

Abstract
Steep rectification in IRK1 (Kir2.1) inward-rectifier K+ channels reflects strong voltage dependence (valence of ∼5) of channel block by intracellular cationic blockers such as the polyamine spermine. The observed voltage dependence primarily results from displacement, by spermine, of up to five K+ ions across the narrow K+ selectivity filter, along which the transmembrane voltage drops steeply. Spermine first binds, with modest voltage dependence, at a shallow site where it encounters the innermost K+ ion and impedes conduction. From there, spermine can proceed to a deeper site, displacing several more K+ ions and thereby producing most of the observed voltage dependence. Since in the deeper blocked state the leading amine group of spermine reaches into the cavity region (internal to the selectivity filter) and interacts with residue D172, its trailing end is expected to be near M183. Here, we found that mutation M183A indeed affected the deeper blocked state, which supports the idea that spermine is located in the region lined by the M2 and not deep in the narrow K+ selectivity filter. As to the shallower site whose location has been unknown, we note that in the crystal structure of homologous GIRK1 (Kir3.1), four aromatic side chains of F255, one from each of the four subunits, constrict the intracellular end of the pore to ∼10 Å. For technical simplicity, we used tetraethylammonium (TEA) as an initial probe to test whether the corresponding residue in IRK1, F254, forms the shallower site. We found that replacing the aromatic side chain with an aliphatic one not only lowered TEA affinity of the shallower site ∼100-fold but also eliminated the associated voltage dependence and, furthermore, confirmed that similar effects occurred also for spermine. These results establish the evidence for physically separate, sequential ion-binding loci along the long inner pore of IRK1, and strongly suggest that the aromatic side chains of F254 underlie the likely innermost binding locus for both blocker and K+ ions in the cytoplasmic pore.
INTRODUCTION
Inward-rectifier K+ (Kir) channels are so named because they can conduct much larger inward currents at membrane voltages (Vm) negative to the K+ equilibrium potential (EK) than outward currents at Vm positive to EK, even as the K+ concentrations ([K+]) on both sides of the membrane are made equal experimentally (Katz, 1949; Hodgkin and Horowicz, 1959; Hagiwara and Takahashi, 1974; Hagiwara et al., 1976). Physiologically, since Vm is usually positive to EK, net K+ currents through Kir channels are outward. The small outward currents enable Kir channels to accomplish numerous important biological tasks (Hille, 2001).
Rectification in Kir channels reflects voltage dependence of pore block by intracellular blockers such as Mg2+ and polyamines (Horie et al.,1987; Matsuda et al., 1987; Vandenberg, 1987; Ficker et al., 1994; Lopatin et al., 1994; Fakler et al., 1995). The voltage dependence of IRK1 (Kir2.1) block by the long polyamine spermine is so strong that the associated overall valence (Z) is about five (Lopatin and Nichols, 1996; Guo and Lu, 2000a,b; Guo and Lu, 2003; Xie et al., 2003; Shin and Lu, 2005). This remarkably large valence primarily results from displacement, by spermine traveling along the inner pore, of up to five K+ ions across the narrow K+ selectivity filter (Guo and Lu, 2003; Guo et al., 2003; Shin and Lu, 2005; for review see Lu, 2004), where the drop of Vm mainly takes place. The 50–70-Å-long inner pore, extending from the intracellular end of the pore to the intracellular end of the selectivity filter, is formed by cytoplasmic termini and the second transmembrane (M2) segment (Doyle et al., 1998; Nishida and MacKinnon, 2002; Kuo et al., 2003; Pegan et al., 2005). Recently, our group showed that spermine and decane-bis-trimethylammonium bind to IRK1 in (at least) two sequential steps, with increasing numbers of K+ ions displaced (Shin and Lu, 2005). Consequently, the apparent voltage dependence of channel block increases with membrane depolarization: the (limiting) weakly and strongly voltage-dependent blocking phases reflect the properties of channels with a blocker located at the shallow and deep locus, respectively. At the shallower site whose location has been unknown, spermine blocks ion conduction with modest voltage dependence as it encounters/displaces the innermost K+ ion. To identify the residues that form the shallower site we used TEA, for its relatively structural simplicity, as an initial probe, and then further examined the identified residues with spermine. Spermine progresses from the shallower to the deeper location so that its leading end reaches the so-called “cavity” region, interacting with residue D172, displacing several K+ ions across the selectivity filter, and thereby producing the observed strong voltage dependence (Guo and Lu, 2003; Guo et al., 2003; Lu, 2004; Shin and Lu, 2005). In addition to that of D172 (Lu and MacKinnon, 1994; Stanfield et al., 1994; Wible et al., 1994), mutations of E224 and E299 are also known to affect spermine block (Taglialatela et al., 1995; Yang et al., 1995; Kubo and Murata, 2001; Xie et al., 2002, 2003; Guo and Lu, 2003; Guo et al., 2003). The distance between D172 and E224/E299, estimated from the KirBac1.1 crystal structure (Kuo et al., 2003), is ∼35 Å, which is significantly greater than the length of spermine (∼20 Å; the center-to-center distance between the nitrogen atoms at the two ends of an extended spermine molecule is ∼17 Å). Relatively modest effects on various amine blockers of replacing E224/E299 (versus D172) with neutral residues are consistent with a model where the blockers interact with the two glutamate residues (at least in part) via a through-space electrostatic mechanism (Guo and Lu, 2003; Guo et al., 2003; Lu, 2004; see Introduction of Shin and Lu, 2005). It is likely that the trailing end of spermine also interacts with other more closely located residues. Since in the deeper bound state the leading amine of spermine is located near D172, the trailing one is expected to be near M183, located three α-helical turns more intracellularly (each 3.6-residue turn results in 5.4-Å displacement). Therefore, we also tested whether a mutation at M183 affected spermine block in the deeper blocked state.
