

Abstract
δ-Catenin was first identified through its interaction with Presenilin-1 and has been implicated in the regulation of dendrogenesis and cognitive function. However, the molecular mechanisms by which δ-catenin promotes dendritic morphogenesis were unclear. In this study, we demonstrated δ-catenin interaction with p190RhoGEF, and the importance of Akt1-mediated phosphorylation at Thr-454 residue of δ-catenin in this interaction. We have also found that δ-catenin overexpression decreased the binding between p190RhoGEF and RhoA, and significantly lowered the levels of GTP-RhoA but not those of GTP-Rac1 and -Cdc42. δ-Catenin T454A, a defective form in p190RhoGEF binding, did not decrease the binding between p190RhoGEF and RhoA. δ-Catenin T454A also did not lower GTP-RhoA levels and failed to induce dendrite-like process formation in NIH 3T3 fibroblasts. Furthermore, δ-catenin T454A significantly reduced the length and number of mature mushroom shaped spines in primary hippocampal neurons. These results highlight signaling events in the regulation of δ-catenin-induced dendrogenesis and spine morphogenesis.
δ-Catenin was first identified by yeast two-hybrid screening as a molecule that interacts with Presenilin-1 (PS-1), which is the most prominently mutated gene in familial Alzheimer's disease (FAD) patients (1, 2). The interaction of δ-catenin with PS-1, along with its abundant expression in neurons, suggests that δ-catenin has specialized neuronal functions (3, 4). Indeed, δ-catenin deficient mice showed severe learning deficits and abnormal synaptic plasticity, suggesting a special role of δ-catenin at the synapse (5). Furthermore, the hemizygous loss of the chromosomal 5p15.2 region, which contains the human δ-catenin gene, results in the severe mental retardation associated with Cri du Chat syndrome. This chromosomal abnormality may account for 1% of all mentally retarded individuals (6).
Structural analysis indicated that δ-catenin is a member of the p120-Catenin (hereafter, p120ctn) subfamily of armadillo proteins and has a DSWV sequence at the carboxyl terminus that binds to the PDZ (PSD-95/Disc-larg/ZO-1) domain-containing proteins (7). δ-Catenin also contains SH3 binding domains at the N-terminus (4, 8), a GKKKKKKK sequence (putative NLS) that can potentially promote lipid intermixing (9), and a proline-rich domain that is likely to be involved in the interaction with the actin-binding protein, Profilin (4). The presence of 10 Arm repeats in δ-catenin suggests its potential participation in various protein-protein interactions. In addition to PS-1, the δ-catenin-associated proteins identified thus far include E-cadherin (4), S-SCAM (7), p0071 (10), Densin-180 (11), PSD-95, Abl (8), Cortactin (12), Sphingosine kinase (13), and Kaiso (14), suggesting its many possible roles in cells. Our previous reports demonstrated that the overexpression of δ-catenin induces the branching of dendrite-like processes in both NIH 3T3 fibroblasts and primary hippocampal neurons (15). We have also reported that an E18 hippocampal neuron overexpressing δ-catenin demonstrates enhanced arborization of dendrites, swelling, and maturation of dendritic spines (15, 16). δ-Catenin induced dendrogenesis is quite similar in shape to the morphological changes induced by treatment with C3 exotoxin, which is a specific Rho inhibitor derived from Clostridium botulinum (12, 17). The inhibition of Rho signaling also enhances not only the formation of the p120ctn induced branch (18) but also δ-catenin-induced dendrogenesis (12). However, the underlying mechanism for how δ-catenin overexpression affects Rho signaling is unclear.
The Rho family of small GTPases, consisting of Cdc42, Rac, and Rho, has been implicated in the control of numerous cellular processes including cytoskeletal reorganization, transcriptional activation, cell cycle progression, cell fate determination, and synaptic plasticity (17, 19, 20). Of the Rho GTPase family members, Cdc42, Rac1, and RhoA have been studied most extensively. The activation of the Rho family proteins requires GDP-GTP exchange that is catalyzed by various guanine nucleotide exchange factors (GEFs), and the cessation of their actions is regulated by GTPase-activating proteins (GAPs) and guanine nucleotide dissociation inhibitors (GDIs) (21-23). Among the Rho-specific GEFs, p190RhoGEF was first identified as a brain-enriched GDP/GTP exchange factor that activates RhoA and contains the predicted α-helical coiled-coil domain in the C-terminal (24, 25). The overexpression of p190RhoGEF mimics activated RhoA in stimulating cytoskeletal contraction and preventing neurite outgrowth (25). The C-terminal of p190RhoGEF has been shown to interact with microtubules (25), JIP-1 (26), a destabilizing element in the 3′UTR of neurofilament light (NF-L) mRNA (27), FAK (focal adhesion kinase) (28) and 14-3-3ζ (29), which is important for its regulatory function in cells. Furthermore, activation of Rho GTPases by p190RhoGEF leads to a decrease in dendritic branching, whereas GDP-Rho (inactive) induces dendrogenesis (24). Among the binding partners of p190RhoGEF, 14-3-3 was recently identified as a new binding partner of δ-catenin as well (30). Most 14-3-3 binds to the phospho-serine/threonine residue on a partner protein when it is phosphorylated by various kinases including protein kinase A, protein kinase C and Akt (31-33). Akt/PKB, a serine/threonine protein kinase, regulates multiple biological processes including synaptic strength, cell survival, proliferation, growth and glycogen metabolism (34-36). In mammals, Akt/PKB is composed of three Akt family members such as Akt1/PKBα, Akt2/PKBβ and Akt3/PKBγ, and is activated by various growth and survival factors through a pathway requiring PI3K-dependent generation of phosphatidylinositol (3, 4, 5) trisphosphate (37, 38).
In this study, we found that δ-catenin interacts with p190RhoGEF and significantly lowered the levels of GTP-RhoA, for which Akt1 plays an important role through the Thr-454 phosphorylation on δ-catenin. By introducing a substitution of Thr-454 residue on δ-catenin to Ala, we demonstrated that δ-catenin T454A, a defective form in binding p190RhoGEF, significantly reduced the length and number of mature mushroom shaped spines in primary hippocampal neurons. Our results suggest the interaction between δ-catenin and p190RhoGEF can act as a key modulator in regulating δ-catenin induced dendrogenesis and spine formation.
Experimental Procedures
Plasmids and antibodies
The construction of δ-catenin full-length (FL-) and ΔC207- in pEGFP-C1 has been previously described (15, 44). The δ-catenin T454A mutant, in which the Thr-454 residue was substituted to Ala, was generated by site-directed mutagenesis. The expression plasmid for p190RhoGEF (pcDNA3-HA) was kindly provided by W. Moolenaar (The Netherlands Cancer Institute, Amsterdam, The Netherlands); myc-RhoA by K.Y. Lee (Chonnam National University, Gwanju, Korea).
The antibodies were obtained as follows: anti-δ-catenin (#07-259) and anti-Rac1 (#05-389) (Upstate biotechnology); anti-GFP (632376), anti-δ-catenin (C98320) (BD Biosciences); anti-β-tubulin and anti-phospho-threonine (P6623, Sigma); and anti-RhoA (sc-418n), anti-Cdc42 (sc-87), and anti-phospho-serine (sc-57555, Santa Cruz Biotechnology). The antibody for p190RhoGEF was kindly provided by D. Schlaepfer and B. Margolis. HA or myc epitopes were detected using media from 12CA5 or 9E10 hybridoma, respectively.
Cell culture and transfection
The NIH 3T3, mouse embryonic fibroblast (MEF), and Bosc23 cells were grown in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin at 37°C with 5% CO2. The cells were transfected using calcium phosphate or Lipofectamine Plus reagent (Invitrogen), according to the manufacturer's instructions.
Immunoblotting and Akt kinase assay
Immunoprecipitation/Immunoblotting was performed as previously described (39). For the kinase assays, the full-length and mutant GFP-δ-catenin constructs were transiently overexpressed in MEF cells or Bosc23 cells. To collect P1 mouse brain lysates, 1 day post-natal mouse were sacrificed. δ-Catenin proteins were purified by immunoprecipitation using the specific antibody and protein G sepharose (BD Biosciences). The immune-complexes were washed three times with lysis buffer and twice with kinase buffer, and then incubated for 30 min at 30°C in a 20 μl reaction mixture supplemented with recombinant Akt protein and either 10μM cold ATP or 10 μCi [γ-32P]-ATP Akt, cold ATP and kinase buffer were purchased from Cell Signaling Technology and [γ-32P]-ATP was purchased from BMS. Radio-active phosphorylation status of δ-catenin was detected by autoradiograph and non radio-active phosphorylation status was determined by immunoblotting using the phospho-Ser or phospho-Thr antibodies. Equal loading of proteins was confirmed using EZ staining kits (EZBiopaQ, Co., Ltd.).
