

Abstract
CLC-ec1 is a prokaryotic CLC-type Cl−/H+ exchange transporter. Little is known about the mechanism of H+ coupling to Cl−. A critical glutamate residue, E148, was previously shown to be required for Cl−/H+ exchange by mediating proton transfer between the protein and the extracellular solution. To test whether an analogous H+ acceptor exists near the intracellular side of the protein, we performed a mutagenesis scan of inward-facing carboxyl-bearing residues and identified E203 as the unique residue whose neutralization abolishes H+ coupling to Cl− transport. Glutamate at this position is strictly conserved in all known CLCs of the transporter subclass, while valine is always found here in CLC channels. The x-ray crystal structure of the E203Q mutant is similar to that of the wild-type protein. Cl− transport rate in E203Q is inhibited at neutral pH, and the double mutant, E148A/E203Q, shows maximal Cl− transport, independent of pH, as does the single mutant E148A. The results argue that substrate exchange by CLC-ec1 involves two separate but partially overlapping permeation pathways, one for Cl− and one for H+. These pathways are congruent from the protein's extracellular surface to E148, and they diverge beyond this point toward the intracellular side. This picture demands a transport mechanism fundamentally different from familiar alternating-access schemes.
INTRODUCTION
Cl−-transporting membrane proteins of the CLC family fall into two distinctly different mechanistic subclasses: Cl−-selective ion channels and H+-coupled secondary active Cl− transporters (Accardi and Miller, 2004; Jentsch et al., 2005; Picollo and Pusch, 2005; Scheel et al., 2005). The former catalyze transmembrane movement of small inorganic anions via passive electrodiffusion through an aqueous pore, while the latter mediate stoichiometric exchange of Cl− for H+. Of the nine CLCs in the human genome, four (CLC-1, -2, -Ka, and -Kb) are plasma membrane ion channels, and three (CLC-3, -4, and -5) are exchangers residing in intracellular membranes that bound acidified compartments; the remaining two homologues, CLC-6 and -7, have not been characterized functionally, but are likely to be exchange transporters as well since they are found in acid-transporting intracellular membranes (Kida et al., 2001; Kornak et al., 2001; Jentsch et al., 2005).
The structure of CLC-ec1, a CLC homologue from Escherichia coli, has been determined at high resolution by x-ray crystallography (Dutzler et al., 2002, 2003). This protein belongs to the Cl−/H+ exchanger subclass (Accardi and Miller, 2004) and participates in the extreme acid-resistance response that enables the organism to survive in the acid environment of the stomach (Iyer et al., 2002). The x-ray structure has proven useful in guiding electrophysiological studies of eukaryotic CLC channels, for which no direct structural information exists, but its greatest potential utility lies in gaining understanding of the exchange transport mechanism.
We know very little about the molecular details by which this protein brings about transmembrane Cl− movement coupled to H+ transport in the opposite direction, beyond an initial estimate of the exchange stoichiometry, 2 Cl−/1 H+ (Accardi and Miller, 2004). One key mechanistic insight is that an extracellular-facing glutamate residue, E148, specifically mediates the transfer of H+ between external solution and protein interior. Removal of this carboxylate group by mutation leads to complete loss of H+ movement while preserving Cl− permeation (Accardi et al., 2004). Moreover, the strong stimulation of Cl− flux rates by low pH, a characteristic feature of the wild-type protein, is totally absent in E148 mutants, for which Cl− flux takes on maximal rates across the entire pH range of 3–7 (Iyer et al., 2002; Accardi et al., 2004). Thus, in the absence of a carboxyl group at this position, protons are no longer coupled to the transport of Cl− nor do they control the anion's flux rate. The critical function of E148 also shows itself in the Cl− channel subclass. Substitution of neutral residues for the E148 equivalents in several eukaryotic CLC channels abolishes extracellular pH dependence of gating and produces constitutively open channels (Dutzler et al., 2003; Estevez et al., 2003; Niemeyer et al., 2003), mimicking the opening of wild-type channels at low extracellular pH (Chen and Chen, 2001).
These phenomena are readily comprehensible from the crystal structure of CLC-ec1 (Dutzler et al., 2003). In wild-type protein, the E148 side chain lies close to a bound Cl− ion and blocks the anion's access to the extracellular solution. In the E148Q mutant, the side chain rotates away from the bound Cl− and thereby opens an extracellular aqueous pathway. For this reason, E148 is thought to act as a proton-linked “gate” in CLC channels; in the transporter subclass, such a movement provides an obvious means for linking H+ and Cl− binding at some point in the transport cycle.
The existence of a specific extracellular proton transfer group suggests that a similar site might be present on the intracellular side of this protein. In this work we search for such a residue by mutating inward-facing glutamate and aspartate side chains in CLC-ec1, identifying E203 as a likely inward-facing proton transfer group.
