

Abstract
Major advances have been made on the inhibition gate and ATP site of the Kir6.2 subunit of the KATP channel, but little is known about conformational coupling between the two. ATP site mutations dramatically disrupt ATP-dependent gating without effect on ligand-independent gating, observed as interconversions between active burst and inactive interburst conformations in the absence of ATP. This suggests that linkage between site and gate is conditionally dependent on ATP occupancy. We studied all substitutions at position 334 of the ATP site in Kir6.2ΔC26 that express in Xenopus oocytes. All substitutions disrupted ATP-dependent gating by 10-fold or more. Only positive-charged arginine or lysine at 334, however, slowed ligand-independent gating from the burst, and this was in some but not all patches. Moreover, the polycationic peptide protamine reversed the slowed gating from the burst of 334R mutant channels, and speeded the slow gating from the burst of wild-type SUR1/Kir6.2 in the absence of ATP. Our results support a two-step ligand-dependent linkage mechanism for Kir6.2 channels in which ATP-occupied sites function to electrostatically dissociate COOH-terminal domains from the membrane, then as in all Kir channels, free COOH-terminal domains and inner M2 helices transit to a lower energy state for gate closure.
INTRODUCTION
The ATP-inhibited potassium (KATP) channel couples energy metabolism to membrane electrical activity in a variety of cells and is important in several physiological systems (Noma, 1983; Ashcroft et al., 1984; Cook and Hales, 1984; Jovanovic et al., 1998; Aguilar-Bryan and Bryan, 1999). The mechanism by which ATP site occupancy couples to inhibition gate closure is critical to our understanding of the signaling that coordinates cell physiology throughout our bodies (Ashcroft, 1988; Babenko et al., 1998; Aguilar-Bryan et al., 2001). Typically, elevated energy metabolism, signaled by an increase in the ATP/ADP ratio, inhibits KATP channel activity, which triggers cell excitability and Ca2+ influx, leading to muscle contraction or secretion. In the β cell of the endocrine pancreas, for example, the signal flow stimulates insulin secretion in response to high glucose levels in the blood. The importance of this dynamic signal flow has been recently underscored by the discovery of mutations that by disrupting the ATP binding site of the KATP channel cause hypo-insulinemia, leading to permanent neonatal diabetes (Gloyn et al., 2004a,b; Sagen et al., 2004; Zung et al., 2004).
The KATP channel is assembled from two types of subunit. A potassium pore-forming subunit, Kir6.x (Inagaki et al., 1995), is the primary seat of inhibition gating by ATP (Drain et al., 1998; John et al., 1998; Tucker et al., 1997, 1998). A sulfonylurea receptor subunit, SURx (Aguilar-Bryan et al., 1995), mediates inhibition by sulfonylureas and activation by MgADP and potassium channel openers (Nichols et al., 1996; Gribble et al., 1997, 1998; Shyng et al., 1997; Tucker et al., 1997; Babenko et al., 1999a, 2000; Ribalet et al., 2000; Zingman et al., 2001). Investigation of the molecular mechanisms underlying inhibition gating has focused on ligand binding involving the cytoplasmic NH2 and COOH termini of Kir6.2 (Tucker et al., 1997, 1998; Drain et al., 1998; Takano et al., 1998; Koster et al., 1999; Reimann et al., 1999a,b; John et al., 2001; Enkvetchakul and Nichols, 2003; Antcliff et al., 2005).
KATP channel gating occurs in bursts of brief openings that alternate with briefer closings, and these active bursts are separated by long-lived inactive interbursts (Ashcroft et al., 1984; Cook and Hales, 1984; Gillis et al., 1989; Qin et al., 1989; Nichols et al., 1991; Alekseev et al., 1998; Drain et al., 1998; Lorenz et al., 1998; Trapp et al., 1998; Babenko et al., 1999b,c; Li et al., 2002). In the context of ATP inhibition gating, the KATP channel may be viewed simply as having two major functional states, the active burst and inactive interburst states. At a relatively slow rate in the absence of ligand (ligand-independent gating) or at a greatly accelerated rate in the presence of ATP (ligand-dependent gating) this burst gate occludes the conduction pathway for potassium ion current flow. ATP binds to active channels, speeding the transitions to the inactive burst state (Li et al., 2002), as well as binds to the inactive interburst state further stabilizing the already shut gate. Although ligand-independent and ATP-dependent gating differ in rate and by the presence of bound ATP, the two processes share mechanisms of pore occlusion (Drain et al., 1998, 2004; Trapp et al., 1998; Tucker et al., 1998; Loussouarn et al., 2000; Li et al., 2002; Phillips and Nichols, 2003; Phillips et al., 2003).
Structural models of the cytoplasmic COOH terminus of the Kir6.2 subunit, including residues 182 (Li et al., 2000), 185 (Tucker et al., 1997, 1998), and 334 (Drain et al., 1998), have demarcated the likely beginning, middle, and end of an inhibitory ATP site groove, respectively (Enkvetchakul and Nichols, 2003; John et al., 2003; Trapp et al., 2003). At the 182 end, we found that each of 14 substitutions profoundly disrupted ATP-dependent gating, with exceptional effects on ligand-independent gating (Li et al., 2000). Only positive-charged substitutions at 182, in some but not all patches, slowed gating from the burst to the interburst. The slowed gating from the burst indicated that positive-charged substitutions at 182 can alter, and therefore must provide linkage to, the gating mechanism. We proposed that uniquely positive charges at 182, through electrostatic interactions, likely move into proximity of charged or polar environments, to account for the dramatically slowed gating. The communication between charged residues at 182 and gating in the absence of ATP might reflect conformational coupling between I182 and the gating mechanism that is normally only engaged during occupancy of the binding site by ATP. Perhaps, only upon ATP binding do the gating and site domains of the KATP channel undergo tight conformational coupling to speed gate closure, implying induced-fit mechanisms (Koshland et al., 1966; Eigen, 1968; Hammes and Wu, 1971).
So far, the conditional gating effect of positive-charged substitutions on ligand-independent gating distinguishes 182 from other positions of the ATP site of Kir6.2. Although mutational analysis is not often as complete at these positions, such mutations studied that increased the Ki for ATP inhibition do so without effect on ligand-independent gating. For example, all substitutions that express at position 185 in ΔC26 without SUR1 increased Ki (Tucker et al., 1997, 1998), without a change in ligand-independent gating (Tucker et al., 1998). Other neighboring mutations E179Q and Q173A increased the Ki modestly but with little or no change in ligand-independent gating.
Ligand-dependent linkage can explain two additional features of the Kir6.2 subunit of KATP channels. First, mutations including those in the 182–185 region (Tucker et al., 1997; 1998; Reimann et al., 1999a; Li et al., 2000) and the 334 region (Drain et al., 1998) can strongly disrupt ligand-dependent gating as measured by the Ki,ATP, but consistently with no effect on ligand-independent gating. This is unexpected of strict allosteric mechanisms (Monod et al., 1965) and suggests that in the absence of ligand the gating between burst and interburst can operate largely independently of changes at the ATP binding site. Second, Kir6.2 expression without SUR (Tucker et al., 1997; Alekseev et al., 1998; Drain et al., 1998; John et al., 1998; Babenko et al., 1999a) shows that the gating equilibrium in the absence of ligand can be changed to greatly favor the interburst with a 10-fold decrease, not an increase, in the apparent affinity for ATP binding. Thus, ligand-dependent linkage, by having gate and site conformationally coupled in the presence but not absence of ATP site occupancy, better accommodates the nonreciprocal effects on ligand-independent gating and apparent ATP binding affinity observed in many KATP channel mutants.
Based on the slowed gating effects of the positive-charged substitutions at 182, and of membrane anionic lipids, we hypothesized that native positive-charged residues in the 176 region or substituted positive-charged residues at 182 interact with the membrane anionic lipids in the same linkage mechanism coupling conformational changes accompanying ATP binding and inhibition gating (Li et al., 2000). The hypothesis raises the critical question of the role of long-range electrostatic interactions underlying the linkage mechanism not only at the 182 end but also at other positions along the ATP site groove. In this study, we therefore specifically tested whether positions 334 and 333, in addition to 182, can be mutated to provide a linkage to the gating mechanism, and if so, whether properties of the linkage effects are shared with those identified at 182.
MATERIALS AND METHODS
Mutagenesis
The truncated ΔC26 channel (Tucker et al., 1997) and the substitutions at position 334 in this background were constructed by a saturation mutagenesis technique (Reidhaar-Olson et al., 1991), using PCR overlap extension and silent sites, as previously described (Li et al., 2000). 50 random, independent mutagenic primer-containing clones were selected. 15 different substitutions at 334 were obtained in this way. The remaining four substitutions at 334 and an additional four substitutions at 333 were constructed separately using QuickChange Site-Directed Mutagenesis Kit (Stratagene). All the mutants were confirmed by DNA sequencing.
Oocyte Expression and Electrophysiology
Preparation and injection of Xenopus oocytes, patch pipette fabrication, solutions, and inside-out patch excised recording techniques were as previously described (Drain et al., 1994, 1998). Macroscopic and single-channel currents were recorded with the inside-out patch configuration at −80 mV with the following pipette and bath solutions, unless indicated otherwise (in mM): pipette solution (in mM): 150 KCl, 10 NaCl, 1 CaCl2, 10 EGTA, and 10 HEPES, pH 7.4; bath solution: same as pipette solution but with 1.3 MgATP. Recordings including those to determine K i,ATP for ATP inhibition of 334 mutant channels were obtained using different ATP concentrations applied to the patches by constant superfusion of the cytoplasmic face of patches using a Biologic RSC-160 nine-sewer pipe syringe-pressurized system (Molecular Kinetics). Recordings were always begun within 1 min after excision. ATP was added in the superfusate and was also present in the bath as the magnesium salt to minimize rundown (Trube and Hescheler, 1984; Drain et al., 1998, 2004; Li et al., 2000). Experiments showing rundown, characterized by a sudden significant decrease in open probability PO in the absence of ATP were discarded. Patch-clamp currents were amplified with Axopatch 200A (Axon Instruments) or EPC-9 (HEKA Elektronik) instruments, low-pass filtered with an eight-pole Bessel filter (Frequency Devices) at a corner frequency of 4 kHz, and sampled at 20 kHz using HEKA PULSE v.8.0 (HEKA Elektronik). For the protamine experiment, 50 μM protamine (Sigma-Aldrich) was applied in the absence of ATP to either the slow burst exit gating Kir6.2/ΔC26/G334R channel patches or the normally wild-type slow burst exit gating SUR1/Kir6.2 channels.
