

Abstract
The methyl 6-hydroxyhexanoyl glycoside of lactose was treated with each of 1,2-diaminoethane or hydrazine hydrate, and the corresponding amino amide 4 and acyl hydrazide 13, were treated with each of squaric acid dimethyl, diethyl, dibutyl, and didecyl esters. The monoesters were conjugated to bovine serum albumin (BSA) at different concentrations of hapten using 0.05 and 0.5 M pH 9 borate buffer. Maximum loading was achieved faster, and the conjugation efficiency was higher, when the conjugation was conducted at higher concentrations of both hapten and buffer. Conjugations involving haptens 14–17 prepared from hydrazide 13 were generally slower and less efficient than those with compounds 5–8, which were made from amino amide 4. Maintaining pH 9 during conjugation was found to be the most important factor in ensuring that the conjugation was a fast, highly efficient and reproducible process. When the pH of the conjugation mixture fell during the reaction, resulting in decreased reaction rate or even termination of the conjugation process, the normal course of the conjugation process could be restored by addition of buffer salts. Hydrolysis studies with monoesters formed from amino amide 4 under conjugation conditions showed that the decyl ester 8 was the most stable and that the methyl compound 5 was the one most readily hydrolyzed. The stability of monoesters prepared form hydrazide 13 were similar and comparable to the decyl ester prepared from 4. No definite advantage was found for the use any of the four dialkyl squarate reagents (methyl-, ethyl-, butyl-, and decyl-) for conversion of carbohydrate derivatives to species amenable for conjugation. Nevertheless, dimethyl squarate seemed to be the most convenient reagent because it is a crystalline, easy to handle, and commercially available material with very good reactivity.
Keywords: Conjugate, Neoglycoconjugate, Squaric acid diester, 5-Methoxycarbonyl-β-lactoside, SELDI-TOF MS
1. Introduction
Synthetic oligosaccharides and their conjugates to other molecules are indispensable probes in the life sciences.1–3 One of the methods that allow conjugation by a single-point attachment4 to form well-defined neoglycoconjugates without cross-linking is based on squaric acid chemistry.5,6,7 In general, conjugations are often carried out with a large excess of synthetic oligosaccharides, to ensure reasonable reaction rates. Thus, there is a need to develop new, highly efficacious methods, or improve reaction efficiency of existing methods for conjugation, so that the labor-intensive, synthetic oligosaccharides can be used more efficiently. Earlier work involving squaric acid diesters was done with dimethyl8,9 and diethyl esters,5 and more recently Bergh et al.10 described the use of didecyl squarate. The popularity of this class of reagents in synthesizing glycoconjugates increased after Kamath et al.11 reported conjugation of oligosaccharides to protein on a very small scale using squaric acid amide ethyl esters, and showed that carbohydrate–protein ratio (loading) in glycoconjugates thus formed could be objectively established by MALDI-TOF mass spectrometry.
We have studied this conjugation method in detail,12 and have also developed a protocol that allows the one-pot preparation of a series of neoglycoconjugates with predetermined carbohydrate–protein ratios.13 An attractive feature of conjugation by squaric acid chemistry is that the excess hapten used at the onset of the conjugation that remained unchanged during the reaction can be recovered.10,14–16 Izumi et al.17 compared the efficiency of conjugation of the ethyl squarate derivative of mannose to BSA with that by six other, unrelated reagents and found the protocol involving squaric acid chemistry was one of the three most potent methods. The diethyl ester has been the most popular reagent, although squaric acid methyl15,18 and decyl10 diesters have also been used in squaric acid ester-mediated conjugations, and dibutyl squarate is commercially available. There is no obvious rational for this practice, as no systematic study has been carried out to establish advantage of that reagent to form neoglycoconjugates.
Based on previous observations,9,19 Kamath et al. suggested11 that it should be possible to prepare squaric acid derivatives of saccharides from hydrazides and, indeed, linker-equipped carbohydrates have been conjugated to proteins in this way.15,18 We have not found20 a significant difference in immunogenicity of conjugates from the hexasaccharide fragment of Vibrio cholerae O:1, serotype Ogawa that were made by squaric acid chemistry involving either 1,2-diaminoethane or hydrazine hydrate. This finding, showing that both types of squaric acid derivatives can be useful in conjugate vaccine development, creates a need to evaluate their conjugation properties in more detail.
Data on hydrolysis of some individual squaric acid diesters are available6,8 but a stability study with amide esters derived from carbohydrates under the conditions of conjugation to proteins has not been undertaken. Clearly, there is a need for systematic studies in these areas to establish general guidelines for efficient conjugation of carbohydrates to proteins by this method. The objective of this work was to evaluate the stability of amide monoesters made from amine amides (compounds 5–8) and hydrazides (compounds 14–17) under identical conditions and to determine the efficiency of conjugation to a model carrier (BSA) to give conjugates 9–12 and 18–21 (Scheme 1). Aside from obvious reasons, information about the rate of hydrolysis of the squaric acid monoesters at pH 9 is important, e.g., when poorly reactive carriers are being conjugated, as it can indicate the time at which point continuing conjugation becomes futile because virtually no active ester would be present in the reaction mixture.
Scheme 1.
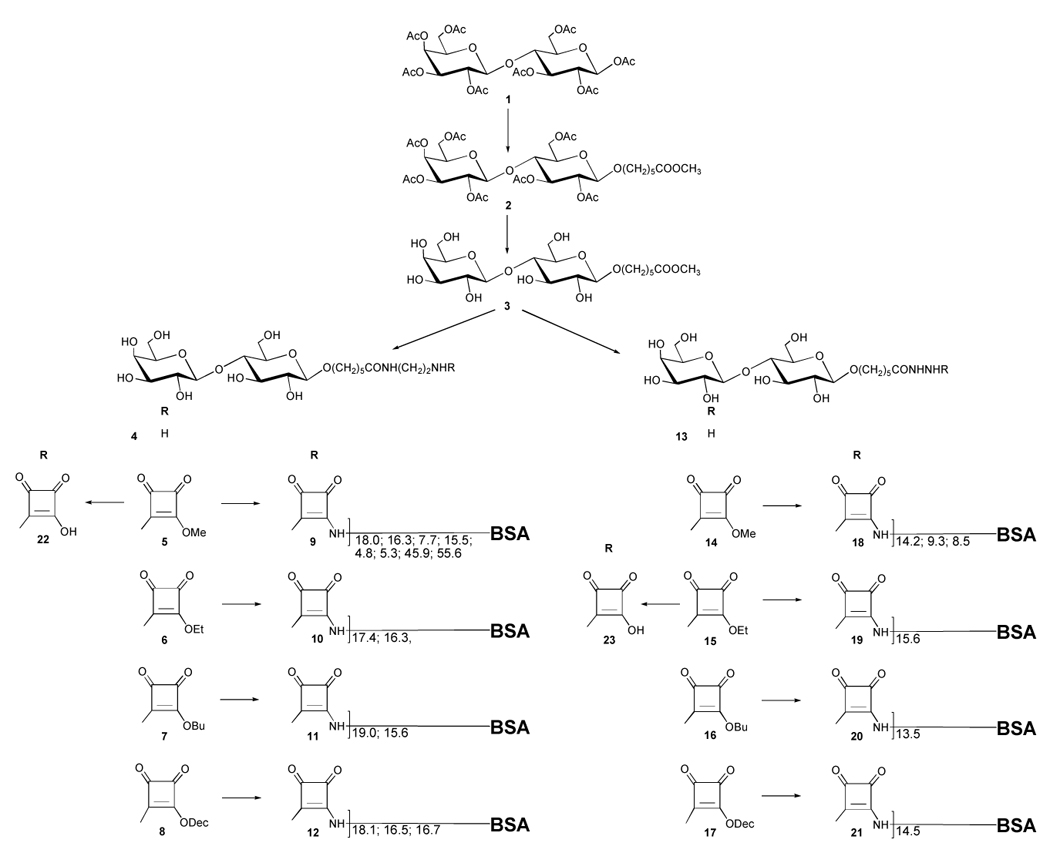
Numerous data on conjugation from different laboratories are available but not readily comparable because it appears that no two laboratories have conjugated saccharides to proteins under identical, or at least very similar, conditions (the same linker and carrier, concentration of reactants and buffers, and initial carbohydrate–protein ratio). Furthermore, the results of conjugations to BSA performed in different laboratories cannot be readily compared because the commercial source and history of isolation and purification of the carrier is often not disclosed and, as we show here, this can have a profound effect on the outcome of conjugation. A comparison of the outcome of earlier conjugations with more recent ones can be misleading because in the more distant past, carbohydrate–protein ratios in neoglycoconjugates were determined by various colorimetric methods and not by mass spectrometry. The reliability of calibration curves required by colorimetry is questionable when they are to be used to analyze mixtures of oligosaccharides that mimic the structure of rare or unstable sugar-containing bacterial polysaccharides with variable amounts of protein carriers. In addition to comparing rates and efficiency of conjugation of squaric acid derivatives made from the amino amide 4 and hydrazide 13, compounds 5–8 vs. 14–17, we have evaluated the importance of concentration and excess of hapten as well as of buffer concentration upon the conjugation reaction.