MATERIALS AND METHODS
Molecular Biology and Oocyte Preparation
The cDNA of IRK1 (Kubo et al., 1993) was subcloned in pGEM-HESS plasmid (Liman et al., 1992). All mutant cDNAs were obtained through PCR-based mutagenesis and confirmed by DNA sequencing. The cRNA was synthesized with T7 polymerase (Promega) using linearized cDNA as a template. Oocytes harvested from Xenopus laevis (Xenopus One) were incubated in a solution containing 82.5 mM NaCl, 2.5 mM KCl, 1.0 mM MgCl2, 5.0 mM HEPES (pH 7.6), and 2–4 mg/ml collagenase. The oocyte preparation was agitated at 80 rpm for 60–90 min. It was then rinsed thoroughly and stored in a solution containing 96 mM NaCl, 2.5 mM KCl, 1.8 mM CaCl2, 1.0 mM MgCl2, 5 mM HEPES (pH 7.6), and 50 μg/ml gentamicin. Defolliculated oocytes were selected and injected with RNA at least 2 and 16 h, respectively, after collagenase treatment. All oocytes were stored at 18°C.
Recordings and Solutions
Macroscopic currents were recorded from inside-out membrane patches of Xenopus oocytes heterologously expressing IRK1 channels using an Axopatch 200B amplifier (Axon Instruments, Inc.), filtered at 10 kHz, and sampled at 100 kHz using an analogue-to-digital converter (Digidata 1322A; Axon Instruments, Inc.) interfaced with a personal computer. pClamp8 software was used for amplifier control and data acquisition. Unless specified otherwise, during current recordings, the voltage across the membrane patch was first hyperpolarized from the 0 mV holding potential to −100 mV, and then stepped to various test voltages between −100 and 100 mV in 10-mV increments and back to 0 mV. Background leak current correction was performed as previously described (Lu and MacKinnon, 1994; Guo and Lu, 2000b). The recording solution contained (in mM): 5 K2EDTA, 10 “K2HPO4 + KH2PO4” in a ratio yielding pH 8.0, and sufficient KCl to bring total K+ concentration to 100 mM (Guo and Lu, 2002; see also Guo and Lu, 2000b). To reduce channel rundown, the intracellular solution contained 5 mM fluoride, 10 mM pyrophosphate, and 0.1 mM vanadate (Huang et al., 1998). All chemicals were purchased from Fluka Chemical Corp.
RESULTS
Variation of the Apparent Voltage Dependence of TEA Block with Membrane Voltage
Fig. 1 shows currents of IRK1 channels recorded in the absence or presence of two representative concentrations of intracellular TEA. For a given TEA concentration, TEA inhibits the outward current more than the inward current at the corresponding positive and negative voltages. The voltage dependence of channel inhibition renders the I-V curve inwardly rectifying (Fig. 2 A). To determine the apparent equilibrium dissociation constant (appKd), we plotted the normalized current versus the TEA concentration for four representative voltages (Fig. 2 B). The curves through the data are fits of an equation assuming that one TEA blocks one channel, yielding appKd. To quantify the voltage dependence of appKd, we plotted its natural logarithm (ln) against Vm (Fig. 2 C). The plot is clearly nonlinear: its slope increases with membrane depolarization. The increase with voltage of the voltage dependence itself is consistent with a model in which TEA also (like long-chain blockers) first binds at a shallower site, impeding K+ conduction, before reaching a deeper site favored by depolarization (Shin and Lu, 2005).
Figure 1.

Inhibition of IRK1 currents by TEA. Current traces were recorded from a single patch in the absence (control) or presence of TEA at the concentrations indicated. Currents were elicited by stepping membrane voltage from the 0 mV holding potential to −100 mV and then to test voltages between −100 mV and +100 mV in 10-mV increments before returning it to the holding potential. Dotted line indicates the zero current level.
Figure 2.
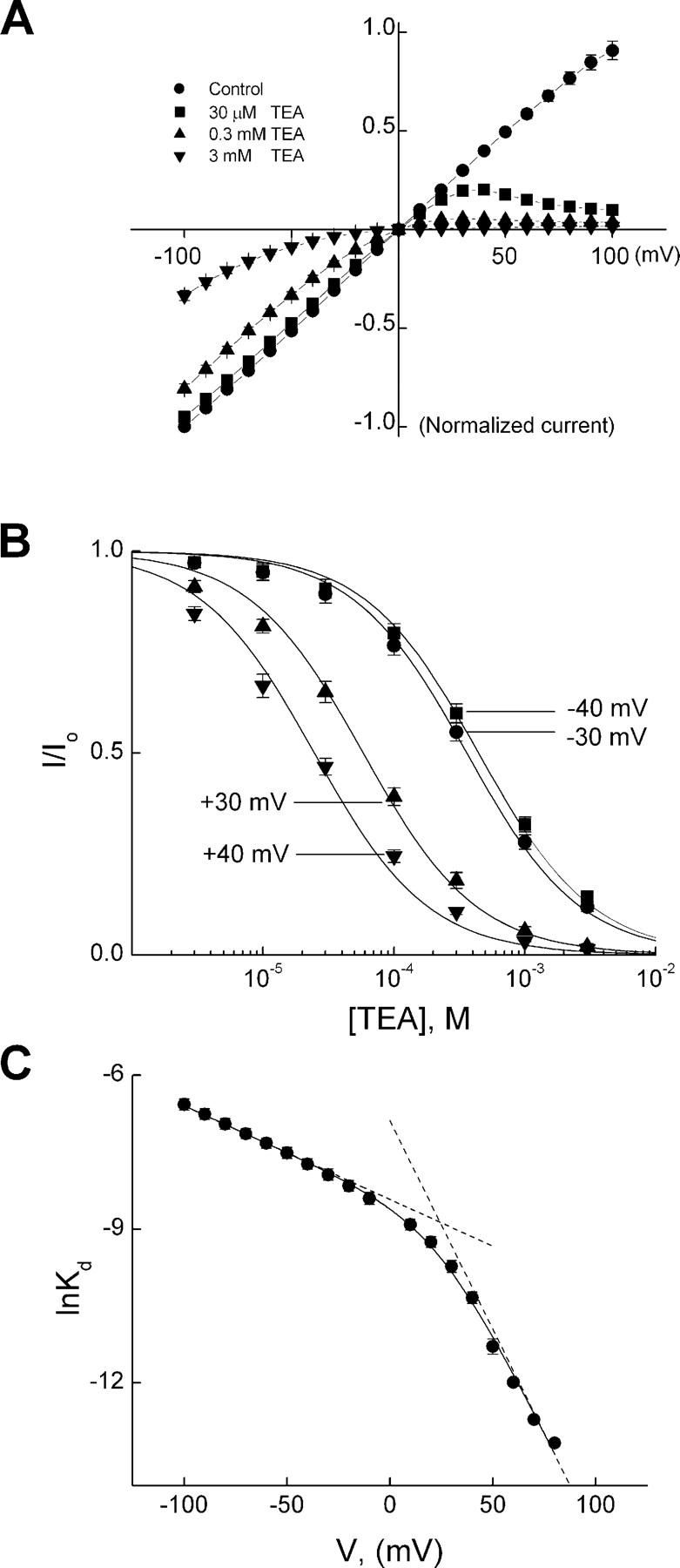
Voltage dependence of steady-state IRK1 block by TEA. (A) Averaged I-V curves (mean ± SEM; n = 6) determined at the end of test pulses in the absence (control) or presence of various concentrations of TEA. (B) The fraction of current not blocked is plotted against TEA concentration at the four representative voltages indicated. Curves through the data represent the equation I/Io = appKd/(appKd + [TEA]). (C) The natural logarithm of appKd is plotted against membrane voltage. The curve through the data is a fit of Eq. 1a, yielding K1 = 2.21 (±0.18) × 10−4 M and K2 = 4.65 ± 1.41 with valences Z1 = 0.47 ± 0.03 and Z2 = 1.61 ± 0.08 (mean ± SEM; n = 6). The dashed lines indicate the limiting slopes.
