

Abstract
Dendritic cells (DC) play a critical role in initiating and directing adaptive immune responses against pathogens and tumours. Immature DC are thought to act as sentinels in peripheral tissues where their main function is to capture antigen at sites of infection, whereas mature DC are highly efficient at priming T-cell-mediated immune responses against infectious pathogens. The DC maturation process is thought to be an important step in the efficient generation of cytotoxic T lymphocytes (CTL). It is well established that many aspects of immune function, including CTL-mediated antiviral immunity, are modulated by neuroendocrine-derived products. Corticosterone (CORT), an adrenal hormone produced at increased concentrations during a stress response, has been shown to play a role in impaired CTL responses in stressed animals, leading to high mortality in mice normally resistant to viral infection. While direct effects of neuroendocrine mediators on CTL have been studied, little is known about their effects on DC that are critical for CTL priming. Here, we found that physiologically relevant concentrations of CORT, acting via the glucocorticoid receptor, functionally compromise DC maturation. DC exposed to CORT remained phenotypically and functionally immature after stimulation with lipopolysaccharide and were impaired for the production of interleukin (IL)-6, IL-12, and tumour necrosis factor-α. These effects were biologically significant, as CORT treatment resulted in a marked reduction in the ability of DC to prime naive CD8+ T cells in vivo. These findings offer a potential mechanism underlying stress-associated immunosuppression.
Keywords: dendritic cells, glucocorticoids, neuroimmunology
Introduction
Immature dendritic cells (DC) are highly efficient at sampling their antigenic environment but are inefficient at T-cell priming and express low levels of surface major histocompatibility complex (MHC) class II and costimulatory molecules B7.1 and B7.2.1–3 Upon encounter with a pathogen or inflammatory signal, DC undergo maturation, a process that is associated with a change in function from antigen uptake to antigen presentation.4,5 Maturing DC up-regulate surface expression of MHC class II and costimulatory molecules.5 Maturation eventually results in a decrease in antigen uptake3,6 and is accompanied by the production of pro-inflammatory cytokines.7,8 The net result of these changes is that mature DC possess a potent ability to prime naive T lymphocytes.1,2,5 The maturation process is likely to be one of the earliest critical steps in the initiation of many adaptive immune responses.
It is well established that products and processes of the nervous and endocrine systems can have substantial effects on both innate and adaptive immunity.9,10 In humans and animals, external stimuli can initiate a ‘stress response’ involving activation of the hypothalamic–pituitary–adrenal axis, resulting in increased production of corticosterone (CORT, cortisol in humans) by the adrenal glands, which is released into circulation.11 We and others have shown that CORT plays an important role in the stress-induced impairment of cytotoxic T lymphocyte (CTL)-mediated antiviral immunity12–14 leading to a high death rate from viral infection in normally resistant mice.15 In addition to direct effects on T cells, stress-induced CORT may modulate DC function. Because DC play a crucial role in priming CTL-mediated immune responses16,17 it is essential to understand the role and mechanisms of neuroendocrine mediators in modulating DC function.
Glucocorticoids, including endogenously produced CORT, have long been known to possess immunosuppressive properties.18 Our previous studies have shown that CORT, at physiologically relevant concentrations, impairs the generation of antigenic peptide leading to a functional decrease in antigen processing and presentation on MHC class I by virus-infected DC.19,20 Here we extend these studies to examine the effects of CORT on DC maturation and function using primary bone marrow-derived DC. We have determined that physiologically relevant ‘stress levels’ of CORT21,22 acting through the glucocorticoid receptor, significantly impeded or completely blocked a number of phenotypic and functional changes associated with lipopolysaccharide-induced maturation of primary DC. These effects of CORT were functionally relevant, as they resulted in impaired priming of naive CD8+ T cells in vivo. Together, these findings demonstrate that CORT impairs DC maturation and function, lending new insights into potential mechanisms underlying immunosuppression resulting from interactions of the nervous, endocrine, and immune systems. Overall, these studies further elucidate the complex role of endogenously produced, stress-associated hormones in regulating immune responses against infectious pathogens.
Materials and methods
Mice
Male C57BL/6 mice (Jackson Laboratories, Bar Harbor, ME) were housed under specific pathogen-free conditions. All experiments were performed according to the guidelines of the American Association for Laboratory Animal Care International and the National Institutes of Health.
Generation of bone marrow-derived dendritic cells (BMDC)
BMDC were generated as described previously, with modifications.23 Bone marrow was washed from the femurs and tibiae of mice using sterile Hanks' balanced salt solution. Approximately 107 bone marrow cells were plated in Petri dishes containing Iscove's modified Dulbecco's medium (IMDM) with 5% fetal bovine serum (FBS), 2 mm glutamine, 200 U/ml penicillin, 100 µg/ml streptomycin sulphate, 10 ng/ml granulocyte–macrophage colony-stimulating factor (GM-CSF, PeproTech, Rocky Hill, NJ), and 2 ng/ml interleukin (IL)-4 (Sigma, St. Louis, MO). Every 2 days, cultures were gently swirled to dislodge loosely adherent cells, and half of the media was replaced with fresh media.
Corticosterone (CORT)/lipopolysaccharide (LPS) treatment
Unless otherwise indicated, on day 6 of BMDC culture, at which time dendritic cells (DC) were differentiated but predominantly immature, CORT (98% pure; MP Biomedicals, Solon, OH) or vehicle (VEH, 0·1% ethanol) was added to the culture media for 48 hr. During the final 12 hr, cells were stimulated with 100 ng/ml Escherichia coli 055:B5 LPS (Sigma).
Glucocorticoid receptor (GR) antagonist treatment
BMDC were generated as described above. On day 6, cells were pretreated with 10−6m RU-486 (Sigma) for 2 hr prior to the addition of 10−6m CORT. Thirty-six hr later, cells were stimulated with 100 ng/ml LPS for 12 hr before harvesting.