Data Analysis
Analysis and display were done using TAC v.4.1.5 (Bruxton, Inc.), IGOR Pro v.5.02 (WaveMetrics, Inc.), and Illustrator v.9.0 (Adobe Systems, Inc.). Dose–response measurements were fit to the Hill equation, 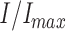 =
=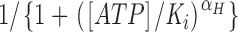 , where [ATP] is the concentration of ATP, I/I
max the fractional current at the indicated [ATP] relative to that in the same solution in the absence of added ATP (I
max was defined as the average of measurements taken before and after current measurements in the presence of [ATP]), K
i the [ATP] at which inhibition is half-maximal, and αH the slope factor. Data are presented as mean ± SEM. For Hill equation fits to the data from the mutant KATP channels with very large K
i values, we set αH = 1.0, a reasonable constraint given that when αH was treated as a free variable, we found αH = 1.0 ± 0.1 for the wild-type channel and αH = 1.0 ± 0.2 for all our less severely affected K
i mutant channels. Single-channel current events were detected using the time of the half-amplitude of transitions between current levels with TAC v.4.1.5 (Bruxton, Inc.). Durations were corrected for missed events during construction of duration histograms based on the filter corner frequency of the recording by the method of Colquhoun and Sigworth (1995). Duration analysis was done with TAC-FIT v4.1.5 (Bruxton, Inc.), which uses the transformations of Sigworth and Sine (1987) to construct and fit duration histograms.
, where [ATP] is the concentration of ATP, I/I
max the fractional current at the indicated [ATP] relative to that in the same solution in the absence of added ATP (I
max was defined as the average of measurements taken before and after current measurements in the presence of [ATP]), K
i the [ATP] at which inhibition is half-maximal, and αH the slope factor. Data are presented as mean ± SEM. For Hill equation fits to the data from the mutant KATP channels with very large K
i values, we set αH = 1.0, a reasonable constraint given that when αH was treated as a free variable, we found αH = 1.0 ± 0.1 for the wild-type channel and αH = 1.0 ± 0.2 for all our less severely affected K
i mutant channels. Single-channel current events were detected using the time of the half-amplitude of transitions between current levels with TAC v.4.1.5 (Bruxton, Inc.). Durations were corrected for missed events during construction of duration histograms based on the filter corner frequency of the recording by the method of Colquhoun and Sigworth (1995). Duration analysis was done with TAC-FIT v4.1.5 (Bruxton, Inc.), which uses the transformations of Sigworth and Sine (1987) to construct and fit duration histograms.
Structural Homology Modeling
The sequence of Kir6.2 between residues 177 and 357 was aligned with that of Kir3.1 (1N9P) between residues 190 and 370 using CLUSTALW, T-Coffee (http://us.expasy.org/tools/#align), and ALIGN (http://umber.sbs.man.ac.uk/dbbrowser/ALIGN/). The aligned sequence exhibited amino acid residue identity over 49% so that the quality of homology modeling was highly ensured. The structure alignment was achieved using Swiss-Model 3.5 (http://swissmodel.expasy.org//SWISS-MODEL.html). The resulting model was further energy minimized with NAMD 2.5 (http://www.ks.uiuc.edu/Research/namd). The modeled structure was compared with the structure of Kir3.1 and visualized using Deep View Swiss-PDB Viewer 3.7 (http://www.expasy.org/spdbv/). The RMSD of Cα backbone of the modeled Kir6.2 structure is ∼2.4 Å. An extended conformer of ATP extracted from 1BUO was set as a flexible ligand and docked into Kir6.2 using the Lamarckian genetic algorithm in the program AUTODOCK 3.0 (Goodsell et al., 1996; Morris et al., 1996). Docking parameters were selected based on optimized results from a previous blind docking study (Hetenyi and van der Spoel, 2002).
RESULTS
In the absence of ATP and at −80 mV, the wild-type KATP channel interconverts between two major gating conformations, an active bursting state and an inactive interburst state, and we refer to these conformational transitions as ligand-independent gating (Drain et al., 1998; Li et al., 2000). The ΔC26 truncated channel (Tucker et al., 1997), when expressed in the absence of SUR and ATP, exhibits a kinetically similar inactive interburst state, however, the truncated channel's burst and open times are significantly shorter, because of dramatically speeded burst exit rates (Drain et al., 1998; John et al., 1998; Lorenz et al., 1998; Tucker et al., 1998; Babenko et al., 1999b,c; Li et al., 2000, 2002). Point mutations of Kir6.2 that dramatically decrease ATP-dependent gating, as measured by increases in Ki,ATP, can be classified into candidates for gate versus ATP site residue by their effects on the speeded gating from the burst in the ΔC26 channels without SUR. For example, the T171A gating mutation dramatically slows, and the G334D ATP site mutation fails to alter, the speeded burst exit gating in ΔC26, even though both mutations dramatically decrease ATP-dependent gating as measured by the Ki,ATP. In the present study, we used this ΔC26 ligand-independent gating to classify all substitutions at the G334 position in the ΔC26 background that express functional channels.
ATP Inhibition of Substitutions at Position 334 of Kir6.2
Fig. 1 shows the effect of bath-applied ATP to 334 substituted channels representative of the range of results obtained. The current responses from each of the 334 substitutions showed blunted responses to ATP, compared with the control wild-type G334 channels. Even at 10 mM ATP the 334 substitution mutants exhibited substantial activity, compared with the complete inhibition of the control wild-type G334 channels. The results suggest that the 334 substitutions disrupt ATP inhibition gating. Fig. 2 shows fits of the Hill equation to the dose–response data used to quantify the effects of the substitutions on the ATP inhibition gating, by the Ki for ATP inhibition. The Ki,ATP values for G334 (wild type), 334R, 334A, 334F, and 334E were 0.19 ± 0.04, 2.0 ± 0.04, 2.1 ± 0.05, 3.0 ± 0.03, and 5.9 ± 0.02 mM ATP, respectively. Each was significantly increased compared with the parent control values. The Ki,ATP for the 334R and 334A channels were not significantly different from another, but were each significantly different from the Ki,ATP for the 333F and 334E channels (P < 0.05; one-way ANOVA). Thus, the positive-charged arginine and alanine substitutions at 334 had the least effect of all substitutions, disrupting ATP inhibition by just over 10-fold, whereas the negative-charged glutamate at 334 had the most dramatic effect, disrupting ATP inhibition by over 30-fold. The small hydrophobic alanine (11-fold) and the large hydrophobic phenylalanine (16-fold) also did not substitute well, suggesting that the absence of a side chain of the wild-type glycine residue along the 334 loop of Kir6.2 is important to binding or linkage of the ATP site.
Figure 1.
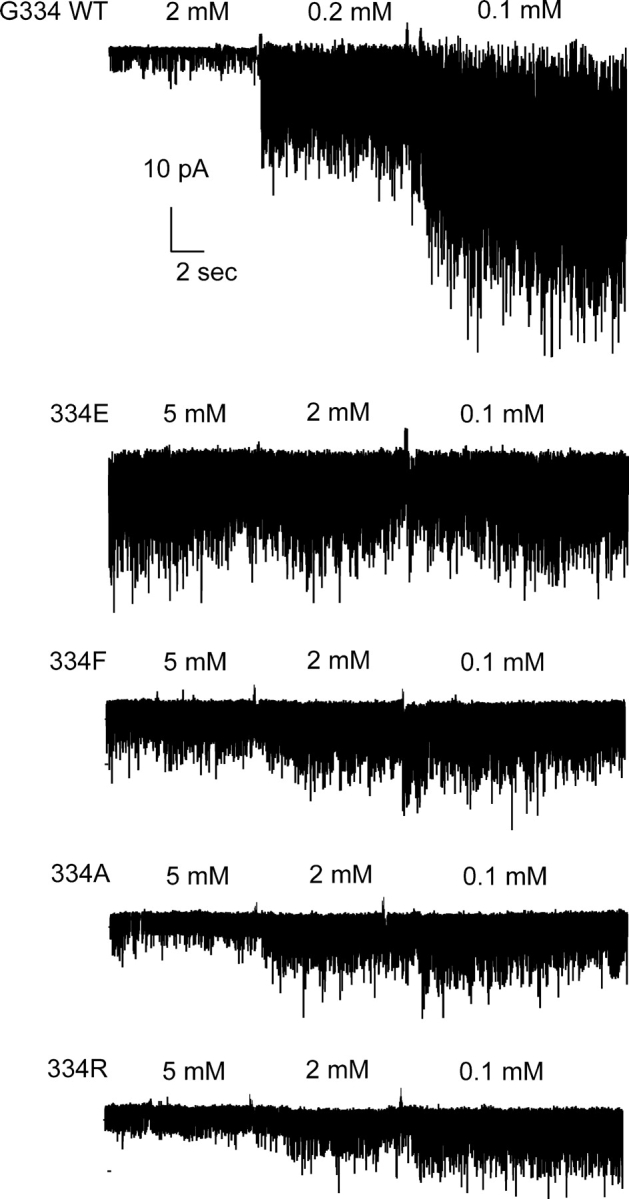
Mutations at position 334 disrupt ATP inhibition of Kir6.2ΔC26 channels. Response to indicated concentration of ATP of channels representative of strong and moderate disruption of ATP sensitivity is shown. NPO was determined at the different concentrations of ATP including 0 ATP. The strongest effects were obtained for the negative-charged aspartate and glutamate. The weakest effects were observed with the positive-charged arginine and lysine.