2. Results and discussion
β-Lactose octaacetate (1) was converted (Scheme 1), through 5-methoxycarbonylpentyl β-lactoside (3), to squaric acid monoesters 5–8 and 14–17 and conjugated to a model carrier bovine serum albumin (BSA). Compound 3 was prepared by boron trifluoride etherate-mediated glycosidation of 121,22 with methyl 6-hydroxyhexanoate (24),23 to give β-glycoside 2 in 60–70% yield. In addition, small amounts of the 2I-hydroxy derivative (27), as well as 6-O-acetyl derivative of 24, compound 25, were isolated as side products. Analogous compounds were isolated from stannic tetrachloride catalyzed glycosylation of 8-ethoxycarbonyloctanol with a number of oligosaccharides.24 Formation of such by products can be expected based on the mechanism25–28 of glycosylation with donors having a participating group at position 2 vicinal to the anomeric position involved in the glycosidation. Deacetylation (Zemplén) of 2 gave crystalline, linker-equipped lactoside 3, which, in turn, was converted to the corresponding amino amide 4 and hydrazide 13 by reaction with 1,2-diaminoethane and hydrazine hydrate, respectively. Each of the products 4 and 13 were then treated with dimethyl, diethyl, dibutyl, and didecyl10 squarates to give monoalkyl squarates (squarate monoesters) 5–8 and 14–17. The conversion of 3 with 1,2-diaminoethane was much slower than that with hydrazine hydrate. To be able to follow the hydrolysis of squaric acid monoesters confidently by HPLC, acids 22 and 23 were also prepared.
Conjugation of carbohydrates to proteins by squaric acid chemistry with different squaric acid reagents has been carried out at different, largely arbitrarily chosen conditions, namely different concentration of hapten and buffer and initial hapten–carrier ratio. We have observed some irregularities in the outcome of our previous conjugations, such as occasional poor reproducibility,29 faster conjugation of a higher compared to a lower oligosaccharide13,30 or very different reaction rates of conjugation of the same size of oligosaccharides.31 We could offer no other explanation for these irregularities except that, perhaps, conjugation was affected by factors of which we were unaware.
Our past conjugations were performed using 0.05 M borate buffer, and the same buffer was used for the first conjugation experiment in this series (See Section 3.8.1). We have previously shown12 that conjugation efficiency increased with hapten concentrations between 5 and 25 mM. We hypothesized that using still higher concentrations might further increase the rate and efficiency and, therefore, the first conjugation with methyl squarate 5 was performed at a hapten concentration of 40 mM in 0.05 M borate buffer. The reaction was carried out at a molar ratio 5/BSA = 20 and the progress of the conjugation was monitored by SELDI TOF MS. This showed that only a small fraction of the hapten used at the onset of the conjugation was attached to BSA (see Section 3.8.1), and that the reaction virtually stopped after four hours. A check with Hydrion MicroFine pH Paper showed the pH of the mixture to be 7.2 ± 0.2. Considering the previous16 results of stability, which we largely confirmed (see Fig. 3 and 4), and the loading achieved, most of the hapten used at the onset of the reaction must have been unchanged after 25 hours. Thus, the lowered pH of the mixture, which caused the termination of conjugation, could not be due to the small amount of acid that could have been formed as a result of ester hydrolysis. When a small amount of buffer salts was added, the pH raised to ~8.8 ± 0.2, and after additional 12 hours the carbohydrate–protein ratio was 18:1, as determined by SELDI TOF MS analysis. When the reaction was repeated using 0.5 M buffer, the latter loading was reached after 8 hours (Table 1). This confirmed the hypothesis formulated above, i.e., that increasing the concentration of the hapten increases the rate and efficiency of conjugations by squaric acid chemistry.
Fig. 3.

Hydrolysis of squaric acid monoesters 5–8 made from amino amide 4 and squaric acid monoesters 14–17 made from hydrazide 13 at pH 9 Buffer B.
Fig. 4.

Hydrolysis of squaric acid monoesters 5–8 made from amino amide 4 in Buffer C at hapten concentration 4 mM with that of 5 in Buffer B at hapten concentration 0.4 mM.
Table 1.
Conjugation of squaric acid monoesters 5–8 made from amino amide 4 and monoesters 14–17 made from hydrazide 13 to BSAa
| Hapten/Conjugateb | Concentration Hapten [mM]/Buffer [M] | Reaction Time [h] | Hapten/BSA [M/M (n)b] | Yieldc [%] | Conjugation Efficiency |
|---|---|---|---|---|---|
| 5/9 | 40/0.5 | 1 | 8.9 | ||
| 2 | 13.5 | ||||
| 3 | 15.4 | ||||
| 6 | 17.9 | ||||
| 8 | 18.0 | 91 | 90 | ||
| 5/9 | 4/0.5 | 1 | 4.0 | ||
| 3 | 8.6 | ||||
| 6 | 12.0 | ||||
| 8 | 13.6 | ||||
| 24 | 16.3 | 92 | 82 | ||
| 5/9 | 0.4/0.5 | 1 | 1.5 | ||
| 3 | 3.2 | ||||
| 8 | 3.8 | ||||
| 24 | 6.8 | ||||
| 48 | 7.7 | 89 | 39 | ||
| 5/9 | 4/0.05 | 1 | 3.0 | ||
| 3 | 6.4 | ||||
| 6 | 9.5 | ||||
| 7 | 10.1 | ||||
| 8 | 10.5 | ||||
| 27 | 14.3 | ||||
| 48 | 15.5 | 91 | 78 | ||
| 6/10 | 40/0.5 | 1 | 7.8 | ||
| 2 | 10.3 | ||||
| 3 | 12.3 | ||||
| 6 | 15.3 | ||||
| 7 | 15.7 | ||||
| 8 | 16.0 | ||||
| 24 | 17.4 | 86 | 87 | ||
| 6/10 | 4/0.5 | 1 | 2.4 | ||
| 3 | 4.7 | ||||
| 8 | 8.4 | ||||
| 24 | 13.2 | ||||
| 48 | 15.5 | ||||
| 72 | 16.3 | 92 | 82 | ||
| 7/11 | 40/0.5 | 1 | 7.4 | ||
| 2 | 10.2 | ||||
| 3 | 12.3 | ||||
| 7 | 15.2 | ||||
| 8 | 16.1 | ||||
| 24 | 19.0 | 88 | 95 | ||
| 7/11 | 4/0.5 | 1 | 2.4 | ||
| 3 | 4.9 | ||||
| 6 | 8.1 | ||||
| 8 | 9.2 | ||||
| 24 | 13.9 | ||||
| 48 | 15.6 | 93 | 78 | ||
| 8/12 | 40/0.5 | 1 | 8.8 | ||
| 2 | 10.5 | ||||
| 3 | 13.2 | ||||
| 6 | 14.4 | ||||
| 8 | 15.2 | ||||
| 24 | 17.6 | 66 | 91 | ||
| 8/12 | 4/0.5 | 1 | 9.7 | ||
| 2 | 12.3 | ||||
| 3 | 13.6 | ||||
| 6 | 15.5 | ||||
| 8 | 16.5 | ||||
| 24 | 16.5 | 93 | 83 | ||
| 8/12 | 0.4/0.5 | 1 | 8.5 | ||
| 2 | 11.0 | ||||
| 3 | 11.1 | ||||
| 6 | 14.1 | ||||
| 8 | 16.1 | ||||
| 24 | 16.7 | 91 | 84 | ||
| 14/18 | 40/0.5 | 1 | 3.1 | ||
| 3 | 5.7 | ||||
| 8 | 8.2 | ||||
| 24 | 12.5 | ||||
| 48 | 14.2 | 90 | 71 | ||
| 14/18 | 4/0.5 | 1 | 0.6 | ||
| 3 | 1.2 | ||||
| 6 | 1.6 | ||||
| 8 | 2.5 | ||||
| 24 | 4.7 | ||||
| 48 | 7.1 | ||||
| 96 | 8.5 | ||||
| 120 | 9.3 | 92 | 47 | ||
| 14/18 | 4/0.05 | 1 | 0.6 | ||
| 3 | 1.5 | ||||
| 8 | 2.8 | ||||
| 24 | 4.3 | ||||
| 48 | 5.6 | ||||
| 96 | 7.5 | ||||
| 120 | 7.4 | ||||
| 144 | 8.0 | ||||
| 168 | 8.5 | 92 | 43 | ||
| 15/19 | 40/0.5 | 1 | 0.6 | ||
| 2 | 2.0 | ||||
| 3 | 2.1 | ||||
| 6 | 3.9 | ||||
| 8 | 4.6 | ||||
| 24 | 9.2 | ||||
| 48 | 12.2 | ||||
| 72 | 13.9 | ||||
| 120 | 15.6 | 80 | 78 | ||
| 16/20 | 40/0.5 | 1 | 0.6 | ||
| 2 | 1.3 | ||||
| 3 | 2.1 | ||||
| 6 | 4.2 | ||||
| 8 | 5.4 | ||||
| 24 | 10.8 | ||||
| 72 | 13.5 | 85 | 68 | ||
| 17/21 | 40/0.5 | 1 | 2.9 | ||
| 2 | 4.3 | ||||
| 3 | 5.3 | ||||
| 7 | 7.3 | ||||
| 24 | 11.5 | ||||
| 48 | 13.9 | ||||
| 72 | 14.5 | 91 | 73 |
All conjugations were carried out at hapten–BSA ratio 20:1 and ambient temperature (22–24°C) at pH 9 (Buffer B) using BSA (Sigma Cat. Number A-0281) as carrier. The final time noted is the one when the hapten–carrier ratio leveled off; Progress of conjugation was monitored by SELDI-TOF MS.
