

Abstract
Ionizing radiation (IR) began to be a powerful medical modality soon after Wilhelm Röntgen’s discovery of X-rays in 1895. Today, more than 50% of cancer patients receive radiotherapy at some time during the course of their disease. Recent technical developments have significantly increased the precision of dose delivery to the target tumor, making radiotherapy more efficient in cancer treatment. However, tumor cells have been shown to acquire a radioresistance that has been linked to increased recurrence and failure in many patients. The exact mechanisms by which tumor cells develop an adaptive resistance to therapeutic fractional irradiation are unknown, although low-dose IR has been well defined for radioadaptive protection of normal cells. This review will address the radioadaptive response, emphasizing recent studies of molecular-level reactions. A prosurvival signaling network initiated by the transcription factor NF-κB, DNA-damage sensor ATM, oncoprotein HER-2, cell cyclin elements (cyclin B1), and mitochondrial functions in radioadaptive resistance is discussed. Further elucidation of the key elements in this prosurvival network may generate novel targets for resensitizing the radioresistant tumor cells.
Keywords: Ionizing radiation, NF-κB, Signal transduction, Adaptive radiation resistance, Free radicals
Does ionizing radiation (IR) induce radioadaptive resistance?
Acquired tumor radioresistance can be induced during radiotherapy due to tumor repopulation [1]. Although tumor radioresistance stands as an fundamental barrier limiting the effectiveness of radiation therapy, the exact molecular mechanisms underlying the radioadaptive response are largely unknown. The term radioadaptive response was originally described as a reduced cell sensitivity to a higher challenging dose when a smaller inducing radiation dose had been applied earlier [2]. Olivieri et al. [3] first described an adaptive response of human lymphocytes to ionizing radiation. Since then a substantial number of reports make a strong case for the existence of cellular radioprotective mechanisms that can be activated in response to a small dose of ionizing radiation. It is assumed that a specific prosurvival signaling network is induced in irradiated mammalian cells. Such prosurvival molecular networks, especially induced in the tumor under clinical fractional irradiation, have not yet been fully elucidated. New therapeutic means including manipulating the radioadaptive response require further knowledge of the consequences of the activation of such prosurvival pathways.
Exposure to low levels of IR is evidenced to generate beneficial effects for mammalian cells with respect to the maintenance of genomic integrity and the ability to repair damaged DNA [4,5]. Although the IR doses applied vary on a large scale, a similar radioadaptive resistance is significantly induced in many other species, including Escherichia coli, protozoa, algae, higher plant cells, and insect cells. For example, fruit flies experience far fewer mutations when hit by high-dose IR if they are first exposed to a low level of IR [6]. Bhattarcharjee and Ito report that whole-body preirradiation of Swiss mice with five repeated exposures to small doses of 1 cGy per day reduces the incidence of thymic lymphoma from 46 to 16% with a challenge dose of 2 Gy [7]. Likewise, Liu et al. demonstrate radioprotection in rabbits exposed to low-dose γ-irradiation [8]. In addition, human lymphocytes exposed to low-level IR show fewer chromatid breaks when later exposed to large radiation doses [9]. The above data and many other reports clearly suggest that a radioadaptive response conserved in all species is involved in tumor acquired radioresistance.
Does repopulation/selection of cancer stem cells cause radioadaptive resistance?
Although there is a lack of convincing clinical data supporting tumor radioadaptive resistance, xenograft tumors generated either from human or from mouse tumor cell lines show an enhanced survival with an accelerated cell repopulation and doubling time under fractional irradiation [1,10,11]. Similar adaptive resistance has been induced in cultured tumor cells [12,13]. Recent evidence suggests that only a certain type of tumor cell, cancer stem cells (CSCs) or cancer-initiating cells, harbor tumorigenic potential [14], and these cells may be responsible for therapeutic failure [15]. In the early 1990s, the concept of CSCs was tested by John Dick’s group at the University of Toronto [16,17] and further tested and identified by Michael Clarke’s group at the University of Michigan. Several aspects about the characteristics of CSCs continue to be tested and confirmed. Nevertheless, accumulating reports tend to indicate that there are CSCs in almost all tumor types [18]. Recently, using the CD133 as the brain stem cell marker, Bao et al. at Duke University described an increased proportion of brain CSCs, from about 2 to about 8% in control versus irradiated tumors, which was associated with tumor radioresistance [19]. The group at UCLA showed that breast cancer-initiating cells displaying the marker of breast CSCs (CD24−/low/CD44+) are radioresistant and the cells with these markers increase after short courses of fractional irradiation [20]. All of these findings shed new light on the mechanisms of an accelerated tumor proliferation with a repopulation of radio-resistant CSCs. We have reported that fractional irradiation with a protocol of 2 Gy per fraction, five times per week for 6 weeks, enhances the radioresistance of human breast cancer MCF-7 cells [21]. Because CSCs have been shown to be radioresistant, it needs to be clarified whether radioresistant clones isolated by our group after therapeutic radiation doses [22] are CSCs or a population enriched with CSCs.
The findings by the Duke and UCLA groups [19,20] suggest that radioresistance may be a general property of CSCs and thus raise three important questions that need to be clarified in future studies: (1) Can the accumulation of CSCs after radiotherapy seen in vitro and in xenografts be documented in patients? (2) Does the percentage of CSCs within human tumors predict radiosensitivity? (3) Are CSCs of all tumor types radioresistant? Nevertheless, the two studies have important clinical implications. In light of these studies, it is even clearer that identifying and characterizing CSCs for every tumor possible is of paramount importance and will likely lead to new therapeutic avenues. In the case of radiotherapy, it seems possible that assays that capture the response of CSCs rather than the bulk of a tumor could prove to be more sensitive predictors of treatment outcome than traditional measures of treatment response. Also, work on radiosensitizers should begin to focus on preferentially affecting CSCs compared with normal tissues and normal tissue stem cells.
The theory of therapy-mediated cancer stem cell repopulation, however, seems not to be able to explain all the features of radioadaptive resistance induced in both normal and tumor cells by exposure to various levels of radiation doses. This is especially true when the adaptive resistance is induced in cells after exposure to a sublethal low dose of IR that is not capable of repopulating or selecting CSCs. By this reasoning, specific molecular pathways activated in radioadaptive response need to be elucidated in a population with stem and nonstem cells. In the following, to further understand the mechanism of tumor radioadaptive resistance, we will discuss the evidence for a prosurvival network induced by the transcription factor NF-κB that can be induced by as low as 5 cGy X-radiation and can offer a significant survival advantage.
NF-κB protein structure
NF-κB was originally identified as a protein bound to a sequence in the immunoglobulin κ light chain enhancer in B cells [23]. NF-κB can activate a great number of genes involved in stress responses, inflammation, and programmed cell death (apoptosis). In mammals, the NF-κB family consists of five members of the Rel family: RelA (also called p65), RelB, c-Rel, p50/p105 (also called NF-κB1), and p52/p100 (also called NF-κB2). Although the heterodimer of p50 and p65 is shown to be the most abundant form of NF-κB [24], different combinations of homo- or heterodimers can be formed that are thought to determine the intrinsic NF-κB specificity and its regulation [25–28]. NF-κB DNA binding sites in the promoter regions of many stress-responsive genes are capable of binding p50 homodimers or p50/p65 or p50/c-Rel heterodimers, suggesting a complex gene and physiological regulation controlled by NF-κB in stress response [29].
Under nonstimulated conditions, the NF-κB complex, formed mainly by a p50/p65 heterodimer, binds to a member of the NF-κB inhibitors (the IκB family) [29,30], and the nuclear localization signal of NF-κB is effectively hidden through the noncovalent binding with IκB. The mammalian IκB family has been identified to include IκB-α, IκB-β, IκB-γ, IκB-ε, Bcl-3, p105, and p100, of which IκB-α is the most studied prominent inhibitor. Two protein kinases with high sequence similarity, IKK-α (IκB kinase-α) and IκB-β, can phosphorylate and activate IκB. Most of these kinases are part of IKK complexes that also contain a regulatory subunit, IKK-γ (IκB kinase-γ) or NEMO (NF-κB essential modulator). Therefore, the trimolecular complex, i.e., IKK–IκB–NF-κB, which may contain an additional substrate-targeting subunit named ELKS (a protein rich in glutamate, leucine, lysine, and serine) [31], is referred to as the IKK complex. After genotoxic stimulation, DNA-binding subunits p50 and p52 that carry a Rel homology domain (RHD) are proteolytically released from p105 and p100, respectively. The RHD with a nuclear localization sequence functions in dimerization, sequence-specific DNA binding, and interaction with the inhibitory IκB proteins. In addition, Bcl-3 can act as a transcriptional activator when binding to the p50 or p52 homodimers. Based on the published data, we provide a summary of the structures of the mammalian NF-κB, IκB, and IKK family members with posttranslational (phosphorylation and acetylation) modification sites of the NF-κB major subunit p65, shown in Fig. 1.
Fig. 1.
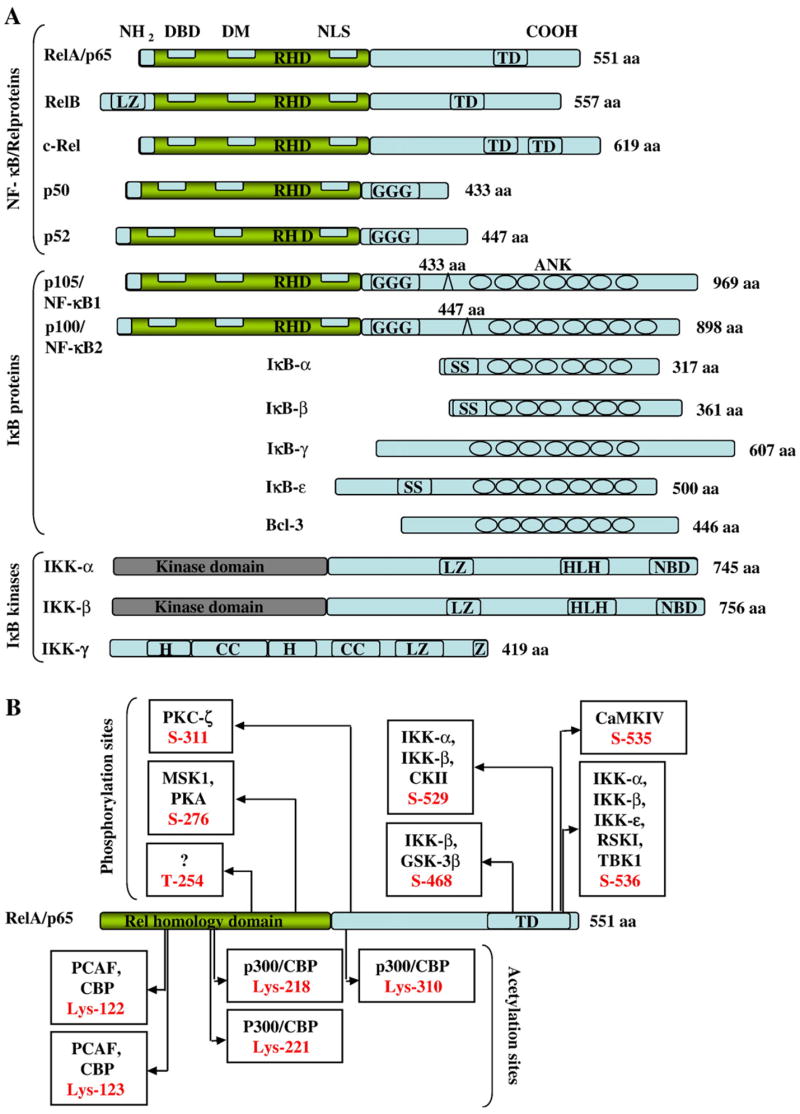
Schematic representations of NF-κB/Rel proteins, their regulators, and posttranslational modification sites of NF-κB p65/RelA. (A) Structures of the mammalian NF-κB, IκB, and IKK proteins. The number of amino acids in each protein is indicated on the right. Presumed sites of cleavage for p105/NF-κB1 (amino acid 433) and p100/NF-κB2 (amino acid 447) are shown on the top of each protein. The positions of functional domains are indicated, including the Rel homology domain (RHD), DNA binding domain (DBD), dimerization domain (DM), nuclear localization signal (NLS), transactivation domains (TD), glycine-rich hinge region (GGG), ankyrin repeats (ANK), double serine phosphorylation sites (SS), leucine zipper (LZ), helix-loop-helix (HLH), NEMO-binding domain (NBD), α-helix (H), coiled coil (CC), and zinc finger (Z). (B) Phosphorylation and acetylation sites within NF-κB p65. Six inducible phosphorylation and five acetylation sites have been identified in the NF-B p65 subunit. The known kinases and target residues include PKC-ζ (protein kinase C-ζ), MSK1 (mitogen or stress-activated kinase 1), PKA (protein kinase A), CKII (casein kinase II), GSK-3β (glycogen synthase kinase-3β), CaMKIV (calmodulin-dependent kinase IV), RSKI (ribosomal S6 kinase), TBK1 [TANK (TRAF family member-associated NF-κB activator)-binding kinase 1], PCAF (p300/CBP-associated factor), CBP (CREB-binding protein).