Figure 2.

Dose–response data fit to the Hill equation. To measure the effect of substitutions at position 334 we fit the fractional current as a function of ATP concentration to the Hill equation. The channels show varying changes in the Ki,ATP from 11-fold to over 30-fold, compared with the 0.19 mM value obtained for the wild-type control. The Ki,ATP values for 334 substitutions 334R, 334A, 334F, and 334E were determined in five or more independent experiments and averaged 2.0 ± 0.04, 2.1 ± 0.06, 3.1 ± 0.03, and 5.9 ± 0.02 mM ATP, respectively. Each was significantly increased compared with the parent control values. Although Ki,ATP for the 334R and 334A channels were not significantly different from another, they were each significantly different from the Ki,ATP for the 333F and 334E channels (P < 0.05; one-way ANOVA). In the fits to wild-type Kir6.2ΔC26 channels the slope factor was 1.0 ± 0.1. For the mutant channels, the fits exhibited greater variation in the slope factor, when both the slope factor and Ki,ATP were free parameters. However, previously (Drain et al., 1998; Li et al., 2000) we have found with all wild-type and mutant channels with less mild disruptions in ATP inhibition that the slope factor from fits was 1.0 ± 0.1, as well. We therefore assume that the slope factor does not change by mutations and constrain it to 1.0 ± 0.1 so that we can rank the channels on the basis of the subsequent changes from the fit in the Ki,ATP.
Ligand-independent Gating Kinetics of 334 Substitution Channels
Fig. 3 shows results indicating that all 13 substitutions at position 334 had little or no effect on ligand-independent gating despite dramatic effects on ATP-dependent gating measured by the Ki. Here, the ligand-independent gating of the mutant channels was quantified by the open probability PO in the absence of ATP. Except for the positive-charged arginine and lysine substitutions, the 334 substitutions had no effect at all on ligand-independent gating. The results indicate that the 334 region of Kir6.2 is excluded from an essential role in the burst-to-interburst gating step of the overall ATP-dependent inhibition gating mechanism.
Figure 3.
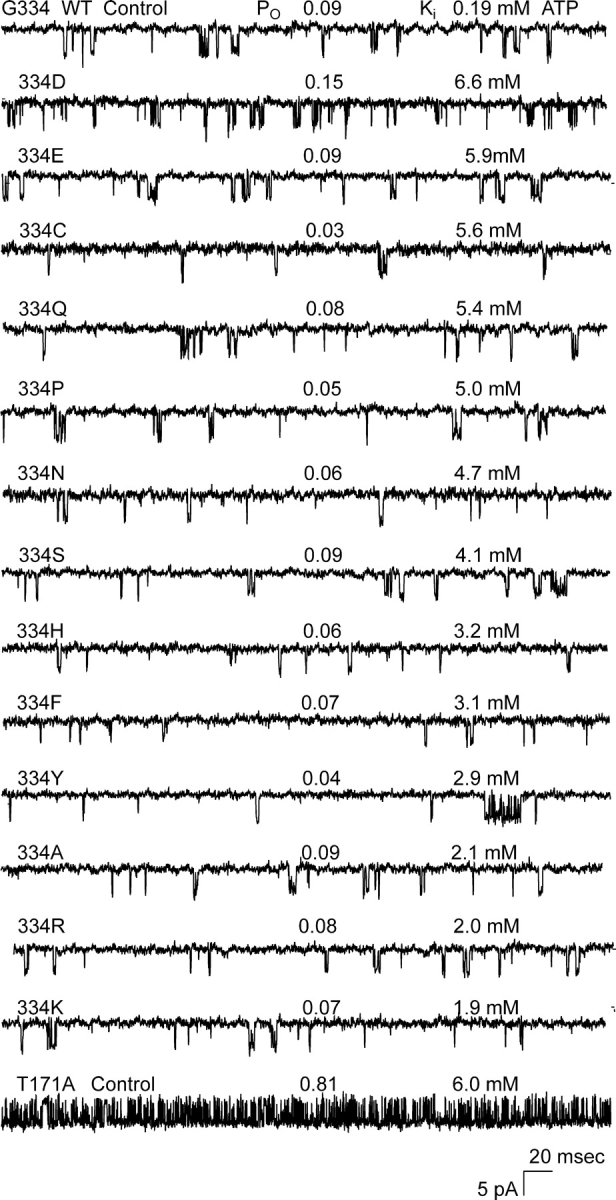
Ligand-independent gating of Kir6.2ΔC26 334 substituted channels. A total of 13 different substitutions at position 334 gave rise to functional channels. Ligand-independent gating was assessed by measuring open probability PO in the absence of ATP. 10 independent patches were analyzed for each 334 substitution and the average PO is indicated together with the most representative single-channel current record obtained. The current records are listed in descending order of their Ki,ATP. Although the Ki,ATP changed significantly across the mutant channels, the PO remained the same low value measured for the parental wild-type control. This low PO is known to result from dramatically speeded gating from the burst.
Gating from the Burst Dramatically Slowed by Arginine and Lysine at Position 334
Fig. 4 shows that in some but not all patches, the arginine and lysine substitutions at position 334 dramatically slowed gating from the burst. The slow and fast gating states of 334R and 334K channels were not observed to interconvert spontaneously, which indicates that they were quite stable. For the arginine mutants, 5 of 11 patches showed the fast gating of the parent ΔC26 channels, and 6 of 11 patches exhibited the slowed gating. Similarly, for the lysine mutants, 6 of 10 patches showed the fast gating, and 4 of 10 patches the slowed gating. Thus, the fast and slow gating states were also about evenly split and depended, apparently randomly, upon the particular patch of membrane excised. The slowed gating was limited to the positive-charged substitutions and was not observed in at least 10 patches for each of the other substitutions at 334 that expressed. The results indicate that mutant channels with positive-charged substitutions at position 334 can conditionally transit to a state of slow gating from the burst.
Figure 4.

Exceptional slowed gating kinetics in only the positive-charged 334R and 334K channels. (A) Parental control ΔC26 channels without SUR show fast burst exit gating, observed as very short bursts of one to several openings and a PO ∼0.08. (B) For arginine at position 334, 6 of 11 patches exhibited dramatically slowed gating. (C) For the lysine mutant, 6 of 10 patches showed the fast gating, and 4 of 10 patches the slowed gating. On average, the exceptional slowed gating for either positive-charged substitution at 334 was characterized by approximately fivefold increase in PO, from ∼0.1 to ∼0.5. During these initial experiments, interconversions between the fast and slow gating conformations were not observed and could not be induced by a variety of bath solutions.
Gating from the Burst Dramatically Slowed by Lysine at Adjacent Position 333
The slowed gating of positive-charged substitutions at positions 182 and 334 at either end of the ATP site groove are consistent with long-range electrostatic interactions with the transmembrane gating domain. To test the hypothesis further, we expressed K, L, S, and C substitutions at position 333, adjacent to 334, in the Kir6.2ΔC26 channel. All four substitution mutants showed high cell-attached activity, compared with wild-type controls, reflecting decreased sensitivity to ATP. Only 333K showed altered ligand-independent gating. Fig. 5 shows that in 6 of 12 patches, 333K dramatically slowed gating from the burst, compared with the other 6 patches with fast gating, which were indistinguishable from parent ΔC26 control channels. In 10 or more patches, 333L, 333S, and 333C each showed only the fast exit gating indistinguishable from the parent control channel. The Ki,ATP value for 333K and 333L were 2.2 ± 0.30 (n = 6) and 4.2 ± 0.60 mM (n = 6), respectively, compared with 0.19 ±0.04 for F333 (wild type). The 333K and 333L were significantly different from control and from each other (P < 0.05; one-way ANOVA). Three additional mutations were constructed, positive-charged 333R and negative-charged 333D and 333E, but failed to express. The results indicate that a positive-charged substitution, but not three noncharged substitutions, at position 333, conditionally allows transitions to a state of slow gating from the burst.
Figure 5.

Positive-charged 333K channels also show exceptional slowed gating from the burst. (A) Parental control ΔC26 channels without SUR exhibited fast gating, observed as very short bursts of one to several openings and PO of 0.05. (B) Similar to positive-charged substitutions at 334, lysine at position 333 exhibited the slowed gating in 6 of 12 patches, with PO increased from 0.05 to 0.65.
Homology Models of the Cytoplasmic COOH-terminal Domain of Kir6.2
Fig. 6 shows the results of our homology modeling based on the crystal structure of the COOH-terminal crystal of Kir3.1 (Nishida and MacKinnon, 2002; see also John et al., 2003; Trapp et al., 2003). The extended ATP conformer used is consistent with results where AMP-PNP, AMP-PCP, and ATP with and without Mg2+ (Ashcroft and Kakei, 1989; Hehl and Neumcke, 1994) all comparably inhibit the KATP channel. ATP associated most frequently with a groove in the upper half of the COOH-terminal homology structure that stretched from the G334 to the I182 region of Kir6.2 and included the K185 region. Extensive mutational and gating analysis of each of these positions (Drain et al., 1998; Tucker et al., 1998; Reimann et al., 1999a; Li et al., 2000), including here, have demonstrated a role in ATP binding steps and excluded a role in the gating steps of the overall ATP inhibition mechanism. Most importantly, the model shows F333 and G334 amides at the protein surface in proximity to the γ-phosphate of ATP docked to the ATP site. Positive-charged substitutions at these positions are unlikely to be buried but rather likely to be extended outward where they could interact with other protein surface charges and anionic membrane lipids involved in conformational coupling with the transmembrane gating domain.
Figure 6.