See Scheme 1.
The amount of material used up for analysis during monitoring of the conjugation has been accounted for.
The cause of the observed lowering of the pH of the conjugation mixture was found when a sample of BSA used in the preliminary experiment (533 mg, Sigma Cat. No. A-4503, purified32) or another sample of commercially available BSA (Sigma Cat. Number A-0281) were dissolved in pH 9 buffer (0.05 M, 4 mL, to mimic solution concentration of the conjugation described above); the pH of the solutions were found to be 7.46 and 8.0, respectively. On the other hand, the pH of solutions of these proteins prepared in the same way in 0.5 M buffer was lowered from 9.0 by only ~0.2 pH units. It appears that when highly concentrated solutions of BSA are prepared in low concentration buffers, the pH of these solutions is lowered depending on the mode of preparation and purification of the protein. The same, with similar consequences concerning conjugation, may be the case with other carriers.
Unfortunately, it is not customary to report detailed information concerning the nature and purification of the BSA used, although this protein in its many, but seldom specified forms, is the most commonly used carrier in synthesizing experimental tools for the life sciences. The above results prompted us to ensure that the pH of reaction mixtures in further experiments persisted close to 9.0 during the whole duration of the conjugation. This was most conveniently realized by conducting the conjugation in more concentrated, higher capacity, 0.5 M buffer. The use of still more concentrated buffer (1 M; this experiment is not described in the Experimental) did not provide additional benefit for the outcome of conjugation. Concentration of the buffer and its capacity, an important variable in application of squaric acid chemistry, has not been duly emphasized, and information that would make it possible to identify technical specifications of buffers is often not available from the description of conjugation experiments. On the other hand, the improved efficiency of the conjugations carried out in buffers slightly more basic than pH 9 has been noted,14,33,34 despite pH 9.0 being generally considered optimal for squaric acid chemistry conjugations.5,11 Because the lowered pH of the reaction media following solubilization of carriers, or other concrete reasons for the use of more basic buffers, had not been mentioned in that context, the more basic media used for successful conjugation in those situations must have serendipitously compensated for the acidity of the carrier, which then resulted in conjugation proceeding normally.
Based on the observations described above, a series of conjugations of squarates 5–8 and 14–17 to BSA were carried out at a hapten concentration 40 mM, buffer concentration 0.5 and 0.05 M, and an initial carbohydrate protein ratio of 20:1 (Table 1–3, Fig. 1–3). Because conjugations have also been carried out successfully at much lower concentrations of hapten,7,10,17,35,36 we also performed some conjugations at hapten concentrations of 4 and 0.4 mM (Table 1, Fig. 2). We ensured that the pH of the reaction medium in all conjugations persisted between 8.5–9.0. The objective was to compare conjugations using different types of alkyl squarate (made from amino amide or hydrazide) as the reactive species, and to examine the effect of buffer and hapten concentration on the rate and efficiency of conjugation. Aided by the results of the study, we aimed to identify structural features in tethers that make conjugation more efficient. When applied in future conjugations of different haptens and carriers, the best conditions found here with BSA and haptens derived from lactose may have to be adjusted, to account for reactivity of other reagents and the desired hapten–carrier ratios.
Table 3.
Conjugations of the di- (5), tri- (29), tetra- (30) and hexasaccharide (31) to BSA at different initial hapten–BSA ratiosa
| Hapten/Conjugate | Initial hapten/BSA | Reaction Time [h] | Hapten/BSA in product | Yield [%] | Conjugation Efficiency [%] |
|---|---|---|---|---|---|
| 5/9 | 6:1 | 2 | 5.3 | 88 | 88 |
| 5/9 | 6:1b | 4 | 4.8 | 93 | 80 |
| 5/9 | 60:1 | 72 | 45.9 | 88 | 76.5 |
| 5/9 | 120:1 | 72 | 55.6 | 91 | 46 |
| 29/32 | 6:1 | 8 | 5.3 | 80 | 88 |
| 30/33 | 6:1 | 8 | 4.9 | 85 | 81.6 |
| 31/34 | 7:1 | 5 | 5.4 | 91 | 77 |
| 31/34 | 15:1 | 2.5 | 5.4 | 88 | Irrelevantc |
| 31/34 | 15:1 | 48 | 12 | NDc | 80 |
Conjugation carried out at 22–24°C and concentration of hapten 40 mM in 0.5 M buffer
Hapten concentration, 4 mM
Two conjugates prepared in one pot (see text); ND: Not determined.
Fig. 1.

Conjugation of squaric acid monoesters 5–8 made from amino amide 4 and squaric acid monoesters 14–17 made from hydrazide 13 and BSA. All conjugations were done in Buffer B at hapten concentration 40 mM.
Fig. 2.

Conjugation of squaric acid monoesters 5 and 8 made from amino amide 4 and BSA in Buffer B at different concentration of hapten.
At a hapten concentration 40 mM (Table 1, Fig. 1), conjugations involving haptens 5–8 made from amino amide 4 were faster and afforded conjugates with higher final loading than those with haptens 14–17 made from hydrazide 13. That squarates prepared from the hydrazide react with lesser efficiency manifests itself most clearly at low concentrations of hapten in a low concentration buffer (Table 1). At 40 mM concentration of hapten, the four squarates within the two groups (5–8 and 14–17) reacted at a comparable rate and, when the reaction within the two groups was complete, afforded two groups of products (9–12 and 18–21) with similar hapten–BSA ratios (Table 1, Fig. 1). Conjugation at three concentrations of hapten (40, 4 and 0.4 mM) was compared with the methyl (5) and the decyl (8) esters. The reaction rate and the final loading was much more concentration dependent with the methyl derivative 5 than with its decyl counterpart 8 (Table 1, Fig. 2), which showed practically no dependence on concentration.
We have previously12 investigated the effect of concentration of hapten (c = 5–25 mM) upon conjugation efficiency with an ethyl squarate derivative. When 0.05 M borate buffer was used, the conjugation was substantially more efficient when carried out at c = 15 mM (66%) than at c = 5 mM (45.6%). A further increase in hapten concentration was much less beneficial (67.6% at 20 mM), and the conjugation efficiency actually decreased to 63.4% at a concentration of 25 mM. We did not ascribe any significance to this moderate decrease in efficiency at the time those studies were carried out. However, in view of the now observed lowering of the pH of the reaction medium upon solubilization of BSA in 0.05 M buffer (see above), it is reasonable to conclude, in retrospect, that in that past conjugation at 25 mM concentration, which was also conducted in a 0.05 M buffer, the buffer may have just started to break at that hapten concentration. At those conditions, i.e., when only a relatively small volume of buffer was used (because the concentration of the hapten was relatively high), the buffer capacity was low because only small amount of buffer salts was present, and the rate and efficiency of conjugation decreased when the reaction medium became less basic. Support for this conclusion was found when the conjugation of 5 was conducted in the same buffer but at a lower concentration of hapten (4 mM). Here, a proportionally larger volume of buffer was used, which contained a larger total amount of salts – in fact the same amount of salts as when the conjugation is done in 0.5 M buffer at a hapten concentration 40 mM – and the buffer had, therefore, a larger capacity. The conjugation was somewhat slower, as could be expected due to the lower concentration of hapten (Table 1), but the product obtained was very similar to the one formed in 0.5 M buffer (Table 1). Monitoring the pH of the reaction mixtures showed that the pH persisted during both conjugations.