Analysis of protein expression by flow cytometry
BMDC were harvested, and Fc receptors and non-specific binding sites were blocked with supernatant from the anti-CD16/32 hybridoma (2.4G2)24 containing 20% mouse serum. Cells were then stained with various combinations of directly labeled antibodies: fluorescein isothiocyanate (FITC)-conjugated anti-I-Ab (AF6-120.1; BD Biosciences, San Jose, CA), FITC-conjugated anti-CD40 (HM40-3; eBioscience, San Diego, CA), FITC-conjugated anti-B7.1 (CD80, 16-10A1; BD Biosciences), FITC-conjugated anti-B7.2 (CD86, GL1; BD Biosciences), phycoerythrin (PE)-Cy5-conjugated anti-CD11c (N418; eBioscience), and PE-conjugated anti-Toll-like receptor (TLR)-4 (MTS510; eBioscience). In some experiments, cells were stained for surface CD11c prior to fixation with 2% paraformaldehyde and permeabilization with 0·5% saponin (Sigma). These cells were then stained for I-Ab, B7.1, or B7.2 to measure total (surface and intracellular) expression of each of these proteins. Flow cytometry was performed on a FACSCanto (Becton Dickinson, San Diego, CA), and data were analysed using FlowJo software (TreeStar, Ashland, OR). DC were identified as CD11c+ for analysis.
Detection of apoptosis
BMDC were harvested and stained with FITC-conjugated anti-CD11c (N418; eBioscience), PE-annexin-V (BD Biosciences), and 7-amino-actinomycin D (7-AAD; BD Biosciences). Flow cytometry was performed on a FACScan (Becton Dickinson), and data were analysed as described above.
Real-time polymerase chain reaction (PCR)
The effects of CORT/LPS treatment on the transcription of genes encoding I-Ab, B7.1, and B7.2 were analysed by real-time PCR. DC were purified from BMDC cultures using magnetic CD11c microbeads with an AutoMACS cell sorter (Miltenyi Biotec, Auburn, CA) according to the manufacturer's instructions. RNA was isolated using TRI reagent (Sigma), and cDNA was synthesized using Omniscript reverse transcriptase (Qiagen, Valencia, CA). Real-time PCR was performed on an Applied Biosystems 7900 HT Fast Real-Time PCR System (Applied Biosystems, Foster City, CA) with TaqMan Universal PCR Master Mix (Applied Biosystems) and prevalidated TaqMan Gene Expression Assays Mm00439216_m1 (H2-Ab1), Mm00444543_m1 (B7.1), Mm00711660_m1 (B7.2), and Mm00446973_m1 (TATA-binding protein (TBP), all Applied Biosystems). Relative changes in gene expression were calculated using the ΔΔCT method, computed by the SDS software package v2.2.2 (Applied Biosystems) normalizing to TBP expression as the endogenous control.
Fluorescence microscopy
DC in the same cultures were stained for both cell-surface and intracellular ligands by indirect immunofluorescence. DC were cultured from bone marrow on glass Lab-Tek II eight-well chamber slides (Nalge Nunc International, Naperville, IL) and treated with CORT and/or LPS as described above. Non-specific binding sites were blocked with 10% goat serum (Sigma) in Hanks, balanced salt solution/bovine serum albumin on ice for 20 min. For colocalizing intracellular MHC class II (I-Ab) and lysosomes (lysosome-associated membrane protein (LAMP)-1), the cells were first incubated with anti-CD11c antibody clone N418 (monoclonal antibody supernatant from the hamster hybridoma) for 1–3 hr on ice. Following washes with phosphate-buffered saline (PBS), FITC-conjugated anti-hamster secondary antibody (diluted 1 : 200, eBioscience) was applied for 30 min. This approach allowed for visualization of surface CD11c to identify DC in the cultures. The cells were washed, fixed with 4% paraformaldehyde, then permeabilized with 0·05% saponin for 20 min at room temperature. Following washes, intracellular staining of the same cells was performed using a rat primary antibody against mouse LAMP-1 (CD107α, BD Pharmingen) and primary anti-mouse I-Ab (Y3P monoclonal antibody supernatant from mouse HB-183 hybridoma, American Type Culture Collection, Rockville, MD) for 1 hr at room temperature. Following washes, Alexa 546-conjugated anti-rat and Alexa 647-conjugated anti-mouse secondary antibodies (both diluted 1 : 200; Molecular Probes, Carlsbad, CA) were applied for 30 min. The slides were mounted with ProLong Gold antifade reagent (Molecular Probes), and examined under an Olympus IX81 Deconvolution microscope with Slidebook 4.0 software. All images within a given experiment were acquired using identical exposure times.
Antigen uptake
The uptake of soluble protein by BMDC was determined by measuring the rates of uptake of FITC-conjugated ovalbumin (FITC-OVA, Molecular Probes). DC were purified from BMDC cultures using magnetic CD11c microbeads with an AutoMACS cell sorter according to the manufacturer's instructions. CD11c+ cells were incubated in prewarmed (37°) complete phenol red-free IMDM (Invitrogen) for 30 min. FITC-OVA was added at a final concentration of 10 µg/ml. Cells were incubated at 37°, and 3 × 105 cells were removed at 20 min intervals and transferred to ice-cold PBS 1% FBS. Cells were transferred to black 96-well plates (Dynex Technologies, Chantilly, VA), washed extensively, and lysed in PBS containing 0·3% Triton-X-100. Lysates were analysed for fluorescence using an XFluor4 Safire II plate reader (Tecan, Research Triangle Park, NC). Samples were excited at a wavelength of 490 nm, and emissions were read at 520 nm. Cells incubated with FITC-OVA on ice served as negative controls.
In vivo T-cell priming
DC were purified from BMDC cultures using magnetic CD11c microbeads with an AutoMACS cell sorter as described. Cells were incubated with the immunodominant herpes simplex virus (HSV)-1 gB498−505 peptide (100 nm, SSIEFARL) for 40 min, washed and resuspended in PBS containing 1% FBS. Mice received 5 × 105 CORT-treated or VEH-treated cells intravenously in a volume of 500 µl. One week later, the mice were euthanized and their spleens were removed. Spleens were homogenized by passage through 60-gauge stainless steel mesh screens. The resulting cell suspension was purified using Lymphocyte Separation Media (Cambrex Bio Science, Walkersville, MD). Cells were then used for intracellular cytokine staining or degranulation assays.