Is NF-κB required for radioadaptive resistance?
The elevated basal NF-κB activity in certain cancers has been linked with tumor resistance to chemotherapy and radiation [32]. The work reported by Donald Kufe’s group first demonstrated that IR activates DNA binding of NF-κB [33]. Blocking NF-κB activation increases apoptotic response and decreases growth and clonogenic survival of several human cancer cell lines [34–36], although not all experiments show an enhanced radiosensitivity by NF-κB inhibition [37]. In androgen-independent prostate cancer cells, inhibition of NF-κB by a dominant negative superrepressor IκB mutant enhances apoptosis in DU145 [38] but not in PC3 cells [37], suggesting that NF-κB affects cell sensitivity to radio/chemotherapy in a cell-type-specific manner. However, NF-κB in adaptive radioresistance is evidenced in mouse epidermal cells [39] and human keratinocytes (unpublished data), and inhibition of NF-κB blocks the adaptive radioresistance [39]. Human breast cancer cells treated with fractional γ-irradiation show an enhanced clonogenic survival and NF-κB activation [21,40]. Blocking NF-κB inhibited the adaptive radioresistance. These results provide the first evidence that activation of NF-κB is required for signaling the radio-adaptive resistance by exposure to a radiation dose equivalent to medical diagnostic use of radioactivity. Together with the assumption that NF-κB is able to regulate more than 150 effector genes, these results suggest that NF-κB plays a key role in tumor radioadaptive resistance under fractional ionizing radiation. This prosurvival network initiated by NF-κB and linked with the DNA-damage sensor protein ATM, EGFR family HER-2, and mitochondrial antioxidant MnSOD [21,39,41–43] will be discussed in the following sections.
How is NF-κB activated?
NF-κB can be activated via two different pathways, i.e., classical (also termed canonical) and alternative [44]. The IKK-β-dependent pathway responsible for the rapid degradation of IκB-α, IκB-β, and IκB-ε is referred to as the classical NF-κB pathway, whereas the IKK-α-dependent pathway leading to processing of p100 and activation of p52/RelB is defined as the alternative pathway described by Senftleben et al. [45]. It has been well established that NF-κB activation typically occurs via the classical pathway, in which phosphorylation of IκB-α at Ser-32 and Ser-36, or IκB-β at Ser-19 and Ser-23, takes place through the function of ubiquitin-dependent protein kinase after stimulation by IR, TNF-α (tumor necrosis factor-α), PMA (phorbol 12-myristate 13-acetate), LPS (lipopolysaccharide), or interleukins. Phosphorylated IκB proteins are then ubiquitinated at nearby lysine residues (lysines 21 and 22 of IκB-α and lysine 9 of IκB-β), and this triggers a rapid degradation of IκB proteins by 26S proteasome [46,47]. Upon IκB degradation, NF-κB is able to quickly translocate to the nucleus where it either binds to a specific 10-bp consensus site, GGGPuNNPyPyCC (Pu, purine; Py, pyrimidine; and N, any base) or interacts with other transcription factors, thereby regulating the transcription of various genes. Although it has been suggested that the degraded IκB may still be associated with NF-κB in mammalian cells, activated NF-κB typically exists as a dimeric protein, and this transcriptionally active form possesses both DNA-binding and transactivation domains. In the nucleus, NF-κB activates a wide variety of gene promoters. Interestingly, it can up-regulate the transcription of its own inhibitor, IκB-α, indicating a feedback control of NF-κB regulation. The newly synthesized IκB-α is able to enter the nucleus and remove NF-κB from its DNA-binding sites and transport it back to the cytoplasm, thereby terminating the NF-κB-dependent gene transcription [48,49]. The alternative pathway, which is completely independent of IKK-β and IKK-γ, can be typically triggered by ligands to particular members of the TNF receptor superfamily that include the B-cell-activating factor of the TNF family, CD40, or lymphotoxin β. Upon receptor triggering, adaptor proteins of the TRAF family enable the recruitment of NF-κB-inducing kinase (NIK; a kinase for the phosphorylation of IKK-α; described in the following section). Then IKK-α targets p100 for phosphorylation and ubiquitination, leading to the limited proteolysis of its ankyrin-like C-terminus and the generation of a functional p52/RelB heterodimer to activate gene regulation.
The NF-κB-dependent transcription also requires multiple coactivators (p300/CBP, P/CAF, and SRC-1/NcoA-1) possessing histone acetyltransferase (HAT) activity [50–52]. The interactions between NF-κB and these HATs indicate a link between acetylation events and NF-κB-mediated gene transactivation. In fact, deacetylase inhibitors (such as trichostatin A or sodium butyrate) have been shown to enhance NF-κB-dependent gene expression in the presence of TNF-α [53–55]. Most importantly, the p50/p65 heterodimer can be acetylated at multiple lysine residues [56], which is believed to change its transcriptional function, DNA-binding affinity, and IκB-α affinity. On the other hand, the p50 subunit, which does not possess a transactivation domain, can be acetylated by p300/CBP [57], and the enhanced p50 acetylation is correlated with increased p50 binding to the cyclooxygenase-2, an important NF-κB-regulated effector [58]. Acetylation of p65 is also detected in vivo [55] with stimulation of TNF-α and PMA [59]. In both cases, p65 is acetylated after overexpression of p300/CBP and p65 is deacetylated through a specific interaction with histone deacetylase-3 [55,59,60]. These results strongly suggest that acetylation of NF-κB subunits negatively regulates their ability to bind to DNA and transactivate genes. Further studies need to address the question of whether the status of posttranslational modification, i.e., acetylation, contributes to NF-κB activation and cell radioresistance.
The antiapoptotic function of NF-κB has been linked with the TNF receptor I pathway [61]. This receptor is coupled via the Fas-associated death domain (FADD) to initiator caspase 8 or 10. The NF-κB survival pathway may function as a substrate for caspases at different levels. The p65 and p50 subunits are cleaved by caspase 3, and c-Rel has three cleavage sites for caspase 3 [62,63]. The TNF-induced NF-κB activity induces transcription of a set of genes coding for antiapoptotic proteins, e.g., c-IAP-1 and c-IAP-2, which can block caspase functions [64]. Most of the NF-κB-regulated promotion of cell death is induced by genes that code for the death receptor Fas or its ligand FasL [62]. Therefore, IR-induced NF-κB activation could be involved in cell cycle arrest and prevention of apoptosis to allow cells to repair damaged DNA [65,66]. An NF-κB binding site was identified in the cyclin D1 promoter region [67,68], and NF-κB is able to regulate the cell cycle via induction of G1/S protein cyclin D1, and a sustained activation of NF-κB could permit cells with accumulated DNA damage to escape apoptosis, and a constitutive activation of NF-κB has been shown to prevent cancerous cells from apoptosis [65,69]. These results reflect a complex regulation of NF-κB under the stress of fractional IR. Based on the published results and our own data, we provide the following putative prosurvival signaling pathways centered on NF-κB activation in the FIR-induced radioadaptive response (Fig. 2). The linkage of NF-κB and other key signal elements and NF-κB-related effector genes in adaptive radioresistance will be addressed in the following sections.
Fig. 2.

Schematic presentation of the NF-κB signaling network in radiation-induced adaptive radioresistance. Fractional ionizing radiation can directly induce DNA damage causing double-strand breaks (DSB) and single-strand breaks (SSB). The damaged DNA activates nuclear ATM, which in turn translocates to the cytoplasm to activate NF-κB via regulation of IKK activity, resulting in the dissociation of IκB from the complex and then activation of NF-κB. The reactive oxygen species (ROS) generated in cells by IR not only induce DNA damage in the nucleus but also activate NF-κB via the TRAFs pathway. Therefore, NF-κB activation by both nuclear and cytoplasmic pathways seems to be necessary for up-regulation of IR-effector genes that include at least partial antiapoptotic and cell cycle elements. The NF-κB effector genes have been shown to be necessary for an enhanced cell survival when the irradiated cells are exposed again to IR. In addition, the mitochondrial antioxidant enzyme MnSOD, which detoxifies superoxide free radicals in mitochondria, is regulated by NF-κB, which may play a key role in the regulation of the cell cycle and apoptosis, although the exact mechanism is to be elucidated.
NF-κB activation by IR-induced cytokines
As illustrated in Fig. 2, IR-induced ROS may play an active role in the radioadaptive response because IR-induced cytokines affect the overall radiosensitivity [70,71]. Here we focus on the reports of TNF-α, a cytokine produced originally by activated T cells and macrophages with an ability to induce NF-κB via receptor activation [72]. Details of the NF-κB pathways responding to the cytokines TNF-α and IL-1 have been described [73,74] and numerous immune and inflammatory response genes can be up-regulated [29,75]. As such, a mutual activation of NF-κB and TNF-α may be required for the inflammatory response induced by radiation. TNF-α can activate NF-κB and JNK, which requires TRAFs [76] that in turn interact with the downstream NIK [77]. NIK was originally identified as a TRAF2-interacting protein and now is a member of the mitogen-activating protein kinase kinase kinase family. As described above, there are two different NF-κB activating pathways (classical and alternative) and NIK seems to be an integral component of the alternative NF-κB pathway [78]. The primary site of IKK-α phosphorylation by NIK (Ser-176) has been linked with the activation of IKK-α, which phosphorylates and inactivates IκB for NF-κB activation. Thus, NIK plays a key role in cytokine-induced NF-κB activation in irradiated cells and may be targeted to inhibit radiation-induced inflammatory response.