The results of conjugations of methyl squarates 5 and 14 at different concentrations of buffer (0.5 and 0.05 M, i.e., the buffer capacity in both cases was sufficient to keep the pH close to 9) allowed us to conclude (Table 1) that the concentration of buffer is another variable that affects the conjugation process. This finding widens choices of conditions suitable for a specific conjugation. There are situations when it may be beneficial to conduct conjugations at a lower concentration of haptens or buffers, or both, or not with the most powerful reagent. Such is the case, for example, when a series of conjugates are being prepared in one pot,13 and some conjugates in the series are targeted to have low loading, because a slower reaction may be easier to terminate when the desired, low carbohydrate–protein ratio has been reached.
The high reaction rate and efficiency of conjugation observed when using 0.5 M pH 9 buffer at an initial hapten–BSA ratio 20:1 prompted us to examine, with different oligosaccharides, the minimum excess of hapten necessary to attach ~5 hapten residues per carrier. That loading was targeted because a conjugate from hexasaccharide 3120 (Scheme 2) and BSA with loading ~4.5 was previously37 found to be a more potent immunogen for anti Vibrio cholerae O:1, serotype Ogawa antibodies than similar conjugates containing ~10 or ~15 hapten residues/BSA. In addition to hapten 5, conjugation of the tri- (29),13 tetra- (30)13 and hexasaccharide (31) fragments of the O-PS of Vibrio cholerae O:1, serotype Ogawa was also examined. The original preparations13 of conjugates from BSA and the Ogawa tri- and the tetrasaccharides were aimed at preparations of three conjugates in one pot, with loadings of 5, 10, and 15, hence the high initial hapten–BSA ratio of 75:1. Under those conditions (0.05 M pH 9 buffer, 15 mM concentration of hapten), the first desired loading was reached in both cases after 3 hours. As we show here, when the 0.5 M buffer was used at the hapten concentration of 40 mM, a conjugate with loading ~5 could be obtained within an acceptable time of 8 hours (Table 3), while only slight, 20% excess of hapten was used (initial hapten–BSA ratio, 6:1).
Scheme 2.

Because conjugation of larger oligosaccharides is a slower process compared to that of smaller molecules, the bulkier hexasaccharide 31 was conjugated in 0.5 M buffer at the initial hapten–BSA ratio of 7:1. The desired conjugate with ~5 haptens attached to BSA was obtained after 5 hours (Table 3), when the reaction was virtually complete. For comparison, the conjugation was also carried out in 0.5 M buffer, at the hapten–BSA ratio of 15:1 as in the past.20 At that time, the conjugation was carried out in 0.05 M buffer at a hapten concentration of 20 mM, and 5 hapten residues were attached after 24 hours. The increasing molecular mass of the conjugate leveled off after 14 days with 11 moles of hapten/BSA. In the present experiment, 5 hapten residues/BSA were attached after 2.5 hours, and the increasing hapten/BSA ratio leveled off after 2 days, when the conjugate formed showed hapten/BSA ratio of 12.
The efficiency of the new protocol (Section 3.8.2) was further tested and verified when hapten 5 was conjugated to BSA under conditions when the amount of hapten used was not the limiting factor. There are 59 lysine residues38 in BSA, each bearing a terminal amino group but not all are equally accessible to chemical transformations. If all engaged in the formation of a conjugate, the primary amino groups would be transformed into the same number of secondary amino groups, which could further react.6 Thus, it should be theoretically possible to attach many more than 59 hapten residues/BSA. Conjugations with the disaccharide 5 were carried out at the initial hapten–BSA ratio 60:1 and 120:1. The products formed after 3 days, when the loading no longer increased, contained (Table 3) on average, ~46 and ~56 hapten residues/BSA, respectively. Experiments aimed at determining if any tertiary amines had been formed are in progress. Elsewhere,18 human serum albumin (HSA, 58 lysine residues) was treated with a methyl squarate monoester prepared from d-glucose, at the initial hapten–HSA ratio of 197:1. The hapten was equipped with the same linker as the one present in 5, and 32 sugar residues were attached after 7 days (conjugation efficiency 17%). More recently, Izumi17 conjugated to BSA ethyl squarate derivative of d-mannose equipped with a different spacer at a hapten–BSA ratio of 60 and 120, and obtained conjugates with ~25 and ~39 hapten residues, respectively, after 5 days (conjugation efficiency, 41.6 and 32.5 %).
The results of the above experiments showed that conjugates could be prepared very efficiently by applying the new protocol (Section 3.8.2), with an excess of labor-intensive synthetic oligosaccharides as low as that used in organic synthesis of small molecular mass substances (~20%). Thus, recovery of the unchanged hapten from the conjugation reaction15,16 becomes virtually a non-issue.
To refine and expand on our previous fragmentary data29 regarding the stability of squarate monoesters obtained by TLC analysis, the hydrolysis of 5–8 and 14–17 under the conjugation conditions but in the absence of BSA was monitored by HPLC. In Buffer B (Section 3.1) at a 40 mM hapten concentration, alkyl squarates prepared from hydrazide 13 (squarates 14–17) were more stable than compounds prepared from amino amide 5, except for the decyl compound 8, whose stability was similar to those of 14–17. The hydrolysis of 5–8 followed a similar pattern in Buffer C at 4 mM hapten concentration. Because of their poor reactivity at low concentrations of hapten, the hydrolysis of squarates made from hydrazide 13, was not examined with the latter conditions. The relative propensity of monoalkyl squarates to hydrolysis is shown in Fig. 3 and 4. The relatively high rate of hydrolysis of squarate 5 is particularly noteworthy, and it explains the low efficiency of conjugation at these conditions (Table 1). Nevertheless, because at the conditions of the new protocol almost all hapten is conjugated within 24 hours, the commercially available, crystalline squaric acid dimethyl ester is a very useful reagent to transform carbohydrates into derivatives amenable to conjugation.
3. Experimental
3.1 General Methods
Unless stated otherwise, optical rotations were measured at ambient temperature with a Perkin-Elmer automatic polarimeter, Model 341. All reactions were monitored by thin-layer chromatography (TLC) on Silica gel 60 coated glass slides. Column chromatography was performed by elution from columns of silica gel with the CombiFlash Companion Chromatograph (Isco, Inc.). Solvent mixtures for TLC were more polar than those used for preparative separation. Nuclear Magnetic Resonance (NMR) spectra were measured at 300 MHz (¹H) and 75 MHz (13C) with a Varian Gemini or Varian Mercury spectrometer, or at 600 MHz (¹H) and 150 MHz (13C) with a Bruker Avance 600 spectrometer. Assignments of NMR signals were made by homonuclear and heteronuclear 2-dimensional correlation spectroscopy, run with the software supplied with the spectrometers. When reporting assignment of NMR signals, nuclei associated with the spacer (linker) are denoted with a prime. Sugar residues in oligosaccharides are serially numbered, beginning with the one bearing the aglycone, and are identified by a Roman numeral superscript in listings of signal assignments. Liquid Chromatography–Electron Spray-Ionization Mass Spectrometry (ESI-MS) was performed with a Hewlett-Packard 1100 MSD spectrometer. Attempts have been made to obtain correct combustion analysis data for all new compounds. However, some compounds tenaciously retained traces of solvents, despite exhaustive drying, and analytical figures for carbon could not be obtained within ± 0.4%. Structures of these compounds follow unequivocally from the mode of synthesis, NMR data and m/z values found in their mass spectra, and their purity was verified by TLC, HPLC and NMR spectroscopy. HPLC was performed with Agilent 1100 Series Chromatography System, using a 150 × 4.5 mm column with Ultra IBD RP packing, particle size, 5 µm (Restek Corporation, Bellefonte, PA 16823). A mobile phase of 30% MeOH–70% 10 mM monobasic potassium phosphate (1.0 mL/min) was used for quantitative analysis of methyl and butyl squarates 5, 7, 14 and 16. For ethyl and decyl squarates (6, 8, 15 and 17), 10% MeCN–90% 10 mM monobasic potassium phosphate (1.0 mL/min) was used as the mobile phase. Compounds were detected at 280 nm. BSA was purchased from Sigma Chemical Company: A. Cat. Number A-0281; B. Fraction V, Sigma Cat. No. A-4503, from which fatty acids were removed by charcoal treatment.32 Buffers used were as follows. A, BuffAR pH 7.0 Reference solution (Mallincrodt, Cat. No. 0031-04), concentrated to 1/5 volume; B, 0.5 M Borate buffer pH 9, made in house (1 L) from boric acid (30.9 g), KCl (26.1 g) and KOH (8.42 g), and final adjustment to pH 9.0 by addition of solid KOH; C, 0.05 M pH 9.0 buffer (RICCA chemical company, Cat. No. 1590-16; information concerning the concentration of this buffer was provided by the company’s technical assistance service). Buffer salts were obtained by freeze-drying Buffer B. It was experimentally determined that 27 mL of Buffer A had to be added to 2.5 mL Buffer B to bring pH to 7. Carbohydrate–protein conjugates were rid of low-molecular mass materials by filtration through either Amicon Ultra-15 Centrifugal Filter (Milipore Corporation) or similar Vivaspin 15 R (Sartorius Group) devices.31 Measurement of pH was done with Corning pH Meter, Model 440 and a semi-micro refillable glass electrode. For quick check of pH of reaction mixtures run in a very small volume (75–150 µL), samples (~0.5–1 µL) were withdrawn with a glass capillary and applied onto small strips of Hydrion MicroFine pH Paper (Micro Essential Laboratory, Inc., Cat. No. MF-1616 and MF-1608), whose accuracy is described as ±0.2–0.3 pH units. Dimethyl, diethyl and dibutyl squarates were purchased from Aldrich Chemical Company. Didecyl squarate was prepared as described.10 Aqueous solutions were made using HPLC grade water. Solutions in organic solvents were dried with anhydrous Na2SO4, and concentrated at 40°C/2 kPa.
