β site. Arrowhead marks the dimer-binding complex and arrow depicts the monomer-binding complex. Underlined sequences represent core binding sites for the transcription factors. Competition and supershift assays using unlabeled cold probes and antibodies against AP2α, AP2β and HA are shown in lanes 5-10. Experiments were repeated two times. (B) P19 cells were transfected with siRNAs for Tcfap2b and treated with RA and NGF as described above. Isolated RNAs were analyzed by RT-PCR to assess the effectiveness of RNA interference of Tcfap2b. (C) The expression of endogenous KOR c and actin in P19 cells transfected with the Tcfap2b siRNA. Amplified KOR c was detected by Southern blot. siRNA experiments were performed two times.
To confirm the functional roles for endogenous AP2β in NGF-stimulated P2 activity in P19, we employed a RNA interference strategy to knockdown endogenous AP2β, Tcfap2b. Effective silencing of Tcfap2b (encoding AP2β) was validated as shown in Fig. 3B. The effect of silencing AP2β was determined as shown in Fig. 3C. NGF-stimulated expression of KOR c in scrambled RNA treated (control) culture was validated as shown in the top panel. Importantly, in culture treated with Tcfap2b specific siRNA, NGF could no longer stimulate KOR c expression. This result confirms the functional role for AP2β in NGF-stimulated P2 activation.
NGF induces rapid recruitment of endogenous AP2β to P2
To examine the kinetics of endogenous AP2β recruitment to P2 chromatin, we performed Western blot analyses to monitor the expression level of endogenous AP2β in RA-induced, NGF-stimulated P19 (Fig. 4A), and conducted ChIP assays to assess its recruitment to the endogenous P2 chromatin (Fig. 4B). Based upon the data of Western blot analysis, it appeared that the expression of AP2β remained relatively constant during the course of NGF treatment. Interestingly, ChIP data showed that AP2β was rapidly recruited to the endogenous P2 after NGF stimulation (Fig. 4B, as early as 7 h) and stayed on P2 even at 24 hrs. These results further support that, in response to NGF signal, AP2β is rapidly recruited to its endogenous chromatin target on P2 of KOR gene.
Fig. 4.
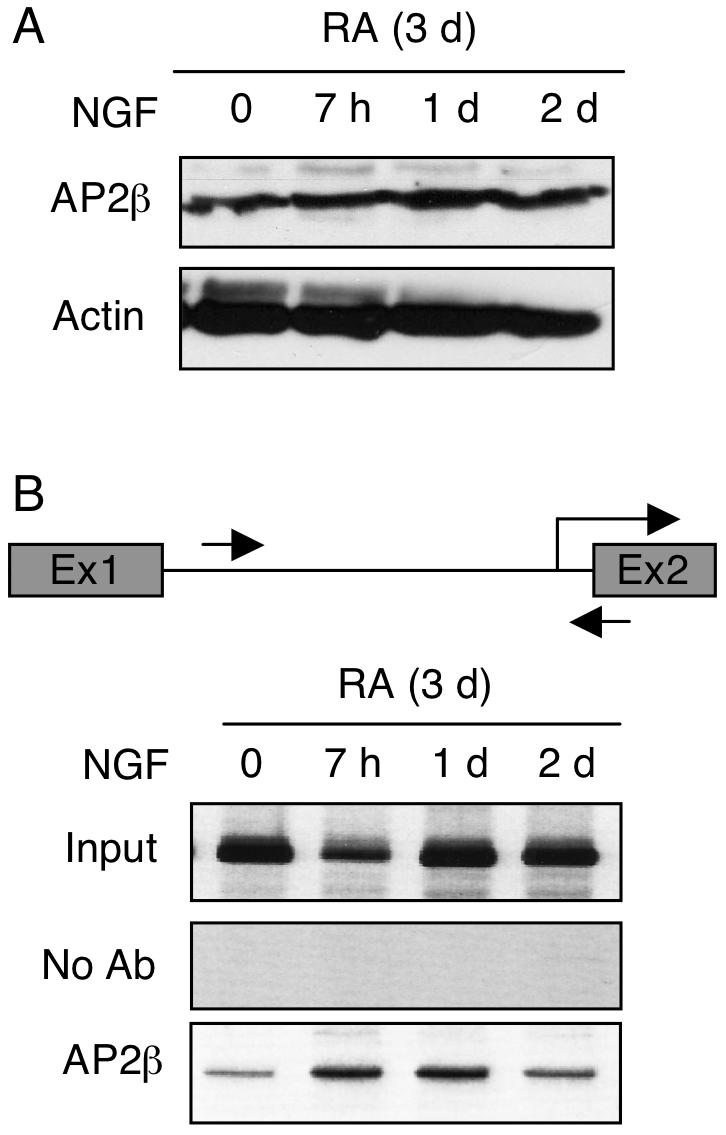
NGF enhances recruitment of AP2β to the chromatin of endogenous KOR P2. P19 cells were treated with RA for 3d and followed by NGF stimulation for the indicated periods of time, and were subjected to Western blot analyses to monitor protein expression levels (A), and in ChIP assays (B). DNA precipitated with antibodies against AP2β was amplified with primers specific to KOR P2, depicted with arrows (B, top panel). The images shown were the representative of three experiments.
