

Abstract
Phosphatidylinosital-4,5-bisphosphate (PIP2) acts as an essential factor regulating the activity of all Kir channels. In most Kir members, the dependence on PIP2 is modulated by other factors, such as protein kinases (in Kir1), Gβγ (in Kir3), and the sulfonylurea receptor (in Kir6). So far, however, no regulator has been identified in Kir2 channels. Here we show that polyamines, which cause inward rectification by selectively blocking outward current, also regulate the interaction of PIP2 with Kir2.1 channels to maintain channel availability. Using spermine and diamines as polyamine analogs, we demonstrate that both spontaneous and PIP2 antibody–induced rundown of Kir2.1 channels in excised inside-out patches was markedly slowed by long polyamines; in contrast, polyamines with shorter chain length were ineffective. In K188Q mutant channels, which have a low PIP2 affinity, application PIP2 (10 μM) was unable to activate channel activity in the absence of polyamines, but markedly activated channels in the presence of long diamines. Using neomycin as a measure of PIP2 affinity, we found that long polyamines were capable of strengthening either the wild type or K188Q channels' interaction with PIP2. The negatively charged D172 residue inside the transmembrane pore region was critical for the shift of channel–PIP2 binding affinity by long polyamines. Sustained pore block by polyamines was neither sufficient nor necessary for this effect. We conclude that long polyamines serve a dual role as both blockers and coactivators (with PIP2) of Kir2.1 channels.
INTRODUCTION
Inward rectifier potassium (Kir) channels readily conduct inward currents at membrane potentials negative to the K+ reversal potential (EK), but pass progressively less outward current as membrane potential becomes more positive than EK. This inward rectifying property plays an essential role in stabilizing resting membrane potential and regulating excitability (Doupnik et al., 1995; Hille, 2001). The mechanism of inward rectification involves voltage-dependent block of outward currents by polyamines (Ficker et al., 1994; Lopatin et al., 1994; Fakler et al., 1995) and Mg2+ (Matsuda et al., 1987; Vandenberg, 1987). All seven subfamilies (Kir1-7) share a common structure consisting of intracellular NH2 and COOH termini and two membrane spanning segments (M1 and M2) flanking a pore-forming P-loop with a signature K conductance sequence. Recently published crystal structures (Doyle et al., 1998; Nishida and MacKinnon, 2002; Kuo et al., 2003) show that the Kir pore consists not only of the classic “transmembrane pore” (∼35 Å long, spanning the plasma membrane), which is formed by the P-loop (containing the selectivity filter ∼12 Å and the M1 and M2 helices [∼20 Å inner part]), but also of a “cytoplasmic pore” formed by regions of the cytoplasmic NH2 and COOH termini. The cytoplasmic pore extends the total pore length intracellularly by ∼30 Å, with a width varying from 7 to 15 Å. In Kir2.1 channels, negatively charged residues in both the transmembrane pore at D172 (Lu and MacKinnon, 1994; Stanfield et al., 1994; Wible et al., 1994; Yang et al., 1995) and in the cytoplasmic pore at E224 and E299 (Yang et al., 1995; Kubo and Murata, 2001) confer strong inward rectification by interacting with polyamines.
In addition to polyamines, Kir2.1 channels, as well as other Kir channels, are regulated by membrane phosphoinositides such as PIP2. The direct interaction between the negative phosphate head group of PIP2 and several positively charged residues in NH2 and COOH termini (e.g., R67, K188, R189, R218, and R312 in Kir2.1) are essential for activation of channels (Fan and Makielski, 1997; Shyng et al., 2000; Lopes et al., 2002; Schulze et al., 2003; Zeng et al., 2003). Moreover, in other Kir family members, different signaling partners appear to influence Kir channel activity by modulating their interaction with PIP2. For example, PKA phosphorylation enhance Kir1.1 (ROMK1)–PIP2 interaction (Liou et al., 1999). G protein βγ subunits (Gβγ) stabilize the Kir3.1/4 (GIRK1/4)–PIP2 interaction (Huang et al., 1998; Ho and Murrell-Lagnado, 1999; Zhang et al., 1999). The PIP2 interaction with the KATP (Kir6.x) channels is regulated by SUR and relates to ATP sensitivity (Baukrowitz et al., 1998; Shyng and Nichols, 1998; Song and Ashcroft, 2001). So far, however, no regulator of PIP2's interaction with the strong inward rectifier Kir2.1 channel has been identified (Rohacs et al., 1999; Zhang et al., 1999; Hilgemann et al., 2001; Soom et al., 2001). In this study, we present evidence that polyamines play this role. We show that polyamines act as cofactors in PIP2 regulation of Kir2.1 channel activity. Long polyamines, such as spermine and 1,10-decanediamine (DA10) or 1,12-dodecanediamine (DA12), are capable of stabilizing the channel in an open configuration, enabling an apparent increase of channel–PIP2 binding affinity. The mechanism involves polyamines interacting with negative charges at D172 in the transmembrane pore, without requiring sustained pore block. Thus, polyamines serve dual roles in Kir2.1 channels, by both inducing inward rectification and regulating channel availability.
MATERIALS AND METHODS
Molecular Biology
Kir channel cDNAs were subcloned into pBluescript KS (Stratagene). “Quikchange” mutagenesis (Stratagene) was used to construct individual mutants. cRNAs were synthesized using T7 polymerase (Ambion) and injected into stage IV–V Xenopus oocytes.
Oocyte Preparation and Channel Expression
Xenopus oocytes were isolated by partial ovariectomy in mature female Xenopus and then defolliculated by treatment for 1 h with 1 mg/ml collagenase in the Barth's solution. The day after collagenase treatment, selected oocytes were pressure-injected with ∼50 nl RNA (1–100 ng/ml) and kept at 18°C in Barth's solution. Electrophysiological studies were conducted 1–3 d later. The vitelline membrane was removed immediately before patching.