Data further indicate that the protein kinase cascades activated by TNF-α are able to phosphorylate IκB kinases and c-jun N-terminal kinase, which would induce opposite cell fates due to the activation of anti- versus proapoptosis pathways associated with NF-κB and JNK activation. There are three mitogen-activated protein kinases (MAPKs), i.e., extracellular signal-regulated kinase (ERK), JNK, and p38, which are all closely related to NF-κB activation [79–81]. ERK, which is also able to activate NF-κB, tends to induce antiapoptosis, whereas the JNK and p38 pathways promote apoptosis. Therefore, the anti- and proapoptotic response induced by different doses of radiation may result from an imbalance of NF-κB versus JNK pathways induced by TNF-α. Blocking of NF-κB by mutant IκB-α sensitizes Ewing sarcoma cells to TNF-α-induced killing with activation of JNK-mediated apoptosis [82,83]. These results strongly suggest that both NF-κB-mediated antiapoptotic actions and JNK-mediated apoptosis can be induced by radiation-induced cytokines, e.g., TNF-α, although the exact imbalances under the stress of different radiation doses are not yet identified. Interestingly, JNK inhibition can be induced by the NF-κB target genes [34,84]. These include GADD45β (growth arrest and DNA damage-inducing protein β) and XIAP (X-chromosome-linked inhibitor of apoptosis); both participate in the cross talk between NF-κB and JNK pathways. A recent work by Papa et al. [85] made significant progress toward understanding how GADD45β blocks JNK activation. They demonstrated that overexpression of GADD45β blocks TNF-α-induced activation of mitogen-activated protein kinase kinase 7 and its downstream target, JNK. We have recently shown that antisense blocking of GADD45β inhibits NF-κB and ERK activity [81], supporting the concept that GADD45β acts as an essential factor in the balance of anti- versus proapoptosis due to radiation-induced cytokine activation. In addition, Tang et al. show that NF-κB-induced XIAP negatively modulates TNF-α-mediated JNK activation [34]. In summary, IR activation of cytokines (e.g., TNF receptors) can trigger downstream molecules with the activation of NF-κB and JNK. The balance/imbalance between the anti- and proapoptosis pathways due to the activation of NF-κB and JNK in radioadaptive resistance is largely unknown.
NF-κB activation by IR-induced MEK/ERK pathways
The MAPKs, i.e., ERK, JNK, and p38, are sensitive to IR stress [86,87]. ERK is strongly induced by high doses of IR via membrane-associated tyrosine kinase. The prosurvival function of ERK1/2 is demonstrated by the fact that inhibition of ERK signaling leads to increased sensitivity of ovarian cancer cell lines to cisplatin-induced apoptosis [88]. The results reported by Suzuki et al. have shown that IR in a range of very low doses (2–5 cGy) activates ERK and enhances proliferation of normal human diploid cells as well as tumor cells [89]. Recent data further indicate that the ERK-mediated antiapoptotic response is dependent upon its cellular locations and interaction with NF-κB/IκB complexes [82]. After exposure to a single high dose of IR (5 Gy), ERK is activated along with an enhancement of the NF-κB transactivation in human breast cancer cells [81]. This coactivation of NF-κB and ERK occurs in a pattern of mutual dependence and is involved in activating GADD45β to defend cells against the cytotoxicity induced by IR. These results suggest that NF-κB and ERK are able to coordinate the increase in cell survival after the lethal damage caused by the acute response to a single dose of IR. Although the ERK pathway has been reported to contribute to radioresistance [90], treatment of melanoma cells with a MEK/ERK inhibitor, PD98059, shows either little [91] or no effect on apoptosis [92]. Therefore, the role of ERK activation in cell radiosensitivity has not been clarified. In addition, unlike coactivation of NF-κB and ERK in response to a single dose (acute response) of IR, inhibition of ERK phosphorylation and NF-κB activation is associated with the development of radioadaptive resistance induced in a fraction of clones of MCF-7 cells derived from therapeutic doses of IR [22]. These results provide insights on how ERK activity is differentially regulated by NF-κB in acute and chronic radiation. Thus, targeting NF-κB with the activation of the MEK/ERK pathways may promise a new approach to preventing therapy-associated tumor radioresistance.
Mutual activation of NF-κB and HER-2
Many breast cancer patients benefit from radiotherapy combined with chemotherapeutic agents. Most importantly, therapy resistance is strikingly increased when tumor cells are HER-2 (also called ErbB2 or Neu) positive. For instance, overexpression of HER-2 has been related to an increased risk of local relapse in breast cancer patients who received conservative surgery and radiation therapy [93]. Recently, HER-2 level has been suggested as a predictive marker for the diagnosis of metastatic breast cancer for patient treatment plans [93,94]. These results suggest that HER-2-mediated therapy resistance involves an anti-radiation signaling network. HER-2 tyrosine kinase, one of the four members of the ErbB receptor family (ErbB1, i.e., EGFR; ErbB2; ErbB3; and ErbB4), plays a critical role in the control of diverse cellular functions involved in differentiation, proliferation, migration, and cell survival via multiple signal transduction pathways. Although a normal level of HER-2 is required for the regulation of normal breast growth and development [95], amplification and overexpression of HER-2 cause the disruption of normal cellular control and the formation of aggressive breast tumor cells [96,97]. Over-expression of HER-2, observed in HER-2-positive breast cancer patients, is believed to make the tumor resistant to an array of anti-cancer agents and indicate a poor prognosis. The molecular mechanisms underlying HER-2-mediated tumor resistance, especially the connections between HER-2 and therapy-resistant signaling networks, need to be further investigated.
It has been well documented that overexpression of HER-2 increases cell proliferation and survival [98], which causes NF-κB activation [99]. As shown in Fig. 2, the PI3-kinase/Akt pathway is involved in HER-2-mediated NF-κB activation [100]. Both IKK-dependent and -independent pathways contribute to the deregulation of NF-κB in breast cancers. The IKK-independent pathway involves calpain-mediated IκB-α degradation [100]. This pathway also requires PI3K and its downstream kinase Akt, which is subject to inhibition by the tumor suppressor phosphatase PTEN. In another study, Akt-mediated NF-κB activation blocked apoptosis in HER-2-expressing cells [101]. It is therefore highly possible that the PI3K/Akt pathway mediated by HER-2 expression is involved in NF-κB activation that regulates downstream effector genes required for HER-2-mediated tumor resistance against therapeutic regimens. As illustrated in Fig. 2, HER-2 overexpression can activate NF-κB. Recently, we observed that NF-κB may also activate HER-2 expression. The 10-bp NF-κB DNA binding sequence, i.e., consensus site (GGGACGACCC; located between −364 and −355), is located in the promoter region of HER-2. Using a luciferase reporter assay, we observed that 5-Gy IR induced HER-2 activation in breast cancer MCF-7 and MDA-MB-231 cells (unpublished data). The increased reporter gene transcription was reduced to control (sham-irradiated cells) levels if the NF-κB binding sequence was deleted. These results strongly suggest that NF-κB induces HER-2 gene transcription in HER-2-negative cancer cells. This is an important finding and worthy of further investigation. Overall, published results and our own data indicate that HER-2 and NF-κB may function in a mutually dependent pattern to enhance cell survival.
NF-κB activation by IR-induced ATM
It is generally believed that a balance in the degree of DNA damage and repair decides the fate of an irradiated cell [102–106], in that cells may increase their survival by reducing or repairing DNA damage via activation of stress-responsive genes [2,21,40,107–109]. Therefore, if DNA repair pathways are activated by preexposure, cells become tolerant to subsequent IR. The mechanism by which IR activates NF-κB is unclear because signals of IR-induced nuclear DNA damage (mainly double-strand breaks, DSBs) must be carried to the cytoplasm, the only place where NF-κB can be activated. ATM-independent activation of NF-κB has been reported by several investigators. Jung et al. has demonstrated a correlation of ATM with NF-κB in cell radiosensitivity [110]. Their data indicate that the loss of ATM function promotes radiosensitivity by activation of NF-κB [110]. In a recent report Wu et al. [111] demonstrated that a cytosolic signaling complex containing ATM, NEMO, IKK catalytic subunits, and ELKS (an IKK regulatory subunit) is activated, and this links nuclear DNA damage to NF-κB activation. This model was based on their findings that ATM interacts with NEMO and phosphorylates Ser-85 of NEMO after DSBs. This event is required for monoubiquitination of NEMO, followed by nuclear export of NEMO and ATM and subsequent interaction with an IKK subunit to activate NF-κB. For instance, normal diploid cells derived from ataxia-telangiectasia patients do not exhibit constitutive activation of NF-κB [112]. Another study also demonstrates that ATM is essential for activation of the entire NF-κB pathway by DSBs in both cultured human cells and mouse tissues, including IKK activation, IκB-α degradation, and induction of NF-κB DNA binding activity [113], but ATM is not required for activation of this pathway by pro-inflammatory stimuli, such as TNF-α, PMA, or LPS. In addition, the DNA-dependent protein kinase, a member of the PI3K-like family of protein kinases, has also been implicated in the activation of NF-κB after exposure to IR [114]. Although the role of ATM in NF-κB activation is confirmed, there is no direct evidence that ATM-mediated NF-κB activation is required for tumor adaptive radioresistance.
NF-κB effector genes in radioadaptive resistance
Two stress-responsive proteins, metallothionein and Kuautoantigen, activated in cells with a radioresistant phenotype, are regulated by NF-κB [115,116]. IR also induces other NF-κB target genes, including IL-1β, IL-6, TNF-α [117], intercellular adhesion molecule-1 [118], MnSOD [119,120], galectin [121], P-selectin [122], and γ-glutamylcysteine synthetase (the rate-limiting enzyme of GSH synthesis involved in radioprotection) [123]. With microarray analysis, Amundson et al. showed that of 1238 human genes, 48 (3.87%) are inducible by a single dose of irradiation [124]. A similar fraction of gene expression is observed in array profiles of irradiated MCF-7 cells (3.9%) [40] and human keratinocytes (4.4%) [35]. Interestingly, gene expression profiles of the radioresistant human keratinocyte cell line HK18-IR demonstrate a specific group of stress-responsive genes of which 10–25% are linked with NF-κB activation [35]. Although exact functions of these NF-κB-associated genes are unknown, they are able to influence cell fate through regulating cell cycle and DNA damage repairs [125,126]. Investigation of these NF-κB target genes is important for elucidation of radiation-induced adaptive resistance. Therefore, studies continue to address the question of whether NF-κB downstream target genes are responsible for tumor radioresistance.
The effector genes of NF-κB rather than NF-κB itself may provide more efficient drug targets to enhance radiosensitivity because such effector genes may be predominant in radio-resistant tumor cells. Among the genes up-regulated in human keratinocytes derived from long-term irradiation [35], six are identified as putative target genes of NF-κB. Of these six genes, cyclin B1, cyclin D1, and HIAP-1 are down-regulated by inhibiting NF-κB with mutant IκB, but the other three up-regulated genes, BAG-1, TTF, and fibronectin, are not down-regulated by mutant IκB. Because the genes down-regulated by inhibition of NF-κB correlate well with a significantly decreased cell survival, any or all of these NF-κB effector genes, i.e., cyclin B1, cyclin D1, and HIAP-1, may play a role in radioresistance. In radiation-derived MCF-7 cells that show enhanced cell survival, cyclin B1 is induced, and when expression of cyclin B1 is inhibited with antisense transfection, radioresistance is reduced significantly [34]. These results provide evidence that cyclin B1 is among the NF-κB effector genes, as shown in Fig. 2, responsible for radioadaptive resistance.