NGF activates P2 through PI3K
To identify the specific signaling pathway of NGF that mediated the activation of P2, we employed reporter analysis and inhibitors of specific signaling enzymes. To efficiently compare the effects in the presence of signaling inhibitors, these reporter assays were conducted in PC12 cells that expressed abundant NGF receptors and proper down stream signaling enzymes for NGF. As shown in Fig. 5, the activating effect of NGF on both the full-length P2 reporter (K18, panel A) and the minimal AP2 reporter (Kd50, panel B) was significantly inhibited by LY294002, a PI3K inhibitor, but not by U73122, a PLC inhibitor. This result suggests that NGF activates P2 via the PI3K pathway. Therefore, NGF stimulation of KOR P2 is mediated by the NGF-coupled NGFR/PI3K pathway that activates AP2β to bind to P2 chromatin, thereby stimulating its transcription.
Fig. 5.

NGF signals are transmitted through PI3K to enhance AP2 reporter activity. PC12 cells were transfected with reporter constructs of the full length P2 K18 (A) and AP2 Kd50 (B), and treated with NGF for 24 h, or NGF in the presence of U73122 (U73) or LY294002 (LY29). Each luciferase reading was normalized to the internal control reading. The value of the control (con) group was arbitrarily set at 1 and the value of each experimental group was plotted as fold induction and SD by comparing to the control. The data were collected from three independent experiments. Column NGF *p<0.01 versus control, column LY294002 **p<0.05 versus NGF for both constructs.
NGF triggers chromatin modification on, and RNA polymerase recruitment to, P2
Since NGF rapidly stimulated P2 activity by inducing AP2β recruitment to P2 chromatin, it was suspected that epigenetic changes might occur on P2 chromatin, which could efficiently activate the transcription machinery on this promoter. ChIP assays were performed to test this possibility (Fig. 6). As shown on the left panel, in RA-induced differentiating P19 cells without NGF treatment, histone H3 dimethylation at Lys9 (H3-K9-me2) was apparent on P2 chromatin. Following NGF stimulation, this promoter was then dimethylated mostly at Lys4 (H3-K4-me2), demonstrating that NGF signal switched the otherwise repressive chromatin (H3-K9-me2) of P2 to a de-repressed state (H3-K4-me2). Interestingly, pan acetylation of histone H4 was not affected by NGF (AcH4, Fig. right panel). Further, the heterochromatin marker (HP1α), initially appeared on this promoter in RA-induced cells without NGF treatment, was completely removed from this promoter in cells further stimulated with NGF. Consistently, RNA polymerase II (polII) was increasingly recruited to P2 following NGF stimulation. These results show that the chromatin of P2 is altered from a repressed to an active state by NGF stimulation. This occurs in RA-primed, differentiating P19 cells when they begin to express NGF receptors and are able to transmit NGF signals to activate transcription factors for binding to specific chromatin targets. As a result, epigenetic changes occur on this chromatin region, allowing RNA polymerase II to be recruited to this promoter to activate transcription in late differentiating cells.
Fig. 6.