Electrophysiology and Data Analysis
Membrane currents were recorded from excised inside-out giant patches from injected oocytes with an Axopatch 200A or B amplifier (Axon Instruments) at room temperature. Patch electrodes were pulled from thin-wall borosilicate glass (Garner Glass) and had a tip diameter of 20–30 μm after fire polishing. The patch electrode solution contained (in mM) 85 KCl, 1.8 CaCl2, 5 K2HPO4, 5 KH2PO4 (pH 7.4 adjusted with KOH). The standard bath solution contained (in mM) 75 KCl, 5 K2EDTA, 5 K2HPO4, 5 KH2PO4 (pH 7.2 adjusted with KOH). The MgATP solution used to reactivate channels contained (in mM) 85 KCl, 2 MgATP, 5 K2HPO4, 5 KH2PO4 (pH 7.2 adjusted with KOH). PIP2 (dipalmitoyl-, pentaammonium salt) was purchased from Calbiochem and was prepared in bath solution (10 μM) and sonicated for ∼20 min in ice water before using. PIP2 antibody was purchased from Assay Designs, Inc. and prepared by 20-fold dilution into the bath solution. TEA, spermine, and neomycin were added to the bath solution with pH readjusted. Data were filtered with an 8-pole Bessel filter (Frequency Devices) at 2 kHz and digitized at 5 kHz via a DigiData 1200 interface (Axon Instruments). Data acquisition and analysis were performed using an Axopatch 200A amplifier (Axon Instruments) and pCLAMP 7 or 9 software (Axon Instruments). Continuous I-V relations were recorded using ramp pulses generated from a holding potential of 0 mV and ramped between −100 and +100 mV at a rate of 400 mV/s or 100 mV/s. Current amplitudes measured at −100 mV were used to evaluate channel activity. The outward current at +100 mV in the presence of 100 μM DA10, DA12, and 30 mM TEA or after complete rundown was considered as leak current, and the same amount of inward current was subtracted from the current at −100 mV. In some experiments, continuous inward currents were recorded by holding patches constantly at −80 mV.
All experiments were conducted at room temperature (20–24°C). Data were presented as mean ± SEM. The unpaired Student's t test was used to assess statistical significance.
RESULTS
Rundown and Recovery of the Kir2.1 Channel Activity Depend on Membrane PIP2 Level
Kir2.1 channels were expressed in Xenopus oocytes and macroscopic currents recorded in giant inside-out membrane patches. Upon excising and perfusing the patch in a Mg2+- and polyamine-free solution, the outward current increased gradually, as endogenous Mg2+ and polyamines were washed out (Fig. 1, A, D, and E). Since outward current through Kir2.1 channels is very sensitive to contamination by residual polyamines and other cations, we used the inward current at −100 mV to index channel activity. In most patches, channel activity started to rundown after the outward current reached a maximal level, until eventually all channels closed. The average half-time for spontaneous rundown was 4.8 ± 0.4 min (n = 27). Channel activity could be recovered by applying exogenous PIP2 (10 μM), without further rundown thereafter (Fig. 1, A and B). Early application of PIP2 prevented rundown completely (Fig. 1 C). (The inhibition of the outward current during PIP2 perfusion might be caused by ammonium ions present in the PIP2 formulation.) Fig. 1 D shows that after spontaneous rundown, channels could also be reactivated by 2 mM MgATP, but not with K2ATP or AMP-PNP (not depicted). This reactivating effect was blocked by wortmannin (100 μM), an inhibitor of phosphatidylinositol kinases (Fig. 1 E), which prevents resynthesis of PIP2 by MgATP-sensitive PI kinases. These results are reminiscent of Kir6.2 channels (Xie et al., 1999a,b) and indicate that membrane PIP2 levels are also the main determinant of the Kir2.1 channel activity.
Figure 1.

Stabilization of Kir2.1 channel activity by PIP2. Representative current traces elicited by the ramp pulses between −100 mV and +100 mV from a holding potential of 0 mV are shown. (A) After patch excision into Mg- and polyamine-free bath solution, inward rectification disappeared and current transiently increased before running down. Application of phosphatidylinositol-4,5-bisphosphate (PIP2, 10 μM) after rundown recovered channel activity. (B) Current–voltage (I-V) relationships obtained at times a–d in A. (C) Application of PIP2 (10 μM) immediately after patch excision prevented rundown. (D) MgATP (2 mM), but not K2ATP (2 mM), reactivated current after rundown. (E) After rundown, the PIP2 synthesis inhibitor wortmanin (WMN, 100 μM) prevented reactivation by MgATP (2 mM). The triangle to the right of each trace indicates zero current level. The upward arrow under each trace indicates the patch excision from cell-attached to inside-out configuration. Applications of reagents are indicated by the horizontal bars. The same markings are applicable to subsequent figures.
Long Polyamines Slow the Rundown Process of Kir2.1 Channels
When long polyamines such as spermine, DA12, or DA10 were included in the bath solution, rundown was markedly slowed (Fig. 2, A–C). In the presence of 100 μM spermine, DA12, or DA10, the residual current at 5 min after excising the patch remained at 90 ± 7%, 113 ± 5%, or 93 ± 4%, respectively, of the initial level, compared with 35 ± 7% in the absence of polyamines (ctl), as summarized in Fig. 2 E. Fig. 2 C shows that channel activity remained stable for ∼4 min in the presence of 100 μM DA10, but ran down rapidly to ∼8% over ∼2 min once DA10 was removed. Reapplication of DA10 did not recover the channel activity, indicating that DA10 could not directly reactivate channels once they had closed due to PIP2 depletion. However, subsequent application of MgATP (2 mM) to resynthesize PIP2 reactivated channel activity. Short diamines such as DA4 (putrescine) did not prevent rundown (Fig. 2, D and F).