Intracellular cytokine staining
Splenocytes were incubated in 96-well plates (4 × 106 cells/well) in the presence of 10 µm HSV-1 gB498−505 or an irrelevant peptide (OVA257−264) at 37°. After 2 hr, 5 µg/ml brefeldin A (Sigma) was added to the cells. After an additional 4 hr, the cells were stained with PE-Cy5-conjugated anti-CD8α (53–6·7; eBioscience). Cells were then fixed with 2% paraformaldehyde, permeabilized with 0·5% saponin, and stained intracellularly with FITC-conjugated anti-interferon (IFN)-γ (XMG1.2; eBioscience) and PE-conjugated antitumour necrosis factor (TNF)-α (MP6-XT22; eBioscience).
BMDC were treated with CORT and/or LPS as described above. Four hrs prior to harvesting, the cells were treated with 5 µg/ml brefeldin A to inhibit cytokine secretion. BMDC were harvested and stained with FITC-conjugated anti-CD11c. Cells were fixed with 2% paraformaldehyde, permeabilized with 0·5% saponin, and stained intracellularly with PE-conjugated anti-IL-6 (MP5–20F3; eBioscience), PE-conjugated anti-IL-12 p40 (C17·8; eBioscience), or PE-conjugated anti-TNF-α. Flow cytometry was performed on splenocytes and BMDC as described above.
Degranulation assay
The regulated secretion of lytic granules from cytotoxic T lymphocytes (CTL) is triggered by T-cell receptor recognition of a target cell. CD107a (LAMP-1) is found on lysosomal membranes. These membranes are transiently exposed to the extracellular media during CTL degranulation. By incubating cells with antibody against CD107a, we were able to measure degranulation in response to a specific peptide. Splenocytes were incubated in 96-well plates (4 × 106 cells/well) in the presence of 10 µm HSV-1 gB498−505 or an irrelevant peptide (OVA257−264) at 37°. Wells also contained FITC-conjugated anti-CD107a (1D4B; BD Pharmingen). After 1 hr, 10 mm NH4Cl was added to cells to prevent endosome acidification. Three hrs later, the cells were stained with PE-conjugated anti-I-Ab (AF6-120·1; BD Pharmingen) and PE-Cy5-conjugated anti-CD8α. Flow cytometry was performed using a FACSCanto, and data were analysed using FlowJo software. Cells were stained for I-Ab to exclude CD8α+ DC that constitutively undergo endocytosis and take up anti-CD107a. Analyses were performed on the CD8+, 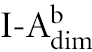 population of cells.
population of cells.
Statistical analyses
Statistical analyses were performed by applying Student's t-test. Significance was determined as P < 0·05. Error bars in all figures represent standard error of the mean.
Results
Generation of BMDC
Bone marrow-derived cells were cultured in the presence of GM-CSF and IL-4 for 8 days. At the end of this time, the cultures predominantly contained DC (80–90%) that were CD11c+, CD11b+, CD8α–, resembling myeloid DC described in vivo.25,26 Most of these cells (90%) were immature, expressing low levels of MHC class II, B7.1, and B7.2 as measured by mean fluorescence intensity (MFI) of antibody staining (Fig. 1a, shaded histogram).
Figure 1.

Effects of CORT and/or LPS on DC maturation-associated marker surface expression. (a) Representative histograms depicting the surface staining profile for VEH (shaded) and CORT-treated (dashed line) DC for the expression of each marker. CD11c+ cells were gated for analysis. (b) Representative histograms depicting the surface staining profile for VEH-treated (shaded), LPS-treated (thin line), and CORT/LPS treated (bold line) DC for the expression of each marker. CD11c+ cells were gated for analysis. Left panels (a, b), thin dashed line is unstained cells. (c) Surface expression of maturation-associated markers was determined by flow cytometry. MFI were normalized to VEH-treated cells (100% Baseline MFI) for seven separate experiments. *P < 0·05.
CORT impairs LPS-induced up-regulation of maturation-associated markers
DC maturation is associated with increased surface expression of MHC class II and the costimulatory molecules, B7.1, B7.2 and CD40. To determine the effects of CORT on DC maturation, we treated BMDC with CORT (10−6m) or VEH for 48 hr. This concentration of CORT is similar to endogenous concentrations that we and others have measured in mice undergoing a stress response (1–3 × 10−6m).21,22 During the final 12 hr of CORT treatment, cells were also treated with LPS (100 ng/ml) to induce maturation. After LPS stimulation, the cells were harvested and stained for CD11c, MHC class II, B7.1, B7.2, and CD40 for analysis by flow cytometry. On CD11c+ cells, MHC class II, B7.1, B7.2, and CD40 each showed substantial increases in surface expression after LPS stimulation (averaging 46%, 90%, 102%, and 194% increases in MFI, respectively, Fig. 1b, c). The effects of LPS on MHC class II and B7.2 expression were completely blocked when the cells were pretreated with CORT, while the effect on B7.1 was reduced by 50%, and CD40 up-regulation was only slightly attenuated (Fig. 1b, c). These results suggest that CORT impairs DC maturation. Although CORT treatment alone slightly reduced the expression of each marker, this treatment did not result in a substantial change of DC staining profiles (Fig. 1a), because unstimulated cells were mostly immature. Time-course studies demonstrated that cells required 12 hr of exposure to CORT to maximally impair DC maturation. CORT-treatment of DC for 6 hr or less failed to substantially affect LPS-induced expression of MHC class II, B7.1 or B7.2.
DC maturation is impaired by physiological concentrations of CORT
CORT is constitutively produced in vivo at low levels but is up-regulated under conditions of stress. At higher concentrations, CORT mediates its effects by binding the GR. Because DC maturation was impaired by 10−6m CORT, we examined the effects of a range of concentrations of CORT (10−9−10−6m) on DC maturation. A low dose of CORT (10−9m), as compared to no CORT (VEH), slightly enhanced the effect of LPS on BMDC. However, concentrations of CORT > 10−8m, saturating the GR (GR Kd = 0·5–1 × 10−8m), significantly impaired or blocked the LPS-induced up-regulation of MHC class II (Fig. 2a), B7.1 (Fig. 2b), and B7.2 (Fig. 2c).
Figure 2.