Effects of Mutations at E224 or E299 on TEA Block
Four pairs of COOH-terminal residues (E224 and E299) in IRK1 form a “ring” near the outer end of the “cytoplasmic” pore (Fig. 3; Nishida and MacKinnon, 2002; Kuo et al., 2003; Pegan et al., 2005). Since replacement of these glutamate residues with neutral ones is known to affect channel block by various cationic blockers (Taglialatela et al., 1995; Yang et al., 1995; Kubo and Murata, 2001; Guo and Lu, 2003; Guo et al., 2003; Xie et al., 2002, 2003), we examined its effect on TEA block. Fig. 4 A shows macroscopic currents of the E224 or E299 mutant channels, each recorded in the absence or presence of two representative concentrations of TEA. Both mutant channels, as previously noted, exhibit modest inward rectification even in the absence of intracellular blockers. To quantify the effects of the mutations on TEA block, we plotted lnappKd of the mutant (as well as wild-type) channels against Vm (Fig. 4 B). Since the mutant E224 and E299 channels exhibit only a shallow phase comparable to that of the wild type, the mutations must have dramatically disrupted the interaction of TEA with the deeper site.
Figure 3.

Crystal structure of cytoplasmic termini of GIRK1 (Nishida and MacKinnon, 2002). Shown on the left are two subunits of the cytoplasmic pore whose external end is on top. Side chains are highlighted for residues selenomethionine (Mse)308 (top, yellow), Ser225/Glu300 (middle, red), and Phe255 (bottom, pink), corresponding to Met307, Glu224/Glu299, and Phe254 in IRK1. Shown on the right are the center regions of cross sections through the tetrameric structure at the three levels indicated.
Figure 4.

Effects of E224G or E299S mutations on voltage-dependent channel block by TEA. (A) Current traces for each type of mutant channel were recorded from a single patch in the absence (control) or presence of TEA at the concentrations indicated. Dotted lines indicate the zero current level. (B) The natural logarithm of appKd for the mutant channels (determined as shown in Fig. 2 B) is plotted against membrane voltage. The data for wild-type channels (taken from Fig. 2 C) are plotted here for comparison. The lines through the data for mutant channels are fits of a Boltzmann function. The fits yield appKd = 2.47 (±0.57) × 10−4 M and valence appZ = 0.63 ± 0.01 (mean ± SEM; n = 5) for E224G, and appKd = 2.51 (±0.56) × 10−4 M and valence appZ = 0.60 ± 0.04 for E299S (n = 5).
Effects of Mutations at Residue F254 or D255 on TEA Block
Searching for the shallow site, we noticed that the intracellular end of GIRK1's cytoplasmic pore is constricted by four loops, one each from the four subunits. For example, the closest distance between the Van der Waals surfaces of opposite aromatic side chains of F255 within the loop is ∼10 Å (Fig. 3; Nishida and MacKinnon, 2002). We therefore tested whether the corresponding aromatic residue in IRK1, F254, is critical for TEA binding at the shallow site by replacing it with Ala, Val, or Tyr, and also the adjacent residue D255 with Ala. Fig. 5 shows currents of the mutant channels recorded in the absence or presence of intracellular TEA. Like wild-type channels, all four mutant channels are blocked by TEA in a voltage-dependent manner. However, unlike the wild type (Fig. 2 A), the F254A or F254V mutant channels exhibit little block of inward currents even in the presence of the same (3 mM) TEA concentration. The apparent absence of current block at negative voltages seems to be caused specifically by removal of F254's aromatic side chain, because the inward currents of the F254Y, or adjacent D255A, mutant channels remain blocked by TEA in a manner comparable to that of wild-type channels. (Kinetic consequences of the mutations may not be straightforward and are beyond the scope of the present study.)
Figure 5.

Inhibition by TEA of currents through channels with a mutation at F254 or D255. Current traces for each type of mutant channel were recorded from a single patch in the absence (control) or presence of TEA at the concentrations indicated. Dotted lines indicate the zero current level.