Figure 2.
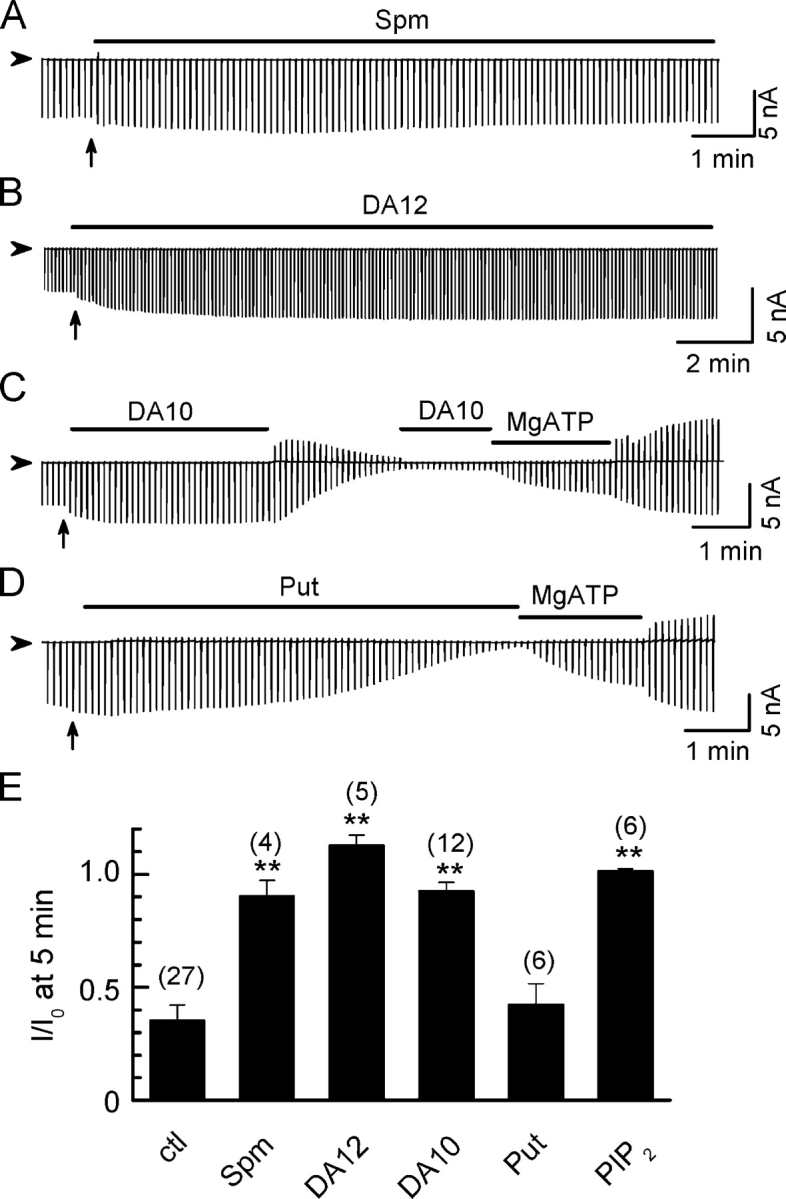
Stabilization of Kir2.1 channel activity by long diamines. Ramp pulse–elicited current traces showing that spontaneous rundown after patch excision was prevented by (A) spermine (Spm), (B) diamine 12 (DA12), and (C) diamine 10 (DA10), but not (D) putrescine (Put, DA4), all at 100 μM concentration, holding potential between ramps 0 mV. (E). Normalized current (I/I0) 5 min after excising the patch into control solution (ctl), or solution containing 100 μM Spm, DA12, DA10, Put, or 10 μM PIP2. I0 represents current level at −100 mV before rundown. Numbers of patches are indicated in parentheses above each bar. Data are shown and mean ± SEM. **, P < 0.05 compared with ctl.
These results indicate that two factors contribute to spontaneous rundown of Kir2.1 channels after patch excision into MgATP- and polyamine-free solution: (1) gradual depletion of membrane PIP2 (due to dephosphorylation by inositol-polyphosphate phosphatase or hydrolysis by PLC [Huang et al., 1998]); (2) washout of long polyamines from the patch.
Long Polyamines Stabilize Channel Activity by Strengthening the PIP2–Channel Interaction
Channel activity showed minimal rundown after reactivation by MgATP (Fig. 3 A). The subsequent experiments were performed after spontaneous rundown was halted by treating patches with 1 mM MgATP for ∼1 min. Under these conditions, channel activity could be inhibited by exposure to PIP2 antibodies (PIP2-Ab) (Fig. 3, B and D). The half-time for PIP2-Ab–induced rundown was ∼4 min. Fig. 3 (C and D) shows that DA12 (100 μM) inhibited the ability of PIP2-Ab to cause channel rundown, prolonging the half-time to ∼9 min, suggesting that polyamines stabilize channel activity by increasing channel–PIP2 binding affinity.
Figure 3.

Long polyamines strengthen the PIP2–channel interaction in wild-type Kir2.1 channels. (A) Channel activity shows minimal rundown after reactivated by treatment with 2 mM MgATP. (B) After exposure to 2 mM MgATP to minimize spontaneous rundown, application of PIP2 antibody (PIP2-Ab) induced rundown with a half-time of ∼4 min. (C) DA12 (100 μM) slowed PIP2-Ab–induced rundown. (D) Average time course of rundown induced by PIP2-Ab under control (ctl) and in the presence of DA12 (100 μM). Currents (I) are normalized to the value before PIP2 was added (I0). (E) Ramp pulse–elicited current traces showing differential inhibitory effect of 100 μM neomycin (Neo) in the absence and presence of 100 μM DA10. (F) Neomycin sensitivities in the absence and presence of 100 μM spermine. (G) Neomycin sensitivities in the absence and presence of 100 μM putrescine (Put). (H) Dose–response curves of normalized current (I/I0) versus neomycin concentration in the absence and presence of 100 μM Put, DA10, or spermine. I0 represents current level at −100 mV before perfusion of neomycin. Numbers of patches and mean IC50 are indicated in the legends.