Affinity-precipitation of cellular GTPases
The cellular RhoA activity was determined using GST-RBD as previously described by Ren et al. (40). The cells were lysed in lysis buffer (50 mM Tris, pH 7.4, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 500 mM NaCl, 10 mM MgCl2, and protease inhibitor mixture). The lysates were incubated with the GST-RBD beads (Cytoskeleton Inc.) at 4°C for 1 hr. The beads were then washed four times with washing buffer (50 mM Tris, pH 7.4, 1% Triton X-100, 150 mM NaCl, 10 mM MgCl2, and protease inhibitor mixture). The bound RhoA proteins were detected by immunoblotting using a monoclonal antibody against RhoA.
The cellular Rac1 and Cdc42 activities were determined using GST-PBD as described in other studies (41). The cDNA of the p21-binding domain (PBD) from PAK1 (amino acids 67-150) was cloned into the bacterial expression vector pGEX-4T3 and expressed as a GST fusion protein. The purified fusion proteins were isolated from glutathione-Sepharose 4B beads. The lysates were incubated with the GST-PBD beads, and the beads were washed twice with 25 mM Tris, pH 7.4, 1 mM DTT, 30 mM MgCl2, 40 mM NaCl, 1% Nonidet P-40 and twice with the same buffer without Nonidet P-40. The bound Rac1 and Cdc42 proteins were detected by immunoblotting using their specific antibodies.
The relative activity of each GTPase was determined by quantifying each band of GTP-bound GTPase and the total amount of GTPase using the TINA 2.09 software program (Raytest), and the values of the GTP-bound bands were normalized to the value of the total amount. All results were determined using three different film exposures from at least three independent experiments.
Hippocampal neuron culture and transfection
Cultured hippocampal neurons were prepared from embryonic day 18 (E-18) fetal Sprague-Dawley rats and plated on poly-D-lysine coated five 18 mm glass coverslips at a density of 200,000 cells/60 mm dish. The cultures were grown in Neurobasal medium (Gibco) supplemented with 2% B-27, and 0.5 mM L-Glutamine. The neurons were transfected at 16 DIV using the calcium-phosphate mediated method (42). Briefly, 8–10 μg of cDNA and 7.5 μl of 2M CaCl2 were mixed in distilled water to a total volume of 75 μl, and same volume of 2X BBS was added. The cell culture medium was completely replaced by transfection medium (MEM, 1 mM pyruvate, 0.6% glucose, 10 mM glutamine, and 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), pH 7.65), and the cDNA mixture was added to the cells, which were then incubated in a 5% CO2 incubator for 90 min. They were washed twice with transfection medium (pH 7.35) and then returned to the original culture medium. Tranfection efficiency was about 1-5%.
Image acquisition and data analysis
The cells were fixed in 4% parafirmaldehyde/4% sucrose/PBS for 15 min, washed 2 × 5min, and permeabilized for 5 min in 0.25% Triton X-100/PBS. The images were obtained using an Olympus IX71 microscope (Olympus) with 40x N.A. 1.0 or 60x N.A. 1.4 oil lens using a CoolSNAP-Hq CCD camera (Roper Scientific) driven by MetaMorph imaging software (Universal Imaging Co.). Light from a mercury lamp was shuttered using a VMM1 Uniblitz shutter (Vincent Associates). The number of dendritic branches was analyzed with a sphere, 100 μm in radius, centered at the soma. The number of intersections between the dendritic branches and a sphere was averaged. Statistical analysis was performed using ANOVA and Tukey's HSD post hoc test. The analysis and quantification of the data were performed using MetaMorph software and SigmaPlot 8.0 (Systat Software). The expression of each construct was confirmed by the retrospective immunostaining using specific antibodies, and only immunopositive neurons were included in the analysis. Data analysis was performed in a blinded manner. The data is presented as the mean ± SE.
Results
δ-Catenin interacts with p190RhoGEF, for which Thr-454 residue of δ-catenin is indispensable
The published results have shown that 14-3-3 can interact with either p190RhoGEF (29) or δ-catenin (30), but association between p190RhoGEF and δ-catenin has not been demonstrated until now. Therefore, the interaction between δ-catenin and p190RhoGEF was examined by co-transfecting HA tagged p190RhoGEF into wild type MEFs with GFP tagged δ-catenin, followed by immunoprecipitation analysis. The results indicated that δ-catenin specifically binds to the p190RhoGEF (Fig. 1A). The interaction between δ-catenin and p190RhoGEF was also confirmed using endogenous proteins. As shown in Figure 1B, mouse brain expressed both endogenous δ-catenin and p190RhoGEF, and endogenous p190RhoGEF was specifically co-immunoprecipitated with δ-catenin. In order to map the region responsible for the interaction between δ-catenin and p190RhoGEF, we used a site-directed δ-catenin mutant and a deletion mutant as shown in a schematic diagram in Figure 1C. The immunoprecipitation assay showed that p190RhoGEF could interact with δ-catenin FL wt and ΔC207 but not with the T454A mutant (Fig. 1D).
Fig. 1. δ-Catenin interacts with p190RhoGEF, for which Thr-454 residue of δ-catenin is indispensable.
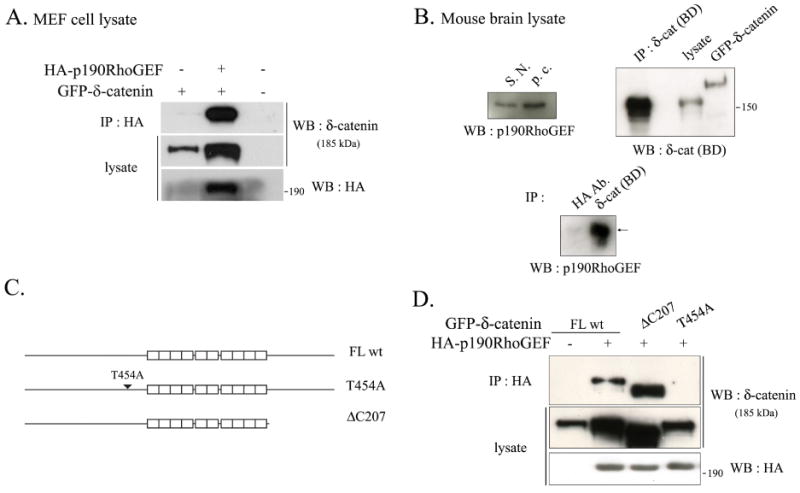
(A) The MEF cells were transfected with GFP-tagged δ-catenin with or without HA-p190RhoGEF. The binding of δ-catenin and p190RhoGEF was examined by immunoprecipitation with the anti-HA antibody, and western blotting was performed with the anti-δ-catenin antibody (upper panel). The expression of δ-catenin or p190RhoGEF in the cell lysates were detected using the anti-δ-catenin antibody (Middle panel), or the anti-HA antibody (bottom panel), respectively. (B) Adult mouse brain lysates were used to examine the endogenous interaction between δ-catenin and p190RhoGEF (Bottom panel). HA antibody was used as a negative control for immunoprecipitation assay. Endogenous expression of p190RhoGEF in the adult mouse brain region of substantia nigra (indicated as S.N.) and pars compacta (indicated as p.c.) were shown (Upper left panel). Endogenous expression of δ-catenin in the whole adult mouse brain lysate was shown, and the efficiency of immunoprecipitation using the anti-δ-catenin antibody was determined with the same adult mouse brain lysates (Upper right panel). (C) Schematic illustration of δ-catenin constructs used throughout experiments. (D) The MEF cells were transfected with either GFP-δ-catenin FL wt or a mutant of δ-catenin (ΔC207 and T454A) together with or without HA-p190RhoGEF as indicated in the figure. The binding of δ-catenin and p190RhoGEF was examined by immunoprecipitation with the anti-HA antibody, and bloted with the anti-δ-catenin antibody (upper panel). The expression of δ-catenin or p190RhoGEF in the cell lysates was detected with the anti-δ-catenin antibody (middle panel) or the anti-HA antibody (bottom panel), respectively.
Our protein motif scan analysis revealed that mouse δ-catenin contains 3 putative 14-3-3 binding sites (Ser-282, Thr-454, Ser-1094). We thus investigated the possibility of 14-3-3 requirement for the interaction between δ-catenin and p190RhoGEF. The deletion constructs were specifically designed to remove the putative 14-3-3 binding residue on δ-catenin as follows: △N85-325-(deletion of Ser-282), △C207- (deletion of Ser-1094), site-directed mutant, δ-catenin T454A. As shown in Figure S1A, all of these δ-catenin deletion mutants were specifically immunoprecipitated by 14-3-3ε, but δ-catenin △N85-325 bound to 14-3-3ε to a lesser extent (Fig. S1A). Interestingly, FL wt, ΔN85-325, and δ-catenin T454A had a noticeable interaction with 14-3-3ζ, whereas δ-catenin ΔC207 barely interacted (Fig. S1B), suggesting a 14-3-3 isoform-specific interaction with δ-catenin and 14-3-3. Exceptionally, δ-catenin T454A, a defective form in p190RhoGEF binding, retained the capability to bind both 14-3-3 isoforms, suggesting that 14-3-3 does not mediate the interaction between δ-catenin and p190RhoGEF.