CORT acts at concentrations that saturate the GR and is blocked by pretreatment with GR antagonist. (a–c) Dose–response relationship between CORT and surface expression of maturation-associated markers. DC were treated with various concentrations of CORT for 48 hr and stimulated with LPS during the last 12 hr. Surface expression was analysed by flow cytometry for (a) MHC class II (b) B7.1, and (c) B7.2. Normalized MFI relative to VEH-treated cells without LPS are displayed for the average of three separate experiments. *P < 0·05. (d) GR specificity of CORT. DC were pretreated with RU-486 for 2 hr before subsequent treatment with CORT and/or LPS. Surface expression of maturation-associated markers was analysed by flow cytometry. Data are expressed as MFI normalized from three separate experiments.
Effects of CORT are mediated through the glucocorticoid receptor (GR) and are not caused by apoptosis
To determine whether the effects of CORT on DC maturation were mediated through the GR, we used the GR antagonist RU-486. BMDC were pretreated with RU-486 for 2 hr prior to treatment with CORT and/or LPS. While RU-486 alone slightly reduced the LPS-induced up-regulation of MHC class II, B7.1, and B7.2, the GR antagonist completely prevented CORT from having any additional effects on the LPS-induced up-regulation of these markers (Fig. 2d). These results suggest that the impairment of DC maturation by CORT is mediated through the GR.
Glucocorticoids have been reported to induce apoptosis in DC under certain conditions.27 It was possible that CORT had induced apoptosis in DC that otherwise would have undergone maturation. To address this issue, we stained CORT/LPS-treated BMDC with annexin-V and 7-AAD to detect early apoptotic and dead cells. However, in three separate experiments, CORT did not increase the percentage of early apoptotic (annexin-V+, 7-AAD–) or apoptotic/dead (annexin-V+, 7-AAD+) cells over the 48 hr treatment period examined (Fig. 3a), suggesting that the observed CORT-induced impairment of DC maturation was not caused by increased apoptosis or cell death.
Figure 3.

CORT did not induce apoptosis or down-regulate TLR-4 on DC. (a) Scatterplots depicting Annexin V/7-AAD staining of CD11c+ cells treated with CORT or VEH. Scatterplots are representative of results from three separate experiments. (b) Surface expression of TLR-4 was determined by flow cytometry. MFI were normalized to VEH-treated cells (100% baseline MFI) for three separate experiments.
Because LPS induces DC maturation through binding to TLR-428 it was possible that CORT reduced the surface expression of TLR-4, which would make DC resistant to LPS-induced maturation. However, we did not observe any reduction in surface TLR-4 expression in CORT-treated DC. Furthermore, LPS-induced TLR-4 down-regulation was not modulated by CORT (Fig. 3b).
CORT impairs transcription of B7.1 and B7.2 and causes intracellular retention of MHC class II
To determine the mechanism by which CORT impaired the up-regulation of DC maturation markers, we examined mRNA and total protein levels of MHC class II, B7.1, and B7.2 in CORT/LPS-treated DC. Total protein expression (surface and intracellular) for each of these molecules was determined by staining permeabilized cells for flow cytometry. Staining profiles for total B7.1 and B7.2 (Fig. 4a, b) were similar to those obtained from surface-stained cells (Fig. 1a, b). LPS-treated DC contained significantly more B7.1 and B7.2 than VEH-treated cells (average of 2·0- and 2·4-fold increases, respectively), and these increases were partially (B7.1) or completely (B7.2) inhibited in cells that were pretreated with CORT (Fig. 4b, c).
Figure 4.

Effects of CORT and/or LPS on protein and mRNA expression of DC maturation-associated markers. CORT/LPS treated cells were fixed and permeabilized prior to staining for flow cytometry. (a) Representative histograms depicting the total protein staining profile for VEH-treated (shaded) and CORT-treated (dashed line) DC for the expression of each marker. (b) Representative histograms depicting the total protein staining profile for VEH-treated (shaded), LPS-treated (thin line), and CORT/LPS treated (bold line) DC for the expression of each marker. (c) MFI were normalized to VEH-treated cells for three separate experiments. (d) Relative mRNA expression of each gene as determined by real-time RT-PCR. Data represent the mean of three separate experiments. *P < 0·05.
Quantitative real-time PCR was used to measure the transcripts encoding these proteins. LPS treatment resulted in significantly increased transcription of B7.1 and B7.2 (average of 4·3- and 3·8-fold, respectively, Fig. 4d). Furthermore, CORT-treatment significantly impaired this increase, with a greater effect on B7.2 than on B7.1. These results correlate well with our observations that CORT impairs the LPS-induced expression of B7.1 and B7.2 protein (Figs 1c and 4c), demonstrating that CORT modulated the expression of B7.1 and B7.2 through effects on the transcription or stability of RNA.
Although total protein staining for B7.1 and B7.2 resulted in expression profiles similar to those of non-permeabilized cells, intracellular staining of MHC class II did not resemble MHC class II surface expression. Recall that surface expression of MHC class II was low in VEH-treated cells and increased upon LPS-stimulation, but remained low when cells were pretreated with CORT (Fig. 1b). Permeabilized DC stained strongly for MHC class II, regardless of whether they were treated with CORT and/or LPS (Fig. 4a–c). When MHC class II transcripts were measured, the amounts of mRNA did not change in DC that had been treated with CORT and/or LPS (Fig. 4d). These data indicate that CORT did not have substantial effects on MHC class II at the transcriptional or translational levels, indicating that the effect of CORT on MHC class II was post-translational.
Besides transcriptional and translational regulation, cell surface proteins can be regulated at the level of trafficking. The localization of MHC class II in CORT/LPS-treated cells was examined by deconvolution microscopy (Fig. 5). In VEH- and CORT-treated cells, MHC class II was retained intracellularly and colocalized with the lysosomal marker LAMP-1. Upon LPS treatment, MHC class II was expressed predominantly at the cell surface. However, when cells were treated with CORT prior to LPS stimulation, MHC class II was retained intracellularly and colocalized with LAMP-1. These studies indicate that the effects of CORT on surface expression of MHC class II are mediated by inhibiting LPS-induced trafficking to the plasma membrane.
Figure 5.