NF-κB-mediated cyclin B1 activation
Cyclins are a group of stress-sensitive proteins in controlling cell death and survival in DNA damage response. The B-type cyclin, i.e., cyclin B1, is an essential cell cycle component in the regulation of transition from G2 to M phase [127–130]. Cyclin B1 and the phosphorylated Cdc2 form a complex with 14-3-3 proteins [131] that accelerates cyclin B1/Cdc2 translocation into the nucleus to participate in cell cycle regulation. Cyclin B1 is rapidly accumulated in the nucleus of cells sensitive but not resistant to IR-induced apoptosis [132]. Increased expression of cyclin B1 coincides with diminished radiation-induced G2-phase arrest [133], and G2 delay is decreased in IR-exposed cells when cyclin B1 is overexpressed by gene transfection [129]. On the other hand, delayed expression of cyclin B1 was observed during the G2 arrest [134]. These results indicate that cyclin B1 plays a key role in IR-initiated cell cycle arrest. A linkage between NF-κB and cyclin B1 in the radioadaptive response was found recently by our group in mouse [39] and human (unpublished data) skin epithelial cells. The accumulation of cyclin B1 and 14-3-3ζ immunoreactive proteins was observed in the same time frame as adaptive radioresistance was being induced by low-dose IR (LDIR) in mouse skin epithelial JB6P+ cells [39]. The nuclear cotranslocation of the cyclin B1/14-3-3ζ complex was also enhanced by LDIR, suggesting that the interaction may contribute to the signal communication required for LDIR-induced adaptive responses. Inhibition of NF-κB reduced the expression of cyclin B1 and 14-3-3ζ and diminished LDIR-induced adaptive resistance. Clinically, cyclin B1 was shown to be associated with the radioresistant phenotype observed in patients with squamous cell carcinoma [135,136], and overexpression of cyclin B1 has been linked with the regional recurrence of head and neck cancer treated by radiotherapy [137]. Using DNA microarray and Western blotting analysis, we demonstrated an induction of cyclin B1 activity in radioadaptive cancer cells derived from therapeutic fractionated radiation [21,35,40]. Antisense blocking of cyclin B1 enhanced the radiosensitivity of radiation-derived radioresistant MCF+FIR cells [40]. Therefore, the clinical results and the data from our laboratory provide direct evidence suggesting that cyclin B1 is involved in the signaling radioprotection. However, the mechanisms of cyclin B1 activation in radiation-adapted radioresistance have not been identified. Cyclin B1-mediated radioadaptive resistance and cyclin B1-targeted drugs need to be studied further.
NF-κB and mitochondrial antioxidants
Mitochondria in mammalian cells not only provide cellular energy, but also serve as the major site for initiating a cascade network causing programmed cell death (apoptosis) [138,139]. The mitochondrial electron transport chain is a principal source of endogenous ROS production during normal metabolism [140]. Although ROS are required for normal physiologic functions in cells, excessive ROS, e.g., generated by IR, cause mitochondrial apoptosis [141–145]. Among various intracellular targets of ROS-mediated injury, mitochondria are particularly prone to ROS-induced damage [146]. Mitochondrial DNA (mtDNA) has no introns, which makes its coding sequence more likely subject to random mutation, and no protective histones or effective DNA repair system [147]; thus, mtDNA is highly sensitive to mutations caused by endogenous ROS. Under normal circumstances, ROS are eliminated by antioxidant enzymes. The superoxide dismutase (SOD) scavenger enzymes, i.e., MnSOD (mitochondrial antioxidant manganese-containing superoxide dismutase; SOD2) or Cu/ZnSOD (cytoplasmic anti-oxidant copper/zinc-containing superoxide dismutase; SOD1), convert superoxide anions to H2O2, which is subsequently detoxified by catalase or glutathione peroxidase [140,148].
The connection between NF-κB and MnSOD has been observed in several studies on radiation protection compounds. Radioprotective thiols can modulate TNF-α-induced MnSOD gene expression with NF-κB activation [149,150]. A delayed radioresistance was recently reported by the Grdina group at the University of Chicago, who indicated that NF-κB mediated MnSOD expression in SA-NH tumor cells after treatment with thiol-containing drugs [151]. Overexpression of MnSOD protects cells from apoptosis [152,153], which was highlighted by another experiment in which MnSOD was significantly induced by radiation in the heart and gut [154,155]. Over-expression of MnSOD also maintains the mitochondrial membrane potential with reduced apoptosis [156,157]. Moreover, MnSOD overexpression can restore cell resistance to TNF-α-induced cell death. Using MnSOD-knockout mice (Sod2−/−), MnSOD was shown to protect against ROS-induced injury during O2 metabolism [158]. Therefore, MnSOD-mediated redox regulation is essential for protecting cells from IR-mediated toxicity. Recent data also suggest that MnSOD affects the release of proapoptotic cytochrome c from mitochondria due to alterations in ROS levels in mitochondria [159]. NF-κB binding sites were found to be located in the regulatory regions of the SOD2 gene, which encodes MnSOD [160–162], providing direct evidence of the NF-κB–MnSOD connection. The enhanced radioresistance in radiation-derived radioresistant breast carcinoma MCF-7 cells [21] and human keratinocytes [21] is related to NF-κB-activated MnSOD. Therefore, MnSOD seems to be a key NF-κB effector gene in radioadaptive resistance. NF-κB is actively involved in the regulation of MnSOD expression in mouse skin cells after exposure to low-dose IR [39]. Inactivation of NF-κB with an IKK-β inhibitor (IMD-0354) suppressed LDIR-induced expression of MnSOD and diminished the adaptive radioresistance. In addition, treatment with small interfering RNA against mouse MnSOD was shown to inhibit the development of LDIR-induced radioresistance. These results support the concept that NF-κB-mediated induction of mitochondrial MnSOD contributes to the scavenging of radiation-induced ROS and the signaling network leading to adaptive radioresistance.
Conclusions
The mechanisms causing tumor radioadaptive resistance are attracting a great deal of interest because of its essential role in the efficacy of clinical anti-cancer radiotherapy. Accumulating evidence suggests that repopulation with radioresistant cancer stem cells may significantly contribute to tumor radioresistance. However, as shown in Fig. 2, a prosurvival pathway initiated by NF-κB seems to be responsible for the radioadaptive response in all cells. In addition, NF-κB is able to inhibit JNK-mediated proapoptosis, indicating that a balance of anti- versus proapo-ptosis under different cytotoxic stresses needs to be explored further. Taken together, the promise of NF-κB and its associations as an anti-tumor target could not be fulfilled without a deeper understanding of the mechanisms that regulate specific NF-κB networks in adaptive radioresistance. Tumor radio-resistance may also be linked to heterogenic NF-κB activation due to tumor repopulation and/or cancer stem cell selection by radiotherapy, which is currently unknown.
Acknowledgments
The authors thank Dr. Larry W. Oberley and Dr. William C. Dewey for reading the manuscript. This work was supported by the National Institutes of Health, USA (RO1 CA101990), and the Office of Science (BER), Department of Energy, USA, low-dose radiation research program (Grant DE-FG02-05ER63634).
Abbreviations
- ATM
ataxia-telangiectasia mutated
- ERK
extracellular signal-regulated kinase
- FIR
fractionated ionizing radiation
- GADD45β
growth arrest DNA damage gene 45β
- IκB
inhibitor-κB
- IKK
IκB kinase
- IL-1
interleukin-1
- IR
ionizing radiation
- JNK
c-Jun N-terminal kinase
- LPS
lipopolysaccharide
- MnSOD
manganese-containing superoxide dismutase
- NEMO
NF-κB essential modulator
- NF-κB
nuclear factor-κB
- NIK
NF-κB-inducing kinase
- PMA
phorbol 12-myristate 13-acetate
- redox
oxidation/reduction reaction
- RHD
Rel homology domain
- ROS
reactive oxygen species
- TRAF
TNF receptor-associated factor
- TNF-α
tumor necrosis factor-α
- XIAP
X-chromosome-linked inhibitor of apoptosis
References
- 1.Kim JJ, Tannock IF. Repopulation of cancer cells during therapy: an important cause of treatment failure. Nat Rev, Cancer. 2005;5:516–525. doi: 10.1038/nrc1650. [DOI] [PubMed] [Google Scholar]
- 2.Stecca C, Gerber GB. Adaptive response to DNA-damaging agents: a review of potential mechanisms. Biochem Pharmacol. 1998;55:941–951. doi: 10.1016/s0006-2952(97)00448-6. [DOI] [PubMed] [Google Scholar]
- 3.Olivieri G, Bodycote J, Wolff S. Adaptive response of human lymphocytes to low concentrations of radioactive thymidine. Science. 1994;223:594–597. doi: 10.1126/science.6695170. [DOI] [PubMed] [Google Scholar]
- 4.Kelsey KT, Memisoglu A, Frenkel D, Liber HL. Human lymphocytes exposed to low doses of X-rays are less susceptible to radiation-induced mutagenesis. Mutat Res. 1991;263:197–201. doi: 10.1016/0165-7992(91)90001-k. [DOI] [PubMed] [Google Scholar]
- 5.Feinendegen LE, Bond VP, Sondhaus CA, Muehlensiepen H. Radiation effects induced by low doses in complex tissue and their relation to cellular adaptive responses. Mutat Res. 1996;358:199–205. doi: 10.1016/s0027-5107(96)00121-2. [DOI] [PubMed] [Google Scholar]