To quantitatively evaluate whether long polyamines might stabilize channel activities by strengthening the PIP2–channel interaction, we used neomycin sensitivity to estimate PIP2 binding affinity as previously reported (Huang et al., 1998; Schulze et al., 2003; Ribalet et al., 2005). Neomycin is a polycation that binds PIP2 (Arbuzova et al., 2000), preventing it from interacting with the channel. Channel activity was inhibited by applying neomycin in a dose-dependent manner (Fig. 3 F), and the concentration causing 50% inhibition (IC50) was used to indicate the strength of channel–PIP2 interaction (or PIP2-binding affinity), i.e., low neomycin sensitivity indicates a strong channel–PIP2 interaction, and high neomycin sensitivity a weak channel–PIP2 interaction.
As shown in Fig. 3 E, 100 μM neomycin caused ∼60% inhibition in the absence of polyamines. However, in the presence of DA10 (100 μM), the same concentration of neomycin caused only ∼5% inhibition unless DA10 was removed. Under control conditions (i.e., in the absence of polyamines), neomycin inhibited inward current with an IC50 of ∼33 ± 5 μM. In the presence of 100 μM DA10, however, neomycin sensitivity decreased 14-fold (IC50 = 487 ± 134 μM) (Fig. 3 H). Spermine (100 μM) had similar effects, producing a 25-fold decrease in neomycin sensitivity (IC50 = 834 ± 142 μM) (Fig. 3, E and H). Unlike polyamines with alkyl chain lengths ≥10 (DA10, DA12, and spermine), however, 100 μM putrescine (DA4) did not shift neomycin sensitivity, with an IC50 of 38 ± 11 μM (Fig. 3, G and H). Other shorter diamines, such as 1,6-hexanediamine (DA6) and 1,8-octanediamine (DA8), did not significantly decrease neomycin sensitivity either (unpublished data), although they blocked outward currents, indicating that block of the pore per se was not sufficient to increase PIP2 affinity. To determine whether block of the pore by long polyamines was necessary for their effects on neomycin sensitivity, we also measured neomycin sensitivity in the absence and presence of spermine when the patch was held continuously at −80 mV. Under these conditions, the channels passed inward current continuously, and so were unblocked by spermine most, if not all, of the time. Neomycin sensitivity was still reduced by spermine under these conditions (IC50 of 32 ± 11 μM vs. 503 ± 130 μM in the absence and presence of spermine, respectively, n = 3 patches).
Fig. 4 provides further evidence that long polyamines stabilize channel activity by strengthening the PIP2–channel interaction. The PIP2 binding site(s) in Kir2.1 involve positively charged residues in the cytoplasmic NH2 and COOH termini, putatively located just underneath the inner leaflet of the plasma membrane (e.g., K188, R216, also see Fig. 7). Mutation of these residues reduces channel–PIP2 binding affinity and channel activity. Fig. 4 (A–C) shows that K188Q mutant channels exhibited low channel activity under control conditions. The channel activity ran down with a faster time course (half-time 1.3 ± 0.1 min) compare with WT channels (half-time 4.8 ± 0.4 min) and could not activated by exogenous application of 10 μM PIP2, consistent with its known low PIP2 affinity. However, application of DA12 (100 μM) in the presence of PIP2 activated channels dramatically, although with a slow time course, suggesting DA12 enhanced PIP2 affinity. DA12 maintained its activating effect even after withdrawal of exogenous PIP2 from the perfusate, presumably because the membrane level of PIP2 remained high (Fig. 4 A). In contrast, DA12 alone did not stimulate channel activity before application of PIP2 (Fig. 4 B). Thus, channel activation required the simultaneous presence of DA12 and abundant membrane PIP2. Similar results were obtained in patches treated with MgATP (2 mM) to elevate membrane PIP2 levels (Fig. 4, C and D). After MgATP, both DA10 and DA12 dramatically increased the channel activity, whereas shorter diamines such as DA8 had no effect (Fig. 4, C and D). Neomycin inhibited K188Q channels with a much faster time constant (Fig. 4 E) and lower IC50 (∼3 ± 1 μM), reconfirming that neomycin is a reliable indicator of channel–PIP2 binding affinity (Fig. 4, F and H). In the presence of 100 μM DA12, neomycin sensitivity decreased 23-fold (IC50 of 69 ± 29 μM; Fig. 4, E–H). These results indicate that long polyamines enhance PIP2's ability to interact with the channel and stabilize the open state.
Figure 4.

Long polyamines strengthen the PIP2–channel interaction in Kir2.1/K188Q channels. (A and B) Current traces recorded from the K188Q channels, which have reduced PIP2 binding affinity. Strong channel activation was observed only in the presence of both PIP2 (10 μM) and long diamine (100 μM DA12). The inset in each panel shows the current traces with expanded ordinate scale for clarity. (C) Long polyamines (DA12 and DA10) enhanced channel activity after MgATP treatment. Application of 2 mM MgATP and 100 μM diamines of various lengths are indicated by the horizontal bars above the current traces. (D) Summary of the data shown in the C. The currents are normalized to that in the presence of DA12 (IDA12). **, P < 0.05 compared with ctl. (E) Comparison of time course of neomycin (10 μM) inhibition on Kir2.1 wild type (WT) versus K188Q mutation. Currents were recorded at a holding potential of −80 mV. (F and G) Current traces recorded from K188Q channels showing sensitivities to neomycin under control conditions (ctl, in the absence of any polyamine) and in the presence of 100 μM DA12, respectively. (H) Dose–response curve of normalized current (I/I0) versus neomycin concentration in the absence and presence of 100 μM DA12 in K188Q channels. I0 represents current level at −100 mV before perfusion of neomycin. Number of patches and mean IC50 are indicated in the legends.