3.2 2,3,4,6-Tetra-O-acetyl-β-d-gactopyranosyl-(1→4)-1,2,3,6-tetra-O-acetyl-β-d-glucopyranose (1)
This compound was prepared as described.21,22 Traces of the α anomer, which was impractical to remove by multiple recrystallization, were removed by chromatography on silica gel, mp 91–95 °C (CH2Cl2–MeOH), [α]d −4.3 (c 6, CHCl3). The compound is polymorphous. Lit.21 m.p. 140–143 °C, [α]d −4.0 (c 3.6, CDCl3), lit.22 mp 90 °C, [α]d −4.7 (c 10.3, CHCl3). ¹H NMR data agreed with those reported.21 13C NMR (CDCl3, 150 MHz): δ 100.9 (C-1II), 91.5 (C-1I), 75.6 (C-4I), 73.5 (C-5I), 72.6 (C-3I), 70.9 (C-3II), 70.7 (C-5II), 70.5 (C-2I), 69.0 (C-2II), 66.6 (C-4II), 61.7 (C-6I), 60.8 (C-6II).
3.3 5-Methoxycarbonylpentyl 2,3,4,6-tetra-O-acetyl-β-d-galactopyranosyl-(1→4)-2,3,6-tri-O-acetyl-β-d-glucopyranoside (2)
Methyl 6-hydroxyhexanoate23 (745 mg, 5.10 mmol) and BF3•Et2O (0.5 mL, 2.08 mmol) was added to a solution of 1 (1.38 g, 2.04 mmol) in CH2Cl2 (10 mL) and the mixture was stirred at room temperature overnight, when TLC showed that the reaction was virtually complete. After neutralization with Et3N (2 mL) and concentration, chromatography (4:1 hexane–acetone) gave first methyl 6-O-acetylhexanoate 25 (0.12 g, 13%). ¹H NMR (CDCl3): δ 4.06 (t, 2 H, J = 7.0 Hz, H-1), 3.67 (s, 3 H, OCH3), 2.32 (t, 2 H, J = 7.4 Hz, H-5), 1.68–1.62 (m, 4 H, H-2 and H-4), 1.39 (m, 2H, H-3); 13C NMR (CDCl3): δ 173.7 (CO), 170.9 (CO), 64.0 (C-1), 51.3 (OCH3), 33.7(C-5), 28.1(C-2), 25.3 (C-3), 24.3 (C-4), 20.8 (CH3CO); TOF-MS: 189 [M + H]+, 129 [M−HOAc]+.
Eluted next was the title compound 2 (970 mg, 62%), [α]d −12.7 (c 1.1, CHCl3). ¹H NMR (CDCl3): δ 5.34 (dd, 1 H, J3,4 = 3.5 Hz, J4,5 = 1.1 Hz, H-4II), 5.19 (t, 1 H, J = 9.3 Hz, H-3I), 5.10 (dd, 1 H, J1,2 = 8.0Hz, J2,3 = 10.4 Hz, H-2II), 4.96 (dd, 1 H, J3,4 = 3.5 Hz, H-3II), 4.87 (dd, 1 H, J1,2 = 8.0 Hz, J2,3 = 9.6 Hz, H-2I), 4.50 (d, 1 H, J1,2 = 7.9 Hz, H-1I), 4.47 (dd, 1 H, J5,6 = 2.1 Hz, J6a,6b = 12.0 Hz, H-6aI), 4.45 (d, 1 H, J1,2 = 8.0 Hz, H-1II), 4.14–4.07 (m, 3 H, H-6bI, H-6a,bII), 3.88 (m, 1 H, H-5II), 3.83 (m, 1 H, H-1′a), 3.79 (t, 1 H, J = 9.3 Hz, H-4I), 3.66 (s, 3 H, OCH3), 3.60 (m, 1 H, H-5I), 3.45 (m, 1 H, H-1′b), 2.29 (t, J = 7.6 Hz, 2 H, H-5′), 2.17, 2.15, 2.12, 2.06, 2.04, 2.03, 1.96 (7 s, 21 H, 7 COCH3), 1.64–1.54 (m, 4 H, H-2′ and H-4′), 1.35 (m, 2 H, H-3′); 13C NMR (CDCl3): δ 173.8, 170.2, 170.1, 169.9, 169.8, 169.6, 169.4, 168.9, 100.9 (C-1II), 100.4 (C-1I), 76.1 (C-4I), 72.6 (C-3I), 72.4 (C-5I), 71.5 (C-2I), 70.8 (C-3II), 70.5 (C-5II), 69.6 (C-1′), 68.9 (C-2II), 66.4 (C-4II), 61.9 (C-6I), 60.6 (C-6II), 51.3 (OCH3), 33.7 (C-5′), 28.9 (C-2′), 25.2 (C-3′), 24.4 (C-4′), 20.7, 20.6, 20.5, 20.4 (3 C), 20.3; TOF-MS: [M + Na]+ calcd for C33H48O20Na, 787.2637, found, 787.2640. Anal. Calcd for C33H48O20: C, 51.83; H, 6.33. Found C, 52.08; H, 6.38.
Continued elution gave the amorphous 5-methoxycarbonylpentyl 2,3,4,6-tetra-O-acetyl-β-d-galactopyranosyl-(1→4)-3,6-di-O-acetyl-α-d-glucopyranoside (27, 170 mg, 12%): ¹H NMR (CDCl3): δ 5.35 (dd, 1 H, J3,4 = 3.8 Hz, J4,5 = 1.0 Hz, H-4II), 5.21 (t, 1 H, J = 9.5 Hz, H-3I), 5.12 (dd, 1 H, J1,2 = 8.0 Hz, J2,3 = 10.5 Hz, H-2II), 4.96 (dd, 1 H, J3,4 = 3.4 Hz, J2,3 = 10.4 Hz, H-3II), 4.83 (d, 1 H, J1,2 = 3.9 Hz, H-1I), 4.50 (d, 1 H, J1,2 = 7.9 Hz, H-1II), 4.42–4.39 (dd, 1 H, J5,6 = 2.1 Hz, J6a,6b = 11.9 Hz, H-6aII), 4.19–4.16 (dd, 1 H, J5,6 = 6.0 Hz, J6a,6b = 11.2 Hz, H-6aI), 4.14–4.11 (dd, 1 H, J5,6 = 5.0 Hz, J6a,6b = 12.0 Hz, H-6bII), 4.09–4.06 (dd, 1 H, J5,6 = 7.7Hz, J6a,6b = 11.1Hz, H-6bI), 3.89–3.85 (m, 2 H, H-5I and H-5II), 3.74–3.70 (m, 1 H, H-1′a), 3.67 (s, 3 H, OCH3), 3.66 (t, 1 H, J = 10.4Hz, H-4I), 3.56–3.52 (m, 1 H, H-2I, shifts after acetylation to δ 4.78), 3.48–3.44 (m, 1 H, H-1′b), 2.35 (t, 2 H, H-5′), 2.16 (s, 3 H, COCH3), 2.12 (2 s, 6 H, 2 COCH3), 2.06 (s, 6 H, 2 COCH3), 1.96 (s, 3 H, COCH3), 1.69–1.61 (m, 4 H, H-2′ and H-4′), 1.40–1.36 (m, 2 H, H-3′); 13C NMR (CDCl3): δ 173.9, 170.6, 170.3, 170.2, 170.1, 170.0, 169.0, 101.7 (C-1II), 98.0 (C-1I), 76.2 (C-4I), 73.4 (C-3I), 71.2 (C-2I), 71.0 (C-3II), 70.4 (C-5I), 69.1 (C-2II), 68.3 (2 C, overlap, C-5II and C-1′), 66.5 (C-4II), 62.1 (C-6II), 60.7 (C-6I), 51.4 (OCH3), 33.7 (C-5′), 28.8 (C-2′), 25.4 (C-3′), 24.4 (C-4′), 21.0, 20.7, 20.5, 20.4; TOF-MS: 740.3 [M + NH4]+, 745.2 [M + Na]+, 761.2 [M + K]+.