δ-Catenin undergoes Akt1-mediated phosphorylation at Thr-454 residue
As our protein motif scan analysis showed that the Thr-454 residue in mouse δ-catenin could also be a putative target phosphorylation site for Akt, we investigated the possibility of Akt1-mediated δ-catenin phosphorylation. An in vitro Akt1 kinase assay was performed using endogenous δ-catenin in 1 day post-natal mouse brain lysates and GFP-tagged exogenous δ-catenin (Fig. 2A and 2B, respectively). As shown in Figure 2A and 2B, both endogenous and exogenous wild type δ-catenin were specifically phosphorylated by Akt1. δ-Catenin T454A, however, was not phosphorylated by Akt1, indicating that the Thr-454 residue in mouse δ-catenin is a target site for Akt1-phosphorylation. In order to confirm these results, immunoblotting with specific anti-phosphor-Ser or -Thr antibodies was performed (Fig. 2C and 2D, respectively). 14-3-3ζ, a well-known target protein that can be phosphorylated by Akt on its Ser-58 residue, was used as a positive control to certify the activity of anti-phospho-Ser antibody, and no phosphoryled δ-catenin band by Akt1 was detected (Fig. 2C). This demonstrates that Akt1 is not responsible for phosphorylation of the Ser-282 or Ser-1094 residues in δ-catenin. In contrast, phosphorylated δ-catenin FL wt, but not T454A mutant band, was detected by antibody against phospho-threonine residues (Fig. 2D). This result again proved the principal role of Thr-454 residue on δ-catenin in Akt1-mediated phosphorylation. Akt1 is inferred as an important regulator for p190RhoGEF interaction with δ-catenin from a series of experiments. The increased amounts of either wild type or dominant negative p190RhoGEF increased the levels of δ-catenin (Fig. S2). But only wild type Akt1 showed dose-dependent increases in the level of δ-catenin, whereas kinase dead form of Akt1 slightly decreased the level of δ-catenin (Fig. S3). These results suggest that association of p190RhoGEF with δ-catenin followed by Akt1-mediated phosphorylation enhances the stability of δ-catenin and GEF activity of p190RhoGEF is not a critical factor for the association.
Fig. 2. δ-Catenin undergoes Akt1-mediated phosphorylation at Thr-454 residue.
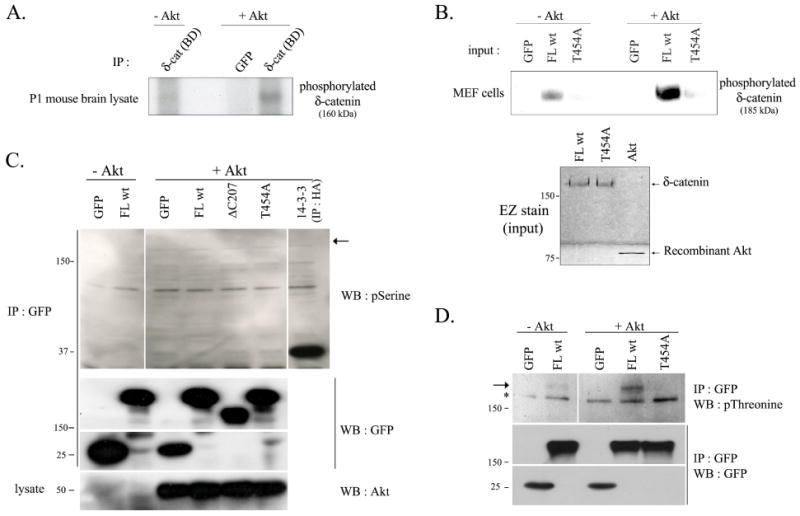
An Akt1 kinase assay was performed as described in Materials and Methods. GFP, mock transfected proteins were used as a negative control in the assay, and GFP-immunocomplex was obtained by immunoprecipitating with GFP antibody. (A) Purified immunocomplex of 1 day post-natal mouse brain lysates with δ-catenin (BD) antibody was used as a substrate for an Akt kinase assay. [γ-32P]-ATP was supplemented with a reaction buffer, and phosphorylation status was detected by autoradiograph. (B) GFP-tagged δ-catenin wt (FL wt) and T454A mutant were transfected in MEF cells, and purified immunocomplex with δ-catenin (BD) antibody was used as a substrate for an Akt kinase assay. [γ-32P]-ATP was supplemented with a reaction buffer, and phosphorylation status was detected by autoradiograph (Upper panel). The level of δ-catenin FL wt and T454A mutant existing in immunocomplex and of recombinant Akt1 were confirmed by EZ staining kit (Bottom panel). (C-D) GFP-tagged full length (FL wt) and mutant δ-catenin (ΔC207 and T454A) were overexpressed in Bosc23 cells, and purified immunocomplex with GFP antibody was used as a substrate for Akt kinase assay. Radio-inactive cold ATP was supplemented with a reaction buffer, and phosphorylation status on serine and threonine residue in the wild type or mutant type δ-catenin was detected using anti-phosphoserine antibody (C) or with anti-phosphothreonine antibody (D) (upper panel). HA-14-3-3 was immunoprecipitated with HA antibody and was used as a positive control (C). An arrow indicates the predicted site (C) or an actual band (D) of phosphorylated δ-catenin, and an asterisk indicates a nonspecific band. The immunocomplexes were sub sequentially reprobed with anti-GFP antibody (middle panels), and the added recombinant Akt1 was also confirmed using anti-Akt antibody (C, bottom panel).
Overexpression of δ-catenin significantly reduces the level of GTP-RhoA, while the level of GTP-Rac1 and -Cdc42 are not affected
Even though there are many reports showing that small GTPase family proteins including Rac1, Cdc42, and RhoA affect dendrogenesis (17, 19), there is no evidence showing how the activities of these proteins are affected by δ-catenin overexpression. In this report, we propose a model showing that overexpressed δ-catenin can lower GTP-RhoA levels by sequestering p190RhoGEF from its substrate RhoA, thereby attenuating RhoA activation. GST-PBD (p21-binding domain) and GST-RBD (Rho-binding domain) were used, and GST pull down assays were performed to determine the levels of the GTP bound Rac1 and Cdc42, and RhoA, respectively, using their specific antibodies. δ-Catenin overexpression significantly decreased the level of GTP-RhoA by ∼40% compared with those in the control cells (Fig. 3A and 3B). As a comparison, overexpression of δ-catenin ΔC207 significantly decreased the level of GTP-RhoA by ∼20% compared with those in the GFP overexpressed control cells. δ-Catenin T454A, however, did not affect the level of GTP-RhoA (Fig. 3E and 3F). In contrast, the levels of GTP-Rac1 and -Cdc42 were not affected by δ-catenin overexpression (Fig. 3A and 3C-3D).
Fig. 3. Overexpression of δ-catenin results in a decrease in the level of GTP-RhoA, while the Rac1 and Cdc42 activity are unaffected.