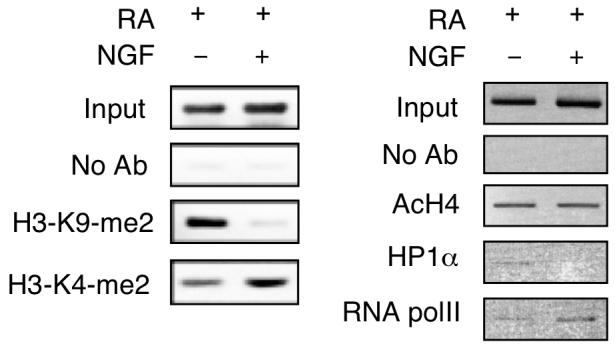
NGF triggers epigenetic changes on P2. P19 cells were induced by RA for 3 days, followed by NGF treatment for 24 h. Cells were subjected to ChIP assays with antibodies against dimethyl Lys9 of histone H3 (H3-K9-me2), dimethyl Lys4 of histone H3 (H3-K4-me2), acetylated histone H4 (AcH4), heterochromatin protein 1α (HP1α) and RNA polymerase II (RNA polII). The images shown were the representative of two experiments.
Discussion
P2 of the mouse KOR gene is silenced in P19 stem cells and during embryonic development when most neuronal maturation is not yet completed. We are able to detect transcripts from P2 only in very limited brain stem areas of adult animals (Bi et al., 2001), suggesting that more maturation in neuronal differentiation is required for the activation of P2. To this end, it has been puzzling to us whether and how P2 of the mouse KOR gene might be activated in later stages of development and in more mature neurons. This study provides the first evidence that P2 activation requires epigenetic changes on its chromatin, which is brought about by the signals of neurotrophin NGF.
In P19 stem cells, neither the pan-neurotrophin receptor p75 nor TrkA NGF receptor is expressed, suggesting that these are specialized receptors that need to be induced such as by differentiation agent RA. As expected, in RA-induced differentiating P19 cells, p75 begins to be expressed to mediate NGF signal. This is consistent with the expression of p75 paralleling that of the early neuronal marker BM88 in differentiating P19 cells. p75 (possibly also TrkA) then stimulates binding or recruitment of AP2β to KOR P2, resulting in the switch from repressive chromatin (H3-K9-me2) to active chromatin (H3-K4-me2). How AP2β is activated and recruited to P2 is a topic of future investigation. The effects of neurotrophins on KOR gene activity prompted us to explore possible effects of opioid peptides or ligands on the activity of KOR gene in differentiating P19. Interestingly, KOR ligands, including dynorphin, exert little effects in terms of KOR gene expression In P19 neurons. However, since P19 neurons may not recapitulate the full capacity of primary neurons, it remains to be determined whether the lack of effects of KOR ligands on its gene activity holds true in the context of whole animals.
In studies using PC12 cells that are widely used for examining NGF, others have shown that NGF increases the expression of certain genes by augmenting DNA binding or activity of transcription factors such as Sp1, c-fos, c-jun, Egr-1, and STAT3 etc. (Pierchala et al., 2004; Sobue et al., 2005; Ng et al., 2006; Pelligrino and Stork, 2006; Rojo et al., 2006; Tai et al., 2006). NGF signal is transduced through various pathways to activate transcription factors. For example, NGF can activate the Ras/Raf/MEK1/Erk1 pathway to enhance Sp1 function on the N-methyl-D-aspartate receptor 1 promoter, but not Erk2 (Liu et al., 2001). On the heme oxygenase-1 gene promoter, NGF induces phosphorylation of Sp1 through a pathway that involves PI3K, PKC-ζ, MEK, and Erk (Rojo et al., 2006). PLCγ has been shown to be required for Raf/MEK/Erk activation (Rong et al., 2003). Recently, it has been suggested that NGF stimulates matrix metalloproteinase-2 via the PI3K/Akt signaling pathway and AP-2 (Park et al., 2007). To regulate P2 in P19, NGF activates AP2β, but not AP2α, through a pathway involving PI3K, but not PLCγ (Fig. 5). PKC inhibitor calphostin C did not change NGF effect on the reporters of full length P2 and AP2 binding (data not shown), ruling out the involvement of PKC in this pathway. The roles of Erk1/2 in this signaling pathway remain unclear. Fig. 7 briefly depicts NGF-elicited pathways triggering epigenetic changes on P2 of the mouse KOR gene in RA-induced P19 neuorns.