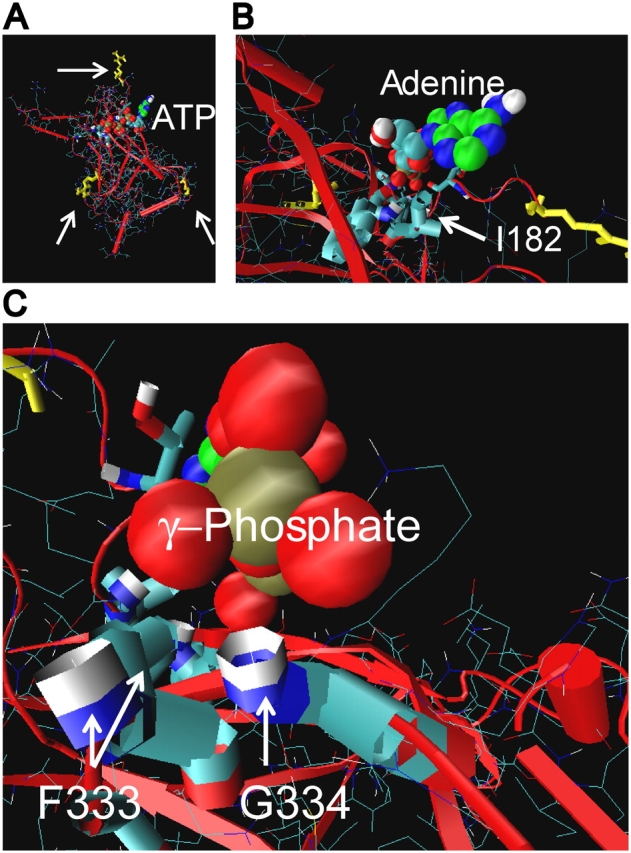
Relationship between membrane, ATP site groove, and cytoplasmic COOH-terminal domain of Kir6.2. (A) Structural model of cytoplasmic COOH-terminal domain of Kir6.2. Arrow at top points to R177, which is thought to interact with the inner leaflet of the membrane. Arrow at lower left points to R314, and at right points to E229, which form ion pairs between adjacent subunit domains (Lin et al., 2003). Downward from R177, I182 is at the right end of the putative ATP site groove and G334 is at the left end. A favorable crystallographic ATP conformer complexed onto the Kir6.2/3.1 homology structure by AUTODOCK is shown. The triphosphate (red oxygen and brown phosphorus balls) extends leftward along the groove. The adenine (green carbon and blue nitrogen balls) extends up and out of the groove. The binding site groove, therefore, appears to be relatively high on the cytoplasmic domain, and facing out to the cytoplasm toward the inner leaflet of the membrane. (B) Close-up view of the adenosine moiety binding near the hydrophobic side chain and hydrophilic peptide bond amide of I182. (C) Close-up view of the oxygens of the γ-phosphate of ATP approaching the backbone amides of F333 and G334, where favorable hydrogen bonding could occur. The proximity of the two amides and γ-phosphate of ATP, together with the opposite effects of positive-charged side-chain substitutions at 333 and 334 versus ATP occupancy of the site, suggests an electrostatic environment of this end of the ATP site that can alter gating at the transmembrane domain.
The Orientation of ATP in the Site Groove Is Not Discernible by Modeling
In our molecular dynamics modeling, the ribose-adenine base orientation can flip with the ribose approaching I182 and the adenine base approaching G334, or vice versa, with either orientation equally favorable in energetic terms. K185 extends outward from the ATP site groove, within a physiological Debye length (7–12 Å) of the β phosphate, regardless of the ATP orientation. The homology modeling indicates, however, in one but not the other ATP orientation, that the γ phosphate of ATP and the G334 likely come into close approach where they could interact. This is supported by the electrophysiological results indicating that at 334, positive-charged substitutions have the weakest loss in ATP inhibition gating out of 13 substitutions that express at the position. The results at 333 are consistent with this model of ATP binding. In this orientation, the adenosine moiety of ATP also could undergo entropic interactions with the hydrophobic side chain and hydrophilic bonding with the side chain and amide of I182, respectively. A close up of this orientation of ATP in the site groove shows oxygen atoms of the γ-phosphate approaching the backbone amides of G334 and F333, where favorable bonding interactions could occur.
Protamine Reverses Slowed Gating of the 334R Substituted Channel
The presence or absence of long-range electrostatic interactions, between the positive-charged arginine side chain at 334 and either anionic lipids at the cytoplasmic leaflet of the membrane or negative-charged side chains of Kir6.2, might determine whether 334R mutants exhibit slowed or speeded gating from the burst. We reasoned that the same interactions between the COOH-terminal arginines and membrane anionic lipids that prolong the active burst state of Kir channel activity in general (Hilgemann and Ball, 1996; Fan and Makielski, 1997; Baukrowitz et al., 1998; Shyng and Nichols, 1998; Fan and Makielski 1999; Lopes et al., 2002; Rohacs et al., 2003; Du et al., 2004) might be involved in the prolongation of the burst in the mutant channels. We might be able to use protamine, a polycationic peptide comprised of mostly arginine residues, to screen out the electrostatic interactions accounting for the slowed gating in the mutant channels. Fig. 7 shows a representative effect of 50 μM protamine on the slowed gating of a 334R channel. In all 12 patches with 334R channels exhibiting the slow burst exit gating studied, 50 μM protamine appeared to fully restore the fast gating from the burst. Washing out the protamine by superfusion reversed the protamine effect completely in four and partially in eight of the patches studied. Quantitative analysis demonstrates that the effect of 50 μM protamine is to completely restore the fast gating rate. Dwell time analysis showed that mean burst times changed from 80.3 ± 10.7 to 2.6 ± 1.8 ms and mean open times from 1.9 ± 0.2 ms to 1.1 ± 0.2 ms (n = 5). The post-protamine burst and open time durations are each indistinguishable from those of Kir6.2ΔC26 channels. These results indicate that polycationic protamine can fully prevent the interactions required for arginine at position 334 to slow gating from the burst.
Figure 7.
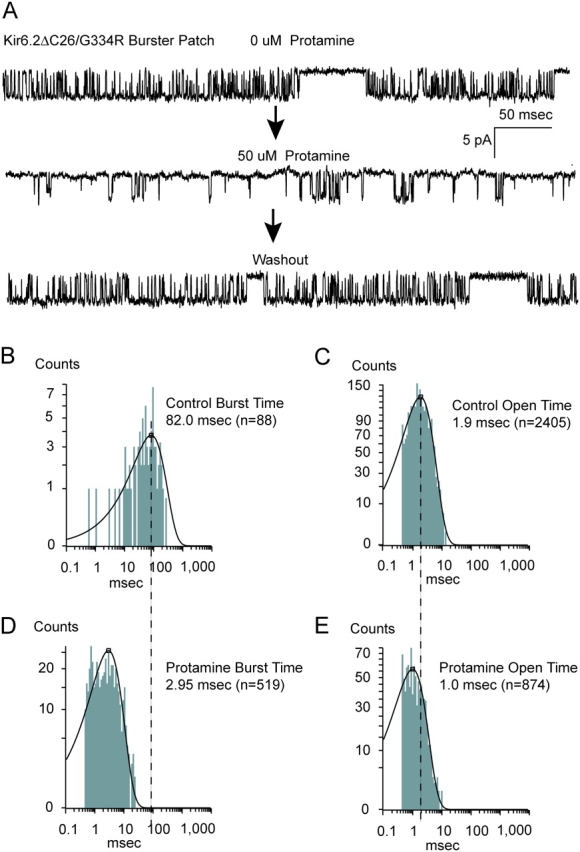
Slow burst exit gating caused by the 334R substitution can be speeded by large polycation protamines applied to the cytoplasmic face. (A) Single-channel currents showing the slow burst exit gating (long bursts of openings) before and after bath application of 50 μM protamine to the inside-out patch. Although requiring extensive washing the protamine effect can be washed out completely. Mean burst duration analysis of slowed gating in the 334R channel before (B) and after (D) the protamine effect. On average the protamine decreased mean burst times from 80.3 ± 10.7 to 2.6 ± 1.8 ms. Mean open duration analysis of the slowed gating in the 334R channel before (C) and after (E) the protamine effect. On average the protamine decreased mean open times from 1.9 ± 0.2 ms to 1.1 ± 0.2 ms (n = 5).
Protamine Polycations Speed Burst Exit Gating of the Wild-type SUR1/Kir6.2 Channel
We recall that SUR1/Kir6.2 wild-type channels in the absence of ATP naturally exhibit slow gating from the burst, similar to that of the slowed gating of Kir6.2ΔC26/334R in some patches. This raises the question whether protamine would speed burst exit gating in SUR1/Kir6.2 wild-type channels as well. Fig. 8 shows that this holds true. The 50 μM protamine applied to the cytoplasmic face of SUR1/Kir6.2 wild-type channels dramatically speeded gating from the burst. In all six SUR1/Kir6.2 patches studied, protamine dramatically speeded the gating. Dwell time analysis of the bursts before and after the protamine application indicated that mean burst times in the six patches changed from 48.0 ± 7.4 ms to 2.6 ± 0.9 and mean open time from 2.1 ± 0.2 ms to 1.0 ± 0.2 ms (n = 6). These changes are consistent with recent quantitative models of the burst gating of KATP channels, where the effect of ATP on burst times could be fully accounted for by its effect on mean open time (Li et al., 2002). Finally, the effect of protamine on SUR1/Kir6.2 wild-type channels was reversible. Washing out the protamine by superfusion reversed the effect completely in five and partially in one of the patches studied. The ability of protamine to speed gating from the burst of wild-type SUR1/Kir6.2 mimics the actions of ATP binding to the channel, and is consistent with long-range electrostatic interactions conformationally coupling ATP binding and gating domains of Kir6.2.
Figure 8.