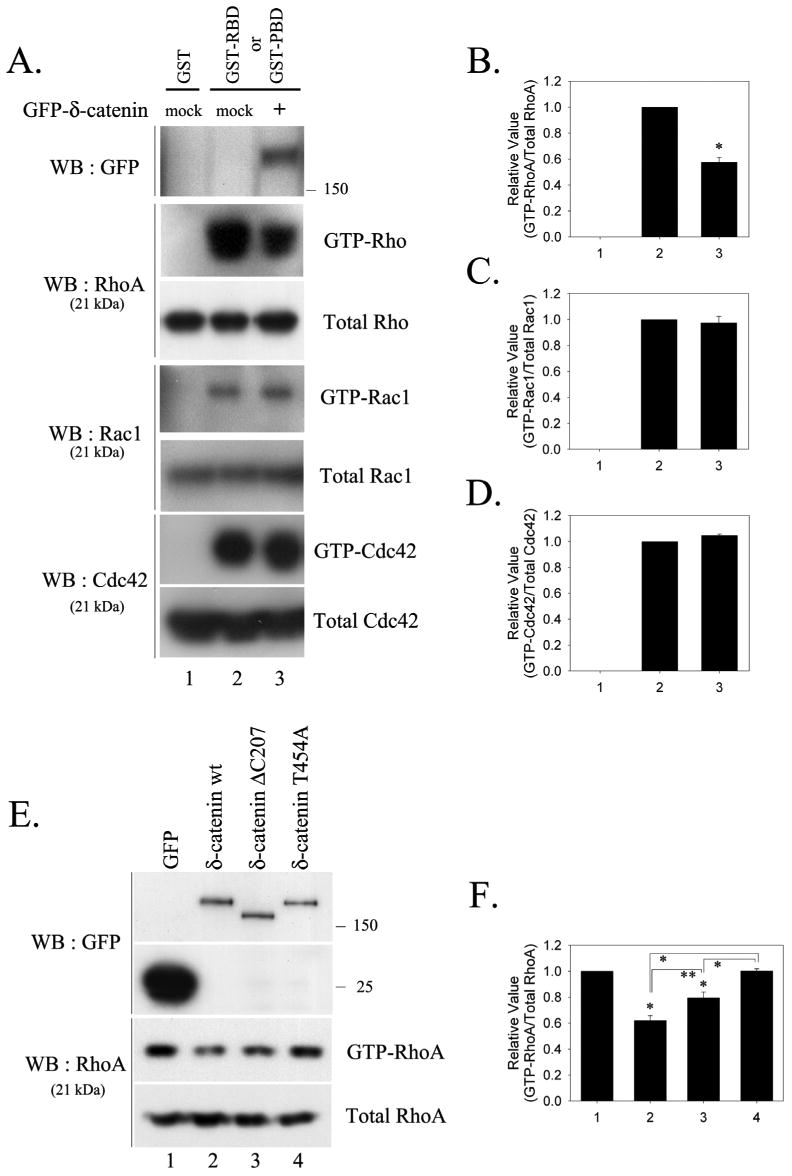
(A) The levels of active GTP-bound RhoA, Rac1 and Cdc42 were measured in MEF cells transfected with either the mock or GFP-δ-catenin. The RhoA activity was measured using a GST-RBD, and the Rac1, Cdc42 activities were measured using a GST-PBD. The total amounts of RhoA, Rac1, and Cdc42 are also shown to compare the endogenous level of each GTPase in the transfected cells. The relative activity of RhoA (B), Rac1 (C), and Cdc42 (D) were determined as described in Materials and Methods. The data is represented as the mean ± SEM (* p<0.01). (E) The level of active GTP-bound RhoA was measured in the MEF cells transfected with GFP or with wt/mutant GFP-δ-catenin constructs using GST-RBD. (F) The relative activity of RhoA was determined. The data is represented as the mean ± SEM (* p<0.01, ** p<0.05). Results are representative of at least three independent experiments.
Effects of the wild type and mutant δ-catenin on the dendrite-like process formation in NIH 3T3 fibroblasts and mature mushroom shaped spine formation in the primary hippocampal neurons
Previously, we reported that δ-catenin expression induces the branching of dendrite-like processes, and that δ-catenin ΔC207 does not induce either dendrite-like branching or significant extension of processes in NIH 3T3 fibroblasts and enhances dendritic morphogenesis in primary hippocampal neurons (15). In this study, the ability of δ-catenin T454A to induce dendrite-like processes formation in NIH 3T3 cells was examined in comparison with δ-catenin full-length and ΔC207 mutant. As shown in Figure 4, δ-catenin T454A did not induce any noticeable morphological changes, suggesting that the association of δ-catenin with p190RhoGEF is essential for the δ-catenin induced dendrite-like process formation in fibroblasts. 100% of wild type δ-catenin overexpressing cells showed dendrite-like process formation in quantitative analysis but neither δ-catenin ΔC207 nor T454A overexpressing cells showed any process formation. The co-expression of constitutive active form of RhoA with wild type δ-catenin totally abolished the dendrite-like process formation, and resulted in a rounded morphology (δ-catenin + RhoA CA in Fig. 4). This suggests that the RhoA signaling might be a downstream effecter of δ-catenin.
Fig. 4. Effects of the wild type and mutant δ-catenins on the dendrite-like process formation in NIH 3T3 fibroblasts.
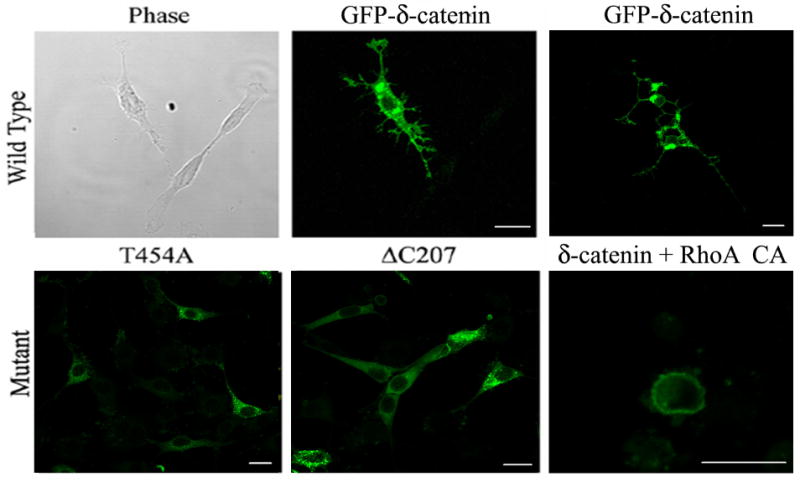
The NIH 3T3 fibroblast cells were transfected with wild type (GFP-δ-catenin) and mutant δ-catenin (T454A and ΔC207) or co-transfected with a constitutive active mutant of RhoA (RhoA CA) as indicated in each image. After 24 h post-transfection, the cells were fixed, and a fluorescent image was taken sometimes together with its corresponding phase contrast image (left and middle, upper panel). GFP-δ-catenin images on top panel indicate typical δ-catenin-induced dendrite-like processes at different stages (upper middle-early stage; upper right-late stage). Scale bars: 20 μm.
We have also previously described that E18 hippocampal neurons overexpressing the full-length δ-catenin demonstrated enhanced arborization of dendrites, swelling, and mature dendritic spines (15). In order to examine the effects of δ-catenin T454A on dendrogenesis and spine formation, δ-catenin and its variants were transfected in cultured hippocampal neurons (Fig. 5). In the δ-catenin transfected neurons, the number of mature spines, either a cotyloid appearance or flat-apex mushroom appearance, was significantly higher compared with the GFP control (0.38 ± 0.03 / μm, n=10, for GFP control; 0.5 ± 0.01 / μm, n=10, for δ-catenin, n=18, p<0.01). The expression of δ-catenin ΔC207 also increased the number of mature spines (0.45 ± 0.02 / μm, n=12) but to a significantly lesser extent than the FL wt δ-catenin (p<0.05). δ-Catenin T454A, which is not able to bind p190RhoGEF, decreased the number of mature spines most significantly compared with the full-length δ-catenin or δ-catenin ΔC207 (0.4 ± 0.04 / μm, n=12, for δ-catenin T454A, p<0.01). Surprisingly, the morphology of the dendritic spines in hippocampal neurons overexpressing δ-catenin T454A dramatically changed. Typical mushroom shaped spines were rare and most spines showed rather long and thin filopodial morphologies. When only the length of mushroom shaped spines was compared, δ-catenin T454A expression significantly reduced the length of the spines when compared with the full-length δ-catenin or its ΔC207 mutant (1.54 ± 0.11 μm, n=10, for δ-catenin T454A; 2.22 ± 0.14 μm, n=10, for full-length δ-catenin; 1.95 ± 0.10 μm, n=10, for δ-catenin ΔC207, p<0.01). Consistent with a previous report (15), the expression of δ-catenin FL also increased the branching of dendrites (13.8 ± 0.8, n=12, for GFP control; 23.2 ± 1.5, n=12, for δ-catenin FL, p<0.01). Unexpectedly, δ-catenin T454A also increased the number of dendrite branches while its ΔC207 mutant did not (19.9 ± 2, n=10, for δ-catenin T454A, p<0.01 compared to GFP control; 10.9 ± 0.7, n=10, for δ-catenin ΔC207).
Fig. 5. Effects of wild type and mutant δ-catenin on the dendrogenesis and spine formation in primary hippocampal neurons.