Effects of CORT/LPS on cellular localization of MHC class II. DC cultures were treated with CORT and/or LPS as described and stained for surface CD11c (green), intracellular LAMP-1 (red), and MHC class II (blue). Deconvolution microscopy was used to visualize cells.
CORT prevents LPS-induced down-regulation of endocytosis
The above studies show that CORT impaired the surface expression of maturation-associated phenotypic markers, including the LPS-induced up-regulation of MHC class II, B7.1 and B7.2. Next, we determined the effects of CORT on DC function, beginning with antigen uptake. Immature DC continuously sample their antigenic environment. Upon encounter with a maturation-inducing stimulus, DC transiently increase their rate of antigen uptake before strongly down-regulating endocytosis.3,6 Rates of uptake of soluble protein (FITC-OVA) were measured in DC that had been treated with CORT and/or LPS. As expected, LPS treatment significantly reduced (47 ± 6%) the rate of antigen uptake compared to VEH-treated cells (Fig. 6). However, cells that were treated with CORT prior to LPS-stimulation endocytosed antigen at a rate similar to VEH-treated cells. Cells treated with CORT alone were also similar to VEH-treated cells. This assay specifically measured active uptake, as control cells incubated on ice did not take up FITC-OVA (Fig. 6a).
Figure 6.

Effect of CORT/LPS on endocytosis of soluble protein. (a) DC were treated with CORT/LPS and incubated with FITC-OVA at 0° (dashed lines) or 37° (solid lines). Aliquots were removed at 20 min intervals, washed extensively, and lysed. FITC-OVA uptake was measured by fluorimetry. Data from a representative time-course are shown, expressed in relative fluorescence units (RFU). (b) The relative rates of uptake from three separate experiments were normalized to VEH-treated cells. *P < 0·05.
CORT impairs cytokine production by DC
Activated DC produce cytokines that regulate the initiation of immune responses.29,30 We determined the effects of CORT on LPS-induced cytokine production by DC using intracellular cytokine staining. LPS stimulation induced the synthesis of IL-6, IL-12, and TNF-α. CORT treatment significantly reduced the percentage of LPS-stimulated cells that produced IL-6 (29 ± 2% versus 45 ± 3%) and IL-12 (21 ± 1% versus 46 ± 2%) (Fig. 7a). In addition, CORT/LPS-treated cells produced significantly less IL-6, IL-12, and TNF-α (averaging 55 ± 0·4%, 58 ± 4%, and 74 ± 3%) decreases in the MFI of cytokine-positive cells, respectively on a per cell basis than cells treated with LPS alone (Fig. 7b).
Figure 7.

Effect of CORT/LPS on cytokine production by DC. Prior to harvesting, DC were treated with CORT/LPS, as described, and brefeldin A was added for 4 hr to inhibit cytokine secretion. Cells were permeabilized and stained for IL-6, IL-12, and TNF-α. (a) Percentages of cells containing each cytokine are displayed. Data were averaged from three separate experiments. (b) Relative amounts of cytokine produced on a per cell basis in cytokine-containing cells are displayed. Data were averaged from three separate experiments and normalized to LPS-treated cells (100%). *P < 0·05.
CORT renders DC inefficient at priming CD8+ T-cell responses
One of the most critical functions of DC is their role in priming T cells to mount an immune response against viral infection. We determined the effects of CORT on the ability of DC to prime CD8+ T-cell responses in vivo by measuring peptide-specific CD8+ T-cell responses. The use of an adoptive transfer strategy enabled us to separate the effects of CORT on the DC from the T cells in vivo. Mice were injected intravenously with DC pulsed with the HSV-1 gB498−505 peptide, resulting in an antigen-specific CD8+ T-cell response as measured by intracellular cytokine staining for IFN-γ and TNF-α. CTL-mediated cytotoxicity was also assessed by measuring degranulation in response to specific peptide. Degranulation of CTL is triggered upon T-cell receptor recognition of a target cell. However, when mice received DC that were treated with CORT prior to peptide-pulsing, the resulting peptide-specific CD8+ T-cell responses were reduced significantly, as measured by IFN-γ(Fig. 8a), TNF-α (Fig. 8b) and degranulation (Fig. 8c). These responses were antigen-specific, as they did not occur when splenocytes were pulsed with an irrelevant peptide (OVA257−264). Mice injected with DC in the absence of peptide resulted in responses similar to naive mice (data not shown).
Figure 8.

Effect of CORT on the ability of DC to prime T cell responses in vivo. CORT or VEH-treated DC were pulsed with HSV-1 gB498−505 peptide, washed and injected into mice intravenously. Seven days later, splenocytes were isolated and assayed for production of (a) IFN-γ, and (b) TNF-α, or (c) degranulation in response to HSV gB498−505 or OVA257−264 (irrelevant peptide). Splenocytes from naive mice were used as negative controls. Data represent the mean responses from groups of four to six mice. *P < 0·05.
Discussion
Many studies have demonstrated that animals undergoing a stress response exhibit reduced CTL function,12–14 which can lead to a high death rate following viral infection in normally resistant mice.15 An important component of this response involves the activation of the hypothalamic–pituitary–adrenal axis resulting in the production of CORT that, in addition to directly impairing T lymphocyte function, may interfere with the ability of DC to prime naive T cells. Most of the studies examining the effects of glucocorticoids on DC function have used the synthetic pharmacological glucocorticoid, dexamethasone.31–35 In our previous work19,20 and current studies, we used the naturally produced glucocorticoid, CORT, which is more appropriate for examining effects of neuroendocrine interactions on immune function.
DC maturation is important for efficient priming of naive T lymphocytes36 and has been shown to be critical in the initiation of an adaptive immune response against viral infection.37 In our studies, the CORT-mediated impairment of DC maturation was associated with a deficiency in priming antigen-specific CD8+ T-cell responses (Fig. 8). Mice receiving CORT-treated DC generated fewer cytokine-producing CD8+ T cells and fewer degranulating cytotoxic CD8+ T cells than mice receiving VEH-treated DC. These data suggest that elevated CORT concentrations found in vivo during a stress-response likely contribute to the inefficient generation of CTL-mediated immunity. A delay or failure to efficiently prime CTL can compromise the successful control of an infection.15
Here, we have demonstrated that CORT-treated DC were deficient in the up-regulation of the costimulatory molecules, B7.1 and B7.2 (Fig. 1). Immature DC express low levels of costimulatory molecules, making them inefficient for T-cell priming.36 Upon maturation, DC normally up-regulate B7.1 and B7.2 to provide the necessary costimulatory signal to naive T cells through CD28. A lack of costimulation can lead to T-cell anergy38 and could contribute to our observed deficiency in T-cell priming in mice that received CORT-treated DC.