To further examine the effects of those mutations, we plotted lnappKd against Vm for wild-type and mutant channels in Fig. 6. As was the case for wild-type channels, the plots for the F254Y and D255A mutant channels exhibit a shallow phase in the negative voltage range and a steep phase in the positive range. For F254A or F254V mutant channels, while appKd at negative voltages becomes too large to be determined, the plot in the positive range is slightly less steep compared with the wild type. To gain further insight into the effects of removal of the aromatic side chain on channel block at negative voltages, we resorted to directly examining the voltage-dependent inhibition curve of the F254A channels by a much higher TEA concentration (30 mM) and at more negative voltages (Fig. 7). At very negative voltages (e.g., −100 mV), the mutation must have lowered the apparent affinity of the channels for TEA ∼100-fold, because 30 mM TEA inhibits the mutant channel current to about the same extent as 0.3 mM TEA inhibits the wild type (Fig. 2 A and Fig. 7). Furthermore, the relatively weak (<20%) inhibition of the mutant channel current by 30 mM TEA at very negative voltages exhibits little voltage dependence. At more depolarized potentials, TEA, as expected from the results in Fig. 6, continues to cause profound, voltage-dependent inhibition of the mutant channel current, which can be reasonably fitted with a single Boltzmann function. Thus, removal of the aromatic side chain of residue 254 appears to dramatically lower TEA binding at the shallower site and essentially eliminate the associated voltage dependence. The latter observation suggests that a K+ ion is normally present at (or near) the shallow TEA site, and that its residence there is also dramatically disrupted by the mutation.
Figure 6.

Voltage dependence of the apparent TEA affinity of channels with a mutation at F254 or D255. The natural logarithm of appKd for the mutant channels (determined as shown in Fig. 2 B) is plotted against membrane voltage. The data for wild-type channels (taken from Fig. 2 C) are plotted for comparison. The curves through the data for the F254Y and D255A mutant channels are fits of Eq. 1a, whereas lines through the data for F254A and F254V mutant channels are fits of a Boltzmann function (with parameters appKd and appZ). The fits yield K1 = 4.38 (±0.39) × 10−4 M and K2 = 2.05 ± 0.82 with valences Z1 = 0.43 ± 0.06 and Z2 = 1.64 ± 0.08 (n = 8) for F254Y; K1 = 2.55 (±0.25) × 10−4 M and K2 = 2.27 ± 1.07 with Z1 = 0.36 ± 0.07 and Z2 = 1.27 ± 0.08 (n = 3) for D255A; appKd = 2.64 (±0.20) × 10−3 M and valence appZ = 1.42 ± 0.03 (n = 5) for F254A; appKd = 2.54 (±0.41) × 10−3 M and appZ = 1.95 ± 0.07 (n = 6) for F254V.
Figure 7.

Inhibition of F254A mutant channels by a high concentration of TEA. (A) Current traces were recorded from a single patch in the absence (control) or presence of 30 mM TEA. Currents were elicited by stepping membrane voltage from the 0 mV holding potential to −200 mV and then to test voltages between −200 mV and +100 mV in 5-mV increments before returning it to the holding potential. Dotted line indicates the zero current level. (B) The (averaged) fraction of current not blocked by 30 mM TEA is plotted against membrane voltage between −200 and 100 mV. The curve though the data is a fit of a Boltzmann function (with an offset to account for the shallow state with much reduced spermine affinity), yielding appKd = 3.41 (±0.19) × 10−3 M and valence appZ = 1.34 ± 0.03 (mean ± SEM; n = 3).
Block of Wild-type and Mutant Channels by Spermine
We now examine the effects of mutations at F254 and D255 on channel block by intracellular spermine. Fig. 8 shows currents of wild-type and mutant (F254A or D255A) channels in the absence or presence of 0.1 mM spermine. At this concentration, spermine almost completely blocks the outward currents of all three mutant channels. It also slightly inhibits inward currents in wild-type channels but imperceptibly in mutant channels. The normalized currents are plotted against membrane voltage for both wild-type and mutant channels in Fig. 9. As shown previously (Lopatin et al., 1995; Xie et al., 2002; Shin and Lu, 2005), the relation for wild-type channels exhibits both a shallow and a steep phase. However, that shallow phase is apparently eliminated by the mutations. Thus, mutations at both F254 and D255 dramatically disrupt spermine interaction with the shallow locus.
Figure 8.

Inhibition by spermine of currents through channels with a mutation at F254 or D255. Current traces for wild-type and F254A and D255A mutant channels, each type recorded from a single patch in the absence (control) or presence of 0.1 mM spermine. During recordings, currents were elicited by stepping membrane voltage from the 0 mV holding potential to −100 mV and then to test voltages between −100 and +100 mV in 10-mV increments before returning it to the holding potential. Dotted lines indicate the zero current level.
Figure 9.

Voltage dependence of spermine inhibition of channels with and without a mutation at F254 or D255. The (averaged) fraction of current not blocked by 0.1 mM spermine is plotted against membrane voltage for wild-type and F254A and D255A mutant channels. The curve through the data for wild-type channels is a fit of Eq. 1; those for the mutant channels are fits of a Boltzmann function (with parameters appKd, appZ, and an offset of ∼5% to account for the shallow state with much reduced spermine affinity), yielding K1 = 1.30 (±0.06) × 10−4 M and K2 = 1.44 (±0.13) × 10−2 with valences Z1 = 0.40 ± 0.02 and Z2 = 4.23 ± 0.13 (mean ± SEM; n = 9) for wild type, appKd = 3.82 (±0.49) × 10−6 M and valence appZ = 3.96 ± 0.15 (n = 5) for F254A, and appKd = 2.45 (±0.28) × 10−6 M and appZ = 4.75 ± 0.14 (n = 8) for D255A.
The outer end of the cytoplasmic pore is also constricted by four additional loops, one from each subunit. The narrowest gap between the Van der Waals surfaces of residue-308 side chains from opposite loops in GIRK1 is ∼7 Å (Fig. 3; Nishida and MacKinnon, 2002). (The narrowest region between the loops in the just published structure for IRK1's cytoplasmic pore is as narrow as ∼3 Å, which, as the authors point out, does not resemble an open state [Pegan et al., 2005].) We tested the effect on spermine block of a double mutation (A306G and M307G; the latter residue corresponding to residue 308 in GIRK1). Fig. 10 shows the currents of the double mutant channels in the absence or presence of 0.1 mM spermine, and the fraction of the mutant (along with wild-type) current not blocked versus membrane voltage. Unlike those mutations in the internal loop, this double mutation in the external loop affects little channel block by spermine.