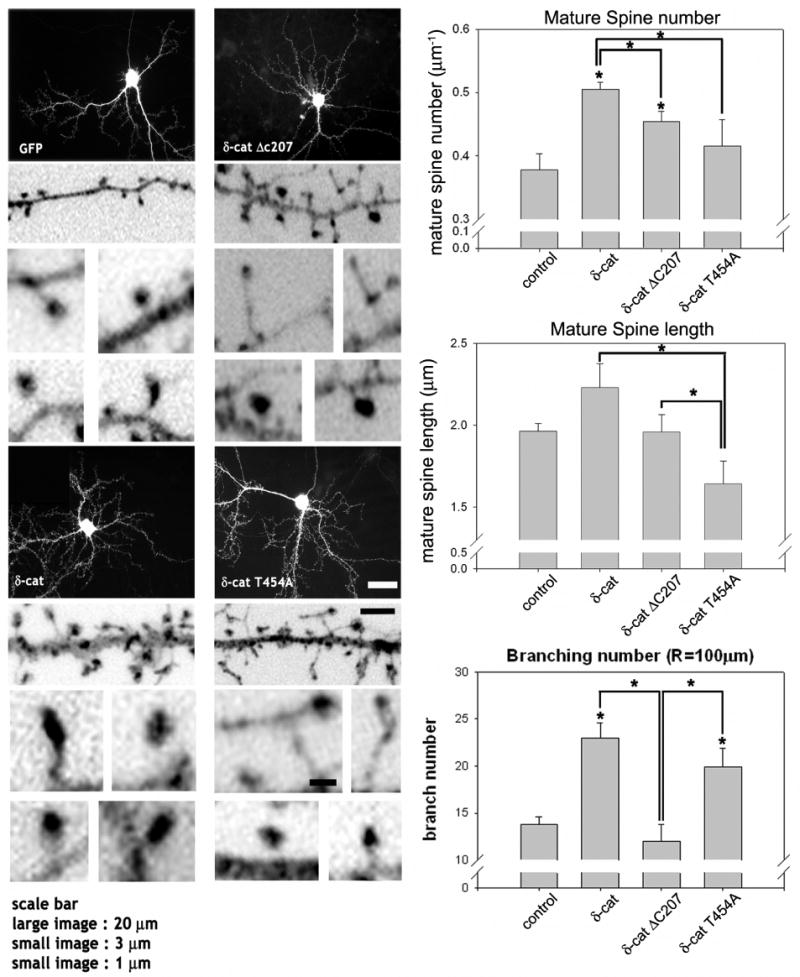
The cultured hippocampal neurons at 16 DIV were transfected with the full-length GFP-δ-catenin or various mutants, fixed and stained with GFP antibody. The high magnification images are inverted for clarity. The average number and length of mature spines, either a cotyloid appearance or flat-apex mushroom appearance, were analyzed and filopodia shaped spines are excluded from analysis (* p<0.01 by ANOVA and Tukey's HSD post hoc test). The number of dendritic branches that intersects a sphere, 100 μm in radius, centered at the soma was added for plotting and statistical analysis using ANOVA and a Tukey's HSD post hoc test. The graphs show the average number of intersections between the dendritic branches and a sphere (* p < 0.01). The study has been repeated with 3 different cultures (3 different litters from 3 different pregnant rats) and from each litter, 3 to 4 coverslips (∼10 neurons per coverslip) have been included in this analysis. Scale bars: 20 μm for low magnification, 3 μm for high magnification, and 1 μm for high magnification of the dendritic spines.
δ-Catenin sequesters p190RhoGEF from RhoA
As shown in Figure 6, we proposed a model illustrating Akt1-mediated Thr-454 phosphorylation on δ-catenin leads to its interaction with p190RhoGEF, which sequesters p190RhoGEF from activating Rho signaling. Thereby, the sequestration of p190RhoGEF by δ-catenin contributes to the attenuated Rho signaling, resulting in enhanced dendrogenesis and spine maturation. In order to prove our proposed model, we performed the immunoprecipitation assays with δ-catenin, p190RhoGEF, and RhoA. If the interaction between δ-catenin and p190RhoGEF indeed sequesters p190RhoGEF from binding to RhoA, the δ-catenin overexpression should decrease the interaction between p190RhoGEF and RhoA. As δ-catenin is known to interact with a cytoplasmic loop of Presenilin-1, HA-tagged Presenilin-1 was used as a positive control for δ-catenin interaction, and as Notch is a well-known substrate for Presenilin-1/γ-secretase complex, myc-tagged Notch was used as a positive control for Presenilin-1 interaction. As shown in Figure 7A, HA-tagged p190RhoGEF was specifically co-immunoprecipitated by RhoA in the absence of δ-catenin (an arrowhead in upper first panel). However, δ-catenin overexpression noticeably decreased the interaction between p190RhoGEF and RhoA (lane 6 vs. 7 in upper first panel). Likewise, RhoA overexpression decreased the interaction between p190RhoGEF and δ-catenin (lane 4 vs. 7 in upper third panel). In contrast to the published result showing p120ctn directly interacts with RhoA, we could not detect such an interaction between δ-catenin and RhoA (upper second panel). Overexpressing δ-catenin T454A mutant did not alter the interaction between p190RhoGEF and RhoA (Fig. 7B). These results strongly support our hypothesis that the overexpressed δ-catenin lowers the active form of RhoA by sequestering p190RhoGEF to prevent it from activating RhoA, rather than by direct interaction between δ-catenin and RhoA.
Fig. 6. A proposed model of δ-catenin-induced morphogenesis.
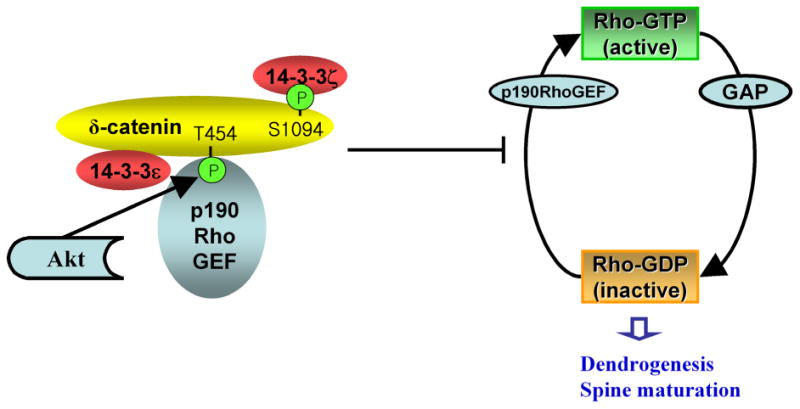
δ-Catenin binds to p190RhoGEF through Akt-mediated Thr-454 phosphorylation, sequesters p190RhoGEF to prevent it from activating RhoA, and thereby reduces the local endogenous RhoA activity. The reduced local GTP-RhoA affects its downstream effectors, which, along with other δ-catenin-induced changes, regulates the dendrogenesis and mature spine formation. The interaction between δ-catenin with the 14-3-3 isoforms occurs through a different binding domain. The C-terminal region of δ-catenin is important for binding to 14-3-3ζ.
Fig. 7. δ-Catenin Overexpression decreases the interaction between p190RhoGEF and RhoA.
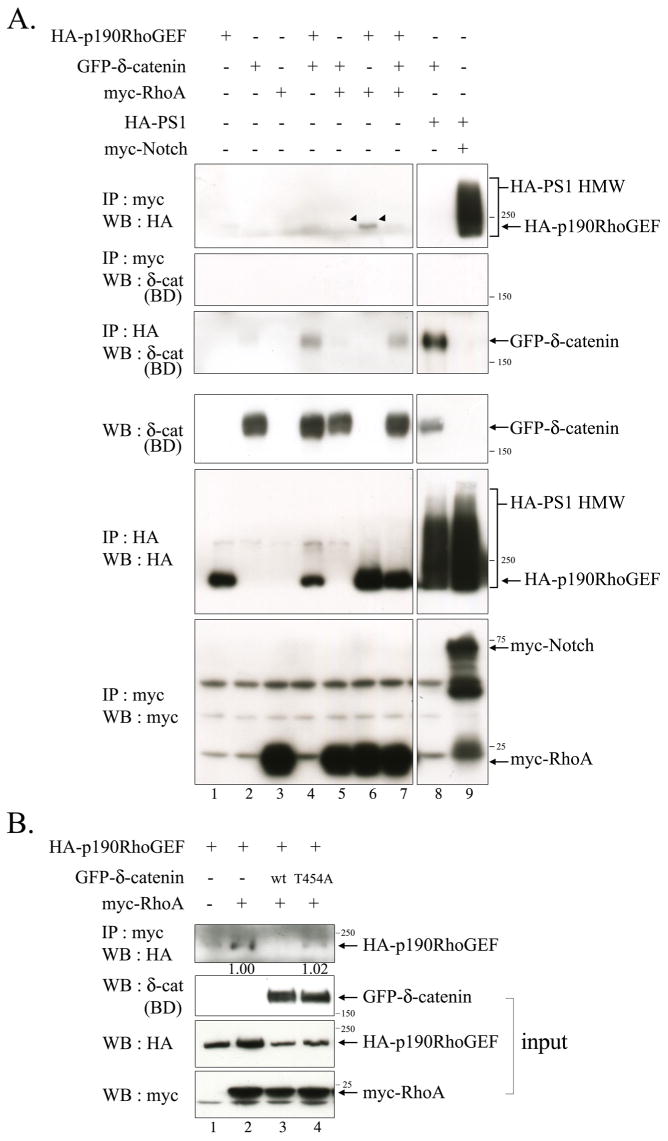
(A) Bosc23 cells were transfected with GFP-δ-catenin, HA-p190RhoGEF, myc-RhoA, HA-PS1 wt, and myc-ΔEN1 in a various combination as indicated in the upper side of the figure. The binding between p190RhoGEF and RhoA was examined by immunoprecipitation with the anti-myc antibody, and western blotting was performed with anti-HA antibody (Upper first panel). The binding between RhoA and δ-catenin was examined by immunoprecipitation with the anti-myc antibody, and western blotting was performed with anti-δ-catenin (BD) antibody (Upper second panel). The bindings between Presenilin-1 with δ-catenin or with Notch were examined for positive control experiments (lane 8, 9 in upper first and third panels). Expression of each protein was shown through the bottom three panels. (B) Bosc23 cells were transfected with either wild type or mutant δ-catenin together with HA-p190RhoGEF and/or myc-RhoA. The binding between p190RhoGEF and RhoA was examined by immunoprecipitation with the anti-myc antibody, and western blotting was performed with anti-HA antibody (Upper first panel). 5% volume of each lysate was subjected to western blot analysis to show the input level of each protein. The relative intensity of the immunoprecipitated HA-p190RhoGEF band in lane 2 vs. 4 was determined by normalization against the input band of HA-p190RhoGEF. 3 μg of each plasmid was used to transfect cells plated in 100 mm dish at 50% confluency.