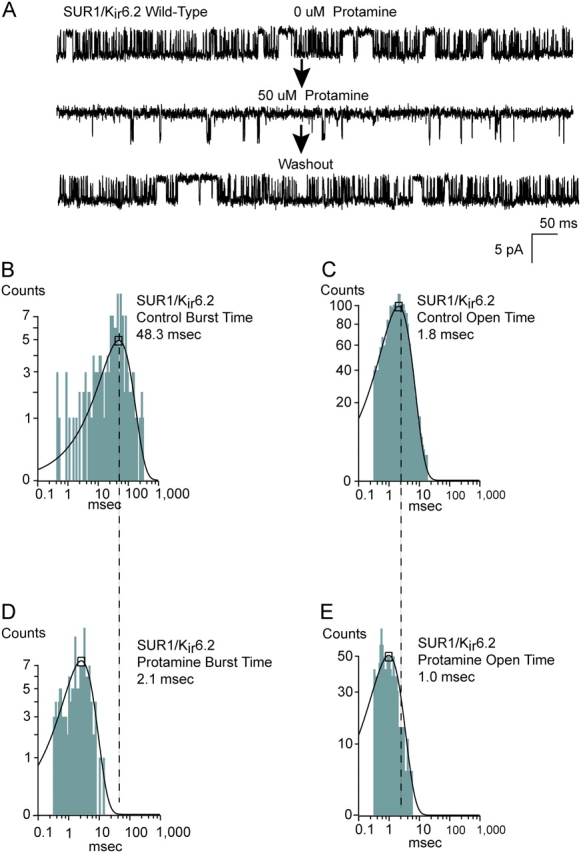
Native slow burst exit gating in wild-type SUR1/Kir6.2 channels also is speeded by protamine. (A) Wild-type SUR1/Kir6.2 channels in the absence of ATP with typical slow gating from the burst, before and after bath application of 50 μM protamine to the inside-out patch. The protamine effect was easily washed out in the patches studied. Mean burst duration analysis of wild-type SUR1/Kir6.2, before (B) and after (D) the protamine effect. On average the protamine decreased mean burst time from 48.0 ± 7.4 ms to 2.6 ± 0.9 (n = 5). Mean open duration analysis of wild-type SUR1/Kir6.2, before (C) and after (E) the protamine effect. On average the protamine decreased mean open time from 2.1 ± 0.2 ms to 1.0 ± 0.2 ms (n = 5).
DISCUSSION
The major point of our findings is that at the 334 end of the ATP site groove of Kir6.2, only positive-charged substitutions can alter and therefore couple to the transmembrane gating domain of the channel. The findings corroborate and extend similar findings at the 182 end of the ATP site groove. The positive-charged substitutions from the ATP site groove slow gating at the transmembrane domain by long-range electrostatic interactions, which is supported by the effect of protamine, as well. The findings are the first to demonstrate linkage specifically between the ATP site groove and gating regions of the KATP channel. Based on our findings of positive-charged substitutions in the ATP site groove and previous results demonstrating that anionic membrane lipids also slow gating from the burst, we propose the ATP site and linkage model for KATP channel gating shown in Fig. 9. Below, we will discuss the model that describes how ATP occupancy of the site groove, by displacing the COOH-terminal Kir6.2 domain from the membrane, indirectly causes inhibition gate closure.
Figure 9.

ATP-dependent linkage via the COOH-terminal domain to the transmembrane gating domain. The model features ATP binding in the site groove, which displaces by electrostatic, and possibly steric, interactions, the Kir6.2 COOH-terminal domain from the membrane. The model fits well with the general Kir1.0-7.0 linkage model, in which membrane anionic lipids bind the COOH-terminal domain to the inner leaflet of the membrane, and accounts for their favoring the burst gating conformation. Our model, however, specifies that ATP couples to the general linkage mechanism via electrostatic interactions based on the ability of only positive-charged substitutions at the 182 and 334 ends of the ATP site to slow gating from the burst, together with the ability of protamine to both reverse this slowing and to speed gating from the burst in wild-type SUR1/Kir6.2. The KATP channel model predicts that the triphosphate moiety of ATP in the site groove is positioned to electrostatically dissociate the COOH-terminal domain from the membrane by disrupting the general Kir electrostatic interactions between the COOH-terminal positive-charged residues and the negative-charged membrane lipids. In the absence of these electrostatic interactions, the associated M2 helices are free to undergo their transitions to the energetically more favorable interburst gating conformation.
Glycine at 334 of Kir6.2 Is Indispensable for Wild-type ATP Inhibition
All tested substitutions at position 334 of ΔC26 decreased ATP-dependent gating. The positive-charged arginine and lysine substitutions had two of the least effects on ATP-dependent gating, but nevertheless decreased it by >10-fold. As originally shown with aspartate (Drain et al., 1998), and shown here with glutamate, a negative-charged substitution at 334 had the strongest effect (31-fold decrease), nearly eliminating ATP-dependent gating. Not even the most closely related alanine (11-fold decrease) substituted well for glycine at the position. Phenylalanine (16-fold) more strongly increased the Ki for ATP inhibition than the alanine substitution, suggesting that the small size of the wild-type glycine residue along the 334 loop is important to the ATP site mechanism. Proline (26-fold), which generally has the least peptide backbone flexibility, more strongly increased the Ki for ATP inhibition than the phenylalanine substitution, suggesting that flexibility afforded by the wild-type glycine residue along the 334 loop of Kir6.2 is important to binding or linkage of the ATP site. Overall, each of the 13 substitutions at 334 increased Ki by 10-fold or more, but none slowed the ligand-independent gating of the channel to the interburst state, with important exceptions discussed below. We conclude that the result of a striking loss in ATP-dependent gating for all 334 substitutions indicates no other residue, including alanine, substituted well and that glycine at 334 is likely indispensable for wild-type inhibition.
Negative-charged Substitutions and the Orientation of Bound ATP
The strongest disruptions of ATP-dependent gating resulted from the negative-charged glutamate and aspartate substitutions at 334. The effects might be explained by an orientation of ATP required for inhibition gating where its triphosphate comes into close approach of 334. The interpretation is consistent with the notion that a negative charge at 334 most strongly would repel the triphosphate, effectively eliminating binding altogether. However, negative-charged substitutions at 182 at the other end of the ATP site groove have two of the strongest effects on ATP-dependent gating, which also might be similarly explained by the triphosphate coming into close approach of 182, rather than 334. At this point, therefore, the effects of negative charges at 182 and 334 do not clearly indicate the orientation of ATP bound to the site. Accordingly, Trapp et al. (2003) suggested that the triphosphate of ATP likely approaches the G334 end of the ATP binding site, whereas John et al. (2003) suggested a distinctly different orientation in which the triphosphate approaches the I182 end of the ATP binding site groove.
Positive-charged Substitutions and the Orientation of Bound ATP
Positive-charged substitutions at 334 had two of the weakest effects on ATP-dependent gating. This sharply contrasts with the positive-charged substitutions at 182, which had two of the strongest effects on ATP-dependent gating. Thus, unlike the effects of negative substitutions, the effects of positive substitutions at the 334 versus the 182 position are strikingly different. The results so far at 333 indicate that the positive-charged lysine substitution had a moderate effect compared with the strong effect of the leucine substitution on ATP-dependent gating, consistent with the 334 results. Direct physical measurements will be needed to definitively know the orientation of ATP in its site groove, but these findings favor the orientation where its triphosphate moiety electrostatically interacts at the 334 end during inhibition gating by ATP (Trapp et al., 2003; Antcliff et al., 2005). This orientation places the 333 and 334 amides in proximity to the γ-phosphate of ATP. Positive-charged substitutions at either 333 or 334 can favor slow gating from the burst, whereas negative-charged ATP favors the fast gating. The model indicates that transmembrane gating domain communicates electrostatically with the 334 end of the ATP site, and is consistent with the critical role of the negative-charged γ-phosphate of bound ATP in the inhibition gating of the KATP channel.
Indirect Conformational Coupling between ATP Binding and Gate Closure
Present data on Kir channels suggest that current flow arises by a conformation of each of the four inner M2 helices maintained by its associated cytoplasmic COOH-terminal domain being bound to the inner leaflet of the membrane (Lopes et al., 2002; Enkvetchakul and Nichols, 2003; Phillips and Nichols, 2003; Phillips et al., 2003; Rohacs et al., 2003; Drain et al., 2004; Du et al., 2004). To inhibit current flow in such a model, a ligand might dissociate the COOH-terminal domain from the membrane, freeing the inner M2 helix for gate closure. In other words, the COOH-terminal domain binding to membrane anionic lipids is conformationally coupled to the gating state of the transmembrane gating domain (Hilgemann and Ball, 1996; Fan and Makielski, 1997, 1999; Baukrowitz et al., 1998; Shyng and Nichols, 1998; Lopes et al., 2002; MacGregor et al., 2002; Enkvetchakul and Nichols, 2003; Rohacs et al., 2003; Du et al., 2004). Stabilization of Kir channel activity by favoring the active burst state of the transmembrane domain is a consequence of associating the COOH-terminal domains to the membrane anionic lipids, e.g., by adding exogenous anionic lipids. The reverse, inhibition of Kir channel activity by favoring the interburst state of the transmembrane domain is a consequence of dissociating the COOH-terminal domains from the membrane, e.g., by adding ATP or protamine, as shown here. Importantly, the conformational changes upon ATP binding to the site need not be directly coupled to the conformational changes accompanying gate closure within the transmembrane domain. Rather, we propose that site and gate coupling might arise in large part from a sequential two-step process in which first the ATP-occupied site groove favors dissociation of the COOH-terminal domain from the membrane, and once dissociated, the COOH-terminal domain and associated inner M2 helices stabilize a lower energy shut gate conformation.