Fig. 7.
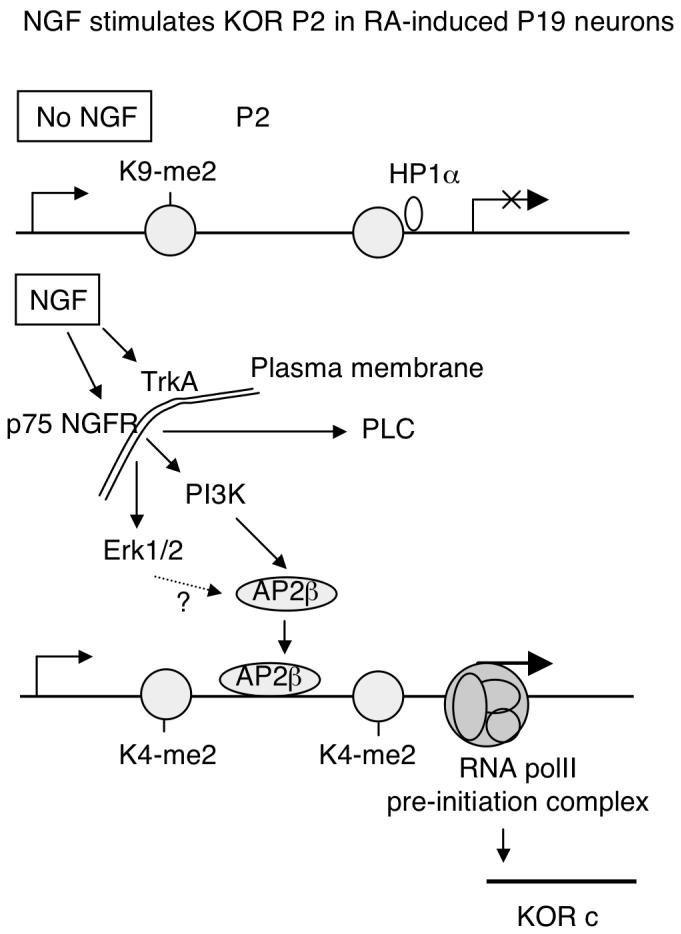
A schematic illustration of NGF activation of KOR P2 during RA-induced neuronal differentiation of P19. In the absence of NGF, P2 is in a heterochromatin conformation marked by histone H3 K9 methylation (K9-me2). NGF can stimulate TrkA receptor and p75 NGFR. The p75 NGFR, but not TrkA, is induced in early differentiating P19, resulting in activation of intracellular signaling molecules such as Erk1/2, PLC, and PI3 kinase. In the P19 system, it is PI3 kinase pathway that is responsible for AP2β activation. Erk1/2 pathway is unclear, depicted with a question mark. PLC is not involved in AP2β. Activated AP2β increasingly binds to KOR P2, triggering epigenetic changes reflected by histone H3 K4 methylation (K4-me2), dissociation of HP1α and formation of RNA polII pre-initiation complex. KOR c is then actively transcribed in NGFR-expressing neurons.
Chromatin remodeling involves, primarily, two events: nucleosome formation/rearrangement and alteration of histone modifications. In the case of P2, histone methylation is altered by NGF treatment, from Lys9 dimethylation of H3 to Lys4 dimethylation. Nucleosome mapping studies, however, have shown that no major changes occur on the nucleosome arrangement of P2 in RA-induced differentiating P19 cells (data not shown). The association of HP1α with P2 in the P2-suppressed condition of P19 cells suggests a heterochromatin feature for P2 in the less differentiated cells. All together, our results provide evidence that, a neuron-specific promoter like KOR P2 can be regulated by epigenetic changes induced only in differentiating neurons that express specific neurotrophin receptors and therefore are responsive to neurotrophin stimulation. The specific epigenetic changes on KOR P2 include switching lysine methylation from Lys9 to Lys4 of H3, as a result of recruitments of specific transcription factors to the specific genomic locus.