Discussion
Because dendritic spines are actin-rich protrusions that form the major postsynaptic sites of the excitatory synaptic input, dendrogenesis and spine morphogenesis are essential for synaptic plasticity and cognitive function. Moreover, spine abnormalities have been shown to be associated with many neurological disorders including most types of mental retardation (19, 43-45). Even though we previously reported that a hippocampal neuron overexpressing δ-catenin demonstrates an elaborate arborization of dendrites, swelling and enhanced dendritic spine maturation (15), a complete picture of how δ-catenin affects dendrogenesis and spine morphogenesis would be very important for understanding its role in synaptic plasticity and cognitive function. In this study, we found that δ-catenin interacts with p190RhoGEF and significantly lowered the level of GTP-RhoA, and that Akt1 mediates the interaction between δ-catenin and p190RhoGEF through Thr-454 phosphorylation of δ-catenin. The formation of δ-catenin-induced dendrite-like process formation in NIH 3T3 fibroblasts was totally abolished by substitution of Thr-454 residue to Ala, which is a defective form in binding p190RhoGEF. Furthermore, δ-catenin T454A significantly reduced the length and number of mature mushroom shaped spines in primary hippocampal neurons. Overall, these results suggest that the interaction between δ-catenin and p190RhoGEF can act as a key modulator in regulating δ-catenin-induced dendrogenesis and spine formation.
Our protein-motif-scan analysis demonstrated 3 putative sites (Ser-282, Thr-454, Ser-1094) in mouse δ-catenin as a putative target phosphorylation sites for Akt and/or 14-3-3 binding sites. The precise mapping for the phosphorylation site by Akt, and subsequently, a binding site for p190RhoGEF and/or 14-3-3, is of great interest because the three molecules, δ-catenin, p190RhoGEF, and 14-3-3, may interact with each other independently. For example, a recent report demonstrated that mouse δ-catenin interacts with 14-3-3ζ through Ser-1094 in a phosphorylation-dependent manner (30). 14-3-3 also binds to p190RhoGEF through a unique phosphorylation-independent binding site (I(1370)QAIQNL) in p190RhoGEF (29). Hence, this study examined the possibility that δ-catenin may bind to p190RhoGEF via 14-3-3 through Ser-1094, forming a trimeric complex. The δ-catenin T454A mutant, unable to bind p190RhoGEF, still interacts with both 14-3-3 isoforms, ε and ζ, suggesting that p190RhoGEF binds directly to the domains containing Thr-454 in δ-catenin in a Akt-phosphorylation-dependent manner rather than by forming a trimeric complex. As shown in Figure 7, the overexpressed δ-catenin noticeably decreased the interaction between p190RhoGEF and RhoA, supporting the direct interaction between δ-catenin and p190RhoGEF as proposed in our model in Figure 6. Similar to the results of Mackie and Aitken (30), δ-catenin ΔC207 (Ser-1094 deleted) showed very little 14-3-3ζ binding. However, we did observe a faint band, possibly due to formation of heterodimers with other 14-3-3 isoforms. In contrast, δ-catenin ΔC207 interacts well with 14-3-3ε, suggesting that δ-catenin can combine with the isoforms of 14-3-3 through different binding domains. Interestingly, p190RhoGEF has been shown to bind to the β, γ, ε and η, isoforms but not to ζ, or τ isoforms of 14-3-3 (29). Therefore, even though our results suggests p190RhoGEF binds directly to δ-catenin in an Akt-dependent manner, the possibility that 14-3-3ε has favorable effects on the association between p190RhoGEF and δ-catenin cannot be totally excluded.
The Rho signaling pathway has attracted considerable attention for several reasons. First, the dendrite-like process formation and cytoskeletal remodeling induced by overexpressed δ-catenin will greatly mimic those shown by the use of the C3 toxin from Clostridium botulinum, in which ADP-ribosylates and specifically inactivates Rho (46, 47). We previously reported that rhodamine phalloidine staining of NIH 3T3 fibroblasts produced lower levels of stress fiber-associated actin filaments (15), which is indicative of reduced Rho signaling in fibroblasts. Even though Martinez et al. (12) reported that δ-catenin enhances the effects of Rho inhibition on neurite branching, there is no evidence showing that δ-catenin reduces the level of GTP-RhoA in cells. This report provides the first biochemical evidence exhibiting that overexpressed δ-catenin indeed reduces the levels of GTP-RhoA but not those of GTP-Cdc42 or -Rac1, along with the mechanism that Akt phosphorylates the Thr-454 residue in δ-catenin and enables its association of p190RhoGEF.
In contrast to the few reports on δ-catenin, studies on p120ctn, a prototype of the p120ctn family protein, have revealed several lines of evidence that may be unique to p120ctn or related to δ-catenin. p120ctn has been shown to primarily enhance the development of spine-like protrusions, which were abolished by deletions in the Arm domain (48, 49). p120ctn overexpression in CHO cells increased the activity of endogenous Cdc42 (∼3.1 fold) and Rac1 (∼1.9 fold) activity, but diminished the Rho activity by ∼45% through the Rho family exchange factor Vav2, which has activity for RhoA, RhoG, Cdc42, and Rac1 (50). Consistent with this data, a deletion of the p120ctn gene in hippocampal pyramidal neurons reduced the GTP-Rac1 level (∼40%) and increased the RhoA level (∼160%) (51). In contrast, another report demonstrated that p120ctn null epidermis showed markedly higher GTP-RhoA levels but similar GTP-Rac1 and -Cdc42 levels (52). Our results demonstrate that overexpressed δ-catenin reduces the level of GTP-RhoA but not those of GTP-Cdc42 or -Rac1. It might be possible because p190RhoGEF is a brain-enriched and RhoA specific GEF (24). In contrast to the published result that p120ctn directly interacts with Rho, we could not observe such interaction between δ-catenin and RhoA, indicating that the interaction between δ-catenin and p190RhoGEF might be a unique mechanism for δ-catenin to lower RhoA activity. The mechanism for δ-catenin binding of p190RhoGEF and subsequent alteration of RhoA activation could be through a sequestration model as suggested in this report. However, we can not yet rule out the possibility that δ-catenin directly alters the exchange activity of the p190RhoGEF.
Compared to the explicit mechanism for δ-catenin T454A inability to lower active RhoA and modulate dendrogenesis, δ-catenin ΔC207 interactions are more complex and needs further investigation. As shown in Figure 1 and 3, δ-catenin ΔC207 binds to p190RhoGEF better than δ-catenin FL wt but displays less capability in lowering RhoA-GTP. This suggests that the C-terminus in δ-catenin also plays a pivotal role in lowering RhoA activity. We speculate two possibilities based on published results. First, in addition to the association between δ-catenin and p190RhoGEF, another protein(s) bound to the C-terminus of δ-catenin can be important for inactivating p190RhoGEF to a full extent. δ-Catenin binds to the last PDZ domain of S-SCAM through its C-terminus, which can affect the association between δ-catenin and NMDA receptors and other glutamate receptors. There was a recent report showing that GRIP, a direct binding protein of AMPA receptor, also binds to the C-terminal region of δ-catenin, whose interaction can affect LTD (53, Ochiishi et al., Proceedings of 36th Annual Neuroscience meeting, 2006). The association between Erbin and the Cadherin and Catenin complex was mediated by an interaction between the C-terminal binding motifs (DSWV-COOH) of δ-catenin and ARVCF (54). Martinez et al. (12) showed that a deletion of 99 amino acid (aa) residues from the δ-catenin COOH terminal (ΔC99), but not a deletion of 205 aa (ΔC205), retains its ability to interact with Cortactin. As RhoA combines with the glutamate receptors at the spine plasma membrane and NMDA receptor activation decreases the RhoA activity (55, 56), the C-terminus of δ-catenin may affect the Rho activity by interacting with one or more of these newly identified proteins. It also would be interesting to examine whether Cortactin or 14-3-3ζ mediates δ-catenin–dependent RhoA inactivation and dendritic spine morphogenesis. Alternatively, the C-terminus in δ-catenin may be important by being specifically localized to a certain membrane junction. Our results showed that δ-catenin ΔC207, which can interact with p190RhoGEF but possess a relatively low level of GTP-RhoA, showed total abolishment of dendrite-like processes in NIH 3T3 fibroblasts and a significantly lower number of dendrites in cultured hippocampal neurons, highlighting the important role of the C-terminus of δ-catenin plus its intact Thr-454 residue in regulating dendrite morphogenesis. Compared with the significantly diminished dendrite formation, δ-catenin ΔC207 has less affect on the formation of mature mushroom shaped spine in terms of the number and length than δ-catenin T454A. This indicates that δ-catenin contributes to dendrogenesis and spine formation through some overlapping and unique signaling pathways for each process. The different contribution of the C-terminus in δ-catenin in dendrogenesis and spine formation will require future study to determine its precise mechanism.