MATERIALS AND METHODS
CLC-ec1 and variants were expressed in E. coli, purified, and crystallized after complexation with a FAB fragment of antibody 10EC3/G4 (National Cell Culture Center), as previously described (Dutzler et al., 2003). Crystals were harvested after 1–3 weeks at 20°C and were frozen in liquid nitrogen. Crystallographic datasets were collected at the Swiss Light Source or the Advanced Photon Source, using radiation at the Br absorption edge (0.919 Å). Diffraction images were indexed and integrated in the HKL program, and electron density and anomalous difference maps were calculated in the CCP4 suite, after molecular replacement with PHASER and refinement with REFMAC5, using the wild-type CLC-ec1-FAB model (accession no. 1OTS) and twofold NCS restraints for the CLC homodimer. The refined model was also minimally rebuilt using sigmaa-weighted 2Fo-Fc and Fo-Fc maps in the COOT program. Model bias was suppressed by prime-and-switch density modification in the SOLVE/RESOLVE program within the PHENIX suite (Terwilliger, 2000).
CLC-ec1 ion transport functions were assayed by 36Cl− and H+ fluxes in liposomes and voltage-clamped currents in planar lipid bilayers as previously described (Accardi et al., 2004; Accardi and Miller, 2004); in preparations used for lipid bilayer experiments, an anion-exchange chromatography step was used in lieu of gel filtration to remove outer membrane porins (Accardi and Miller, 2004). Figures were produced in PyMOL Molecular Graphics (DeLano Scientific, http://www.pymol.org).
RESULTS
We set out to search for an intracellular proton transfer residue by mutagenesis of CLC-ec1, seeking a dissociable side chain whose neutralization would specifically abolish H+ transport coupled to Cl− movement. To make this task tractable in a protein containing 55 protonatable side chains (16 glutamates, 11 aspartates, 6 histidines, 13 lysines, and 9 tyrosines), we limited our search strategy. This transporter operates physiologically under acid conditions, <pH 3.5 extracellular, <pH 5.0 intracellular (Iyer et al., 2002; Foster, 2004; Richard and Foster, 2004), and in vitro attains maximal rates <pH 4.5 (Iyer et al., 2002), and so low-pKa carboxylate residues are likely to be involved in the H+ transport mechanism. Most of the glutamates and aspartates of CLC-ec1 are found in two loci, intracellular and extracellular (Fig. 1), and we accordingly focus mainly on the former set of residues. With these constraints, the initial search reduces to nine candidates, seven glutamates and two aspartates, which we substitute with the isosteric analogues of the protonated side chains, glutamine and asparagine, respectively. The positions of all carboxyl-bearing residues in a single subunit of the homodimeric protein are shown in Fig. 1, accompanied by a CLC alignment in a strongly conserved region commonly known as the “PIGG-pen.” The alignment reveals a glutamate residue, E203 in CLC-ec1, that is uniformly present in the Cl−/H+ exchangers but absent in the Cl− channels, where valine takes its place. This residue is located in an intracellular-facing cluster of glutamates that computations (Yin et al., 2004) have suggested could plausibly contribute to an electronegative H+ pathway through the protein.
Figure 1.
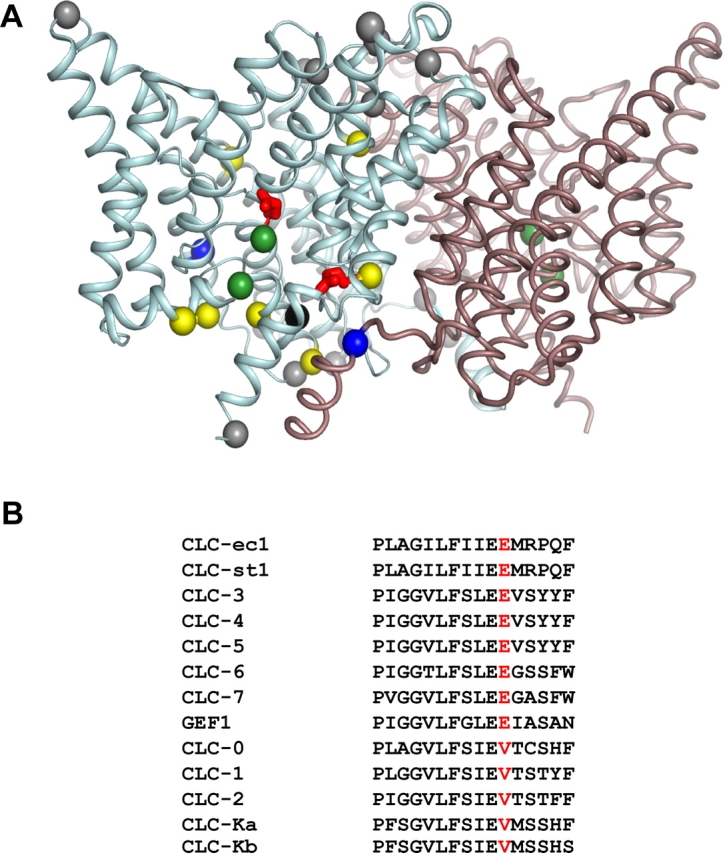
Carboxylates in CLC-ec1. (A) Ribbon representation of the CLC-ec1 dimer, view from within the membrane, with extracellular side up. α carbons of the carboxylate residues mutated here are shown for a single subunit as yellow spheres, except for E148 and E203 (red), and E113 (black); gray spheres mark carboxyl residues that are not studied here, and green spheres designate Cl− ions. Also shown are R28 and K131 (blue). (B) Sequence alignment of the PIGG-pen region, with E203 equivalent indicated in red. Genes listed are of human origin except for CLC-0 (Torpedo marmorata), CLC-ec1 (E. coli), CLC-st1 (Salmonella typhimurium), and GEF1 (Saccharomyces cerevisiae).