Figure 7.

Schematic model of modulation of Kir channels by PIP2 and long polyamines. Only two M2 helices and two cytoplasmic regions of the tetrameric structure are shown for clarity. The channel opens (O state) when the conserved positively charged residues (e.g., K188, R218) in the cytoplasmic region bind to negatively charged heads of PIP2 molecules, and closes (C) when this interaction is lost. The Kir2.1 has a high open probability of >0.9, suggesting a normally high PIP2 binding affinity. When PIP2 is hydrolyzed by PLC or screened by neomycin (Neo), the channel closes (CN state). The absence of new PIP2 to interact causes channel rundown (R state). Long polyamines (PA) interact with D172 in the M2 region to allosterically strengthen the interaction of the cytoplasmic domain with PIP2, thereby locking the channel in the open configuration (OA state) and preventing rundown or inhibition by neomycin. The channel is blocked at positive membrane potential by polyamine plugging the pore near the selectivity filter (BA state).
Negative Charges Inside the Transmembrane Pore Contribute to the Polyamine-induced Stabilization of Channel Open State
To identify the site(s) at which long polyamines interact to strengthen the PIP2–channel interaction, we mutated negatively charged residues known to affect polyamine binding, including D172 in the transmembrane pore (Lu and MacKinnon, 1994; Stanfield et al., 1994; Yang et al., 1995), and E224 and E299 in the cytoplasmic pore (Yang et al., 1995; Kubo and Murata, 2001). Fig. 5 shows that in E224G/E299S channels, the presence of DA10 caused the IC50 for neomycin inhibition to increase approximately sixfold from 21 ± 7 μM to 184 ± 13 μM (Fig. 5, A and B). In contrast, in D172N channels, the effect of DA10 on neomycin sensitivity was abolished (Fig. 5, C and D); the IC50 for neomycin inhibition showed no significant difference between control (11 ± 4 μM) and in the presence of 100 μM DA10 (15 ± 4 μM). These results indicate that D172 plays a major role in strengthening the channel–PIP2 interaction by long polyamines.
Figure 5.

Negative charges at the D172 position, but not E224/E299, are required for the polyamine-induced stabilization of channel open state. (A) Current traces recorded from E224G/E299S mutant channels showing neomycin (Neo) sensitivities in the presence or absence of 100 μM DA10. (B) Normalized current (I/I0)–concentration relationships for neomycin in the E224G/E299S, in the absence (ctl) and presence of 100 μM DA10. Numbers of patches and mean IC50 are indicated in the legends. (C and D) Same as A and B, except for D172N mutant channels.
We also evaluated spontaneous rundown of D172N channels in the absence and presence of 100 μM DA10, respectively. There was no significant difference in the half-times for spontaneous rundown between control (2.1 ± 0.6 min, n = 10) and DA10 group (2.2 ± 0.5, n = 8), although both half-times were shorter than in wild-type channels. These results support the idea that D172 mediates the effects of polyamines on strengthening PIP2 affinity.
Polyamine Effect on PIP2 Affinity in other Kir Channels
Kir1.1 channels are weak inward rectifiers, which lack equivalent rectification sites. As shown in Fig. 6 A, wild-type Kir1.1 channels conducted substantial outward currents either in the cell-attached patches or inside-out patches in the presence of 100 μM DA10. DA10 did not shift neomycin sensitivity in the wild-type Kir1.1 channels. Introducing a negative charge at the equivalent site in the transmembrane pore (N171D) enhanced inward rectification in Kir1.1 channels. However, neomycin sensitivity was not shifted by DA10 (Fig. 6 B). Similar results were obtained with Kir6.2 channels, using either Kir6.2Δ36 or Kir6.2 Δ36/N160D channels. Thus, the presence of a negative charge at the site equivalent to 172 in Kir2.1 channels was not sufficient to confer polyamine-induced enhancement of the channel interaction with PIP2 in Kir1.1 or Kir6.2 channels, indicating that other residues are also important.
Figure 6.

Long polyamines do not strengthen the PIP2–channel interaction in Kir1.1 channels. (A) Ramp pulse–elicited current traces were recorded from wild-type Kir1.1 channels. DA10 (100 μM) did not cause any shift in neomycin (Neo) sensitivity. Note the same degree of inhibition by 1 mM neomycin in the absence or presence of 100 μM DA10. (B) Same as A, except for Kir1.1/N171D mutant channels. The extent of inward rectification was enhanced in this mutation. However, DA10 (100 μM) had no effect on neomycin sensitivity. TEA (30 mM) had less effect on the inward currents.
DISCUSSION
In the present study, we demonstrate that long polyamines, through their interaction with D172 in the transmembrane pore and other as yet unidentified residues, prevent rundown of Kir2.1 channels by enhancing the channel's affinity for membrane-bound PIP2. This conclusion is supported both qualitatively in experiments using direct application of PIP2, PIP2-generating systems, and/or PIP2 antibodies, as well as quantitatively using neomycin sensitivity to index the strength of the channel's interaction with membrane-bound PIP2. Although we cannot exclude the possibility that neomycin may have nonspecific effects in addition to screening PIP2, as reported in the case of polylysine (Lopes et al., 2002), our findings with neomycin are fully consistent with the effects observed by direct application of PIP2, PIP2-generating systems, and/or PIP2 antibodies.