The up-regulation of costimulatory molecules alone is insufficient for T-cell priming.39 Mature DC also secrete many cytokines, including IL-6, IL-12, and TNF-α. IL-6 and TNF-α are generally proinflammatory, and IL-6 renders effector T cells resistant to suppression by regulatory T cells.40 IL-12 is important for promoting cell-mediated T helper 1 (Th1) polarization in CD4+ helper T cells.41 We observed that fewer CORT-treated DC were able to produce IL-6 and IL-12 in response to stimulation with LPS, and that CORT reduced the amount of IL-6, IL-12, and TNF-α on a per cell basis (Fig. 7). The observed reduction in T-cell priming in vivo could be caused by reduced costimulatory molecule expression, cytokine production, or a combination of these factors. In addition, it is possible that these effects could also interfere with priming of CD4+ T cells. Others have previously reported that mice undergoing a stress response exhibit strong Th2 polarization.42,43 These effects could be the result, in part, of reduced production of IL-12 or other cytokines by DC.
The effect of CORT on antigen uptake is further evidence that CORT interferes with DC maturation. While immature DC are efficient at antigen uptake, LPS-stimulation transiently increases endocytosis before strongly inhibiting it.6 This increase is thought to ‘load up’ the DC with the antigen in its immediate environment prior to migration to a lymph node for presentation to T cells. We observed that antigen uptake was reduced after 12 hr of LPS stimulation, as expected. However, CORT treatment prevented this down-regulation (Fig. 6). Although antigen uptake remained high in CORT-treated DC, our previous findings have shown that antigen processing and presentation are impaired in CORT-treated DC.19 Together, these results have significant implications for the effect of CORT on cross-presentation, suggesting that the rate of antigen uptake may not be able to compensate for the effect of CORT on antigen processing and presentation. The effects of CORT and stress on cross-presentation, both in vitro and in vivo, are currently being studied in our laboratory.
Our data suggest that the mechanism of action of CORT is to directly oppose LPS as a DC maturation-inducing stimulus. In LPS-stimulated DC, transcription and expression of B7.1 and B7.2 are increased, while preformed MHC class II is transported from intracellular vesicles to the plasma membrane without any changes in MHC class II transcription.44 CORT treatment impaired both transcription and protein expression of B7.1 and B7.2 in LPS-stimulated cells (Fig. 4). In contrast, surface MHC class II expression is reduced in CORT-treated cells via retention of pre-existing MHC class II molecules within intracellular compartments. (Fig. 5). These results demonstrate that CORT rendered DC resistant to the maturation-inducing effects of LPS. However, the surface expression of TLR-4 did not decrease upon CORT treatment, indicating CORT was not simply reducing the ability of DC to detect LPS. Furthermore, we found that CORT also prevented DC maturation induced by poly (I:C) or CpG DNA (data not shown), which induce maturation through TLR-3 and TLR-9, respectively, suggesting that the effects of CORT are not limited to LPS-induced maturation. Because DC maturation and cytokine production are mediated by distinct pathways downstream of TLR-445 and both were modulated by CORT, it is likely that CORT acts on multiple intracellular targets in DC. Because the effect of CORT on DC maturation was blocked by a GR antagonist (Fig. 2d), it is also likely these effects are mediated via the GR.
We have shown that CORT, a physiologically produced glucocorticoid, functionally impaired DC maturation and cytokine production and reduced the ability of DC to prime naive CD8+ T cells in vivo. This inhibition occurred via the GR with concentrations of CORT similar to those observed in animals undergoing a stress response.22 The deficiencies in DC function we have observed represent a potential mechanism underlying the inefficient generation of antiviral CTL responses observed in stress-associated immunosuppression. These findings illustrate the substantial effect that products of the nervous and endocrine systems have on immune function, and underscore the importance of considering neuroendocrine processes that can influence the outcome of an immune response.
Acknowledgments
The authors thank Dr John Hunzeker and Jennifer Fear for valuable discussions and technical assistance. We also thank Nate Sheaffer of the Cell Science/Flow Cytometry Core Facility of the Section of Research Resources, Penn State College of Medicine for assistance with flow cytometry. This work was supported by PHS Grants AI065702 (M.E.T., C.C.N., R.H.B.) and AI49719 (R.H.B.). M.D.E. is supported by NIH Training Grant 2 T32 CA60395-11. The animal research facility is supported by Research Facilities Improvement Grant Number C06 RR-15428-01 from the NIH.