Figure 10.

Inhibition of A306G-M307G double mutant channels by spermine. (A) Current traces recorded from a single patch in the absence (control) or presence of 0.1 mM spermine. (B) The (averaged) fraction of currents not blocked is plotted against membrane voltage for wild-type (taken from Fig. 9) and mutant channels. The curve through the mutant data is a fit of Eq. 1a, yielding K1 = 9.06 (±0.49) × 10−5 M and K2 = 1.45 (±0.16) × 10−2 with valences Z1 = 0.53 ± 0.02 and Z2 = 4.20 ± 0.16 (mean ± SEM; n = 5).
The length of spermine is ∼20 Å, and the center-to-center distance between the nitrogen atoms at both ends of an extended spermine molecule is ∼17 Å. Since in the deeper blocked state the leading amine of spermine interacts with D172, the trailing one is expected to be near M183, located three α-helical turns more intracellularly (each 3.6-residue turn results in 5.4-Å displacement). We therefore tested the effect of a mutation at M183 on the deeper spermine-bound state. Fig. 11 shows the currents of the M183A mutant channels in the absence or presence of 0.1 mM spermine, and the normalized current versus voltage. The mutation affects little the shallow phase but significantly the steep phase of the blocking curve, as expected if mainly the deeper blocked state was affected.
Figure 11.

Inhibition of M183A mutant channels by spermine. (A) Current traces recorded from a single patch in the absence (control) or presence of 0.1 mM spermine. (B) The (averaged) fraction of currents not blocked is plotted against membrane voltage for wild-type (taken from Fig. 9) and mutant channels. The curve though the mutant data is a fit of Eq. 1a, yielding K1 = 2.11 (±0.39) × 10−4 M and K2 = 9.31 (±1.84) × 10−2 with valences Z1 = 0.21 ± 0.06 and Z2 = 2.20 ± 0.14 (mean ± SEM; n = 6).
DISCUSSION
The voltage dependence of IRK1 channel block by intracellular amine blockers arises primarily from displaced K+ ions, not the blockers themselves, moving across the ion selectivity filter (Guo et al., 2003; Lu, 2004; Shin and Lu, 2005). Depending on the effective number of displaced K+ ions, the apparent (overall) valence for steady-state block varies with blocker type, e.g., it is ∼2 for TEA (Fig. 2 C) and ∼5 for spermine (Fig. 9; Lopatin and Nichols, 1996; Guo and Lu, 2000a,b, 2003; Xie et al., 2003; Shin and Lu, 2005). Binding of a blocker at two locations of different depth in the IRK1 pore produces two serially related blocked states with increasing voltage dependence. As a consequence, the apparent valence itself is voltage dependent (Fig. 2 C and Fig. 9; Shin and Lu, 2005).
For a minimal model (Fig. 12) containing one open (Ch) and two consecutive blocked states (ChB1 and ChB2) the steady-state fraction of current not blocked is given by
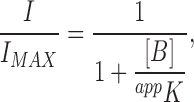 |
(1) |
where
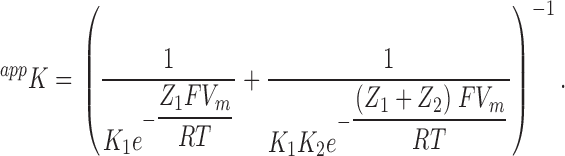 |
(1a) |
Figure 12.

Models for TEA and spermine block of the IRK1 channel. Binding of a TEA (A) or spermine (SPM; B) molecule to a channel (Ch) produces two sequential blocked states (ChTEA1 and ChTEA2 or ChSPM1 and ChSPM2). K1 = [Ch][X]/[ChX1] and K2 = [ChX1]/[ChX2]. Except for the innermost one, K+ ions are positioned arbitrarily in the inner pore of the accompanying diagrams.
Thus, the shallow limiting slope of a plot of lnappKd for TEA against Vm at negative voltages reflects valence Z1 associated with the first blocking transition, whereas the steep limiting slope at positive voltages reflects the sum of Z1 and Z2, informing the overall voltage dependence (Fig. 2 C). Extrapolation of the shallow limiting slope to 0 mV yields the equilibrium dissociation constant for the shallow binding site (K1), whereas that of the steep phase gives the product of K1 and K2 (the notation “0 mV” will henceforth be omitted for simplicity). RTlnK1 and RTln(K1K2) correspond to ΔGs for the shallow and deep bound states, respectively. A fit of Eq. 1a to the plot in Fig. 2 C yields the equilibrium constants for the two transitions K1 = 2.21 × 10−4 M and K2 = 4.65 with valences Z1 = 0.47 and Z2 = 1.61. Thus, at 0 mV, the shallower TEA-bound state has lower free energy than the deeper one that is however favored by depolarization (in the case of spermine, K2 = 1.44 × 10−2, signifying that the deeper blocked state is more stable; see Fig. 9).
If the two TEA-binding sites are physically separate, certain mutations may then differentially perturb one or the other. For example, if a mutation eliminates the interaction of TEA with the deeper site, a single (shallow) Boltzmann function, with appKd (0 mV) = K1 and appZ = Z1, should suffice to account for the blocking properties, and a plot of lnappKd against Vm will be a straight line (Fig. 13 A). This is what we observe with E224G and E299S mutant channels (Fig. 4 B). Thus, the mutations dramatically weaken TEA interaction with the deeper site with little effect on the shallow site.
Figure 13.

Predicted blocking properties of mutant channels lacking the shallow or the deep TEA-binding site. The identical solid curves represent a fit of Eq. 1a to the plot of lnappK versus Vm for wild-type channels, taken from Fig. 2 C. The dotted line in A is a simulation of the model with only the shallow site using the first term of Eq. 1a (K1 = 2.21 × 10−4 M and Z1 = 0.47), whereas the dotted line in B is a simulation of the model with only the deeper site using the second term of Eq. 1a (K1K2 = 1.02 × 10−3 M, Z1 = 0, and Z2 = 1.61). All values of the parameters used in the simulations were taken from Fig. 2 C, except Z1 in B, which was set as zero. The dashed line in B is the limiting slope of the steep phase.