Conserved COOH-terminal Positive Charges Underlie Linkage in Kir Channels In General
Physical features of the model for the general Kir linkage mechanism include positive-charged residues that mediate the association of Kir COOH-terminal domains with the membrane anionic lipids (Hilgemann and Ball, 1996; Fan and Makielski, 1997; Huang et al., 1998; Cukras et al., 2002; Sadja et al., 2001; Lopes et al., 2002; MacGregor et al., 2002; Rohacs et al., 2003; Schulze et al., 2003). The mutagenesis studies have identified multiple arginine or lysine residues conserved on the surface of the COOH terminus of Kir channel subunits, which are required for maintenance or stimulation of activity by the membrane anionic lipids. In the specific case of KATP channels, these cationic residues include R176, R177 (Fan and Makielski, 1997), R192, R201, K222, R301, and R314 (Shyng and Nichols, 1998; John et al., 2001; Cukras et al., 2002; Enkvetchakul and Nichols, 2003). In most cases, no one or two arginine residues appear to be absolutely required for the activation, suggesting that multiple arginine residues incrementally stabilize association of the COOH-terminal domain with the membrane. In addition, KATP channels are activated not only by PI(4,5)P2 (Hilgemann and Ball, 1996), but also by PI(3,4)P2, PI(3,4,5)P3, PI(4)P, PI (Rohacs et al., 2003), phosphatidic acid (Fan et al., 2003), and long-chain acylCoA (Manning-Fox et al., 2004), suggesting avidity rather than affinity properties are important here. The observations have been accounted for by a linkage mechanism general to all Kir channels in which long-range electrostatic interactions of multiple conserved arginine sites along Kir COOH termini associate with membrane anionic lipids of the inner leaflet of the membrane (Fan and Makielski, 1997; Rohacs et al., 1999, 2003; Shyng et al., 2000; Sadja et al., 2001; Cukras et al., 2002; Lopes et al., 2002; Enkvetchakul and Nichols, 2003; Du et al., 2004). The conserved COOH-terminal arginines required for membrane anionic lipid stimulation of all Kir channels can be designated “general” linkage residues of the Kir channel subfamily, to distinguish them from ATP site residues including 182, 333, and 334, required for ATP inhibition, which can be called “specific” linkage residues of Kir6.2.
ATP Site Groove and General Linkage Residues Might Be Structurally Nonoverlapping
In KATP channel gating, the activation by membrane anionic lipids has been shown to reciprocally antagonize the ATP inhibition (Baukrowitz et al., 1998; Shyng and Nichols, 1998; Fan and Makielski, 1999; Wang et al., 2002), reminiscent of ADP antagonism of the ATP-inhibited state of the KATP channel (Alekseev et al., 1998; Ribalet et al., 2000; John et al., 2001; Li et al., 2002). In addition, some arginine residues, for example, R177, R206, and R301 (Baukrowitz et al., 1998; Shyng and Nichols, 1998; Shyng et al., 2000; Cukras et al., 2002) appear to participate in both processes, whereas others are associated with either ATP inhibition or membrane anionic lipid activation. The arginine residues whose mutation disrupts both processes have been taken to indicate functional overlap between ATP binding at the site groove, and COOH-terminal arginine interactions with the anionic membrane lipids (Enkvetchakul and Nichols, 2003). The functional overlap can be explained by these residues being part of the general linkage mechanism. No mutations of ATP site groove positions, however, indicate a role in membrane anionic lipid stimulation, suggesting that the residues of the ATP site groove and the general linkage residues of the COOH-terminal Kir6.2 domain are separate (Enkvetchakul and Nichols, 2003; John et al., 2003; Trapp et al., 2003; this paper). If the ATP site and the general Kir linkage residues are functionally nonoverlapping, then bound ATP itself is expected to directly dissociate the COOH-terminal domain from the membrane.
Effect of Positive Charges in the ATP Site Are Consistent with Ligand-dependent Linkage
Based on our previous results with positive-charged substitutions at the I182 position, we proposed a ligand-dependent linkage model (Li et al., 2000). In ligand-dependent linkage, the unoccupied ATP site groove is decoupled from conformational changes at the transmembrane gating domain. The ligand-dependent linkage is consistent with the lack of strict reciprocity between ligand-independent gating kinetics and the Ki for ATP inhibition gating of numerous mutant KATP channels. The ligand-dependent linkage is also supported by the conditional behavior of positive-charged substitutions to dramatically slow gating from the burst in some but not all patches. An important conclusion from the studies reported here is that positive-charged substitutions now at the 334 end of the ATP site also conditionally engage the gating mechanism through long-range electrostatic interactions. Ligand-dependent linkage in the KATP channel thereby exhibits properties of both conditionality and long-range electrostatics. The physical model predicts that in some but not all patches expressing the SUR-less channels, positive charge at either end of the ATP site groove favors an association of the COOH-terminal domain arginines with the membrane anionic lipids to account for the slowed burst exit gating. In the wild-type channels with SUR in the absence but not in the presence of an ATP-occupied site groove, the association of the general linkage arginines with the membrane is likely favored to account for the slow burst exit gating normally observed there.
The model was further tested by substitution of positive-charged lysine at the adjacent 333 position. With regard to ligand-independent gating, the positive-charged 333K, but not 333L, 333S, or 333C conditionally but dramatically slowed burst exit gating. With regard to ATP-dependent gating, the 333K and 333L substitutions resulted in 12-fold and 22-fold loss, respectively. The results at 333 and 334 are consistent with this end of the ATP site interacting with the triphosphate of ATP, where positive-charged substitutions are better tolerated than hydrophobic or negative-charged substitutions. It is noteworthy that the conserved human F333 of Kir6.2 was recently shown to be naturally mutated to 333I, resulting in loss of glucose-stimulated insulin secretion and permanent neonatal diabetes (Sagen et al., 2004). Taken together, the results indicate that the underlying molecular defect is the disruption of ATP binding and its conformational coupling to gate closure.
Protamine Mimics ATP, Driving the KATP Channel into the Interburst Conformation
Our results also indicate that the polycationic peptide protamine can displace the burst–interburst gating equilibrium of KATP channels to greatly favor the inactive interburst. Because the change in gating occurs in the absence of ATP and in mutant channels with greatly diminished ATP sensitivity, changes in the affinity of the ATP site cannot account for the result. Protamines are polycationic peptides rich in arginines. The findings suggest that protamines likely bind and neutralize anionic phospholipids of the membrane sufficiently to dissociate from the membrane the COOH-terminal domains (Fan and Makielski, 1997; 1999; Shyng et al., 2000; Sadja et al., 2001; Cukras et al., 2002; Lopes et al., 2002; Enkvetchakul and Nichols, 2003; Fan et al., 2003; Rohacs et al., 2003; Du et al., 2004), freeing them and the inner M2 helices for gate closure. ATP and protamines could have additional actions directly on the Kir6.2 protein to speed gate closure.
Electrostatic Interactions between ATP and General Kir Linkage Mechanisms
Taken together, negative-charged ATP at the site and positive-charged protamines at the membrane share the effect of favoring the shut gate state, whereas positive-charged substitutions at 182, 333, and 334 of the site and anionic lipids at the membrane favor the open gate state. The results suggest that when ATP binds to the site groove it interacts with the linkage mechanism through long-range electrostatic interactions. A reasonable hypothesis is that part or all of the four negative triphosphate charges of bound ATP have an effect on the general Kir linkage mechanism that is opposite to the effect of positive-charged substitutions of the ATP site groove observed in the absence of ATP here. Thus, negative-charged phosphates of ATP might repel the membrane anionic lipids, screen the COOH-terminal arginine linkage residues, or sterically block an association of the COOH-terminal domain with the membrane, to favor dissociation and, in turn, burst gate closure. Because the Ki,ATP is far lower than the Ki,ADP then the most effective phosphate in this step of the mechanism is likely to be the γ phosphate.
In summary, the conditional nature of positive-charged substitutions at positions 333 and 334 adds additional support that the conformational coupling between the site groove and the transmembrane gating domain of an individual Kir6.2 subunit occurs only in the presence of ATP bound to that subunit. A given subunit likely can be in the shut gate state of the transmembrane domain without any significant affinity change at its unoccupied site groove because, in the absence of ATP, the two domains are uncoupled. This would explain how a concerted action of the four inner M2 helices to close the gate (Drain et al., 2004) fails to effect a concerted increase in the apparent affinity of all four COOH-terminal ATP sites. The conformational coupling is likely to be a two-step, indirect process in which ATP dissociates the COOH-terminal domain from the membrane as above, and then, the free COOH-terminal domain and inner M2 helices transit to a lower energy interburst conformation stabilized by the COOH-terminal and transmembrane gating domains. The model does not necessarily require the ATP-occupied site groove region to undergo complex and direct peptide–peptide interactions with the transmembrane gating domain. Rather than directly driving gate closure, ATP binding might in large part, simply by dissociating the COOH termini, free the associated inner M2 helices, allowing burst gate closure.
Acknowledgments
The authors thank Ray Frizzell, Bob Bridges, and members of their labs for excellent discussions and oocytes.
This study was supported by National Science Foundation grant MCB 9817116 and National Institutes of Health grant R21 DK068833-01 to P. Drain.
Olaf S. Andersen served as editor.