Abbreviations:
- 7-AAD
7-amino-actinomycin D
- BMDC
bone-marrow-derived DC
- CORT
corticosterone
- CTL
cytotoxic T lymphocyte
- DC
dendritic cell
- FBS
fetal bovine serum
- FITC
fluorescein isothiocyanate
- GM-CSF
granulocyte–macrophage colony-stimulating factor
- GR
glucocorticoid receptor
- HSV
herpes simplex virus
- IFN
interferon
- IL
interleukin
- IMDM
Iscove's modified Dulbecco's medium
- LAMP
lysosome-associated membrane protein
- LPS
lipopolysaccharide
- MFI
mean fluorescence intensity
- OVA
ovalbumin
- PE
phycoerythrin
- TBP
TATA-binding protein
- TLR
Toll-like receptor
- TNF
tumour necrosis factor
- VEH
vehicle
References
- 1.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–52. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 2.Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YJ, Pulendran B, Palucka K. Immunobiology of dendritic cells. Annu Rev Immunol. 2000;18:767–811. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- 3.Sallusto F, Cella M, Danieli C, Lanzavecchia A. Dendritic cells use macropinocytosis and the mannose receptor to concentrate macromolecules in the major histocompatibility complex class II compartment: downregulation by cytokines and bacterial products. J Exp Med. 1995;182:389–400. doi: 10.1084/jem.182.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kitajima T, Caceres-Dittmar G, Tapia FJ, Jester J, Bergstresser PR, Takashima A. T cell-mediated terminal maturation of dendritic cells: loss of adhesive and phagocytotic capacities. J Immunol. 1996;157:2340–7. [PubMed] [Google Scholar]
- 5.De Smedt T, Pajak B, Muraille E, et al. Regulation of dendritic cell numbers and maturation by lipopolysaccharide in vivo. J Exp Med. 1996;184:1413–24. doi: 10.1084/jem.184.4.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.West MA, Wallin RP, Matthews SP, Svensson HG, Zaru R, Ljunggren HG, Prescott AR, Watts C. Enhanced dendritic cell antigen capture via toll-like receptor-induced actin remodeling. Science. 2004;305:1153–7. doi: 10.1126/science.1099153. [DOI] [PubMed] [Google Scholar]
- 7.Macatonia SE, Hosken NA, Litton M, et al. Dendritic cells produce IL-12 and direct the development of Th1 cells from naive CD4+ T cells. J Immunol. 1995;154:5071–9. [PubMed] [Google Scholar]
- 8.Verhasselt V, Buelens C, Willems F, De Groote D, Haeffner-Cavaillon N, Goldman M. Bacterial lipopolysaccharide stimulates the production of cytokines and the expression of costimulatory molecules by human peripheral blood dendritic cells: evidence for a soluble CD14-dependent pathway. J Immunol. 1997;158:2919–25. [PubMed] [Google Scholar]
- 9.Webster JI, Tonelli L, Sternberg EM. Neuroendocrine regulation of immunity. Annu Rev Immunol. 2002;20:125–63. doi: 10.1146/annurev.immunol.20.082401.104914. [DOI] [PubMed] [Google Scholar]
- 10.Ader R, Felten D, Cohen N. Psychoneuroimmunology. 3. San Diego: Academic Press; 2001. [Google Scholar]
- 11.Kant GJ, Leu JR, Anderson SM, Mougey EH. Effects of chronic stress on plasma corticosterone, ACTH and prolactin. Physiol Behav. 1987;40:775–9. doi: 10.1016/0031-9384(87)90282-4. [DOI] [PubMed] [Google Scholar]
- 12.Bonneau RH, Sheridan JF, Feng N, Glaser R. Stress-induced modulation of the primary cellular immune response to herpes simplex virus infection is mediated by both adrenal-dependent and independent mechanisms. J Neuroimmunol. 1993;42:167–76. doi: 10.1016/0165-5728(93)90007-l. [DOI] [PubMed] [Google Scholar]
- 13.Bonneau RH, Zimmerman KM, Ikeda SC, Jones BC. Differential effects of stress-induced adrenal function on components of the herpes simplex virus-specific memory cytotoxic T-lymphocyte response. J Neuroimmunol. 1998;82:191–9. doi: 10.1016/s0165-5728(97)00200-2. [DOI] [PubMed] [Google Scholar]
- 14.Dobbs CM, Vasquez M, Glaser R, Sheridan JF. Mechanisms of stress-induced modulation of viral pathogenesis and immunity. J Neuroimmunol. 1993;48:151–60. doi: 10.1016/0165-5728(93)90187-4. [DOI] [PubMed] [Google Scholar]
- 15.Anglen CS, Truckenmiller ME, Schell TD, Bonneau RH. The dual role of CD8+ T lymphocytes in the development of stress-induced herpes simplex encephalitis. J Neuroimmunol. 2003;140:13–27. doi: 10.1016/s0165-5728(03)00159-0. [DOI] [PubMed] [Google Scholar]
- 16.Norbury CC, Malide D, Gibbs JS, Bennink JR, Yewdell JW. Visualizing priming of virus-specific CD8+ T cells by infected dendritic cells in vivo. Nat Immunol. 2002;3:265–71. doi: 10.1038/ni762. [DOI] [PubMed] [Google Scholar]
- 17.Smith CM, Belz GT, Wilson NS, Villadangos JA, Shortman K, Carbone FR, Heath WR. Cutting edge: conventional CD8alpha (+) dendritic cells are preferentially involved in CTL priming after footpad infection with herpes simplex virus-1. J Immunol. 2003;170:4437–40. doi: 10.4049/jimmunol.170.9.4437. [DOI] [PubMed] [Google Scholar]
- 18.Riccardi C, Bruscoli S, Migliorati G. Molecular mechanisms of immunomodulatory activity of glucocorticoids. Pharmacol Res. 2002;45:361–8. doi: 10.1006/phrs.2002.0969. [DOI] [PubMed] [Google Scholar]
- 19.Truckenmiller ME, Princiotta MF, Norbury CC, Bonneau RH. Corticosterone impairs MHC class I antigen presentation by dendritic cells via reduction of peptide generation. J Neuroimmunol. 2005;160:48–60. doi: 10.1016/j.jneuroim.2004.10.024. [DOI] [PubMed] [Google Scholar]
- 20.Truckenmiller ME, Bonneau RH, Norbury CC. Stress presents a problem for dendritic cells. corticosterone and the fate of MHC class I antigen processing and presentation. Brain Behav Immun. 2006;20:210–8. doi: 10.1016/j.bbi.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 21.Hermann G, Tovar CA, Beck FM, Sheridan JF. Kinetics of glucocorticoid response to restraint stress and/or experimental influenza viral infection in two inbred strains of mice. J Neuroimmunol. 1994;49:25–33. doi: 10.1016/0165-5728(94)90177-5. [DOI] [PubMed] [Google Scholar]