Mechanistic Model
Fig. 7 shows a hypothetical schema, which integrates these new findings with existing information about the structure–function and regulation of Kir2.1 channels. We propose that in the absence of long polyamines, the channel opens (O state) when the negative head charges of PIP2 interacts with positively charged residues in the cytoplasmic domain, such as K188, R218, etc., near the plasma membrane. It has been suggested that PIP2 binding causes conformational changes at bundle crossing formed by M2 helix (Xiao et al., 2003) or the G-loop (girdle) structure near the junction between membrane pore and cytoplasmic pore domain (Pegan et al., 2005), which in turn promotes the open state. The channel closes when the interaction with PIP2 is lost, either transiently (C state), or due to PIP2 depletion (rundown, R state) or PIP2 screening by neomycin (CN state). When long polyamines are present, however, they interact with the channel in a manner dependent on the negative charges at D172 in the M2 region, but also involving other residues that remain to be identified. We speculate that this interaction stabilizes the bundle crossing in its open configuration, allosterically enhancing the interaction with PIP2 at the cytoplasmic side of the membrane (OA state). The bound PIP2 molecules are then protected from hydrolysis or screening by neomycin. This hypothetical schema is consistent with recent observations showing that PIP2 binding to cytoplasmic region links to activation gating involving the M2 region and the selectivity filter (Xiao et al., 2003). The positioning of long polyamines in the transmembrane pore may also facilitate their voltage-dependent entry deeper into the pore-blocking site (OB state) (Xie et al., 2002, 2003). Thus, long polyamines serve dual roles as both coactivators (with PIP2) and pore blockers in Kir2.1 channels.
We suggest that different processes are involved in the channel block (OB) by polyamines and activation (OA) by polyamines, since they were dissociated in the following situations. (a) When Kir2.1 channels were held at −80 mV so that voltage-dependent pore block by polyamines was minimal, long polyamines still prevented rundown fairly effectively. (b) Short polyamines, which could block outward current, had no effect on channel PIP2 affinity as assessed by neomycin sensitivity. (c) In the D172N mutation, 100 μM DA10 did not shift neomycin sensitivity, but still completely blocked the outward current. Thus, block of the pore by polyamines was neither necessary nor sufficient to increase channel PIP2 affinity. We speculate that the effect of long polyamines on PIP2 affinity is mediated by direct electrostatic interaction of their positive headgroups with D172, but cannot exclude the possibility that they interact with other regions of the channel, and that this interaction is allosterically affected by neutralization of D172.
The Specificity of Polyamine and Neomycin Effects
It is likely that polyamines affected PIP2's interaction with single channels, rather than by promoting PIP2 degradation at a global level, since a point mutation inside channel membrane pore (D172N) diminished DA10's effect on neomycin sensitivity (Fig. 5, C and D). We suggest that when polyamines enhance the affinity of the channels for PIP2, the bound PIP2 molecule is less available for degradation by phosphatases and/or PLCs. The shift of neomycin sensitivity to higher IC50 in the presence of polyamines is consistent with this hypothesis. These results suggest that the channel and PIP2 form a functional microdomain regulating local PIP2 metabolism; however, if it exists, this microdomain still interacts with the surrounding environment, since newly synthesized PIP2 after MgATP treatment can still reach and activate channels.
It should be noted that an ionic interaction between polyamines and phospholipids vesicles has been reported (Yung and Green, 1986). Indeed, high concentrations (mM) of spermine have been used to screen PIP2 and inhibit KATP channels (Fan and Makielski, 1997). However, this direct interaction between polyamines and PIP2, which would be expected to reduce channel activity, cannot explain the presently reported effects of lower (100 μM) polyamine concentrations, which stabilized (WT) or increased (K188Q) channel activity.
Neomycin is a polycationic aminoglycoside composed of four alicyclic rings. Unlike polyamines, neomycin inhibited channel activity in a voltage-independent manner similar to PIP2 antibodies (unpublished data), consistent with the idea that neomycin cannot enter into the Kir2.1 channel pore and/or bind to N172 residue in an electrical field. The slow time course of neomycin inhibition also excludes the possibility of the direct occlusion of the intracellular mouth of the channel similar to polyamines. Neomycin has also been used as a PLC blocker to maintain PIP2 levels by attenuating its hydrolysis during PLC (or Gq protein)-linked receptor stimulation (Haruna et al., 2002). However, this effect is not relevant to the present study, which did not involve PLC activation. In addition, it was suggested that neomycin block of PLC is due to the binding to the enzyme's substrate, i.e., PIP2 (Lipsky and Lietman, 1982; Slivka and Insel, 1988), which is consistent with our mechanistic explanation.
Other Kir Channels
The experiments in Kir1.1 and Kir6.2 suggest that the ability of long polyamines to enhance PIP2‘s interaction with the channel is mainly a regulatory feature of strong inward rectifier Kir2.1 channels. Although the negative charges at D172 in the transmembrane pore are critical in Kir2.1 channels, negative charge at the equivalent site in other Kir channels is not sufficient to confer to long polyamines the ability to strengthen channel–PIP2 interaction. It has been reported that multiple sites besides D172 contribute to the polyamine-induce rectification (e.g., E224, E299, D255, D259, M183, and F254) (Yang et al., 1995; Kubo and Murata, 2001; Pegan et al., 2005; Shin et al., 2005), and these sites probably also play a role in mediating the polyamine effect on the channel–PIP2 interaction. This conjecture is supported by observation that the length of polyamine is critical for its effects on PIP2 affinity, suggesting that interaction between the polyamine backbone and channel is also important. Further studies are needed to identify whether other charged residues or hydrophobic interactions are involved. Whether other strong inward rectifiers, such as other members of the Kir2.x subfamily or Kir3 family, share this property also remains to be determined.
Acknowledgments
We thank Dr. L.Y. Jan (University of California San Francisco, CA) for providing the Kir2.1 clone.
This work was supported by National Institutes of Health/NHLBI R37 HL60025 (to J.N. Weiss), by American Heart Association, National Research Center, Scientist Development Grant (0535143N to L.H. Xie), and the Kawata and Laubisch Endowments.
Olaf S. Andersen served as editor.