Conversely, if a mutation eliminates only the shallower TEA-binding site, one expects the following. First, block should vanish at negative Vm where TEA mainly binds at the shallow site. Consistent with this, we found that at very negative voltages, the F254A mutation reduces the channel's apparent affinity for TEA ∼100-fold (Figs. 2 and 7). Second, the stability of the deeper blocked state should be little affected. Indeed, appKd(0 mV) for F254A and F254V is 2.64 mM and 2.54 mM, respectively, comparable to that of wild type (K1K2 = 1.02 mM) (Fig. 2 C and Fig. 6). Third, if the innermost K+ ion also binds at this same locus and that binding is eliminated by the mutations, then Z1 = 0 and appZ = Z2. The slope of lnappKd against Vm for the mutant channels will then be slightly shallower than the steep phase of the wild-type plot (Fig. 13 B), although the two lines should intersect at 0 mV because their overall appKd(0 mV) (or K1K2) remains unchanged. Consequently, at positive voltages, the lnappKd versus Vm plot for the mutant channels will be in the right quadrant with respect to that for wild-type channels. This is essentially what we observe: appZ values for the F254A and F254V channels are 1.42 and 1.95, respectively, compared with wild type's Z2 = 1.61 (Fig. 2 C and Fig. 6). Consistent with Z1 being minimal, at very negative voltages the slight (<20%) inhibition of F254A currents by 30 mM TEA exhibits no significant voltage dependence (Fig. 7). As negative controls, channels with a conservative F254Y or the neighboring D255A mutation behave like the wild type (Fig. 6). These results strongly suggest that removing the aromatic side chain of F254 selectively disrupts the shallower site where either K+ or TEA may bind.
Previously, our group found that the apparent inhibition rate constant (kon) for intracellular symmetric quaternary ammonium (QA) block of IRK1 drops steeply as the blocker becomes larger than tetrapropylammonium (∼9 Å) (Guo and Lu, 2001). This finding strongly implies that the innermost QA site itself, or part of the pore intracellular to it, is ∼10 Å wide. Intriguingly, F255 of GIRK1 (corresponding to F254 of IRK1) is located near the intracellular end of the cytoplasmic pore (Fig. 3; Nishida and MacKinnon, 2002) and, furthermore, the narrowest gap between the Van der Waals surfaces of opposite F255 residues is ∼10 Å, slightly wider than TEA. Here, we found that the existence of a shallow high-affinity TEA-binding site (Kd [0 mV] = 0.2 or 0.4 mM) in IRK1 depends on the presence of either phenylalanine or tyrosine at position 254 (Fig. 2 C and Fig. 6). Replacing those aromatic residues with aliphatic ones reduces the site's TEA affinity ∼100-fold. Given the fact that aromatic side chains form a high-affinity extracellular TEA-binding site in Kv and KcsA channels (MacKinnon and Yellen, 1990; Kavanaugh et al., 1991, 1992; Heginbotham and MacKinnon, 1992; Liman et al., 1992; Heginbotham et al., 1999; Lenaeus et al. 2005), our findings strongly suggest that aromatic side chains of F254 form the high-affinity intracellular TEA site in IRK1 (Fig. 12 A).
Replacing the aromatic side chain with an aliphatic one affects spermine block in a similar manner. The F254A mutation significantly reduces the extent of channel block at very negative voltages and eliminates the modest voltage dependence (Fig. 9). Thus, the aromatic side chains also underlie the shallow spermine-binding locus. In contrast to IRK1 and GIRK1, many Kir channels lack an aromatic residue at the corresponding position (Fig. 14). Spermine block of those channels may not be expected to exhibit the shallow voltage-dependent phase, as indeed appears to be the case for Kir4.1 and Kir6.2 (Shyng and Nichols, 1997; Oliver et al., 1998). We do not imply that merely replacing the relevant residue in those channels with phenylalanine or tyrosine is by itself sufficient to construct the shallow site. Notably, unlike the case of TEA, replacing D255 (nonconserved among Kir channels) with alanine also profoundly affects the interaction of spermine with the shallower site. However, it alters little the affinity of the deeper locus for spermine and the overall voltage dependence. Consequently, removing the acidic side chain of D255 has little effect on spermine block at positive voltages or on the steep steady-state inward rectification. On the other hand, substituting arginine for D255 significantly reduced block (by endogenous blockers) of outward currents at positive voltages in whole oocytes (Pegan et al., 2005). In contrast to the mutations in the internal loops, a double mutation (A306G and M307G) in the loops that constrict the external end of the cytoplasmic pore has little effects on spermine block of the channels (Fig. 10; see also Pegan et al., 2005).
Figure 14.

Comparison of a partial sequence among various Kir channels. Shown is partial sequence alignment among four different Kir channels. Bolded residues correspond to IRK1's F254 and D255.
From the shallower locus, spermine can proceed further toward the selectivity filter and bind at another locus that probably differs from that for TEA (Fig. 12). As already argued in RESULTS, since in the deeper blocked state the leading amine group of spermine reaches into the cavity region and interacts with D172, the trailing end should be near M183. A mutation at M183 may then be expected to affect the deeper blocked state. Indeed, we found that mutation M183A, though having little effect on K1 and Z1, significantly increases K1K2 and decreases the overall valence (Fig. 11), consistent with an effect mainly on the deeper blocked state. The finding is also consistent with the notion that spermine, in the deeper blocked state, mainly resides in the region lined by M2 and not deep in the selectivity filter (Guo and Lu, 2003; Guo et al., 2003; Shin and Lu, 2005).