Even though the potential roles of dendrogenesis by δ-catenin (12, 15) and the promotion of mature spines in cultured hippocampal neurons (15, 16) have been described, the effects of δ-catenin on the number, shape, and length of spines are not completely understood. Interestingly, Elia et al. (51) recently reported a role for p120ctn in regulating spine and synapse formation. Consistent with our previous description, cultured hippocampal neurons expressing full-length δ-catenin showed an increased number of mature mushroom shaped spines compared with the GFP control, whereas δ-catenin T454A showed noticeable increases in the number of long, thin filopodia shaped spines with a concomitant reduction in the number of mature mushroom shaped spines. δ-Catenin ΔC207 showed intermediate changes in the number and length of mature spines between δ-catenin full-length and T454A, which strongly suggests that the C-terminus plus the intact Thr-454 in δ-catenin are essential for its full effects on the head shape and length of the spine, as well as its maturation. Interestingly, the morphological changes in the spine induced by δ-catenin T454A were similar to those induced by the dominant negative form of N-cadherin, alpha N-catenin, and RhoGEF (24, 57-59). There is accumulated evidence suggesting the role of Cadherin complexes and Rho in spine formation (57, 59, 60), whereas the progressive loss of F-actin induced by a treatment with latrunculin B results in a rough spine head with random protrusion (59). Compared with the full-length δ-catenin, δ-catenin T454A showed differences in the recruitment of Akt and p190RhoGEF, and in the levels of GTP-RhoA, suggesting their important roles in δ-catenin-induced spine morphogenesis. The reduced activities of the Rho downstream effectors, ROCK, LIMK and its substrate ADF/cofilin, and Dia, in neurons promotes the formation of dendritic spines or branches (61). Furthermore, the expression of RhoA V14, a constitutively active form, decreased the spine density and length dramatically (62), and the inhibition of Rho using C3 toxin in cortical and hippocampal neurons has also been shown to increase the density and length of some mouse cortical and hippocampal pyramidal neurons in organotypic slices (63). Consistently, we showed that δ-catenin FL wt, which binds to p190RhoGEF and reduces RhoA activation, significantly increased the number and length of spines while δ-catenin T454A, which cannot binds to p190RhoGEF, significantly decreased the number and length of mature spines. Therefore, it is possible to say that δ-catenin exerts its effects on spine formation through very limited spatial sites including junctions containing Cadherins or NMDA and/or AMPA glutamate receptor, which allows for local reduction of RhoA, restricted actin reorganization, and its effects on other potential binding proteins. As the stimuli that produce various forms of long-term potentiation (LTP) cause a rapid local increase in the extension of filopodia and the formation of new spines at the site of stimulation (19, 64), the strong effects of δ-catenin and its mutants on the shape, length, and number of spines suggest its assumptive important role in synaptic plasticity and cognitive function. However, future investigation will be needed to compare the functional roles of δ-catenin full-length, and T454A mutant in cognition using transgenic mice.
Acknowledgments
The authors wish to thank Jong-Kyung Chung for providing the Akt, and W. Moolenaar for p190RhoGEF, and Jong Ran Lee for dominant negative p190RhoGEF. We also wish to thank D. Schlaepfer and B. Margolis for providing antibody for p190RhoGEF. This study was supported by a grant of the Korea Health 21 R&D Project to KK, Ministry of Health & Welfare, Republic of Korea (A040042), and by a grant from the Brain Research Center of 21st Century Frontier Research Program (M103KV010009-06K2201-00910) to SC funded by the Ministry of Science and Technology, Republic of Korea and US National Institutes of Health AG026630 (Q.L.) and CA111891 (Q.L.).
The abbreviations used are
- FL
full length
- GEF
guanine nucleotide exchange factor
- GFP
green fluorescent protein
- MEF
mouse embryonic fibroblast
- PS-1
Presenilin-1
- wt
wild type
References
- 1.Zhou J, Liyanage U, Medina M, Ho C, Simmons AD, Lovett M, Kosik KS. Neuroreport. 1997;8:2085–2090. doi: 10.1097/00001756-199705260-00054. [DOI] [PubMed] [Google Scholar]
- 2.Tanahashi H, Tabira T. Neuroreport. 1999;10:563–568. doi: 10.1097/00001756-199902250-00022. [DOI] [PubMed] [Google Scholar]
- 3.Paffenholz R, Franke WW. Differentiation. 1997;61:293–304. doi: 10.1046/j.1432-0436.1997.6150293.x. [DOI] [PubMed] [Google Scholar]
- 4.Lu Q, Paredes M, Medina M, Zhou J, Cavallo R, Peifer M, Orecchio L, Kosik KS. J Cell Biol. 1999;144:519–532. doi: 10.1083/jcb.144.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Israely I, Costa RM, Xie CW, Silva AJ, Kosik KS, Liu X. Curr Biol. 2004;14:1657–1663. doi: 10.1016/j.cub.2004.08.065. [DOI] [PubMed] [Google Scholar]
- 6.Medina M, Marinescu RC, Overhauser J, Kosik KS. Genomics. 2000;63:157–164. doi: 10.1006/geno.1999.6090. [DOI] [PubMed] [Google Scholar]
- 7.Ide N, Hata Y, Deguchi M, Hirao K, Yao I, Takai Y. Biochem Biophys Res Commun. 1999;256:456–461. doi: 10.1006/bbrc.1999.0364. [DOI] [PubMed] [Google Scholar]
- 8.Lu Q, Mukhopadhyay NK, Griffin JD, Paredes M, Medina M, Kosik KS. J Neurosci Res. 2002;67:618–624. doi: 10.1002/jnr.10151. [DOI] [PubMed] [Google Scholar]
- 9.Bondeson J, Sundler R. Biochim Biophys Acta. 1990;1026:186–194. doi: 10.1016/0005-2736(90)90063-t. [DOI] [PubMed] [Google Scholar]
- 10.Deguchi M, Iizuka T, Hata Y, Nishimura W, Hirao K, Yao I, Kawabe H, Takai Y. J Biol Chem. 2000;275:29875–29880. doi: 10.1074/jbc.M005384200. [DOI] [PubMed] [Google Scholar]
- 11.Izawa I, Nishizawa M, Ohtakara K, Inagaki M. J Biol Chem. 2002;277:5345–5350. doi: 10.1074/jbc.M110052200. [DOI] [PubMed] [Google Scholar]
- 12.Martinez MC, Ochiishi T, Majewski M, Kosik KS. J Cell Biol. 2003;162:99–111. doi: 10.1083/jcb.200211025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fujita T, Okada T, Hayashi S, Jahangeer S, Miwa N, Nakamura S. Biochem J. 2004;382:717–723. doi: 10.1042/BJ20040141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rodova M, Kelly KF, VanSaun M, Daniel JM, Werle MJ. Mol Cell Biol. 2004;24:7188–7196. doi: 10.1128/MCB.24.16.7188-7196.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim K, Sirota A, Chen YH, Jones SB, Dudek R, Lanford GW, Thakore C, Lu Q. Exp Cell Res. 2002;275:171–184. doi: 10.1006/excr.2002.5503. [DOI] [PubMed] [Google Scholar]
- 16.Jones SB, Lanford GW, Chen YH, Moribito M, Kim K, Lu Q. Neuroscience. 2002;115:1009–1021. doi: 10.1016/s0306-4522(02)00532-8. [DOI] [PubMed] [Google Scholar]
- 17.Threadgill R, Bobb K, Ghosh A. Neuron. 1997;19:625–634. doi: 10.1016/s0896-6273(00)80376-1. [DOI] [PubMed] [Google Scholar]