L.-H. Xie and S.A. John contributed equally to this work.
Abbreviations used in this paper: DA10, 1,10-decanediamine; DA12, 1,12-dodecanediamine; PIP2, phosphatidylinosital-4,5-bisphosphate.
References
- Arbuzova, A., K. Martushova, G. Hangyas-Mihalyne, A.J. Morris, S. Ozaki, G.D. Prestwich, and S. McLaughlin. 2000. Fluorescently labeled neomycin as a probe of phosphatidylinositol-4, 5-bisphosphate in membranes. Biochim. Biophys. Acta. 1464:35–48. [DOI] [PubMed] [Google Scholar]
- Baukrowitz, T., U. Schulte, D. Oliver, S. Herlitze, T. Krauter, S.J. Tucker, J.P. Ruppersberg, and B. Fakler. 1998. PIP2 and PIP as determinants for ATP inhibition of KATP channels. Science. 282:1141–1144. [DOI] [PubMed] [Google Scholar]
- Doupnik, C.A., N. Davidson, and H.A. Lester. 1995. The inward rectifier potassium channel family. Curr. Opin. Neurobiol. 5:268–277. [DOI] [PubMed] [Google Scholar]
- Doyle, D.A., J. Morais Cabral, R.A. Pfuetzner, A. Kuo, J.M. Gulbis, S.L. Cohen, B.T. Chait, and R. MacKinnon. 1998. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 280:69–77. [DOI] [PubMed] [Google Scholar]
- Fakler, B., U. Brandle, E. Glowatzki, S. Weidemann, H.P. Zenner, and J.P. Ruppersberg. 1995. Strong voltage-dependent inward rectification of inward rectifier K+ channels is caused by intracellular spermine. Cell. 80:149–154. [DOI] [PubMed] [Google Scholar]
- Fan, Z., and J.C. Makielski. 1997. Anionic phospholipids activate ATP-sensitive potassium channels. J. Biol. Chem. 272:5388–5395. [DOI] [PubMed] [Google Scholar]
- Ficker, E., M. Taglialatela, B.A. Wible, C.M. Henley, and A.M. Brown. 1994. Spermine and spermidine as gating molecules for inward rectifier K+ channels. Science. 266:1068–1072. [DOI] [PubMed] [Google Scholar]
- Haruna, T., H. Yoshida, T.Y. Nakamura, L.H. Xie, H. Otani, T. Ninomiya, M. Takano, W.A. Coetzee, and M. Horie. 2002. α1-Adrenoceptor-mediated breakdown of phosphatidylinositol 4,5-bisphosphate inhibits pinacidil-activated ATP-sensitive K+ currents in rat ventricular myocytes. Circ. Res. 91:232–239. [DOI] [PubMed] [Google Scholar]
- Hilgemann, D.W., S. Feng, and C. Nasuhoglu. 2001. The complex and intriguing lives of PIP2 with ion channels and transporters. Sci. STKE. 2001:RE19. [DOI] [PubMed]
- Hille, B. 2001. Ion Channels of Excitable Membranes. Third edition. Sinauer Associates, Inc., Sunderland, MA. 814 pp.
- Ho, I.H., and R.D. Murrell-Lagnado. 1999. Molecular determinants for sodium-dependent activation of G protein-gated K+ channels. J. Biol. Chem. 274:8639–8648. [DOI] [PubMed] [Google Scholar]
- Huang, C.L., S. Feng, and D.W. Hilgemann. 1998. Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gβγ. Nature. 391:803–806. [DOI] [PubMed] [Google Scholar]
- Kubo, Y., and Y. Murata. 2001. Control of rectification and permeation by two distinct sites after the second transmembrane region in Kir2.1 K+ channel. J. Physiol. 531:645–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo, A., J.M. Gulbis, J.F. Antcliff, T. Rahman, E.D. Lowe, J. Zimmer, J. Cuthbertson, F.M. Ashcroft, T. Ezaki, and D.A. Doyle. 2003. Crystal structure of the potassium channel KirBac1.1 in the closed state. Science. 300:1922–1926. [DOI] [PubMed] [Google Scholar]
- Liou, H.H., S.S. Zhou, and C.L. Huang. 1999. Regulation of ROMK1 channel by protein kinase A via a phosphatidylinositol 4,5-bisphosphate-dependent mechanism. Proc. Natl. Acad. Sci. USA. 96:5820–5825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipsky, J.J., and P.S. Lietman. 1982. Aminoglycoside inhibition of a renal phosphatidylinositol phospholipase C. J. Pharmacol. Exp. Ther. 220:287–292. [PubMed] [Google Scholar]
- Lopatin, A.N., E.N. Makhina, and C.G. Nichols. 1994. Potassium channel block by cytoplasmic polyamines as the mechanism of intrinsic rectification. Nature. 372:366–369. [DOI] [PubMed] [Google Scholar]
- Lopes, C.M., H. Zhang, T. Rohacs, T. Jin, J. Yang, and D.E. Logothetis. 2002. Alterations in conserved Kir channel-PIP2 interactions underlie channelopathies. Neuron. 34:933–944. [DOI] [PubMed] [Google Scholar]
- Lu, Z., and R. MacKinnon. 1994. Electrostatic tuning of Mg2+ affinity in an inward-rectifier K+ channel. Nature. 371:243–246. [DOI] [PubMed] [Google Scholar]
- Matsuda, H., A. Saigusa, and H. Irisawa. 1987. Ohmic conductance through the inwardly rectifying K channel and blocking by internal Mg2+. Nature. 325:156–159. [DOI] [PubMed] [Google Scholar]
- Nishida, M., and R. MacKinnon. 2002. Structural basis of inward rectification: cytoplasmic pore of the G protein-gated inward rectifier GIRK1 at 1.8 A resolution. Cell. 111:957–965. [DOI] [PubMed] [Google Scholar]
- Pegan, S., C. Arrabit, W. Zhou, W. Kwiatkowski, A. Collins, P.A. Slesinger, and S. Choe. 2005. Cytoplasmic domain structures of Kir2.1 and Kir3.1 show sites for modulating gating and rectification. Nat. Neurosci. 8:279–287. [DOI] [PubMed] [Google Scholar]