In the case of TEA, the E224G and E299S mutations dramatically reduce interaction with the deeper site. In principle, this result could reflect the fact that TEA binds near these residues in the cytoplasmic pore (as depicted in Fig. 12 A) or that mutation-induced structural changes prevent TEA from reaching the deeper site such as the M2-lined cavity as in KcsA and Kv channels (e.g., Choi et al., 1993; Zhou et al., 2001; Lenaeus et al., 2005). Nonetheless, the following observations are more readily explained if TEA, unlike spermine, does not reach the cavity within IRK1. (In either case the exact spatial relation between TEA and E224/E299 remains to be determined.) First, replacing IRK1's M2 with the quite different one from ROMK1 has practically no effect on its (steady-state) QA-blocking properties, which are quite different from ROMK1's. Second, in the deeper binding location, the leading end of spermine reaches into the cavity region and displaces up to five K+ ions (Guo et al., 2003; Shin and Lu, 2005), producing an overall valence of ∼5, which far exceeds that (∼2) for TEA block, consistent with TEA not reaching the cavity.
In summary, our results constitute clear evidence for physically separate, sequential ion-binding loci along IRK1's inner pore, and strongly suggest that the aromatic side chains of F254 form the innermost binding locus for blocking ions such as TEA and spermine. The negatively charged side chain of D255 also appears critical for long spermine (not TEA) interaction with the shallow site. Furthermore, the site formed by F254 is likely the innermost site for K+ ions. K+ ions may likewise be present at other blocker-binding loci in the inner pore, which makes the hypothesis that the 50–70-Å-long inner pore can accommodate up to five K+ ions more plausible. It is noteworthy that the presence of (a) narrow region(s) in the inner pore may help prevent K+ ions from bypassing the blocking spermine to “leak” back into the cytoplasm, “forcing” them to move across the narrow selectivity filter, thus engendering the remarkably strong voltage dependence.
Acknowledgments
We thank L.Y. Jan for the IRK1 channel cDNA clone, J. Yang for the cDNA subcloned in the pGEM-HESS vector, and P. De Weer for critical review and discussion of our manuscript.
This study was supported by NIH grant GM55560.
Olaf S. Andersen served as editor.
Abbreviation used in this paper: Kir, inward-rectifier K+.
References
- Choi, K.L., C. Mossman, J. Aubé, and G. Yellen. 1993. The internal quaternary ammonium receptor site of Shaker potassium channels. Neuron. 10:533–541. [DOI] [PubMed] [Google Scholar]
- Doyle, D.A., C.J. Morais, R.A. Pfuetzner, A. Kuo, J.M. Gulbis, S.L. Cohen, B.T. Chait, and R. MacKinnon. 1998. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 280:69–77. [DOI] [PubMed] [Google Scholar]
- Fakler, B., U. Brandle, E. Glowatzki, S. Weidemann, H.P. Zenner, and J.P. Ruppersberg. 1995. Strong voltage-dependent inward rectification of inward rectifier K+ channels is caused by intracellular spermine. Cell. 80:149–154. [DOI] [PubMed] [Google Scholar]
- Ficker, E., M. Taglialatela, B.A. Wible, C.M. Henley, and A.M. Brown. 1994. Spermine and spermidine as gating molecules for inward rectifier K+ channels. Science. 266:1068–1072. [DOI] [PubMed] [Google Scholar]
- Guo, D., and Z. Lu. 2000. a. Mechanism of IRK1 channel block by intracellular polyamines. J. Gen. Physiol. 115:799–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, D., and Z. Lu. 2000. b. Pore block versus intrinsic gating in the mechanism of inward rectification in the strongly-rectifying IRK1 channel. J. Gen. Physiol. 116:561–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, D., and Z. Lu. 2001. Kinetics of inward-rectifier K+ channel block by quaternary alkylammonium ions: dimension and properties of the inner pore. J. Gen. Physiol. 117:395–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, D., and Z. Lu. 2002. IRK1 inward rectifier K+ channels exhibit no intrinsic rectification. J. Gen. Physiol. 120:539–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, D., and Z. Lu. 2003. Interaction mechanisms between polyamines and IRK1 inward rectifier K+ channels. J. Gen. Physiol. 122:485–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, D., Y. Ramu, A.M. Klem, and Z. Lu. 2003. Mechanism of rectification in inward-rectifier K+ channels. J. Gen. Physiol. 121:261–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagiwara, S., and K. Takahashi. 1974. The anomalous rectification and cation selectivity of the membrane of a starfish egg cell. J. Membr. Biol. 18:61–80. [DOI] [PubMed] [Google Scholar]
- Hagiwara, S., S. Miyazaki, and N.P. Rosenthal. 1976. Potassium current and the effect of cesium on this current during anomalous rectification of the egg cell membrane of a starfish. J. Gen. Physiol. 67:621–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heginbotham, L., and R. MacKinnon. 1992. The aromatic binding site for tetraethylammonium ion on potassium channels. Neuron. 8:483–491. [DOI] [PubMed] [Google Scholar]
- Heginbotham, L., M. LeMasurier, L. Kolmakova-Partensky, and C. Miller. 1999. Single Streptomyces lividans K+ channels: functional asymmetries and sidedness of proton activation. J. Gen. Physiol. 114:551–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille, B. 2001. Ion Channels of Excitable Membranes. Sinauer Associates, Inc., Sunderland, MA. 814 pp.