References
- Aguilar-Bryan, L., and J. Bryan. 1999. Molecular biology of adenosine triphosphate-sensitive potassium channels. Endocr. Rev. 20:101–135. [DOI] [PubMed] [Google Scholar]
- Aguilar-Bryan, L., C.G. Nichols, S.W. Weschler, J.P.T. Clement, A.E. Boyd III, G. Gonzalez, H. Herrera-Sosa, K. Nguy, J. Bryan, and D.A. Nelson. 1995. Cloning of the β cell high-affinity sulfonylurea receptor: a regulator of insulin secretion. Science. 268:423–426. [DOI] [PubMed] [Google Scholar]
- Aguilar-Bryan, L., J. Bryan, and M. Nakazaki. 2001. Of mice and men: KATP channels and insulin secretion. Recent Prog. Horm. Res. 56:47–68. [DOI] [PubMed] [Google Scholar]
- Alekseev, A.E., P.A. Brady, and A. Terzic. 1998. A ligand-insensitive state of cardiac ATP-sensitive K+ channels. Basis for channel opening. J. Gen. Physiol. 111:381–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antcliff, J.F., S. Haider, P. Proks, M.S. Sansom, and F.M. Ashcroft. 2005. Functional analysis of a structural model of the ATP-binding site of the KATP channel Kir6.2 subunit. EMBO J. 24:229–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft, F.M. 1988. Adenosine 5′-triphosphate-sensitive potassium channels. Annu. Rev. Neurosci. 11:97–118. [DOI] [PubMed] [Google Scholar]
- Ashcroft, F.M., and M. Kakei. 1989. ATP-sensitive K+ channels in rat pancreatic β-cells: modulation by ATP and Mg2+ ions. J. Physiol. 416:349–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft, F.M., D.E. Harrison, and S.J. Ashcroft. 1984. Glucose induces closure of single potassium channels in isolated rat pancreatic β-cells. Nature. 312:446–448. [DOI] [PubMed] [Google Scholar]
- Babenko, A.P., L. Aguilar-Bryan, and J. Bryan. 1998. A view of SUR/Kir6.x, KATP channels. Annu. Rev. Physiol. 60:667–687. [DOI] [PubMed] [Google Scholar]
- Babenko, A.P., G. Gonzalez, and J. Bryan. 1999. a. The tolbutamide site of SUR1 and a mechanism for its functional coupling to KATP channel closure. FEBS Lett. 459:367–376. [DOI] [PubMed] [Google Scholar]
- Babenko, A.P., G. Gonzalez, and J. Bryan. 1999. b. Two regions of sulfonylurea receptor specify the spontaneous bursting and ATP inhibition of KATP channel isoforms. J. Biol. Chem. 274:11587–11592. [DOI] [PubMed] [Google Scholar]
- Babenko, A.P., G. Gonzalez, and J. Bryan. 1999. c. The N-terminus of Kir6.2 limits spontaneous bursting and modulates the ATP-inhibition of KATP channels. Biochem. Biophys. Res. Commun. 255:231–238. [DOI] [PubMed] [Google Scholar]
- Babenko, A.P., G. Gonzalez, and J. Bryan. 2000. Pharmaco-topology of sulfonylurea receptors. J. Biol. Chem. 275:717–720. [DOI] [PubMed] [Google Scholar]
- Baukrowitz, T., U. Schulte, D. Oliver, S. Herlitze, S.J. Tucker, J.P. Ruppersbrerg, and B. Fakler. 1998. PIP2 and PIP as determinants for ATP inhibition of KATP channels. Science. 282:1141–1144. [DOI] [PubMed] [Google Scholar]
- Colquhoun, D., and F.J. Sigworth. 1995. Fitting and statistical analysis of single-channel records. Single-channel Recording. Second edition. B. Sakmann and E. Neher, editors. Plenum Press, NY. 483–587.
- Cook, D.L., and C.N. Hales. 1984. Intracellular ATP directly blocks K+ channels in pancreatic β-cells. Nature. 311:271–273. [DOI] [PubMed] [Google Scholar]
- Cukras, C.A., I. Jeliazkova, and C.G. Nichols. 2002. Structural and functional determinants of conserved lipid interaction domains of inward rectifying Kir6.2 channels. J. Gen. Physiol. 119:581–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drain, P., A.E. Dubin, and R.W. Aldrich. 1994. Regulation of Shaker K channel inactivation gating by the cAMP-dependent protein kinase. Neuron. 12:1097–1109. [DOI] [PubMed] [Google Scholar]
- Drain, P., L. Li, and J. Wang. 1998. KATP channel inhibition by ATP requires distinct functional domains of the cytoplasmic C terminus of the pore-forming subunit. Proc. Natl. Acad. Sci. USA. 95:13953–13958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drain, P., X. Geng, and L. Li. 2004. Concerted gating mechanism underlying KATP channel inhibition by ATP. Biophys. J. 86:2101–2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du, X., H. Zhang, C. Lopes, T. Mirshahi, T. Rohacs, and D.E. Logothetis. 2004. Characteristic interactions with phosphatidylinositol 4,5-bisphosphate determine regulation of Kir channels by diverse modulators. J. Biol. Chem. 279:37271–37281. [DOI] [PubMed] [Google Scholar]
- Eigen, M. 1968. New looks and outlooks on physical enzymology. Q. Rev. Biophys. 1:3–33. [DOI] [PubMed] [Google Scholar]
- Enkvetchakul, D., and C.G. Nichols. 2003. Gating mechanism of KATP channels: function fits form. J. Gen. Physiol. 122:471–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, Z., and J.C. Makielski. 1997. Anionic phospholipids activate ATP-sensitive potassium channels. J. Biol. Chem. 272:5388–5395. [DOI] [PubMed] [Google Scholar]
- Fan, Z., and J.C. Makielski. 1999. Phosphoinositides decrease ATP sensitivity of the cardiac ATP-sensitive K channel: a molecular probe for the mechanism of ATP-sensitive inhibition. J. Gen. Physiol. 114:251–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, Z., L. Gao, and W. Wang. 2003. Phosphatidic acid stimulates cardiac KATP channels like phosphatidylinositols, but with novel gating kinetics. Am. J. Physiol. Cell Physiol. 284:C94–C102. [DOI] [PubMed] [Google Scholar]
- Gillis, K.D., W.M. Gee, A. Hammoud, M.L. McDaniel, L.C. Falke, and S. Misler. 1989. Effects of sulfonamides on a metabolite-regulated ATPi-sensitive K+ channel in rat pancreatic β-cells. Am. J. Physiol. 257:C1119–C1127. [DOI] [PubMed] [Google Scholar]
- Gloyn, A.L., E.A. Cummings, E.L. Edghill, L.W. Harries, R. Scott, T. Costa, I.K. Temple, A.T. Hattersley, and S. Ellard. 2004. a. Permanent neonatal diabetes due to paternal germline mosaicism for an activating mutation of the KCNJ11 gene encoding the Kir6.2 subunit of the β-cell potassium adenosine triphosphate channel. J. Clin. Endocrinol. Metab. 89:3932–3935. [DOI] [PubMed] [Google Scholar]
- Gloyn, A.L., E.R. Pearson, J.F. Antcliff, P. Proks, G.J. Bruining, A.S. Slingerland, N. Howard, S. Srinivasan, J.M.C.L. Silva, J. Molnes, et al. 2004. b. Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N. Engl. J. Med. 350:1838–1849. [DOI] [PubMed] [Google Scholar]
- Goodsell, D.S., G.M. Morris, and A.J. Olson. 1996. Automated docking of flexible ligands: applications of AutoDock. J. Mol. Recognit. 9:1–5. [DOI] [PubMed] [Google Scholar]
- Gribble, F.M., S.J. Tucker, and F.M. Ashcroft. 1997. The essential role of the Walker A motifs of SUR1 in KATP channel activation by Mg-ADP and diazoxide. EMBO J. 16:1145–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribble, F.M., S.J. Tucker, T. Haug, and F.M. Ashcroft. 1998. MgATP activates the β-cell KATP channel by interaction with its SUR1 subunit. Proc. Natl. Acad. Sci. USA. 95:7185–7190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammes, G.G., and C.-W. Wu. 1971. Regulation of enzyme activity. Science. 172:1205–1211. [DOI] [PubMed] [Google Scholar]
- Hehl, S., and B. Neumcke. 1994. KATP channels of mouse skeletal muscle: mechanism of channel blockage by AMP-PNP. Eur. Biophys. J. 23:231–237. [DOI] [PubMed] [Google Scholar]
- Hetenyi, C., and D. van der Spoel. 2002. Efficient docking of peptides to proteins without prior knowledge of the binding site. Protein Sci. 11:1729–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgemann, D.W., and R. Ball. 1996. Regulation of cardiac Na+, Ca2+ exchange and KATP potassium channels by PIP2. Science. 273:956–959. [DOI] [PubMed] [Google Scholar]
- Huang, C.L., S. Feng, and D.W. Hilgemann. 1998. Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by G beta-gamma. Nature. 391:803–806. [DOI] [PubMed] [Google Scholar]
- Inagaki, N., T. Gonoi, J.P.T. Clement, N. Namba, J. Inazawa, G. Gonzalez, L. Aguilar-Bryan, S. Seino, and J. Bryan. 1995. Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science. 270:1166–1170. [DOI] [PubMed] [Google Scholar]
- John, S.A., J.R. Monck, J.N. Weiss, and B. Ribalet. 1998. The sulphonylurea receptor SUR1 regulates ATP-sensitive mouse Kir6.2 K+ channels linked to the green fluorescent protein in human embryonic kidney cells (HEK293). J. Physiol. 510:333–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John, S.A., J.N. Weiss, and B. Ribalet. 2001. Regulation of cloned ATP-sensitive K channels by adenine nucleotides and sulfonylureas: interactions between SUR1 and positively charged domains on Kir6.2. J. Gen. Physiol. 118:391–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John, S.A., J.N. Weiss, L.H. Xie, and B. Ribalet. 2003. Molecular mechanism for ATP-dependent closure of the K+ channel Kir6.2. J. Physiol. 552:23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic, A., S. Jovanovic, A.J. Carrasco, and A. Terzic. 1998. Acquired resistance of a mammalian cell line to hypoxia-reoxygenation through contransfection of Kir6.2 and SUR1 clones. Lab. Invest. 78:1101–1107. [PubMed] [Google Scholar]
- Koshland, D.E., Jr., G. Nemethy, and D. Filmer. 1966. Comparison of experimental binding data and theoretical models in proteins containing subunits. Biochemistry. 5:365–385. [DOI] [PubMed] [Google Scholar]