Recently, we have also examined the chromatin status of the mouse μ opioid receptor (MOR) gene in P19 that undergoes neuronal differentiation (Hwang et al. 2007) and that of the mouse δ opioid receptor (DOR) gene in Neuro2A (Wang et al. 2005). In the P19 experimental model, like the P2 of the KOR gene, MOR gene is also silenced in the undifferentiated stem cell population and becomes activated in differentiated neurons. It appears that in undifferentiated stem cells, MOR promoter region is highly methylated on CG islands and histone proteins of this chromatin region are mostly deacetylated, indicative of a heterochromatin structure of the MOR promoter. In differentiating neurons, the level of DNA methylation is reduced and histone acetylation is correspondingly increased on MOR promoter, thereby activating its transcription in differentiated neurons. For the DOR gene, repression is mediated by a similar mechanism through DNA-methylation in the Nuron2A model. Therefore, both MOR and DOR genes are repressed, primarily, by DNA methylation, and their activation requires a similar DNA demethylation-elicited epigenetic change. Interestingly, for P2 of the KOR gene, its chromatin is assembled into a compact nucleosome array rich in the heterochromatin marker HP1, and therefore this promoter is repressed, likely also in the form of heterochromatin, in stem cells (Park et al., 2005). This current study extends the previous findings and discovers that activation of possibly heterochromatinized KOR P2 requires a principal epigenetic change elicited by the action of neurotrophins. It would be important to determine whether epigenetic changes on MOR and DOR gene promoters also involve the action of neurotrophins, in addition to DNA demethylation.
Acknowledgements
This work was supported partially by NIH Grants DA11190, DA11806, DK54733, DK60521 and K02-DA13926 to L.N.W., DA000564, DA001583, DA011806, and K05-DA070554 to H.H.L. and by a seed grant of DA011806 to S.W.P.
List of Abbreviations
- KOR
kappa opioid receptor
- NGF
nerve growth factor
- AP2
activation protein 2
- PI3K
phosphatidylinositol 3-kinase
- RA
retinoic acid
- P1
promoter 1
- P2
promoter 2
- PLCγ
phospholipase Cγ
- MAPK
mitogenic activation protein kinase
- EMSA
electrophoretic mobility shift assay
Footnotes
Section editor: Dr. Menahem Segal, Cellular Neuroscience
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Appleyard SM, Patterson TA, Jin W, Chavkin C. Agonist-induced phosphorylation of the κ-opioid receptor. J Neurochem. 1997;69:2405–2412. doi: 10.1046/j.1471-4159.1997.69062405.x. [DOI] [PubMed] [Google Scholar]
- Arden JR, Segredo V, Wang Z, Lameh J, Sadée W. Phosphorylation and agonist-specific intracellular trafficking of an epitope-tagged μ-opioid receptor expressed in HEK 293 cells. J Neurochem. 1995;65:1636–1645. doi: 10.1046/j.1471-4159.1995.65041636.x. [DOI] [PubMed] [Google Scholar]
- Bi J, Hu X, Loh HH, Wei LN. Regulation of mouse κ opioid receptor gene expression by retinoids. J Neurosci. 2001;21:1590–1599. doi: 10.1523/JNEUROSCI.21-05-01590.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi J, Hu X, Loh HH, Wei LN. Mouse κ-opioid receptor mRNA differential transport in neurons. Mol Pharmacol. 2003;64:594–599. doi: 10.1124/mol.64.3.594. [DOI] [PubMed] [Google Scholar]