- Hodgkin, A.L., and P. Horowicz. 1959. The influence of potassium and chloride ions on the membrane potential of single muscle fibres. J. Physiol. 148:127–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horie, M., H. Irisawa, and A. Noma. 1987. Voltage-dependent magnesium block of adenosine-triphosphate-sensitive potassium channel in guinea-pig ventricular cells. J. Physiol. 387:251–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, C.L., S. Feng, and D.W. Hilgemann. 1998. Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gβγ. Nature. 391:803–806. [DOI] [PubMed] [Google Scholar]
- Katz, B. 1949. Les constantes électriques de la membrane du muscle. Arch. Sci. Physiol. (Paris). 3:285–299. [Google Scholar]
- Kavanaugh, M.P., M.D. Varnum, P.B. Osborne, M.J. Christie, A.E. Busch, J.P. Adelman, and R.A. North. 1991. Interaction between tetraethylammonium and amino acid residues in the pore of cloned voltage-dependent potassium channels. J. Biol. Chem. 266:7583–7587. [PubMed] [Google Scholar]
- Kavanaugh, M.P., R.S. Hurst, J. Yakel, M.D. Varnum, J.P. Adelman, and R.A. North. 1992. Multiple subunits of a voltage-dependent potassium channel contribute to the binding site for tetraethylammonium. Neuron. 8:493–497. [DOI] [PubMed] [Google Scholar]
- Kubo, Y., and Y. Murata. 2001. Control of rectification and permeation by two distinct sites after the second transmembrane region in Kir2.1 K+ channel. J. Physiol. 531:645–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo, Y., T.J. Baldwin, Y.N. Jan, and L.Y. Jan. 1993. Primary structure and functional expression of a mouse inward rectifier potassium channel. Nature. 362:127–133. [DOI] [PubMed] [Google Scholar]
- Kuo, A., J.M. Gulbis, J.F. Antcliff, T. Rahman, E.D. Lowe, J. Zimmer, J. Cuthbertson, F.M. Ashcroft, T. Ezaki, and D.A. Doyle. 2003. Crystal structure of the potassium channel KirBac1.1 in the closed state. Science. 300:1922–1926. [DOI] [PubMed] [Google Scholar]
- Lenaeus, M.J., M. Vamvouka, P.J. Focia, and A. Gross. 2005. Structural basis of TEA blockade in a model potassium channels. Nat. Struct. Mol. Biol. 12:454–459. [DOI] [PubMed] [Google Scholar]
- Liman, E.R., J. Tytgat, and P. Hess. 1992. Subunit stoichiometry of a mammalian K+ channel determined by construction of multimeric cDNAs. Neuron. 9:861–871. [DOI] [PubMed] [Google Scholar]
- Lopatin, A.N., E.N. Makhina, and C.G. Nichols. 1994. Potassium channel block by cytoplasmic polyamines as the mechanism of intrinsic rectification. Nature. 372:366–369. [DOI] [PubMed] [Google Scholar]
- Lopatin, A.N., E.N. Makhina, and C.G. Nichols. 1995. The mechanism of inward rectification of potassium channels: “long-pore plugging” by cytoplasmic polyamines. J. Gen. Physiol. 106:923–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopatin, A.N., and C.G. Nichols. 1996. [K+] dependence of polyamine-induced rectification in inward rectifier potassium channels (IRK1, Kir2.1). J. Gen. Physiol. 108:105–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, Z. 2004. Mechanism of rectification in inward-rectifier K+ channels. Annu. Rev. Physiol. 66:103–129. [DOI] [PubMed] [Google Scholar]
- Lu, Z., and R. MacKinnon. 1994. Electrostatic tuning of Mg2+ affinity in an inward-rectifier K+ channel. Nature. 371:243–246. [DOI] [PubMed] [Google Scholar]
- MacKinnon, R., and G. Yellen. 1990. Mutations affecting TEA blockade and ion permeation in voltage-activated K+ channels. Science. 250:276–279. [DOI] [PubMed] [Google Scholar]
- Matsuda, H., A. Saigusa, and H. Irisawa. 1987. Ohmic conductance through the inwardly rectifying K+ channel and blocking by internal Mg2+. Nature. 325:156–159. [DOI] [PubMed] [Google Scholar]
- Nishida, M., and R. MacKinnon. 2002. Structural basis of inward rectification: cytoplasmic pore of the G protein-gated inward rectifier GIRK1 at 1.8 Å resolution. Cell. 111:957–965. [DOI] [PubMed] [Google Scholar]
- Oliver, D., H. Hahn, C. Antz, J.P. Ruppersberg, and B. Fakler. 1998. Interaction of permeant and blocking ions in cloned inward-rectifier K+ channels. Biophys. J. 74:2318–2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegan, S., C. Arrabit, W. Zhou, W. Kwiatkowski, A. Collins, P.A. Slesinger, and S. Choe. 2005. Cytoplasmic domain structures of Kir2.1 and Kir3.1 show sites for modulating gating and rectification. Nat. Neurosci. 8:279–287. [DOI] [PubMed] [Google Scholar]
- Shin, H.-G., and Z. Lu. 2005. Mechanism of voltage sensitivity of IRK1 inward-rectifier K+ channel block by the polyamine spermine. J. Gen. Phyiol. 125:413–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng, S.L., and C.G. Nichols. 1997. Octameric stoichiometry of the KATP channel complex. J. Gen. Physiol. 110:655–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanfield, P.R., N.W. Davies, P.A. Shelton, M.J. Sutcliffe, I.A. Khan, W.J. Brammar, and E.C. Conley. 1994. A single aspartate residue is involved in both intrinsic gating and blockage by Mg2+ of the inward rectifier, IRK1. J. Physiol. 478:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taglialatela, M., E. Ficker, B.A. Wible, and A.M. Brown. 1995. C-terminus determinants for Mg2+ and polyamine block of the inward rectifier K+ channel IRK1. EMBO J. 14:5532–5541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberg, C.A. 1987. Inward rectification of a potassium channel in cardiac ventricular cells depends on internal magnesium ions. Proc. Natl. Acad. Sci. USA. 84:2560–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wible, B.A., M. Taglialatela, E. Ficker, and A.M. Brown. 1994. Gating of inwardly rectifying K+ channels localized to a single negatively charged residue. Nature. 371:246–249. [DOI] [PubMed] [Google Scholar]
- Xie, L.-H., S.A. John, and J.N. Weiss. 2002. Spermine block of the strong inward rectifier potassium channel Kir2.1: dual roles of surface screening and pore block. J. Gen. Phyiol. 120:53–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, L.-H., S.A. John, and J.N. Weiss. 2003. Inward rectification by polyamines in mouse Kir2.1 channels: synergy between blocking components. J. Physiol. 550:67–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, J., Y.N. Jan, and L.Y. Jan. 1995. Control of rectification and permeation by residues in two distinct domains in an inward rectifier K+ channel. Neuron. 14:1047–1054. [DOI] [PubMed] [Google Scholar]
- Zhou, M., J.H. Morais-Cabral, S. Mann, and R. MacKinnon. 2001. Potassium channel receptor site for the inactivation gate and quaternary amine inhibitors. Nature. 411:657–661. [DOI] [PubMed] [Google Scholar]