- 22.Yorty JL, Bonneau RH. Transplacental transfer and subsequent neonate utilization of herpes simplex virus-specific immunity are resilient to acute maternal stress. J Virol. 2003;77:6613–9. doi: 10.1128/JVI.77.12.6613-6619.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Son YI, Egawa S, Tatsumi T, Redlinger RE, Jr, Kalinski P, Kanto T. A novel bulk-culture method for generating mature dendritic cells from mouse bone marrow cells. J Immunol Methods. 2002;262:145–57. doi: 10.1016/s0022-1759(02)00013-3. [DOI] [PubMed] [Google Scholar]
- 24.Unkeless JC. Characterization of a monoclonal antibody directed against mouse macrophage and lymphocyte Fc receptors. J Exp Med. 1979;150:580–96. doi: 10.1084/jem.150.3.580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shortman K, Liu YJ. Mouse and human dendritic cell subtypes. Nat Rev Immunol. 2002;2:151–61. doi: 10.1038/nri746. [DOI] [PubMed] [Google Scholar]
- 26.Pulendran B, Smith JL, Caspary G, Brasel K, Pettit D, Maraskovsky E, Maliszewski CR. Distinct dendritic cell subsets differentially regulate the class of immune response in vivo. Proc Natl Acad Sci USA. 1999;96:1036–41. doi: 10.1073/pnas.96.3.1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abe M, Thomson AW. Influence of immunosuppressive drugs on dendritic cells. Transpl Immunol. 2003;11:357–65. doi: 10.1016/S0966-3274(03)00050-9. [DOI] [PubMed] [Google Scholar]
- 28.Poltorak A, He X, Smirnova I, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–8. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 29.Reis e Sousa C, Sher A, Kaye P. The role of dendritic cells in the induction and regulation of immunity to microbial infection. Curr Opin Immunol. 1999;11:392–9. doi: 10.1016/S0952-7915(99)80066-1. [DOI] [PubMed] [Google Scholar]
- 30.Sparwasser T, Koch ES, Vabulas RM, Heeg K, Lipford GB, Ellwart JW, Wagner H. Bacterial DNA and immunostimulatory CpG oligonucleotides trigger maturation and activation of murine dendritic cells. Eur J Immunol. 1998;28:2045–54. doi: 10.1002/(SICI)1521-4141(199806)28:06<2045::AID-IMMU2045>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 31.Moser M, De Smedt T, Sornasse T, et al. Glucocorticoids down-regulate dendritic cell function in vitro and in vivo. Eur J Immunol. 1995;25:2818–24. doi: 10.1002/eji.1830251016. [DOI] [PubMed] [Google Scholar]
- 32.Piemonti L, Monti P, Allavena P, Sironi M, Soldini L, Leone BE, Socci C, Di Carlo V. Glucocorticoids affect human dendritic cell differentiation and maturation. J Immunol. 1999;162:6473–81. [PubMed] [Google Scholar]
- 33.Matyszak MK, Citterio S, Rescigno M, Ricciardi-Castagnoli P. Differential effects of corticosteroids during different stages of dendritic cell maturation. Eur J Immunol. 2000;30:1233–42. doi: 10.1002/(SICI)1521-4141(200004)30:4<1233::AID-IMMU1233>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 34.Vizzardelli C, Pavelka N, Luchini A, et al. Effects of dexamethazone on LPS-induced activation and migration of mouse dendritic cells revealed by a genome-wide transcriptional analysis. Eur J Immunol. 2006;36:1504–15. doi: 10.1002/eji.200535488. [DOI] [PubMed] [Google Scholar]
- 35.Piemonti L, Monti P, Allavena P, Leone BE, Caputo A, Di Carlo V. Glucocorticoids increase the endocytic activity of human dendritic cells. Int Immunol. 1999;11:1519–26. doi: 10.1093/intimm/11.9.1519. [DOI] [PubMed] [Google Scholar]
- 36.Schuurhuis DH, Laban S, Toes RE, et al. Immature dendritic cells acquire CD8 (+) cytotoxic T lymphocyte priming capacity upon activation by T helper cell-independent or -dependent stimuli. J Exp Med. 2000;192:145–50. doi: 10.1084/jem.192.1.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trevejo JM, Marino MW, Philpott N, Josien R, Richards EC, Elkon KB, Falck-Pedersen E. TNF-alpha-dependent maturation of local dendritic cells is critical for activating the adaptive immune response to virus infection. Proc Natl Acad Sci USA. 2001;98:12162–7. doi: 10.1073/pnas.211423598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gimmi CD, Freeman GJ, Gribben JG, Gray G, Nadler LM. Human T-cell clonal anergy is induced by antigen presentation in the absence of B7 costimulation. Proc Natl Acad Sci USA. 1993;90:6586–90. doi: 10.1073/pnas.90.14.6586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pasare C, Medzhitov R. Toll-dependent control mechanisms of CD4 T cell activation. Immunity. 2004;21:733–41. doi: 10.1016/j.immuni.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 40.Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+ CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299:1033–6. doi: 10.1126/science.1078231. [DOI] [PubMed] [Google Scholar]
- 41.Langenkamp A, Messi M, Lanzavecchia A, Sallusto F. Kinetics of dendritic cell activation. impact on priming of TH1, TH2 and nonpolarized T cells. Nat Immunol. 2000;1:311–6. doi: 10.1038/79758. [DOI] [PubMed] [Google Scholar]
- 42.Iwakabe K, Shimada M, Ohta A, Yahata T, Ohmi Y, Habu S, Nishimura T. The restraint stress drives a shift in Th1/Th2 balance toward Th2-dominant immunity in mice. Immunol Lett. 1998;62:39–43. doi: 10.1016/s0165-2478(98)00021-2. [DOI] [PubMed] [Google Scholar]
- 43.Zhang D, Kishihara K, Wang B, Mizobe K, Kubo C, Nomoto K. Restraint stress-induced immunosuppression by inhibiting leukocyte migration and Th1 cytokine expression during the intraperitoneal infection of Listeria monocytogenes. J Neuroimmunol. 1998;92:139–51. doi: 10.1016/s0165-5728(98)00197-0. [DOI] [PubMed] [Google Scholar]
- 44.Pierre P, Turley SJ, Gatti E, et al. Developmental regulation of MHC class II transport in mouse dendritic cells. Nature. 1997;388:787–92. doi: 10.1038/42039. [DOI] [PubMed] [Google Scholar]
- 45.Kaisho T, Takeuchi O, Kawai T, Hoshino K, Akira S. Endotoxin-induced maturation of MyD88-deficient dendritic cells. J Immunol. 2001;166:5688–94. doi: 10.4049/jimmunol.166.9.5688. [DOI] [PubMed] [Google Scholar]