- Bi J, Tsai NP, Lin YP, Loh HH, Wei LN. Axonal mRNA transport and localized translational regulation of κ-opioid receptor in primary neurons of dorsal root ganglia. Proc Natl Acad Sci USA. 2006;103:19919–19924. doi: 10.1073/pnas.0607394104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi J, Tsai N-P, Lu H-Y, Loh HH, Wei L-N. Copb1 facilitated axonal transport and translation of kappa opioid receptor mRNA. Proc. Natl. Acad. Sci. USA. 2007;104:13810–13815. doi: 10.1073/pnas.0703805104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler CJ, Fraser GL. Mu-, delta-, kappa-opioid receptors and their subtypes. A critical review with emphasis on radioligand binding experiments. Neurochem Int. 1994;24:401–426. doi: 10.1016/0197-0186(94)90089-2. [DOI] [PubMed] [Google Scholar]
- Fukuda K, Kato S, Morikawa H, Shoda T, Mori K. Functional coupling of the δ-, μ-, and κ-opioid receptors to mitogen-activated protein kinase and arachidonate release in Chinese hamster ovary cells. J Neurochem. 1996;67:1309–1316. doi: 10.1046/j.1471-4159.1996.67031309.x. [DOI] [PubMed] [Google Scholar]
- Georgopoulou N, Hurel C, Politis PK, Gaitanou M, Matsas R, Thomaidou D. BM88 is a dual function molecule inducing cell cycle exit and neuronal differentiation of neuroblastoma cells via cyclin D1 down-regulation and retinoblastoma protein hypophosphorylation. J Biol Chem. 2006;281:33606–33620. doi: 10.1074/jbc.M602689200. [DOI] [PubMed] [Google Scholar]
- Henry DJ, Grandy DK, Lester HA, Davidson N, Chavkin C. κ-opioid receptors couple to inwardly rectifying potassium channels when coexpressed by Xenopus oocytes. Mol Pharmacol. 1995;47:551–557. [PubMed] [Google Scholar]
- Hu X, Bi J, Loh HH, Wei LN. An intronic Ikaros-binding element mediates retinoic acid suppression of the κ opioid receptor gene, accompanied by histone deacetylation on the promoters. J Biol Chem. 2001;276:4597–4603. doi: 10.1074/jbc.M005477200. [DOI] [PubMed] [Google Scholar]
- Hu X, Bi J, Loh HH, Wei LN. Regulation of mouse κ opioid receptor gene expression by different 3′-untranslated regions and the effect of retinoic acid. Mol Pharmacol. 2002;62:881–887. doi: 10.1124/mol.62.4.881. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Hwang CK, Song KY, Kim CS, Choi HS, Guo XH, Law PY, Wei LN, Loh HH. Evidence of endogenous Mu opioid receptor regulation by epigenetic control of the promoters. Mol. Cell. Biol. 2007;27:4720–4736. doi: 10.1128/MCB.00073-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson PS, Wang JB, Wang WF, Uhl GR. Expressed mu opiate receptor couples to adenylate cyclase and phosphatidyl inositol turnover. Neuroreport. 1994;5:507–509. doi: 10.1097/00001756-199401120-00035. [DOI] [PubMed] [Google Scholar]
- Koutmani Y, Hurel C, Patsavoudi E, Hack M, Gotz M, Thomaidou D, Matsas R. BM88 is an early marker of proliferating precursor cells that will differentiate into the neuronal lineage. Eur J Neurosci. 2004;20:2509–2523. doi: 10.1111/j.1460-9568.2004.03724.x. [DOI] [PubMed] [Google Scholar]
- Law PY, Loh HH, Wei LN. Insights into the receptor transcription and signaling: implications in opioid tolerance and dependence. Neuropharmacology. 2004;47:300–311. doi: 10.1016/j.neuropharm.2004.07.013. [DOI] [PubMed] [Google Scholar]
- Li LY, Chang KJ. The stimulatory effect of opioids on mitogen-activated protein kinase in Chinese hamster ovary cells transfected to express mu-opioid receptors. Mol Pharmacol. 1996;50:599–602. [PubMed] [Google Scholar]
- Li J, Park SW, Loh HH, Wei LN. Induction of the mouse κ-opioid receptor gene by retinoic acid in P19 cells. J Biol Chem. 2002;277:39967–39972. doi: 10.1074/jbc.M200840200. [DOI] [PubMed] [Google Scholar]
- Liu A, Prenger MS, Norton DD, Mei L, Kusiak JW, Bai G. Nerve growth factor uses Ras/ERK and phosphatidylinositol 3-kinase cascades to up-regulate the N-methyl-D-aspartate receptor 1 promoter. J Biol Chem. 2001;276:45372–45379. doi: 10.1074/jbc.M105399200. [DOI] [PubMed] [Google Scholar]