- 6.Muckerheide J. It’s time to tell the truth about the health benefits of low-dose radiation. 21st Century Sci Technol. 2000 Summer;:43–45. [Google Scholar]
- 7.Bhattacharjee D. Role of radioadaptation on radiation-induced thymic lymphoma in mice. Mutat Res. 1996;358:231–235. doi: 10.1016/s0027-5107(96)00125-x. [DOI] [PubMed] [Google Scholar]
- 8.Liu SZ, Cai L, Sun SQ. Induction of a cytogenetic adaptive response by exposure of rabbits to very low dose-rate gamma-radiation. Int J Radiat Biol. 1992;62:187–190. doi: 10.1080/09553009214552001. [DOI] [PubMed] [Google Scholar]
- 9.Shadley JD, Wolff S. Very low doses of X-rays can cause human lymphocytes to become less susceptible to ionizing radiation. Mutagenesis. 1987;2:95–96. doi: 10.1093/mutage/2.2.95. [DOI] [PubMed] [Google Scholar]
- 10.Begg AC, Hofland I, Kummermehr J. Tumour cell repopulation during fractionated radiotherapy: correlation between flow cytometric and radiobiological data in three murine tumours. Eur J Cancer. 1991;27:537–543. doi: 10.1016/0277-5379(91)90211-u. [DOI] [PubMed] [Google Scholar]
- 11.Rofstad EK. Repopulation between radiation fractions in human melanoma xenografts. Int J Radiat Oncol Biol Phys. 1992;23:63–68. doi: 10.1016/0360-3016(92)90544-r. [DOI] [PubMed] [Google Scholar]
- 12.Russell J, Wheldon TE, Stanton PA. radioresistant variant derived from a human neuroblastoma cell line is less prone to radiation-induced apoptosis. Cancer Res. 1995;55:4915–4921. [PubMed] [Google Scholar]
- 13.Robson T, Price ME, Moore ML, Joiner MC, McKelvey-Martin VJ, McKeown SR, Hirst DG. Increased repair and cell survival in cells treated with DIR1 antisense oligonucleotides: implications for induced radioresistance. Int J Radiat Biol. 2000;76:617–623. doi: 10.1080/095530000138277. [DOI] [PubMed] [Google Scholar]
- 14.Al-Hajj M, Clarke MF. Self-renewal and solid tumor stem cells. Oncogene. 2004;23:7274–7282. doi: 10.1038/sj.onc.1207947. [DOI] [PubMed] [Google Scholar]
- 15.Al-Hajj M, Becker MW, Wicha M, Weissman I, Clarke MF. Therapeutic implications of cancer stem cells. Curr Opin Genet Dev. 2004;14:43–47. doi: 10.1016/j.gde.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 16.Lapidot T, Pflumio F, Doedens M, Murdoch B, Williams DE, Dick JE. Cytokine stimulation of multilineage hematopoiesis from immature human cells engrafted in SCID mice. Science. 1992;255:1137–1141. doi: 10.1126/science.1372131. [DOI] [PubMed] [Google Scholar]
- 17.Dick JE. Normal and leukemic human stem cells assayed in SCID mice. Semin Immunol. 1996;8:197–206. doi: 10.1006/smim.1996.0025. [DOI] [PubMed] [Google Scholar]
- 18.Al-Hajj M. Cancer stem cells and oncology therapeutics. Curr Opin Oncol. 2007;19:61–64. doi: 10.1097/CCO.0b013e328011a8d6. [DOI] [PubMed] [Google Scholar]
- 19.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 20.Phillips TM, McBride WH, Pajonk F. The response of CD24(−/low)/CD44+ breast cancer-initiating cells to radiation. J Natl Cancer Inst. 2006;98:1777–1785. doi: 10.1093/jnci/djj495. [DOI] [PubMed] [Google Scholar]
- 21.Guo G, Yan-Sanders Y, Lyn-Cook BD, Wang T, Tamae D, Ogi J, Khaletskiy A, Li Z, Weydert C, Longmate JA, Huang TT, Spitz DR, Oberley LW, Li JJ. Manganese superoxide dismutase-mediated gene expression in radiation-induced adaptive responses. Mol Cell Biol. 2003;23:2362–2378. doi: 10.1128/MCB.23.7.2362-2378.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ahmed KM, Dong S, Fan M, Li JJ. Nuclear factor-kappaB p65 inhibits mitogen-activated protein kinase signaling pathway in radio-resistant breast cancer cells. Mol Cancer Res. 2006;4:945–955. doi: 10.1158/1541-7786.MCR-06-0291. [DOI] [PubMed] [Google Scholar]
- 23.Sen R, Baltimore D. Inducibility of kappa immunoglobulin enhancer-binding protein NF-kappa B by a posttranslational mechanism. Cell. 1986;47:921–928. doi: 10.1016/0092-8674(86)90807-x. [DOI] [PubMed] [Google Scholar]
- 24.Nabel GJ, Verma IM. Proposed NF-kappa B/I kappa B family nomenclature. Genes Dev. 1993;7:2063. doi: 10.1101/gad.7.11.2063. [DOI] [PubMed] [Google Scholar]
- 25.Kuriyan J, Thanos D. Structure of the NF-kappa B transcription factor: a holistic interaction with DNA. Structure. 1995;3:135–141. doi: 10.1016/s0969-2126(01)00143-5. [DOI] [PubMed] [Google Scholar]
- 26.Ghosh G, van Duyne G, Ghosh S, Sigler PB. Structure of NF-kappa B p50 homodimer bound to a kappa B site. Nature. 1995;373:303–310. doi: 10.1038/373303a0. [DOI] [PubMed] [Google Scholar]
- 27.Gossen M, Freundlieb S, Bender G, Muller G, Hillen W, Bujard H. Transcriptional activation by tetracycline in mammalian cells. Science. 1995;268:1766–1769. doi: 10.1126/science.7792603. [DOI] [PubMed] [Google Scholar]
- 28.Verma IM, Stevenson JK, Schwarz EM, Van Antwerp D, Miyamoto S. Rel/NF-kappa B/I kappa B family: intimate tales of association and dissociation. Genes Dev. 1995;9:2723–2735. doi: 10.1101/gad.9.22.2723. [DOI] [PubMed] [Google Scholar]
- 29.Baeuerle PA, Henkel T. Function and activation of NF-kappa B in the immune system. Annu Rev Immunol. 1994;12:141–179. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- 30.Piette J, Piret B, Bonizzi G, Schoonbroodt S, Merville MP, Legrand-Poels S, Bours V. Multiple redox regulation in NF-kappaB transcription factor activation. Biol Chem. 1997;378:1237–1245. [PubMed] [Google Scholar]
- 31.Sigala JLD, Bottero V, Young DB, Shevchenko A, Mercurio F, Verma IM. Activation of transcription factor NF-κB requires ELKS, an IκB kinase regulatory subunit. Science. 2004;304:1963–1967. doi: 10.1126/science.1098387. [DOI] [PubMed] [Google Scholar]
- 32.Orlowski RZ, Baldwin AS., Jr NF-kappaB as a therapeutic target in cancer. Trends Mol Med. 2002;8:385–389. doi: 10.1016/s1471-4914(02)02375-4. [DOI] [PubMed] [Google Scholar]
- 33.Brach MA, Hass R, Sherman ML, Gunji H, Weichselbaum R, Kufe D. Ionizing radiation induces expression and binding activity of the nuclear factor kappa B. J Clin Invest. 1991;88:691–695. doi: 10.1172/JCI115354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tang G, Minemoto Y, Dibling B, Purcell NH, Li Z, Karin M, Lin A. Inhibition of JNK activation through NF-kappaB target genes. Nature. 2001;414:313–317. doi: 10.1038/35104568. [DOI] [PubMed] [Google Scholar]
- 35.Chen X, Shen B, Xia L, Khaletzkiy A, Chu D, Wong JY, Li JJ. Activation of nuclear factor kappaB in radioresistance of TP53-inactive human keratinocytes. Cancer Res. 2002;62:1213–1221. [PubMed] [Google Scholar]
- 36.Herscher LL, Cook JA, Pacelli R, Pass HI, Russo A, Mitchell JB. Principles of chemoradiation: theoretical and practical considerations. Oncology. 1999;13:11–22. [PubMed] [Google Scholar]
- 37.Pajonk F, Pajonk K, McBride WH. Inhibition of NF-kappaB, clonogenicity, and radiosensitivity of human cancer cells. J Natl Cancer Inst. 1999;91:1956–1960. doi: 10.1093/jnci/91.22.1956. [DOI] [PubMed] [Google Scholar]
- 38.Flynn V, Jr, Ramanitharan A, Moparty K, Davis R, Sikka S, Agrawal KC, Abdel-Mageed AB. Adenovirus-mediated inhibition of NF-kappaB confers chemo-sensitization and apoptosis in prostate cancer cells. Int J Oncol. 2003;23:317–323. [PubMed] [Google Scholar]
- 39.Fan M, Ahmed KM, Coleman MC, Spitz DR, Li JJ. NF-kappaB and MnSOD mediate adaptive radioresistance in low dose irradiated mouse skin epithelial cells. Cancer Res. 2007;67:3220–3228. doi: 10.1158/0008-5472.CAN-06-2728. [DOI] [PubMed] [Google Scholar]
- 40.Li Z, Xia L, Lee ML, Khaletskiy A, Wang J, Wong JYC, Li JJ. Effector genes altered in MCF-7 human breast cancer cells after exposure to fractionated ionizing radiation. Radiat Res. 2001;155:543–553. doi: 10.1667/0033-7587(2001)155[0543:egaimh]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 41.Ahmed KM, Cao N, Li JJ. HER-2 and NF-kappaB as the targets for therapy-resistant breast cancer. Anticancer Res. 2006;26:4235–4243. [PMC free article] [PubMed] [Google Scholar]
- 42.Meyn MS. Ataxia-telangiectasia and cellular responses to DNA damage. Cancer Res. 1995;55:5991–6001. [PubMed] [Google Scholar]
- 43.Guo G, Wang T, Gao Q, Tamae D, Wong P, Chen T, Chen WC, Shively JE, Wong JY, Li JJ. Expression of ErbB2 enhances radiation-induced NF-kappaB activation. Oncogene. 2004;23:535–545. doi: 10.1038/sj.onc.1207149. [DOI] [PubMed] [Google Scholar]
- 44.Hacker H, Karin M. Regulation and function of IKK and IKK-related kinases. Sci STKE. 2006:re13. doi: 10.1126/stke.3572006re13. [DOI] [PubMed] [Google Scholar]
- 45.Senftleben U, Cao Y, Xiao G, Greten FR, Krahn G, Bonizzi G, Chen Y, Hu Y, Fong A, Sun SC, Karin M. Activation by IKKalpha of a second, evolutionarily conserved, NF-kappa B signaling pathway. Science. 2001;293:1495–1499. doi: 10.1126/science.1062677. [DOI] [PubMed] [Google Scholar]
- 46.Chen J-Y, Penco S, Ostrowski J, Balaguer P, Pons M, Starrett JE, Reczek P, Chambon P, Gronemeyer H. RAR-specific agonist/antagonists which dissociate transactivation and AP1 transrepression inhibit anchorage-independent cell proliferation. EMBO J. 1995;14:1187–1197. doi: 10.1002/j.1460-2075.1995.tb07102.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baeuerle PA, Rupec RA, Pahl HL. Reactive oxygen intermediates as second messengers of a general pathogen response. Pathol Biol (Paris) 1996;44:29–35. [PubMed] [Google Scholar]
- 48.Ben-Neriah Y. Regulatory functions of ubiquitination in the immune system. Nat Immunol. 2002;3:20–26. doi: 10.1038/ni0102-20. [DOI] [PubMed] [Google Scholar]