3.4 5-Methoxycarbonylpentyl β-d-galactopyranosyl-(1→4)-α-d-glucopyranoside (28) and 5-methoxycarbonylpentyl 2,3,4,6-tetra-O-acetyl-β-d-galactopyranosyl-(1→4)-α-d-2,3,6-tri-O-acetyl-α-d-glucopyranoside (26)
For characterization, a small amount of compound 27 was treated with NaOMe in MeOH to give 28, mp 149–150 °C (from MeOH); [α]d +87.6 (c 0.9, H2O); ¹H NMR (D2O): δ 4.90 (d, 1 H, J1,2 = 3.9 Hz, H-1I), 4.34 (d, 1 H, J1,2 = 7.8 Hz, H-1II), 3.92 (dd, 1 H, J3,4 = 3.4 Hz, J4,5 = 0.8 Hz, H-4II), 3.90 (dd, 1 H, J5,6 = 2.4 Hz, J6a,6b = 12.2 Hz, H-6aI), 3.84 (dd, 1 H, J5,6 = 4.6 Hz, J6a,6b = 12.2 Hz, H-6bI), 3.81 (dd, 1 H, J2,3 = 9.8 Hz, J3,4 = 8.8 Hz, H-3I), 3.80–3.75 (m, 3 H, H-5I, H-6IIa,b), 3.74–3.71 (m, 2 H, H-5I, H-1′a), 3.66 (dd, 1 H, J2,3 = 10.0 Hz, J3,4 = 3.4 Hz, H-3II), 3.64 (dd, 1 H, J3,4 = 8.8 Hz, J4,5 = 10.0 Hz, H-4I), 3.59 (dd, 1 H, J1,2 = 3.9 Hz, J2,3 = 9.8 Hz, H-2I), 3.54 (dd, 1 H, J1,2 = 7.8 Hz, J2,3 = 10.0 Hz, H-2II), 3.52 (m, 1 H, H-1′b), 2.41 (t, 2 H, J = 7.3 Hz, H-5′), 1.66–1.61 (m, 4 H, H-2′ and H-4′), 1.39 (m, 2 H, H-3′); 13C NMR (D2O): δ 180.4 (CO), 105.6 (C-1II), 100.6 (C-1I), 81.2 (C-4I), 78.1 (C-5II), 75.3 (C-3II), 74.5 (C-3I), 73.8 (2 C, C-2I and C-2II), 73.4 (C-5I), 71.3 (C-4II), 70.9 (C-1′), 63.8 (C-6II), 62.7 (C-6I), 54.9 (OCH3), 36.4 (C-5′), 31.0 (C-2′), 27.7 (C-3′), 26.9 (C-4′); TOF-MS: [M + Na]+ calcd for C19H34O13Na, 493.1897; found, 493.1901. Anal. Calcd for C19H34O13: C, 48.51; H, 7.28. Found: C, 48.46; H, 7.24.
A small amount of 28 was treated with acetic anhydride in pyridine to give amorphous 26, [α]d +7.3 (c 1.7, CHCl3); ¹H NMR (CDCl3): δ 5.46 (dd, 1 H, J2,3 = 10.1 Hz, J3,4 = 9.3 Hz, H-3I), 5.34 (dd, 1 H, J3,4 = 3.5Hz, J4,5 = 1.0Hz, H-4II), 5.11 (dd, 1 H, J1,2 = 7.9 Hz, J2,3 = 10.4 Hz, H-2II), 4.97–4.94 (m, 2 H, H-1I and H-3II), 4.78 (dd, 1 H, J1,2 = 3.7 Hz, J2,3 = 10.3 Hz, H-2I), 4.49 (d, 1 H, J1,2 = 8.0 Hz, H-1II), 4.45–4.42 (dd, J5,6 = 2.1 Hz, J6a,6b = 12.0 Hz, H-6aI), 4.16–4.12 (m, 2 H, H-6bI and H-6aII), 4.08 (dd, 1H J = 7.5 Hz, J = 11.1 Hz, H-6bII), 3.92–3.90 (m, 1 H, H-5I), 3.88–3.86 (m, 1 H, H-5II), 3.72 (t, J = 9.0 Hz, 1 H, H-4I), 3.68–3.64 (m, 4 H, OCH3 and H-1′a), 3.40–3.37 (m, 1 H, H-1′b), 2.32 (t, 2 H, J = 7.2 Hz, H-5′), 2.15 (s, 3 H, COCH3), 2.12 (s, 3 H, COCH3), 2.06 (s, 3 H, COCH3), 2.05 (2 s, 6 H, 2 COCH3), 2.04 (s, 3 H, COCH3), 1.96 (s, 3 H, COCH3), 1.67–1.59 (m, 4 H, H-2′ and H-4′), 1.40–1.36 (m, 2 H, H-3′); 13C NMR (CDCl3): δ 173.9, 170.4, 170.3, 170.2, 170.1, 170.0, 169.4, 169.0, 101.1 (C-1II), 95.5 (C-1I), 76.6 (C-4I), 71.1 (C-2I), 71.0 (C-3II), 70.5 (C-5II), 69.9 (C-3I), 69.1 (C-2II), 68.2 (C-1′), 68.0 (C-5I), 66.5 (C-4II), 61.9 (C-6I), 60.7 (C-6II), 51.4 (OCH3), 33.8 (C-5′), 28.8 (C-2′), 25.4 (C-3′), 24.5 (C-4′), 20.8 (2 C), 20.6, 20.5 (2 C), 20.4; TOF-MS: [M + NH4]+ calcd for C33H52NO20, 782.3083; found, 782.3072. Anal. Calcd for C33H48O20: C, 51.83; H, 6.33. Found: C, 52.10; H, 6.54.
3.5 5-Methoxycarbonylpentyl β-d-galactopyranosyl-(1→4)-β-d-glucopyranoside (3)
A few drops of NaOMe in MeOH (1 M) was added to a solution of 2 (16.59 g, 21.7 mmol) in a mixture of MeOH (300 mL) and CH2Cl2 (50 mL) until strong alkalinity and the mixture was stirred at room temperature for 3 h, when TLC showed that the reaction was complete. After neutralization with Amberite IR-120 (H+) resin, filteration and concentration, the residue was chromatographed (4:1:0.1 CH2Cl2–MeOH–H2O) to give 3 (8.74 g, 86%) as a hemihydrate, mp 159–160 °C (from MeOH); [α]d −3.5 (c 1, H2O); ¹H NMR (D2O): δ 4.46 (d, 1 H, J1,2 = 8.0 Hz, H-1I), 4.43 (d, 1 H, J1,2 = 7.8 Hz, H-1II), 3.95 (dd, 1 H, J5,6 = 2.2 Hz, J6a,6b = 12.3 Hz, H-6aI), 3.91–3.89 (m, 2 H, H-1a′ and H-4II), 3.80–3.72 (m, 3 H, H-6bI, H-6aII, H-6bII), 3.71 (m, 1 H, H-5II), 3.67 (s, 3 H, OCH3), 3.66–3.61 (m, 4 H, H-1b′, H-3I, H-4I, H-3II), 3.56 (m, 1 H, H-5I), 3.52 (dd, 1 H, J1,2 = 7.8 Hz, J2,3 = 9.9 Hz, H-2II), 3.28 (m, 1 H, H-2I), 2,39 (t, 2 H, J = 7.4 Hz, H-5′), 1.64–1.59 (m, 4 H, H-2′ and H-4′), 1.37 (m, 2 H, H-3′); 13C NMR (D2O): δ 180.4 (CO), 105.7 (C-1II), 104.8 (C-1I), 81.2 (C-4I), 78.1 (C-5II), 77.5 (C-5I), 77.2 (C-3I), 75.6 (C-2I), 75.3 (C-3II), 73.7 (C-2II), 73.1 (C-1′), 71.3 (C-4II), 63.8 (C-6II), 62.9 (C-6I), 54.8 (OCH3), 36.3 (C-5′), 31.1 (C-2′), 27.4 (C-4′), 26.8 (C-3′); TOF-MS: [M + H]+ calcd for C19H35O13, 471.208; found, 471.207. Anal. Calcd for C19H34O13•0.5H2O: C, 47.59; H, 7.36. Found: C, 47.60; H, 7.39.