- Ribalet, B., S.A. John, L.H. Xie, and J.N. Weiss. 2005. Regulation of the ATP-sensitive K channel Kir6.2 by ATP and PIP2. J. Mol. Cell. Cardiol. 39:71–77. [DOI] [PubMed] [Google Scholar]
- Rohacs, T., J. Chen, G.D. Prestwich, and D.E. Logothetis. 1999. Distinct specificities of inwardly rectifying K+ channels for phosphoinositides. J. Biol. Chem. 274:36065–36072. [DOI] [PubMed] [Google Scholar]
- Schulze, D., T. Krauter, H. Fritzenschaft, M. Soom, and T. Baukrowitz. 2003. PIP2 modulation of ATP and pH sensitivity in Kir channels: a tale of an active and a silent PIP2 site in the N terminus. J. Biol. Chem. 278:10500–10505. [DOI] [PubMed] [Google Scholar]
- Shin, H.G., Y. Xu, and Z. Lu. 2005. Evidence for sequential ion-binding loci along the inner pore of the IRK1 inward-rectifier K+ channel. J. Gen. Physiol. 126:123–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng, S.L., C.A. Cukras, J. Harwood, and C.G. Nichols. 2000. Structural determinants of PIP(2) regulation of inward rectifier K(ATP) channels. J. Gen. Physiol. 116:599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng, S.L., and C.G. Nichols. 1998. Membrane phospholipid control of nucleotide sensitivity of KATP channels. Science. 282:1138–1141. [DOI] [PubMed] [Google Scholar]
- Slivka, S.R., and P.A. Insel. 1988. Phorbol ester and neomycin dissociate bradykinin receptor-mediated arachidonic acid release and polyphosphoinositide hydrolysis in Madin-Darby canine kidney cells. Evidence that bradykinin mediates noninterdependent activation of phospholipases A2 and C. J. Biol. Chem. 263:14640–14647. [PubMed] [Google Scholar]
- Song, D.K., and F.M. Ashcroft. 2001. ATP modulation of ATP-sensitive potassium channel ATP sensitivity varies with the type of SUR subunit. J. Biol. Chem. 276:7143–7149. [DOI] [PubMed] [Google Scholar]
- Soom, M., R. Schonherr, Y. Kubo, C. Kirsch, R. Klinger, and S.H. Heinemann. 2001. Multiple PIP2 binding sites in Kir2.1 inwardly rectifying potassium channels. FEBS Lett. 490:49–53. [DOI] [PubMed] [Google Scholar]
- Stanfield, P.R., N.W. Davies, P.A. Shelton, M.J. Sutcliffe, I.A. Khan, W.J. Brammar, and E.C. Conley. 1994. A single aspartate residue is involved in both intrinsic gating and blockage by Mg2+ of the inward rectifier, IRK1. J. Physiol. 478:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberg, C.A. 1987. Inward rectification of a potassium channel in cardiac ventricular cells depends on internal magnesium ions. Proc. Natl. Acad. Sci. USA. 84:2560–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wible, B.A., M. Taglialatela, E. Ficker, and A.M. Brown. 1994. Gating of inwardly rectifying K+ channels localized to a single negatively charged residue. Nature. 371:246–249. [DOI] [PubMed] [Google Scholar]
- Xiao, J., X.G. Zhen, and J. Yang. 2003. Localization of PIP2 activation gate in inward rectifier K+ channels. Nat. Neurosci. 6:811–818. [DOI] [PubMed] [Google Scholar]
- Xie, L.H., M. Horie, and M. Takano. 1999. a. Phospholipase C-linked receptors regulate the ATP-sensitive potassium channel by means of phosphatidylinositol 4,5-bisphosphate metabolism. Proc. Natl. Acad. Sci. USA. 96:15292–15297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, L.H., S.A. John, and J.N. Weiss. 2002. Spermine block of the strong inward rectifier potassium channel Kir2.1: dual roles of surface charge screening and pore block. J. Gen. Physiol. 120:53–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, L.H., S.A. John, and J.N. Weiss. 2003. Inward rectification by polyamines in mouse Kir2.1 channels: synergy between blocking components. J. Physiol. 550:67–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, L.H., M. Takano, M. Kakei, M. Okamura, and A. Noma. 1999. b. Wortmannin, an inhibitor of phosphatidylinositol kinases, blocks the MgATP-dependent recovery of Kir6.2/SUR2A channels. J. Physiol. 514:655–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, J., Y.N. Jan, and L.Y. Jan. 1995. Control of rectification and permeation by residues in two distinct domains in an inward rectifier K+ channel. Neuron. 14:1047–1054. [DOI] [PubMed] [Google Scholar]
- Yung, M.W., and C. Green. 1986. The binding of polyamines to phospholipid bilayers. Biochem. Pharmacol. 35:4037–4041. [DOI] [PubMed] [Google Scholar]
- Zeng, W.Z., X.J. Li, D.W. Hilgemann, and C.L. Huang. 2003. Protein kinase C inhibits ROMK1 channel activity via a phosphatidylinositol 4,5-bisphosphate-dependent mechanism. J. Biol. Chem. 278:16852–16856. [DOI] [PubMed] [Google Scholar]
- Zhang, H., C. He, X. Yan, T. Mirshahi, and D.E. Logothetis. 1999. Activation of inwardly rectifying K+ channels by distinct PtdIns(4,5)P2 interactions. Nat. Cell Biol. 1:183–188. [DOI] [PubMed] [Google Scholar]