- 49.Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell. 2002;109(Suppl):S81–96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- 50.Gerritsen ME, Williams AJ, Neish AS, Moore S, Shi Y, Collins T. CREB-binding protein/p300 are transcriptional coactivators of p65. Proc Natl Acad Sci U S A. 1997;94:2927–2932. doi: 10.1073/pnas.94.7.2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Na SY, Lee SK, Han SJ, Choi HS, Im SY, Lee JW. Steroid receptor coactivator-1 interacts with the p50 subunit and coactivates nuclear factor kappaB-mediated transactivations. J Biol Chem. 1998;273:10831–10834. doi: 10.1074/jbc.273.18.10831. [DOI] [PubMed] [Google Scholar]
- 52.Sheppard KA, Rose DW, Haque ZK, Kurokawa R, McInerney E, Westin S, Thanos D, Rosenfeld MG, Glass CK, Collins T. Transcriptional activation by NF-kappaB requires multiple coactivators. Mol Cell Biol. 1999;19:6367–6378. doi: 10.1128/mcb.19.9.6367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Quivy V, Adam E, Collette Y, Demonte D, Chariot A, Vanhulle C, Berkhout B, Castellano R, de Launoit Y, Burny A, Piette J, Bours V, Van Lint C. Synergistic activation of human immunodeficiency virus type 1 promoter activity by NF-kappaB and inhibitors of deacetylases: potential perspectives for the development of therapeutic strategies. J Virol. 2002;76:11091–11103. doi: 10.1128/JVI.76.21.11091-11103.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Adam E, Quivy V, Bex F, Chariot A, Collette Y, Vanhulle C, Schoonbroodt S, Goffin V, Nguyen TL, Gloire G, Carrard G, Friguet B, De Launoit Y, Burny A, Bours V, Piette J, Van Lint C. Potentiation of tumor necrosis factor-induced NF-kappa B activation by deacetylase inhibitors is associated with a delayed cytoplasmic reappearance of I kappa B alpha. Mol Cell Biol. 2003;23:6200–6209. doi: 10.1128/MCB.23.17.6200-6209.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen L, Fischle W, Verdin E, Greene WC. Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science. 2001;293:1653–1657. doi: 10.1126/science.1062374. [DOI] [PubMed] [Google Scholar]
- 56.Chen LF, Greene WC. Regulation of distinct biological activities of the NF-kappaB transcription factor complex by acetylation. J Mol Med. 2003;81:549–557. doi: 10.1007/s00109-003-0469-0. [DOI] [PubMed] [Google Scholar]
- 57.Furia B, Deng L, Wu K, Baylor S, Kehn K, Li H, Donnelly R, Coleman T, Kashanchi F. Enhancement of nuclear factor-kappa B acetylation by coactivator p300 and HIV-1 Tat proteins. J Biol Chem. 2002;277:4973–4980. doi: 10.1074/jbc.M107848200. [DOI] [PubMed] [Google Scholar]
- 58.Deng WG, Zhu Y, Wu KK. Up-regulation of p300 binding and p50 acetylation in tumor necrosis factor-alpha-induced cyclooxygenase-2 promoter activation. J Biol Chem. 2003;278:4770–4777. doi: 10.1074/jbc.M209286200. [DOI] [PubMed] [Google Scholar]
- 59.Kiernan R, Bres V, Ng RW, Coudart MP, El Messaoudi S, Sardet C, Jin DY, Emiliani S, Benkirane M. Post-activation turn-off of NF-kappa B-dependent transcription is regulated by acetylation of p65. J Biol Chem. 2003;278:2758–2766. doi: 10.1074/jbc.M209572200. [DOI] [PubMed] [Google Scholar]
- 60.Chen LF, Mu Y, Greene WC. Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-kappaB. EMBO J. 2002;21:6539–6548. doi: 10.1093/emboj/cdf660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen G, Goeddel DV. TNF-R1 signaling: a beautiful pathway. Science. 2002;296:1634–1635. doi: 10.1126/science.1071924. [DOI] [PubMed] [Google Scholar]
- 62.Ravi R, Bedi A, Fuchs E, Bedi J. CD95 (Fas)-induced caspase-mediated proteolysis of NF-kappaB. Cancer Res. 1998;58:882–886. [PubMed] [Google Scholar]
- 63.Barkett M, Gilmore TD. Control of apoptosis by Rel/NF-kappaB transcription factors. Oncogene. 1999;18:6910–6924. doi: 10.1038/sj.onc.1203238. [DOI] [PubMed] [Google Scholar]
- 64.Deveraux QL, Reed JC. IAP family proteins—suppressors of apoptosis. Genes Dev. 1999;13:239–252. doi: 10.1101/gad.13.3.239. [DOI] [PubMed] [Google Scholar]
- 65.Jung M, Dritschilo A. NF-kappa B signaling pathway as a target for human tumor radiosensitization. Semin Radiat Oncol. 2001;11:346–351. doi: 10.1053/srao.2001.26034. [DOI] [PubMed] [Google Scholar]
- 66.Zhou D, Brown SA, Yu T, Chen G, Barve S, Kang BC, Thompson JS. A high dose of ionizing radiation induces tissue-specific activation of nuclear factor-kappaB in vivo. Radiat Res. 1999;151:703–709. [PubMed] [Google Scholar]
- 67.Guttridge DC, Albanese C, Reuther JY, Pestell RG, Baldwin AS., Jr NF-kappaB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol Cell Biol. 1999;19:5785–5799. doi: 10.1128/mcb.19.8.5785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ozeki M, Tamae D, Hou DX, Wang T, Lebon T, Spitz DR, Li JJ. Response of cyclin B1 to ionizing radiation: regulation by NF-kappaB and mitochondrial antioxidant enzyme MnSOD. Anticancer Res. 2004;24:2657–2663. [PMC free article] [PubMed] [Google Scholar]
- 69.Smirnov AS, Ruzov AS, Budanov AV, Prokhortchouk AV, Ivanov AV, Prokhortchouk EB. High constitutive level of NF-kappaB is crucial for viability of adenocarcinoma cells. Cell Death Differ. 2001;8:621–630. doi: 10.1038/sj.cdd.4400853. [DOI] [PubMed] [Google Scholar]
- 70.Hallahan DE, Spriggs DR, Beckett MA, Kufe DW, Weichselbaum RR. Increased tumor necrosis factor alpha mRNA after cellular exposure to ionizing radiation. Proc Natl Acad Sci U S A. 1989;86:10104–10107. doi: 10.1073/pnas.86.24.10104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen M, Quintans J, Fuks Z, Thompson C, Kufe DW, Weichselbaum RR. Suppression of Bcl-2 messenger RNA production may mediate apoptosis after ionizing radiation, tumor necrosis factor alpha, and ceramide. Cancer Res. 1995;55:991–994. [PubMed] [Google Scholar]
- 72.Blonska M, You Y, Geleziunas R, Lin X. Restoration of NF-kappaB activation by tumor necrosis factor alpha receptor complex-targeted MEKK3 in receptor-interacting protein-deficient cells. Mol Cell Biol. 2004;24:10757–10765. doi: 10.1128/MCB.24.24.10757-10765.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rossi A, Kapahi P, Natoli G, Takahashi T, Chen Y, Karin M, Santoro MG. Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IkappaB kinase. Nature. 2000;403:103–108. doi: 10.1038/47520. [DOI] [PubMed] [Google Scholar]
- 74.Osborn L, Kunkel S, Nabel GJ. Tumor necrosis factor alpha and interleukin 1 stimulate the human immunodeficiency virus enhancer by activation of the nuclear factor kappa B. Proc Natl Acad Sci U S A. 1989;86:2336–2340. doi: 10.1073/pnas.86.7.2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Baeuerle PA, Baltimore D. NF-kappa B: ten years after. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 76.Inoue J, Ishida T, Tsukamoto N, Kobayashi N, Naito A, Azuma S, Yamamoto T. Tumor necrosis factor receptor-associated factor (TRAF) family: adapter proteins that mediate cytokine signaling. Exp Cell Res. 2000;254:14–24. doi: 10.1006/excr.1999.4733. [DOI] [PubMed] [Google Scholar]
- 77.Lee SY, Choi Y. TRAF-interacting protein (TRIP): a novel component of the tumor necrosis factor receptor (TNFR)- and CD30-TRAF signaling complexes that inhibits TRAF2-mediated NF-kappaB activation. J Exp Med. 1997;185:1275–1285. doi: 10.1084/jem.185.7.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ling L, Cao Z, Goeddel DV. NF-kappaB-inducing kinase activates IKK-alpha by phosphorylation of Ser-176. Proc Natl Acad Sci U S A. 1998;95:3792–3797. doi: 10.1073/pnas.95.7.3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bradbury CM, Markovina S, Wei SJ, Rene LM, Zoberi I, Horikoshi N, Gius D. Indomethacin-induced radiosensitization and inhibition of ionizing radiation-induced NF-kappaB activation in HeLa cells occur via a mechanism involving p38 MAP kinase. Cancer Res. 2001;61:7689–7696. [PubMed] [Google Scholar]
- 80.Dent P, Yacoub A, Fisher PB, Hagan MP, Grant S. MAPK pathways in radiation responses. Oncogene. 2003;22:5885–5896. doi: 10.1038/sj.onc.1206701. [DOI] [PubMed] [Google Scholar]
- 81.Wang T, Hu YC, Dong S, Fan M, Tamae D, Ozeki M, Gao Q, Gius D, Li JJ. Co-activation of ERK, NF-kappaB, and GADD45beta in response to ionizing radiation. J Biol Chem. 2005;280:12593–12601. doi: 10.1074/jbc.M410982200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Javelaud D, Besancon F. NF-kappa B activation results in rapid inactivation of JNK in TNF alpha-treated Ewing sarcoma cells: a mechanism for the anti-apoptotic effect of NF-kappa B. Oncogene. 2001;20:4365–4372. doi: 10.1038/sj.onc.1204570. [DOI] [PubMed] [Google Scholar]
- 83.Javelaud D, Wietzerbin J, Delattre O, Besancon F. Induction of p21Waf1/Cip1 by TNFalpha requires NF-kappaB activity and antagonizes apoptosis in Ewing tumor cells. Oncogene. 2000;19:61–68. doi: 10.1038/sj.onc.1203246. [DOI] [PubMed] [Google Scholar]
- 84.Jin R, De Smaele E, Zazzeroni F, Nguyen DU, Papa S, Jones J, Cox C, Gelinas C, Franzoso G. Regulation of the gadd45beta promoter by NF-kappaB. DNA Cell Biol. 2002;21:491–503. doi: 10.1089/104454902320219059. [DOI] [PubMed] [Google Scholar]
- 85.Papa S, Zazzeroni F, Bubici C, Jayawardena S, Alvarez K, Matsuda S, Nguyen DU, Pham CG, Nelsbach AH, Melis T, Smaele ED, Tang WJ, D’Adamio L, Franzoso G. Gadd45 beta mediates the NF-kappa B suppression of JNK signalling by targeting MKK7/JNKK2. Nat Cell Biol. 2004;6:146–153. doi: 10.1038/ncb1093. [DOI] [PubMed] [Google Scholar]
- 86.Tang D, Wu D, Hirao A, Lahti JM, Liu L, Mazza B, Kidd VJ, Mak TW, Ingram AJ. ERK activation mediates cell cycle arrest and apoptosis after DNA damage independently of p53. J Biol Chem. 2002;277:12710–12717. doi: 10.1074/jbc.M111598200. [DOI] [PubMed] [Google Scholar]