- Lu S, Loh HH, Wei LN. Studies of dual promoters of mouse κ-opioid receptor gene. Mol Pharmacol. 1997;52:415–420. doi: 10.1124/mol.52.3.415. [DOI] [PubMed] [Google Scholar]
- Ng YP, Cheung ZH, Ip NY. STAT3 as a downstream mediator of Trk signaling and functions. J Biol Chem. 2006;281:15636–15644. doi: 10.1074/jbc.M601863200. [DOI] [PubMed] [Google Scholar]
- Ozawa T, Nakagawa T, Minami M, Satoh M. Supersensitization of the adenylyl cyclase system in Chinese hamster ovary cells co-expressing cloned opioid receptors and Gz, a PTX-insensitive G protein. Neurosci Lett. 1999;267:117–120. doi: 10.1016/s0304-3940(99)00347-x. [DOI] [PubMed] [Google Scholar]
- Pan YX, Xu J, Mahurter L, Bolan E, Xu M, Pasternak GW. Generation of the mu opioid receptor (MOR-1) protein by three new splice variants of the Oprm gene. Proc Natl Acad Sci. USA. 2001;98:14084–14089. doi: 10.1073/pnas.241296098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan YX. Identification of alternatively spliced variants from opioid receptor genes. Methods Mol Med. 2003;84:65–75. doi: 10.1385/1-59259-379-8:65. [DOI] [PubMed] [Google Scholar]
- Park MJ, Kwak HJ, Lee HC, Yoo DH, Park IC, Kim MS, Lee SH, Rhee CH, Hong SI. Nerve growth factor induces endothelial cell invasion and cord formation by promoting matrix metalloproteinase-2 expression through the phosphatidylinositol-3 kinase/AKT signaling pathway and AP-2 transcription factor. J Biol Chem. 2007 doi: 10.1074/jbc.M701081200. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- Park SW, Li J, Loh HH, Wei LN. Novel signaling pathway of nitric oxide on transcriptional regulation of mouse kappa opioid receptor gene. J Neurosci. 2002;22:7941–7947. doi: 10.1523/JNEUROSCI.22-18-07941.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SW, Huq MD, Loh HH, Wei LN. Retinoic acid-induced chromatin remodeling of mouse κ opioid receptor gene. J Neurosci. 2005;25:3350–3357. doi: 10.1523/JNEUROSCI.0186-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei G, Kieffer BL, Lefkowitz RJ, Freedman NJ. Agonist-dependent phosphorylation of the mouse δ-opioid receptor: involvement of G protein-coupled receptor kinases but not protein kinase C. Mol Pharmacol. 1995;48:173–177. [PubMed] [Google Scholar]
- Pellegrino MJ, Stork PJ. Sustained activation of extracellular signal-regulated kinase by nerve growth factor regulates c-fos protein stabilization and transactivation in PC12 cells. J Neurochem. 2006;99:1480–1493. doi: 10.1111/j.1471-4159.2006.04250.x. [DOI] [PubMed] [Google Scholar]
- Pierchala BA, Ahrens RC, Paden AJ, Johnson EM., Jr Nerve growth factor promotes the survival of sympathetic neurons through the cooperative function of the protein kinase C and phosphatidylinositol 3-kinase pathways. J Biol Chem. 2004;279:27986–27993. doi: 10.1074/jbc.M312237200. [DOI] [PubMed] [Google Scholar]
- Piros ET, Prather PL, Loh HH, Law PY, Evans CJ, Hales TG. Ca2+ channel and adenylyl cyclase modulation by cloned μ-opioid receptors in GH3 cells. Mol Pharmacol. 1995;47:1041–1049. [PubMed] [Google Scholar]
- Prather PL, Loh HH, Law PY. Interaction of δ-opioid receptors with multiple G-proteins: a non-relationship between agonist potency to inhibit adenylyl cyclase and activation of G-proteins. Mol Pharmacol. 1993;45:997–1003. [PubMed] [Google Scholar]
- Rojo AI, Salina M, Salazar M, Takahashi S, Suske G, Calvo V, de Sagarra MR, Cuadrado A. Regulation of heme oxygenase-1 gene expression through the phosphatidylinositol 3-kinase/PKC-zeta pathway and Sp1. Free Radic Biol Med. 2006;41:247–261. doi: 10.1016/j.freeradbiomed.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Rong R, Ahn JY, Chen P, Suh PG, Ye K. Phospholipase activity of phospholipase C-gamma1 is required for nerve growth factor-regulated MAP kinase signaling cascade in PC12 cells. J Biol Chem. 2003;278:52497–52503. doi: 10.1074/jbc.M306744200. [DOI] [PubMed] [Google Scholar]