3.6 (2-Aminoethylamido)carbonylpentyl β-d-galactopyranosyl-(1→4)-β-d-glucopyranoside (4)
A solution of compound 3 (8.08 g, 17.2 mmol) in ethylenediamine (35 mL, 518 mmol) was stirred at 50 °C with the exclusion of atmospheric moisture and CO2. The mixture, which shortly became pale yellow, was stirred at the same temperature for 24 h and concentrated with co-evaporation of water, to remove ethylenediamine. The residue was chromatographed (6:2:1 MeOH–DCM–NH4OH), to give hemihydrate of 4 (7.11 g, 83.5%), mp 168–170 °C (from MeOH); [α]d −2.6 (c 1.1, H2O); ¹H NMR (D2O): δ 4.46 (d, 1 H, J1,2 7.9 Hz, H-1I), 4.43 (d, 1 H, J1,2 = 7.8 Hz, H-1II), 3.96 (dd, 1 H, J5,6 = 2.2 Hz, J6a,6b = 12.3 Hz, H-6aI), 3.92.3–88 (m, 2 H, H-1a′ and H-4II), 3.80–3.72 (m, 3 H, H-6bI, H-6aII, H-6bII), 3.71 (m, 1 H, H-5II), 3.68–3.61 (m, 4 H, H-1b′, H-3I, H-4I, H-3II), 3.59–3.56 (m, 1 H, H-5I), 3.53 (dd, 1 H, J1,2 = 7.8 Hz, J2,3 = 9.9 Hz, H-2II), 3.29 (dd, 1 H, J1,2 = 8.0 Hz, J2,3 = 9.4 Hz, H-2I), 3.27–3.22 (m, 2 H, H-6′), 2.75 (m, 2 H, H-7′), 2.25 (t, 2 H, J = 7.4 Hz, H-5′), 1.65–1.58 (m, 4 H, H-2′ and H-4′), 1.35 (m, 2 H, H-3′); 13C NMR (D2O): δ 105.7 (C-1II), 104.8 (C-1I), 81.2 (C-4I), 78.1 (C-5II), 77.5 (C-5I), 77.2 (C-3I), 75.6 (C-2I), 75.3 (C-3II), 73.7 (C-2II), 73.1 (C-1′), 71.3 (C-4II), 63.8 (C-6II), 62.9 (C-6I), 43.9 (C-6′), 42.6 (C-7′), 38.5 (C-5′), 31.1 (C-2′), 27.7 (C-4′), 27.3 (C-3′); TOF-MS: [M + H]+ calcd for C20H39N2O12, 499.249; found, 499.250. Anal. Calcd for C19H34O13•0.5 H2O: C, 47.33; H, 7.75. Found: C, 47.54; H, 7.75.
3.7 1-[(2-Aminoethylamido)carbonylpentyl β-d-galactopyranosyl-(1→4)-β-d-glucopyranoside]-2-methoxycyclobutene-3,4-dione (5)
A solution of compound 4 (580 mg, 1.16 mmol) and 3,4-dimethoxy-3-cyclobutene-1,2-dione (247 mg, 1.74 mmol) in Buffer A (6 mL) was stirred at room temperature for 3 h, when TLC showed that almost all 4 was consumed. The mixture was concentrated and the residue was chromatographed (8:1 CH2Cl2–MeOH) to give 5 (540 mg, 76%): ¹H NMR (D2O): δ 4.45 (d, 1 H, J1,2 = 8.1 Hz, H-1I), 4.43 (d, 1 H, J1,2 = 7.9 Hz, H-1II), 4.37 and 4.34 (2 s, 3 H, H-8′), 3.96 (dd, 1 H, J5,6 = 2.2 Hz, J6a,6b = 12.2 Hz, H-6aI), 3.91 (d, 1 H, J3,4 = 3.5 Hz, H-4II), 3.87 (m, 1 H, H-1a′), 3.81–3.72 (m, 3 H, H-6bI, H-6aII, H-6bII), 3.73–3.69 (m, 2 H, H-5II and H-6a′), 3.66–3.61 (m, 4 H, H-1b′, H-3I, H-4I, H-3II), 3.59–3.56 (m, 2 H, H-5I and H-6b′), 3.53 (dd, 1 H, J1,2 = 7.8 Hz, J2,3 = 10.0 Hz, H-2II), 3.40 (m, 2 H, H-7′), 3.30–3.27 (m, 1 H, H-2I), 2.21 (m, 2 H, H-5′), 1.63–1.51 (m, 4 H, H-2′ and H-4′), 1.35–1.28 (m, 2 H, H-3′); 13C NMR (D2O): δ 105.7 (C-1II), 104.8 (C-1I), 81.2 (C-4I), 78.1 (C-5II), 77.5 (C-5I), 77.2 (C-3I), 75.6 (C-2I), 75.3 (C-3II), 73.7 (C-2II), 73.0 (C-1′), 71.3 (C-4II), 63.8 (C-6II), 63.7 (C-8′), 62.9 (C-6I), 46.7 (C-6′), 42.0 (C-7′), 38.5 (C-5′), 31.1 (C-2′), 27.8 (C-4′), 27.4 (C-3′); TOF-MS: [M + Na]+ calcd for C25H40N2O12Na, 631.231; found, 631.232.
3.8 Conjugation of squaric acid monoesters 5–8 and 14–17 to BSA
3.8.1 Preliminary conjugation experiment
Compound 5 (3.7 mg, 0.006 mmol) and BSA (Sigma A-4503, purified,32 20 mg, 0.0003 mmol) were allowed to react at a hapten concentration of 40 mM, using 0.05 M pH 9 buffer (Buffer C, 150 µL). The reaction was monitored by SELDI TOF MS, which showed that after 1, 4, and 25 h the hapten–BSA ratio was 1.3, 3.3, and 3.5 respectively. A small amount of buffer salts was added to raise the pH from 7.2 ± 0.2 to ~8.8 ± 0.2 and, after an additional 12 h, the carbohydrate–protein ratio was 18:1, as showed by SELDI TOF MS analysis. When the reaction was repeated using Buffer B, the same loading was reached after 8 h (Table 1).
3.8.2 General (suggested) conjugation protocol
[The protocol described here for conjugation of 5 and BSA at hapten concentration 40 mmol in Buffer B (0.5 M borate buffer, pH 9.0) should be adjusted as required, depending on the nature and concentration of hapten, carrier and buffer. Conjugates listed in Table 1 have been prepared in a similar way]. With the aid of Buffer B (3 × 50 µL), squaric acid derivative 5 (3.7 mg, 0.006 mm) was transferred into a glass vial containing BSA (20 mg, 0.0003 mmol). The reaction was gently stirred and periodically monitored by SELDI- TOF MS. When the increasing molecular mass leveled off, the mixture was transferred into a centrifugal filter device and processed to remove low molecular mass material. A minimum of 8 washes with aq. 10 mM (NH4)2CO3 were applied, to ensure complete solvent exchange. Freeze-drying afforded conjugates as white solids. To determine yields, materials thus obtained were further dried at 35 °C for 48 h/133 Pa. For results, see Discussion, Table 1 and Table 3 and Figure 1 and Figure 2.
When two conjugates were made in one pot13 (as in the conversion 31→34, Table 3), 90% of the reaction mixture was withdrawn when the first target hapten–BSA ratio was reached, and the remaining mixture was stirred and periodically monitored until the molecular mass of the conjugate being formed no longer increased, as determined by SELDI-TOF MS. The withdrawn portion of the mixture was diluted with 11 volumes of Buffer A, and processed as described above.
3.8.3 Reproducibility of conjugation
Hapten 8 (4.42 mg, 0.006 mmol) was treated with BSA (20 mg, 0.0003 mmol) in Buffer B at hapten concentration 40 mM. Hapten 30 (1.9 mg, 0.0018 mmol and 5.7 mg, 0.0054 mmol) was treated in Buffer B with BSA [20 mg (0.0003 mmol) and 60 mg (0009 mmol), respectively] at hapten–BSA ratio of 20:1 following the general (suggested) protocol. For results, see Table 2.
Table 2.