- 87.Hara T, Namba H, Yang TT, Nagayama Y, Fukata S, Kuma K, Ishikawa N, Ito K, Yamashita S. Ionizing radiation activates c-Jun NH2-terminal kinase (JNK/SAPK) via a PKC-dependent pathway in human thyroid cells. Biochem Biophys Res Commun. 1998;244:41–44. doi: 10.1006/bbrc.1998.8210. [DOI] [PubMed] [Google Scholar]
- 88.Persons DL, Yazlovitskaya EM, Cui W, Pelling JC. Cisplatin-induced activation of mitogen-activated protein kinases in ovarian carcinoma cells: inhibition of extracellular signal-regulated kinase activity increases sensitivity to cisplatin. Clin Cancer Res. 1999;5:1007–1014. [PubMed] [Google Scholar]
- 89.Suzuki K, Kodama S, Watanabe M. Extremely low-dose ionizing radiation causes activation of mitogen-activated protein kinase pathway and enhances proliferation of normal human diploid cells. Cancer Res. 2001;61:5396–5401. [PubMed] [Google Scholar]
- 90.Reardon DB, Contessa JN, Mikkelsen RB, Valerie K, Amir C, Dent P, Schmidt-Ullrich RK. Dominant negative EGFR-CD533 and inhibition of MAPK modify JNK1 activation and enhance radiation toxicity of human mammary carcinoma cells. Oncogene. 1999;18:4756–4766. doi: 10.1038/sj.onc.1202849. [DOI] [PubMed] [Google Scholar]
- 91.Mandic A, Viktorsson K, Heiden T, Hansson J, Shoshan MC. The MEK1 inhibitor PD98059 sensitizes C8161 melanoma cells to cisplatin-induced apoptosis. Melanoma Res. 2001;11:11–19. doi: 10.1097/00008390-200102000-00002. [DOI] [PubMed] [Google Scholar]
- 92.Smalley KS, Eisen TG. Farnesyl thiosalicylic acid inhibits the growth of melanoma cells through a combination of cytostatic and pro-apoptotic effects. Int J Cancer. 2002;98:514–522. doi: 10.1002/ijc.10213. [DOI] [PubMed] [Google Scholar]
- 93.Haffty BG, Brown F, Carter D, Flynn S. Evaluation of HER-2 neu oncoprotein expression as a prognostic indicator of local recurrence in conservatively treated breast cancer: a case-control study. Int J Radiat Oncol Biol Phys. 1996;35:751–757. doi: 10.1016/0360-3016(96)00150-2. [DOI] [PubMed] [Google Scholar]
- 94.Hicks DG, Tubbs RR. Assessment of the HER2 status in breast cancer by fluorescence in situ hybridization: a technical review with interpretive guidelines. Hum Pathol. 2005;36:250–261. doi: 10.1016/j.humpath.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 95.Carraway KL, III, Weber JL, Unger MJ, Ledesma J, Yu N, Gassmann M, Lai C. Neuregulin-2, a new ligand of ErbB3/ErbB4-receptor tyrosine kinases. Nature. 1997;387:512–516. doi: 10.1038/387512a0. [DOI] [PubMed] [Google Scholar]
- 96.Yu D, Hung MC. Overexpression of ErbB2 in cancer and ErbB2-targeting strategies. Oncogene. 2000;19:6115–6121. doi: 10.1038/sj.onc.1203972. [DOI] [PubMed] [Google Scholar]
- 97.Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev, Mol Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 98.Kurokawa H, Arteaga CL. Inhibition of erbB receptor (HER) tyrosine kinases as a strategy to abrogate antiestrogen resistance in human breast cancer. Clin Cancer Res. 2001;7:4436s–4442s. discussion 4411s-4412s. [PubMed] [Google Scholar]
- 99.Galang CK, Garcia-Ramirez J, Solski PA, Westwick JK, Der CJ, Neznanov NN, Oshima RG, Hauser CA. Oncogenic Neu/ErbB-2 increases ets, AP-1, and NF-kappaB-dependent gene expression, and inhibiting ets activation blocks Neu-mediated cellular transformation. J Biol Chem. 1996;271:7992–7998. doi: 10.1074/jbc.271.14.7992. [DOI] [PubMed] [Google Scholar]
- 100.Pianetti S, Arsura M, Romieu-Mourez R, Coffey RJ, Sonenshein GE. Her-2/neu overexpression induces NF-kappaB via a PI3-kinase/Akt pathway involving calpain-mediated degradation of IkappaB-alpha that can be inhibited by the tumor suppressor PTEN. Oncogene. 2001;20:1287–1299. doi: 10.1038/sj.onc.1204257. [DOI] [PubMed] [Google Scholar]
- 101.Yang HY, Zhou BP, Hung MC, Lee MH. Oncogenic signals of HER-2/neu in regulating the stability of the cyclin-dependent kinase inhibitor p27. J Biol Chem. 2000;275:24735–24739. doi: 10.1074/jbc.C000147200. [DOI] [PubMed] [Google Scholar]
- 102.Weichselbaum RR, Hallahan D, Fuks Z, Kufe D. Radiation induction of immediate early genes: effectors of the radiation-stress response. Int J Radiat Oncol Biol Phys. 1994;30:229–234. doi: 10.1016/0360-3016(94)90539-8. [DOI] [PubMed] [Google Scholar]
- 103.Maity A, Kao GD, Muschel RJ, McKenna WG. Potential molecular targets for manipulating the radiation response. Int J Radiat Oncol Biol Phys. 1997;37:639–653. doi: 10.1016/s0360-3016(96)00598-6. [DOI] [PubMed] [Google Scholar]
- 104.Wolff S. Are radiation-induced effects hormetic. Science. 1989;245:575–621. doi: 10.1126/science.2762808. [DOI] [PubMed] [Google Scholar]
- 105.Waldman T, Zhang Y, Dillehay L, Yu J, Kinzler K, Vogelstein B, Williams J. Cell-cycle arrest versus cell death in cancer therapy. Nat Med. 1997;3:1034–1036. doi: 10.1038/nm0997-1034. [DOI] [PubMed] [Google Scholar]
- 106.Schmidt-Ullrich RK, Dent P, Grant S, Mikkelsen RB, Valerie K. Signal transduction and cellular radiation responses. Radiat Res. 2000;153:245–257. doi: 10.1667/0033-7587(2000)153[0245:stacrr]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 107.Wolff S. The adaptive response in radiobiology: evolving insights and implications. Environ Health Perspect. 1998;106(Suppl 1):277–283. doi: 10.1289/ehp.98106s1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Feinendegen LE. The role of adaptive responses following exposure to ionizing radiation. Hum Exp Toxicol. 1999;18:426–432. doi: 10.1191/096032799678840309. [DOI] [PubMed] [Google Scholar]
- 109.Feinendegen LE. Reactive oxygen species in cell responses to toxic agents. Hum Exp Toxicol. 2002;21:85–90. doi: 10.1191/0960327102ht216oa. [DOI] [PubMed] [Google Scholar]
- 110.Jung M, Zhang Y, Lee S, Dritschilo A. Correction of radiation sensitivity in ataxia telangiectasia cells by a truncated I kappa B-alpha. Science. 1995;268:1619–1621. doi: 10.1126/science.7777860. [DOI] [PubMed] [Google Scholar]
- 111.Wu ZH, Shi Y, Tibbetts RS, Miyamoto S. Molecular linkage between the kinase ATM and NF-kappaB signaling in response to genotoxic stimuli. Science. 2006;311:1141–1146. doi: 10.1126/science.1121513. [DOI] [PubMed] [Google Scholar]
- 112.Ashburner BP, Shackelford RE, Baldwin AS, Jr, Paules RS. Lack of involvement of ataxia telangiectasia mutated (ATM) in regulation of nuclear factor-kappaB (NF-kappaB) in human diploid fibroblasts. Cancer Res. 1999;59:5456–5460. [PubMed] [Google Scholar]
- 113.Li N, Banin S, Ouyang H, Li GC, Courtois G, Shiloh Y, Karin M, Rotman G. ATM is required for IkappaB kinase (IKKk) activation in response to DNA double strand breaks. J Biol Chem. 2001;276:8898–8903. doi: 10.1074/jbc.M009809200. [DOI] [PubMed] [Google Scholar]
- 114.Basu S, Rosenzweig KR, Youmell M, Price BD. The DNA-dependent protein kinase participates in the activation of NF kappa B following DNA damage. Biochem Biophys Res Commun. 1998;247:79–83. doi: 10.1006/bbrc.1998.8741. [DOI] [PubMed] [Google Scholar]
- 115.Liu JJ, Nakajima K, Hirano T, Yang-Yen HF. Activation of Stat3 by v-Src is through a Ras-independent pathway. J Biomed Sci. 1998;5:446–450. doi: 10.1007/BF02255934. [DOI] [PubMed] [Google Scholar]
- 116.Chu W, Gong X, Li Z, Takabayashi K, Ouyang H, Chen Y, Lois A, Chen DJ, Li GC, Karin M, Raz E. DNA-PKcs is required for activation of innate immunity by immunostimulatory DNA. Cell. 2000;103:909–918. doi: 10.1016/s0092-8674(00)00194-x. [DOI] [PubMed] [Google Scholar]
- 117.Raju U, Gumin GJ, Tofilon PJ. NF kappa B activity and target gene expression in the rat brain after one and two exposures to ionizing radiation. Radiat Oncol Investig. 1999;7:145–152. doi: 10.1002/(SICI)1520-6823(1999)7:3<145::AID-ROI2>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 118.Baeuml H, Behrends U, Peter RU, Mueller S, Kammerbauer C, Caughman SW, Degitz K. Ionizing radiation induces, via generation of reactive oxygen intermediates, intercellular adhesion molecule-1 (ICAM-1) gene transcription and NF kappa B-like binding activity in the ICAM-1 transcriptional regulatory region. Free Radic Res. 1997;27:127–142. doi: 10.3109/10715769709097846. [DOI] [PubMed] [Google Scholar]
- 119.Lin PS, Ho KC, Sung SJ, Tsai S. Cytotoxicity and manganese superoxide dismutase induction by tumor necrosis factor-alpha and ionizing radiation in MCF-7 human breast carcinoma cells. Lymphokine Cytokine Res. 1993;12:303–308. [PubMed] [Google Scholar]
- 120.Kiningham KK, Xu Y, Daosukho C, Popova B, St Clair DK. Nuclear factor kappaB-dependent mechanisms coordinate the synergistic effect of PMA and cytokines on the induction of superoxide dismutase 2. Biochem J. 2001;353:147–156. [PMC free article] [PubMed] [Google Scholar]
- 121.Dumic J, Lauc G, Flogel M. Expression of galectin-3 in cells exposed to stress-roles of jun and NF-kappaB. Cell Physiol Biochem. 2000;10:149–158. doi: 10.1159/000016345. [DOI] [PubMed] [Google Scholar]
- 122.Hallahan DE, Staba-Hogan MJ, Virudachalam S, Kolchinsky A. X-ray-induced P-selectin localization to the lumen of tumor blood vessels. Cancer Res. 1998;58:5216–5220. [PubMed] [Google Scholar]
- 123.Iwanaga M, Mori K, Iida T, Urata Y, Matsuo T, Yasunaga A, Shibata S, Kondo T. Nuclear factor κB dependent induction of γ-glutamylcysteine synthetase by ionizing radiation in T98G human glioblastoma cells. Free Radic Biol Med. 1998;24:1256–1268. doi: 10.1016/s0891-5849(97)00443-7. [DOI] [PubMed] [Google Scholar]
- 124.Amundson SA, Do KT, Fornace AJ., Jr Induction of stress genes by low doses of gamma rays. Radiat Res. 1999;152:225–231. [PubMed] [Google Scholar]