- Santos SD, Verveer PJ, Bastiaens PI. Growth factor-induced MAPK network topology shapes Erk response determining PC-12 cell fate. Nat Cell Biol. 2007;9:324–330. doi: 10.1038/ncb1543. [DOI] [PubMed] [Google Scholar]
- Smart D, Hirst RA, Hirota K, Grandy DK, Lambert DG. The effects of recombinant rat μ-opioid receptor activation in CHO cell on phospholipase C, [Ca2′]i and adenylyl cyclase. Br J Pharmacol. 1997;120:1165–1171. doi: 10.1038/sj.bjp.0701012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobue S, Hagiwara K, Banno Y, Tamiya-Koizumi K, Suzuki M, Takagi A, Kojima T, Asano H, Nozawa Y, Murate T. Transcription factor specificity protein 1 (Sp1) is the main regulator of nerve growth factor-induced sphingosine kinase 1 gene expression of the rat pheochromocytoma cell line, PC12. J Neurochem. 2005;95:940–949. doi: 10.1111/j.1471-4159.2005.03399.x. [DOI] [PubMed] [Google Scholar]
- Spencer RJ, Jin W, Thayer SA, Chakrabarti S, Law PY, Loh HH. Mobilization of Ca2+ from intracellular stores in transfected neuro2a cells by activation of multiple opioid receptor subtypes. Biochem Pharmacol. 1997;54:809–818. doi: 10.1016/s0006-2952(97)00243-8. [DOI] [PubMed] [Google Scholar]
- Tai TC, Wong-Faull DC, Claycomb R, Wong DL. Nerve growth factor regulates adrenergic expression. Mol Pharmacol. 2006;70:1792–1801. doi: 10.1124/mol.106.026237. [DOI] [PubMed] [Google Scholar]
- Tallent M, Dichter MA, Bell GI, Reisine T. The cloned kappa opioid receptor couples to an N-type calcium current in undifferentiated PC-12 cells. Neuroscience. 1994;63:1033–1040. doi: 10.1016/0306-4522(94)90570-3. [DOI] [PubMed] [Google Scholar]
- Tsai NP, Bi J, Loh HH, Wei LN. Netrin-1 signaling regulates de novo protein synthesis of κ opioid receptor by facilitating polysomal partition of its mRNA. J Neurosci. 2006;26:9743–9749. doi: 10.1523/JNEUROSCI.3014-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Liu T, Wei LN, Law PY, Loh HH. DNA Methylation-Related Chromatin Modification in the Regulation of Mouse delta-Opioid Receptor Gene. Mol. Pharmacology. 2005;67:2032–2039. doi: 10.1124/mol.105.011056. [DOI] [PubMed] [Google Scholar]
- Wehrman T, He X, Raab B, Dukipatti A, Blau H, Garcia KC. Structural and mechanistic insights into nerve growth factor interactions with the TrkA and p75 receptors. Neuron. 2007;53:25–38. doi: 10.1016/j.neuron.2006.09.034. [DOI] [PubMed] [Google Scholar]
- Wei LN, Hu X, Bi J, Loh HH. Post-transcriptional regulation of mouse κ-opioid receptor expression. Mol Pharmacol. 2000;57:401–408. [PubMed] [Google Scholar]
- Wei LN, Law PY, Loh HH. Post-transcriptional regulation of opioid receptors in the nervous system. Front Biosci. 2004;9:1665–1679. doi: 10.2741/1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei LN, Loh HH. Regulation of opioid receptor expression. Curr Opin Pharmacol. 2002;2:69–75. doi: 10.1016/s1471-4892(01)00123-0. [DOI] [PubMed] [Google Scholar]
- Wu EH, Wong YH. Pertussis toxin-sensitive Gi/o proteins are involved in nerve growth factor-induced pro-survival Akt signaling cascade in PC12 cells. Cell Signal. 2005;17:881–890. doi: 10.1016/j.cellsig.2004.11.008. [DOI] [PubMed] [Google Scholar]
- Zimprich A, Simon T, Höllt V. Cloning and expression of an isoform of the rat μ opioid receptor (rMOR1B) which differs in agonist induced desensitization from rMOR1. FEBS Lett. 1995;359:142–146. doi: 10.1016/0014-5793(95)00028-8. [DOI] [PubMed] [Google Scholar]