Reproducibility of Conjugation to BSAa
| Reaction | Hapten/BSA | ||
|---|---|---|---|
| Hapten | Time | ||
| [h] | Run 1 | Run 2 | |
| 8b | 1 h | 8.8 | 8.2 |
| 2 h | 10.5 | 10.4 | |
| 4 h | 13.2 | 12.5 | |
| 6 h | 14.4 | 14.4 | |
| 8 h | 15.2 | 14.5 | |
| 24 h | 17.6 | 18.1 | |
| 30c | 0.5 | 1.4 | 1.7 |
| 1 | 1.9 | 2.3 | |
| 1.5 | 2.9 | 2.8 | |
| 2 | 2.9 | 3.4 | |
| 2.5 | 3.9 | 3.8 | |
| 4 | 4.8 | 4.4 | |
| 6 | 5.0 | 4.9 | |
Unless stated otherwise, all conjugations were carried out at ambient temperature (22–24°C) in 0.5 M pH 9 buffer, at hapten concentration 40 mM
Initial hapten–BSA ratio, 20:1
Initial hapten–BSA ratio, 6:1.
3.9 Hydrolysis of squarate monoesters
Solutions of compounds 5–8 and 14–17 in Buffer B or C were kept at room temperature at hapten concentration 40, 4 and 0.4 mM, respectively. The progress of hydrolysis was followed by HPLC, and results were calculated using calibration curves, which were constructed with the aid of standard solutions of pure substances. For results, see Fig. 3 and 4.
Supplementary Material
Acknowledgements
This research was supported by the Intramural Research Program of the NIH, NIDDK. The authors wish to thank Rick Lake of Restek Corporation for his interest in this work, and for expert advice during the quantitative analysis by HPLC.
Footnotes
Presented as a plenary presentation at the 7th International Meeting of the Portuguese Carbohydrate Group (GLUPOR 7), September 12–15, 2007, Oieras, Portugal.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kováč P, editor. Synthetic Oligosaccharides. Indispensable Probes in the Life Sciences. Washington, D.C.: American Chemical Society; 1994. (ACS Symposium Series 560). [Google Scholar]
- 2.Lee YC, Lee RT. In: Synthetic Glycoconjugates. Allen HJ, Kisailus EC, editors. New York: Marcel Dekker, Inc.; 1992. pp. 121–165. [Google Scholar]
- 3.Galonic DP, Gin DY. Nature. 2007;446:1000–1007. doi: 10.1038/nature05813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dick WE, Jr, Beurret M. In: Glycoconjugates of Bacterial Carbohydrate Antigens. Cruse JM, Lewis RE Jr, editors. Vol. 10. Basel: Krager; 1989. pp. 48–114. [PubMed] [Google Scholar]
- 5.Tietze LF, Arlt M, Beller M, Glüsenkamp K-H, Jähde E, Rajewsky MF. Chem. Ber. 1991;124:1215–1221. [Google Scholar]
- 6.Glüsenkamp K-H, Drosdziok W, Eberle G, Jähde E, Rajewsky MFZ. Naturforsch., C: Biosci. 1991;46:498–501. [Google Scholar]
- 7.Tietze LF, Schröter C, Gabius S, Brinck U, Goerlach-Graw A, Gabius H-J. Bioconjugate Chem. 1991;2:148–153. doi: 10.1021/bc00009a003. [DOI] [PubMed] [Google Scholar]
- 8.Cohen S, Cohen SG. J. Am. Chem. Soc. 1966;88:1533–1536. [Google Scholar]
- 9.Grünefeld J, Bredhauer G, Zinner G. Arch. Pharm. (Weinheim) 1985;318:984–988. [Google Scholar]
- 10.Bergh A, Magnusson B-G, Ohlsson J, Wellmar U, Nilsson UJ. Glycoconjugate J. 2001;18:615–621. doi: 10.1023/a:1020639603070. [DOI] [PubMed] [Google Scholar]
- 11.Kamath VP, Diedrich P, Hindsgaul O. Glycoconjugate J. 1996;13:315–319. doi: 10.1007/BF00731506. [DOI] [PubMed] [Google Scholar]
- 12.Zhang J, Yergey A, Kowalak J, Kováč P. Carbohydr. Res. 1998;313:15–20. doi: 10.1016/s0008-6215(98)00261-4. [DOI] [PubMed] [Google Scholar]
- 13.Saksena R, Ma X, Kováč P. Carbohydr. Res. 2003;338:2591–2603. doi: 10.1016/s0008-6215(03)00273-8. [DOI] [PubMed] [Google Scholar]
- 14.Auzanneau F-I, Pinto M. Bioorg. Med. Chem. 1996;4:2003–2010. doi: 10.1016/s0968-0896(96)00183-6. [DOI] [PubMed] [Google Scholar]
- 15.Chernyak A, Karavanov A, Ogawa Y, Kováč P. Carbohydr. Res. 2001;330:479–486. doi: 10.1016/s0008-6215(01)00018-0. [DOI] [PubMed] [Google Scholar]
- 16.Saksena R, Chernyak A, Poirot E, Kováč P. Method Enzymol. 2003;362:140–160. doi: 10.1016/S0076-6879(03)01011-5. [DOI] [PubMed] [Google Scholar]
- 17.Izumi M, Okumura S, Yuasa H, Hashimoto H. J. Carbohydr. Chem. 2003;22:317–329. [Google Scholar]
- 18.Pozsgay V, Dubois E, Pannell L. J. Org. Chem. 1997;62:2832–2846. doi: 10.1021/jo962300y. [DOI] [PubMed] [Google Scholar]
- 19.Seitz G, Morck H. Arch, Pharm. 1972;305:614–618. [Google Scholar]
- 20.Saksena R, Zhang J, Kováč P. Tetrahedron:Asymmetry. 2005;16:187–197. [Google Scholar]
- 21.Hronowski LJJ, Szarek WA, Hay GW, Krebs A, Depew WT. Carbohydr. Res. 1989;190:203–218. [Google Scholar]
- 22.Hudson CS, Johnson JM. J. Am. Chem. Soc. 1915;37:1270–1275. [Google Scholar]
- 23.Solladie G, Ziani-Cherif C. J. Org. Chem. 1993;58:2181–2185. [Google Scholar]
- 24.Banoub J, Bundle D. Can. J. Chem. 1979;57:2085–2090. [Google Scholar]
- 25.Bochkov AF, Betaneli VI, Kochetkov NK. Bioorg. Khim. 1977;3:39–45. [Google Scholar]
- 26.Bochkov AF, Zaikov GE. Book Chemistry of the Glycosidic Bond: Formation and Cleavage. Oxford: Pergamon Press; 1979. Chemistry of the Glycosidic Bond: Formation and Cleavage. [Google Scholar]
- 27.Banoub J, Bundle DR. Can. J. Chem. 1979;57:2091–2097. [Google Scholar]
- 28.Ziegler T, Kováč P, Glaudemans CPJ. Liebigs Ann. Chem. 1990:613–615. [Google Scholar]
- 29.Saksena R, Chernyak A, Karavanov A, Kováč P. In: Conjugating Low Molecular Mass Carbohydrates to Proteins. 1. Monitoring the Progress of Conjugation. Lee YC, Lee R, editors. Vol. 362. Academic Press; 2003. pp. 125–139. [DOI] [PubMed] [Google Scholar]
- 30.Ma X, Saksena R, Chernyak A, Kováč P. Org. Biomol. Chem. 2003;1:775–784. doi: 10.1039/b211660j. [DOI] [PubMed] [Google Scholar]
- 31.Saksena R, Adamo R, Kováč P. Bioorg. Med. Chem. 2007;15:4283–4310. doi: 10.1016/j.bmc.2007.03.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen RF. J. Biol. Chem. 1967;242:173–181. [PubMed] [Google Scholar]
- 33.Chernyak A, Oscarson S, Turek D. Carbohydr. Res. 2000;329:309–316. doi: 10.1016/s0008-6215(00)00189-0. [DOI] [PubMed] [Google Scholar]
- 34.Lefeber DJ, Kamerling JP, Vliegenthart JFG. Chem-Euro. J. 2001;7:4411–4421. doi: 10.1002/1521-3765(20011015)7:20<4411::aid-chem4411>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 35.Ellis LA, Mc Vay CS, Probert MA, Zhang Z, Bundle DR, Appleton JA. Glycobiology. 1997;7:383–390. doi: 10.1093/glycob/7.3.383. [DOI] [PubMed] [Google Scholar]
- 36.Mawas F, Niggemann J, Jones C, Corbel MJ, Kamerling JP, Vliegenthart JFG. Infect. Immun. 2002:5107–5114. doi: 10.1128/IAI.70.9.5107-5114.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chernyak A, Kondo S, Wade TK, Meeks MD, Alzari PM, Fournier J-M, Taylor RK, Kováč P, Wade WF. J. Infect. Dis. 2002;185:950–962. doi: 10.1086/339583. [DOI] [PubMed] [Google Scholar]
- 38.Hirayama K, Akashi S, Furuya M, Fukuhara K-I. Biochem. Biophys. Res. Commun. 1990;173:639–646. doi: 10.1016/s0006-291x(05)80083-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.