- 18.Anastasiadis PZ, Moon SY, Thoreson MA, Mariner DJ, Crawford HC, Zheng Y, Reynolds AB. Nat Cell Biol. 2000;2:637–644. doi: 10.1038/35023588. [DOI] [PubMed] [Google Scholar]
- 19.Carlisle HJ, Kennedy MB. Trends Neurosci. 2005;28:182–187. doi: 10.1016/j.tins.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 20.Govek EE, Newey SE, van Aelst L. Genes Dev. 2005;19:1–49. doi: 10.1101/gad.1256405. [DOI] [PubMed] [Google Scholar]
- 21.Hoffman GR, Nassar N, Cerione RA. Cell. 2000;100:345–356. doi: 10.1016/s0092-8674(00)80670-4. [DOI] [PubMed] [Google Scholar]
- 22.Schmidt A, Hall A. Genes Dev. 2002;16:1587–1609. doi: 10.1101/gad.1003302. [DOI] [PubMed] [Google Scholar]
- 23.Bernards A, Settleman J. Trends Cell Biol. 2004;14:377–385. doi: 10.1016/j.tcb.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 24.Gebbink MF, Kranenburg O, Poland M, van Horck FP, Houssa B, Moolenaar WH. J Cell Biol. 1997;137:1603–1613. doi: 10.1083/jcb.137.7.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Horck FP, Ahmadian MR, Haeusler LC, Moolenaar WH, Kranenburg O. J Biol Chem. 2001;276:4948–4956. doi: 10.1074/jbc.M003839200. [DOI] [PubMed] [Google Scholar]
- 26.Meyer D, Liu A, Margolis B. J Biol Chem. 1999;274:35113–35118. doi: 10.1074/jbc.274.49.35113. [DOI] [PubMed] [Google Scholar]
- 27.Canete-Soler R, Wu J, Zhai J, Shamim M, Schlaepfer WW. J Biol Chem. 2001;276:32046–32050. doi: 10.1074/jbc.M104104200. [DOI] [PubMed] [Google Scholar]
- 28.Zhai J, Lin H, Nie Z, Wu J, Canete-Soler R, Schlaepfer WW, Schlaefer DD. J Biol Chem. 2003;278:24865–24873. doi: 10.1074/jbc.M302381200. [DOI] [PubMed] [Google Scholar]
- 29.Zhai J, Lin H, Shamim M, Schlaepfer WW, Canete-Soler R. J Biol Chem. 2001;276:41318–41324. doi: 10.1074/jbc.M107709200. [DOI] [PubMed] [Google Scholar]
- 30.Mackie S, Aitken A. FEBS J. 2005;272:4202–4210. doi: 10.1111/j.1742-4658.2005.04832.x. [DOI] [PubMed] [Google Scholar]
- 31.Mackintosh C. Biochem J. 2004;381:329–342. doi: 10.1042/BJ20031332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Powell DW, Rane MJ, Chen Q, Singh S, McLeish KR. J Biol Chem. 2002;277:21639–21642. doi: 10.1074/jbc.M203167200. [DOI] [PubMed] [Google Scholar]
- 33.Kovacina KS, Park GY, Bae SS, Guzzetta AW, Schaefer E, Birnbaum MJ, Roth RA. J Biol Chem. 2003;278:10189–10194. doi: 10.1074/jbc.M210837200. [DOI] [PubMed] [Google Scholar]
- 34.Wang Q, Liu L, Pei L, Ju W, Ahmadian G, Lu J, Wang Y, Liu F, Wang YT. Neuron. 2003;38:915–928. doi: 10.1016/s0896-6273(03)00356-8. [DOI] [PubMed] [Google Scholar]
- 35.Goswami A, Burikhanov R, de Thonel A, Fujita N, Goswami M, Zhao Y, Eriksson JE, Tsuruo T, Rangnekar VM. Mol Cell. 2005;20:33–44. doi: 10.1016/j.molcel.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 36.Jiang T, Qiu Y. J Biol Chem. 2003;278:15789–15793. doi: 10.1074/jbc.M212525200. [DOI] [PubMed] [Google Scholar]
- 37.Franke TF, Kaplan DR, Cantley LC. Cell. 1997;88:435–437. doi: 10.1016/s0092-8674(00)81883-8. [DOI] [PubMed] [Google Scholar]
- 38.Vivanco I, Sawyers CL. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 39.Kim H, Ki H, Park HS, Kim K. J Biol Chem. 2005;280:22462–22472. doi: 10.1074/jbc.M502769200. [DOI] [PubMed] [Google Scholar]
- 40.Ren X, Kiosses WB, Schwartz MA. EMBO J. 1999;18:578–585. doi: 10.1093/emboj/18.3.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Benard V, Bohl BP, Bokoch GM. J Biol Chem. 1999;274:13198–13204. doi: 10.1074/jbc.274.19.13198. [DOI] [PubMed] [Google Scholar]
- 42.Chang S, De Camilli P. Nature Neuroscience. 2001;4:787–793. doi: 10.1038/90489. [DOI] [PubMed] [Google Scholar]
- 43.Ziv NE, Smith SJ. Neuron. 1996;17:91–102. doi: 10.1016/s0896-6273(00)80283-4. [DOI] [PubMed] [Google Scholar]
- 44.Fiala JC, Spacek J, Harris KM. Brain Res Rev. 2002;39:29–54. doi: 10.1016/s0165-0173(02)00158-3. [DOI] [PubMed] [Google Scholar]
- 45.Zhou Q, Homma KJ, Poo MM. Neuron. 2004;44:749–757. doi: 10.1016/j.neuron.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 46.Tigyi G, Fischer DJ, Sebok A, Yang C, Dyer DL, Miledi R. J Neurochem. 1996;66:537–548. doi: 10.1046/j.1471-4159.1996.66020537.x. [DOI] [PubMed] [Google Scholar]
- 47.Brouns MR, Matheson SF, Settleman J. Nat Cell Biol. 2001;3:361–367. doi: 10.1038/35070042. [DOI] [PubMed] [Google Scholar]
- 48.Reynolds AB, Daniel JM, Mo YY, Wu J, Zhang Z. Exp Cell Res. 1996;225:328–337. doi: 10.1006/excr.1996.0183. [DOI] [PubMed] [Google Scholar]
- 49.Li W, Li Y, Gao FB. Devel Dynamics. 2005;234:512–522. doi: 10.1002/dvdy.20496. [DOI] [PubMed] [Google Scholar]
- 50.Noren NK, Liu BP, Burridge K, Kreft B. J Cell Biol. 2000;150:567–580. doi: 10.1083/jcb.150.3.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Elia LP, Yamamoto M, Zang K, Reichardt LF. Neuron. 2006;51:43–56. doi: 10.1016/j.neuron.2006.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Perez-Moreno M, Davis MA, Wong E, Pasolli HA, Reynolds AB, Fuchs E. Cell. 2006;124:631–644. doi: 10.1016/j.cell.2005.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Silverman JB, Restituito S, Lu W, Lee-Edwards L, Khatri L, Ziff EB. J Neurosci. 2007;27:8505–8516. doi: 10.1523/JNEUROSCI.1395-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Laura RP, Witts AS, Held HA, Gerstner R, Deshayes K, Koehler MF, Kosik KS, Sidhu SS, Lasky LA. J Biol Chem. 2002;277:12906–12914. doi: 10.1074/jbc.M200818200. [DOI] [PubMed] [Google Scholar]
- 55.Henle F, Fischer C, Meyer DK, Leemhuis J. J Biol Chem. 2006;281:24955–24969. doi: 10.1074/jbc.M604114200. [DOI] [PubMed] [Google Scholar]
- 56.Schubert V, Da Silva JS, Dotti CG. J Cell Biol. 2006;172:453–467. doi: 10.1083/jcb.200506136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Togashi H, Abe K, Mizoguchi A, Takaoka K, Chisaka O, Takeichi M. Neuron. 2002;35:77–89. doi: 10.1016/s0896-6273(02)00748-1. [DOI] [PubMed] [Google Scholar]
- 58.Abe K, Chisaka O, von Roy F, Takeichi M. Nat Neurosci. 2004;7:357–363. doi: 10.1038/nn1212. [DOI] [PubMed] [Google Scholar]
- 59.Okamura K, Tanaka H, Yagita Y, Saeki Y, Taguchi A, Hiraoka Y, Zeng LH, Calman DR, Miki N. J Cell Biol. 2004;167:961–972. doi: 10.1083/jcb.200406030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bamji SX, Shimazu K, Kimes N, Huelsken J, Birchmeier W, Lu B, Reichardt LF. Neuron. 2003;40:719–731. doi: 10.1016/s0896-6273(03)00718-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Meng Y, Zhang Y, Tregoubov V, Falls DL, Jia Z. Rev Neurosci. 2003;14:233–240. doi: 10.1515/revneuro.2003.14.3.233. [DOI] [PubMed] [Google Scholar]
- 62.Tashiro A, Minden A, Yuste R. Cereb Cortex. 2000;10:927–938. doi: 10.1093/cercor/10.10.927. [DOI] [PubMed] [Google Scholar]
- 63.Kumar V, Zhang MX, Swank MW, Kunz J, We GY. J Neurosci. 2005;25:11288–11299. doi: 10.1523/JNEUROSCI.2284-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Engert F, Bonhoeffer T. Nature. 1999;399:66–70. doi: 10.1038/19978. [DOI] [PubMed] [Google Scholar]