- 125.Maity A, McKenna WG, Muschel RJ. The molecular basis for cell cycle delays following ionizing radiation: a review. Radiother Oncol. 1994;31:1–13. doi: 10.1016/0167-8140(94)90408-1. [DOI] [PubMed] [Google Scholar]
- 126.Almasan A. Cellular commitment to radiation-induced apoptosis. Radiat Res. 2000;153:347–350. doi: 10.1667/0033-7587(2000)153[0347:cctria]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 127.Muschel RJ, Zhang HB, Iliakis G, McKenna WG. Cyclin B expression in HeLa cells during the G2 block induced by ionizing radiation. Cancer Res. 1991;51:5113–5137. [PubMed] [Google Scholar]
- 128.Metting NF, Little JB. Transient failure to dephosphorylate the cdc2–cyclin B1 complex accompanies radiation-induced G2-phase arrest in HeLa cells. Radiat Res. 1995;143:286–292. [PubMed] [Google Scholar]
- 129.Kao GD, McKenna WG, Maity A, Blank K, Muschel RJ. Cyclin B1 availability is a rate-limiting component of the radiation-induced G2 delay in HeLa cells. Cancer Res. 1997;57:753–758. [PubMed] [Google Scholar]
- 130.Azzam EI, de Toledo SM, Gooding T, Little JB. Intercellular communication is involved in the bystander regulation of gene expression in human cells exposed to very low fluences of alpha particles. Radiat Res. 1998;150:497–504. [PubMed] [Google Scholar]
- 131.Taylor WR, Stark GR. Regulation of the G2/M transition by p53. Oncogene. :0.803–1815. doi: 10.1038/sj.onc.1204252. [DOI] [PubMed] [Google Scholar]
- 132.Porter LA, Cukier IH, Lee JM. Nuclear localization of cyclin B1 regulates DNA damage-induced apoptosis. Blood. 2003;101:1928–1933. doi: 10.1182/blood-2002-04-1103. [DOI] [PubMed] [Google Scholar]
- 133.Theron T, Bohm L. Cyclin B1 expression in response to abrogation of the radiation-induced G2/M block in HeLa cells. Cell Prolif. 1998;31:49–57. doi: 10.1046/j.1365-2184.1998.00107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Maity A, Hwang A, Janss A, Phillips P, McKenna WG, Muschel RJ. Delayed cyclin B1 expression during the G2 arrest following DNA damage. Oncogene. 1996;13:1647–1657. [PubMed] [Google Scholar]
- 135.Soria JC, Jang SJ, Khuri FR, Hassan K, Liu D, Hong WK, Mao L. Overexpression of cyclin B1 in early-stage non-small cell lung cancer and its clinical implication. Cancer Res. 2000;60:4000–4004. [PubMed] [Google Scholar]
- 136.Hassan KA, El-Naggar AK, Soria JC, Liu D, Hong WK, Mao L. Clinical significance of cyclin B1 protein expression in squamous cell carcinoma of the tongue. Clin Cancer Res. 2001;7:2458–2462. [PubMed] [Google Scholar]
- 137.Hassan KA, Ang KK, El-Naggar AK, Story MD, Lee JI, Liu D, Hong WK, Mao L. Cyclin B1 overexpression and resistance to radiotherapy in head and neck squamous cell carcinoma. Cancer Res. 2002;62:6414–6417. [PubMed] [Google Scholar]
- 138.Wang X. The expanding role of mitochondria in apoptosis. Genes Dev. 2001;15:2922–2933. [PubMed] [Google Scholar]
- 139.Li P, Nijhawan D, Wang X. Mitochondrial activation of apoptosis. Cell. 2004;116:S57–59. doi: 10.1016/s0092-8674(04)00031-5. [DOI] [PubMed] [Google Scholar]
- 140.Adler V, Yin Z, Tew KD, Ronai Z. Role of redox potential and reactive oxygen species in stress signaling. Oncogene. 1999;18:6104–6111. doi: 10.1038/sj.onc.1203128. [DOI] [PubMed] [Google Scholar]
- 141.Hunt CR, Sim JE, Sullivan SJ, Featherstone T, Golden W, Von Kapp-Herr C, Hock RA, Gomez RA, Parsian AJ, Spitz DR. Genomic instability and catalase gene amplification induced by chronic exposure to oxidative stress. Cancer Res. 1998;58:3986–3992. [PubMed] [Google Scholar]
- 142.Wong GH. Protective roles of cytokines against radiation: induction of mitochondrial MnSOD. Biochim Biophys Acta. 1995;1271:205–209. doi: 10.1016/0925-4439(95)00029-4. [DOI] [PubMed] [Google Scholar]
- 143.Sukhatme VP, Cao XM, Chang LC, Tsai-Morris CH, Stamenkovich D, Ferreira PC, Cohen DR, Edwards SA, Shows TB, Curran T, et al. A zinc finger-encoding gene coregulated with c-fos during growth and differentiation, and after cellular depolarization. Cell. 1988;53:37–43. doi: 10.1016/0092-8674(88)90485-0. [DOI] [PubMed] [Google Scholar]
- 144.Gashler AL, Swaminathan S, Sukhatme VP. A novel repression module, an extensive activation domain, and a bipartite nuclear localization signal defined in the immediate-early transcription factor Egr-1. Mol Cell Biol. 1993;13:4556–4571. doi: 10.1128/mcb.13.8.4556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Datta R, Taneja N, Sukhatme VP, Qureshi SA, Weichselbaum R, Kufe DW. Reactive oxygen intermediates target CC(A/T)6GG sequences to mediate activation of the early growth response 1 transcription factor gene by ionizing radiation. Proc Natl Acad Sci U S A. 1993;90:2419–2422. doi: 10.1073/pnas.90.6.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wei YH, Lu CY, Lee HC, Pang CY, Ma YS. Oxidative damage and mutation to mitochondrial DNA and age-dependent decline of mitochondrial respiratory function. Ann NY Acad Sci. 1998;854:155–170. doi: 10.1111/j.1749-6632.1998.tb09899.x. [DOI] [PubMed] [Google Scholar]
- 147.Bandy B, Davison AJ. Mitochondrial mutations may increase oxidative stress: implications for carcinogenesis and aging. Free Radic Biol Med. 1990;8:523–539. doi: 10.1016/0891-5849(90)90152-9. [DOI] [PubMed] [Google Scholar]
- 148.Rhee SG. Redox signaling: hydrogen peroxide as intracellular messenger. Exp Mol Med. 1999;31:53–59. doi: 10.1038/emm.1999.9. [DOI] [PubMed] [Google Scholar]
- 149.Das KC, Lewis-Molock Y, White CW. Thiol modulation of TNF alpha and IL-1 induced MnSOD gene expression and activation of NF-kappa B. Mol Cell Biochem. 1995;148:45–57. doi: 10.1007/BF00929502. [DOI] [PubMed] [Google Scholar]
- 150.Grdina DJ, Murley JS, Kataoka Y, Calvin DP. Differential activation of nuclear transcription factor kappaB, gene expression, and proteins by amifostine’s free thiol in human microvascular endothelial and glioma cells. Semin Radiat Oncol. 2002;12:103–111. doi: 10.1053/srao.2002.31383. [DOI] [PubMed] [Google Scholar]
- 151.Murley JS, Kataoka Y, Cao D, Li JJ, Oberley LW, Grdina DJ. Delayed radioprotection by NFkappaB-mediated induction of Sod2 (MnSOD) in SA-NH tumor cells after exposure to clinically used thiol-containing drugs. Radiat Res. 2004;162:536–546. doi: 10.1667/rr3256. [DOI] [PubMed] [Google Scholar]
- 152.Wong GH, Elwell JH, Oberley LW, Goeddel DV. Manganous superoxide dismutase is essential for cellular resistance to cytotoxicity of tumor necrosis factor. Cell. 1989;58:923–931. doi: 10.1016/0092-8674(89)90944-6. [DOI] [PubMed] [Google Scholar]
- 153.Siemankowski LM, Morreale J, Briehl MM. Antioxidant defenses in the TNF-treated MCF-7 cells: selective increase in MnSOD. Free Radic Biol Med. 1999;26:919–924. doi: 10.1016/s0891-5849(98)00273-1. [DOI] [PubMed] [Google Scholar]
- 154.Oberley LW, St Clair DK, Autor AP, Oberley TD. Increase in manganese superoxide dismutase activity in the mouse heart after X-irradiation. Arch Biochem Biophys. 1987;254:69–80. doi: 10.1016/0003-9861(87)90082-8. [DOI] [PubMed] [Google Scholar]
- 155.Summers RW, Maves BV, Reeves RD, Arjes LJ, Oberley LW. Irradiation increases superoxide dismutase in rat intestinal smooth muscle. Free Radic Biol Med. 1989;6:261–270. doi: 10.1016/0891-5849(89)90053-1. [DOI] [PubMed] [Google Scholar]
- 156.Epperly M, Bray J, Kraeger S, Zwacka R, Engelhardt J, Travis E, Greenberger J. Prevention of late effects of irradiation lung damage by manganese superoxide dismutase gene therapy. Gene Ther. 1998;5:196–208. doi: 10.1038/sj.gt.3300580. [DOI] [PubMed] [Google Scholar]
- 157.Epperly MW, Defilippi S, Sikora C, Gretton J, Kalend A, Greenberger JS. Intratracheal injection of manganese superoxide dismutase (MnSOD) plasmid/liposomes protects normal lung but not orthotopic tumors from irradiation. Gene Ther. 2000;7:1011–1018. doi: 10.1038/sj.gt.3301207. [DOI] [PubMed] [Google Scholar]
- 158.Williams MD, Van Remmen H, Conrad CC, Huang TT, Epstein CJ, Richardson A. Increased oxidative damage is correlated to altered mitochondrial function in heterozygous manganese superoxide dismutase knockout mice. J Biol Chem. 1998;273:28510–28515. doi: 10.1074/jbc.273.43.28510. [DOI] [PubMed] [Google Scholar]
- 159.Hussain SP, Amstad P, He P, Robles A, Lupold S, Kaneko I, Ichimiya M, Sengupta S, Mechanic L, Okamura S, Hofseth LJ, Moake M, Nagashima M, Forrester KS, Harris CC. p53-induced up-regulation of MnSOD and GPx but not catalase increases oxidative stress and apoptosis. Cancer Res. 2004;64:2350–2356. doi: 10.1158/0008-5472.can-2287-2. [DOI] [PubMed] [Google Scholar]
- 160.Wan XS, Devalaraja MN, St Clair DK. Molecular structure and organization of the human manganese superoxide dismutase gene. DNA Cell Biol. 1994;13:1127–1136. doi: 10.1089/dna.1994.13.1127. [DOI] [PubMed] [Google Scholar]
- 161.Duttaroy A, Parkes T, Emtage P, Kirby K, Boulianne GL, Wang X, Hilliker AJ, Phillips JP. The manganese superoxide dismutase gene of Drosophila: structure, expression, and evidence for regulation by MAP kinase. DNA Cell Biol. 1997;16:391–399. doi: 10.1089/dna.1997.16.391. [DOI] [PubMed] [Google Scholar]
- 162.Xu Y, Kiningham KK, Devalaraja MN, Yeh CC, Majima H, Kasarskis EJ, St Clair DK. An intronic NF-kappaB element is essential for induction of the human manganese superoxide dismutase gene by tumor necrosis factor-alpha and interleukin-1beta. DNA Cell Biol. 1999;18:709–722. doi: 10.1089/104454999314999. [DOI] [PubMed] [Google Scholar]