- Koster, J.C., Q. Sha, S. Shyng, and C.G. Nichols. 1999. ATP inhibition of KATP channels: control of nucleotide sensitivity by the N-terminal domain of the Kir6.2 subunit. J. Physiol. 515:19–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, L., J. Wang, and P. Drain. 2000. The I182 region of Kir6.2 is closely associated with ligand binding in KATP channel inhibition by ATP. Biophys. J. 79:841–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, L., X. Geng, and P. Drain. 2002. Open state destabilization by ATP occupancy is mechanism speeding burst exit underlying KATP channel inhibition by ATP. J. Gen. Physiol. 119:105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, Y.W., T. Jia, A.M. Weinsoft, and S.L. Shyng. 2003. Stabilization of the activity of ATP-sensitive potassium channels by ion pairs formed between adjacent Kir6.2 subunits. J. Gen. Physiol. 122:225–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes, C.M.B., H. Zhang, T. Rohacs, T. Jin, J. Yang, and D.E. Logothetis. 2002. Alterations in conserved Kir channel-PIP2 interactions underlie channelopathies. Neuron. 34:933–944. [DOI] [PubMed] [Google Scholar]
- Lorenz, E., A.E. Alekseev, G.B. Krapivinsky, A.J. Carrasco, D.E. Clapham, and A. Terzic. 1998. Evidence for direct physical association between a K+ channel (Kir6.2) and an ATP-binding cassette protein (SUR1) which affects cellular distribution and kinetic behavior of an ATP-sensitive K+ channel. Mol. Cell. Biol. 18:1652–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loussouarn, G., E.N. Makhina, T. Rose, and C.G. Nichols. 2000. Structure and dynamics of the pore of inwardly rectifying K channels. J. Biol. Chem. 275:1137–1144. [DOI] [PubMed] [Google Scholar]
- MacGregor, G.G., K. Dong, C.G. Vanoye, L. Tang, G. Giebisch, and S.C. Hebert. 2002. Nucleotides and phospholipids compete for binding to the C terminus of KATP channels. Proc. Natl. Acad. Sci. USA. 99:2726–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning-Fox, J.E., C.G. Nichols, and P.E. Light. 2004. Activation of adenosine triphosphate-sensitive potassium channels by acyl coenzyme A esters involves multiple phosphatidylinositol 4,5 bisphosphate-interacting residues. Mol. Endocrinol. 18:679–686. [DOI] [PubMed] [Google Scholar]
- Monod, J., J. Wyman, and J.P. Changeux. 1965. On the nature of allosteric transitions: a plausible model. J. Mol. Biol. 12:88–118. [DOI] [PubMed] [Google Scholar]
- Morris, G.M., D.S. Goodsell, R. Huey, and A.J. Olson. 1996. Distributed automated docking of flexible ligands to proteins: parallel applications of AutoDock 2.4. J. Comput. Aided Mol. Des. 10:293–304. [DOI] [PubMed] [Google Scholar]
- Nichols, C.G., W.J. Lederer, and M.B. Cannell. 1991. ATP dependence of KATP channel kinetics in isolated membrane patches from rat ventricle. Biophys. J. 60:1164–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols, C.G., S.L. Shyng, A. Nestorowicz, B. Glaser, J.P.T. Clement, G. Gonzalez, L. Aguilar-Bryan, M.A. Permutt, and J. Bryan. 1996. Adenosine diphosphate as an intracellular regulator of insulin secretion. Science. 272:1785–1787. [DOI] [PubMed] [Google Scholar]
- Nishida, M., and R. MacKinnon. 2002. Structural basis of inward rectification: cytoplasmic pore of the G protein-gated inward rectifier GIRK1 at 1.8 Å resolution. Cell. 111:957–965. [DOI] [PubMed] [Google Scholar]
- Noma, A. 1983. ATP-regulated K+ channels in cardiac muscle. Nature. 305:147–148. [DOI] [PubMed] [Google Scholar]
- Phillips, L.R., and C.G. Nichols. 2003. Ligand-induced closure of inward rectifier Kir6.2 channels traps spermine in the pore. J. Gen. Physiol. 122:795–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips, L.R., D. Enkvetchakul, and C.G. Nichols. 2003. Gating dependence of inner pore access in inward rectifier K+ channels. Neuron. 37:953-962. [DOI] [PubMed] [Google Scholar]
- Qin, D.Y., M. Takano, and A. Noma. 1989. Kinetics of ATP-sensitive K+ channel revealed with oil-gate concentration jump method. Am. J. Physiol. 257:H1624–H1633. [DOI] [PubMed] [Google Scholar]
- Reidhaar-Olson, J.F., J.U. Bowie, R.M. Breyer, J.C. Hu, K.L. Knight, W.A. Lin, M.C. Mossing, K.R. Shoemaker, and R.T. Sauer. 1991. Random mutagenesis of protein sequences using oligonucleotide cassette. Methods Enzymol. 208:564–586. [DOI] [PubMed] [Google Scholar]
- Reimann, F., T.J. Ryder, S.J. Tucker, and F.M. Ashcroft. 1999. a. The role of lysine 185 in the Kir6.2 subunit of the ATP-sensitive channel in channel inhibition by ATP. J. Physiol. 520:661–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimann, F., S.J. Tucker, P. Proks, and F.M. Ashcroft. 1999. b. Involvement of the N-terminus of Kir6.2 in coupling to the sulphonylurea receptor. J. Physiol. 518:325–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribalet, B., S.A. John, and J.N. Weiss. 2000. Regulation of cloned ATP–sensitive K channels by phosphorylation, MgADP, and phosphatidylinositol bisphosphate (PIP2): a study of channel rundown and reactivation. J. Gen. Physiol. 116:391–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohacs, T., J. Chen, G.D. Prestwich, and D.E. Logothetis. 1999. Distinct specificities of inwardly rectifying K+ channels for phosphoinositides. J. Biol. Chem. 274:36065–36072. [DOI] [PubMed] [Google Scholar]
- Rohacs, T., C.M.B. Lopes, T. Jin, P.P. Ramdya, Z. Molnar, and D.E. Logothetis. 2003. Specificity of activation by phosphoinositides determines lipid regulation of Kir channels. Proc. Natl. Acad. Sci. USA. 100:745–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadja, R., K. Smadja, N. Alagem, and E. Reuveny. 2001. Coupling G beta-gamma-dependent activation to channel opening via pore elements in inwardly rectifying potassium channels. Neuron. 29:669–680. [DOI] [PubMed] [Google Scholar]
- Sagen, J.V., H. Raeder, E. Hathout, N. Shehadeh, K. Gudmundsson, H. Baevre, D. Abuelo, C. Phornphutkul, J. Molnes, G.I. Bell, et al. 2004. Permanent neonatal diabetes due to mutations in KCNJ11 encoding Kir6.2: patient characteristics and initial response to sulfonylurea therapy. Diabetes. 53:2713–2718. [DOI] [PubMed] [Google Scholar]
- Schulze, D., M. Rapedius, T. Krauter, and T. Baukrowitz. 2003. Long-chain acyl-CoA esters and phosphatidylinositol phosphates modulate ATP inhibition of KATP channels by the same mechanism. J. Physiol. 552:357–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng, S., T. Ferrigni, and C.G. Nichols. 1997. Regulation of KATP channel activity by diazoxide and MgADP. Distinct functions of the two nucleotide binding folds of the sulfonylurea receptor. J. Gen. Physiol. 110:643–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng, S.L., and C.G. Nichols. 1998. Membrane phospholipid control of nucleotide sensitivity of KATP channels. Science. 282:1138–1141. [DOI] [PubMed] [Google Scholar]
- Shyng, S.L., C.A. Cukras, J. Hardwood, and C.G. Nichols. 2000. Structural determinant of PIP2 regulation of inward rectifier KATP channels. J. Gen. Physiol. 116:599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigworth, F.J., and S. Sine. 1987. Data transformations for improved display and fitting of single-channel dwell time histograms. Biophys. J. 52:1047–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano, M., L.H. Xie, H. Otani, and M. Horie. 1998. Cytoplasmic terminus domains of Kir6.x confer different nucleotide-dependent gating on the ATP-sensitive K+ channel. J. Physiol. 512:395–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp, S., P. Proks, S.J. Tucker, and F.M. Ashcroft. 1998. Molecular analysis of ATP-sensitive K channel gating and implications for channel inhibition by ATP. J. Gen. Physiol. 112:333–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp, S., S. Haider, P. Jones, M.S. Sansom, and F.M. Ashcroft. 2003. Identification of residues contributing to the ATP binding site of Kir6.2. EMBO J. 22:2903–2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trube, G., and J. Hescheler. 1984. Inward-rectifying channels in isolated patches of the heart cell membrane: ATP-dependence and comparison with cell-attached patches. Pflugers Arch. 401:178–184. [DOI] [PubMed] [Google Scholar]
- Tucker, S.J., F.M. Gribble, P. Proks, S. Trapp, T.J. Ryder, T. Haug, F. Reimann, and F.M. Ashcroft. 1998. Molecular determinants of KATP channel inhibition by ATP. EMBO J. 17:3290–3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker, S.J.F., F.M. Gribble, C. Zhao, S. Trapp, and F.M. Ashcroft. 1997. Truncation of Kir6.2 produces ATP-sensitive K+ channels in the absence of the sulphonylurea receptor. Nature. 387:179–183. [DOI] [PubMed] [Google Scholar]
- Wang, C., K. Wang, W. Wang, Y. Cui, and Z. Fan. 2002. Compromised ATP binding as a mechanism of phosphoinositide modulation of ATP-sensitive K+ channels. FEBS Lett. 532:177–182. [DOI] [PubMed] [Google Scholar]
- Zingman, L.V., A.E. Alekseev, M. Bienengraeber, D. Hodgson, A.B. Karger, P.P. Dzeja, and A. Terzic. 2001. Signaling in channel/enzyme multimers: ATPase transitions in SUR module gate ATP-sensitive K conductance. Neuron. 31:233–245. [DOI] [PubMed] [Google Scholar]
- Zung, A., B. Glaser, R. Nimri, and Z. Zadik. 2004. Glibenclamide treatment in permanent neonatal diabetes mellitus due to an activating mutation in Kir6.2. J. Clin. Endocrinol. Metab. 89:5504–5507. [DOI] [PubMed] [Google Scholar]