E203 Mediates H+ Coupling
The obvious suggestion leaping out of the sequence alignment is difficult to resist. Consequently, we first examined the ion transport behavior of the mutant E203Q, which resembles an always-protonated glutamate side chain. The results meet expectations: removing the carboxylate group at E203 completely abolishes H+ coupling to Cl− transport, as can be seen from several functional indicators. Fig. 2 A illustrates E203Q currents across lipid bilayers separating symmetrical Cl− solutions, with or without a pH gradient. With wild-type protein, imposition of a 4-unit pH gradient across the bilayer produces a large shift in reversal potential (62 mV, Fig. 2 B), a direct manifestation of H+ transport (Accardi and Miller, 2004); in contrast, E203Q shows no pH-induced shift whatsoever (Fig. 2 B). If this loss in H+ transport reflects specific abolition of H+ coupling rather than merely protein damage, a gain in Cl− selectivity should appear in the mutant, since the altered CLC would catalyze permeation of only a single ionic species, Cl−. This prediction is validated in Fig. 2 C, where reversal potentials are plotted against Cl− gradient at symmetrical pH. For both wild-type and E203Q, reversal potential varies logarithmically with Cl− gradient, and the mutation raises the subnernstian slope observed for wild type (Accardi and Miller, 2004) close to the ideally Cl−-selective value (53 mV/decade). This result directly demonstrates that the mutant protein is competent in Cl− transport but has lost its ability to move H+.
Figure 2.

Loss of H+ coupling in E203Q. (A) Currents mediated by E203Q in symmetrical 300 mM Cl−, with or without a 4-unit pH gradient. A 3-s voltage pulse (−100 to +100 mV in 10-mV steps) was applied from a holding potential of 0 mV, followed by a 1-s tail pulse to −100 mV. Dashed line marks zero current. (B) I-V curves, normalized to the current in the same bilayer at −100 mV in symmetrical pH 3.0 and 300 mM Cl−. Open circles, wild type, pH 3.0/pH 7; black circles, E203 pH 3/pH 3; red triangles, E203 pH 3.0/pH 7; green squares, E203 pH 7/pH 3; yellow triangles, E203 pH 7.0/pH 7. Gradients are indicated by convention cis/trans sides of planar bilayer, with the trans side defined as zero voltage. (C) Reversal potential, Vrev, as a function of [Cl−] for wild type (black circles) and E203Q (red triangles); dashed line is Nernst potential predicted for ideal Cl− selectivity.
An additional indication of the loss of H+ coupling can be seen in a direct proton-pumping assay. Here, liposomes reconstituted with CLC protein and loaded with high Cl− are exposed to low external Cl−; in a coupled exchanger, Cl− flowing outward down its imposed gradient drives H+ uphill into the liposomes, as is readily seen with wild-type protein (Fig. 3). In the same assay, however, E203Q produces no observable proton flux, while still fully competent to mediate 36Cl− uptake (Fig. 3). It is notable that 36Cl− influx time courses at pH 4.5 are similar for mutant and wild-type protein, a result implying that the maximal Cl− turnover rate is not greatly altered by the mutation. These experiments demonstrate that E203Q conducts Cl− but has completely lost coupling of anion movement to H+. This behavior is similar to that displayed upon mutation of the extracellular H+ transfer residue E148 (Accardi et al., 2004). We also note that substitution by aspartate here does not undermine the transport mechanism, since E203D supports H+ coupling (Fig. 4) as well as Cl−-driven proton pumping (not depicted); this result is somewhat surprising in light of the conservation of glutamate at this position in the exchange transporter subclass (Fig. 1 B).
Figure 3.

Fluxes of Cl− and H+. Liposomes reconstituted with CLC-ec1 (1–5 μg/mg lipid) were assayed for either 36Cl− uptake or Cl−-driven H+ pumping. Top panels, 36Cl− uptake at pH 7.0 (circles) and pH 4.5 (triangles) for WT (A), E203Q (B), and E148A/E203Q (C). Lower traces show H+ flux assays, upward deflection indicating increase in pH of the liposome suspension. Additions of 1 μM valinomycin or FCCP are denoted by * and +, respectively.
Figure 4.
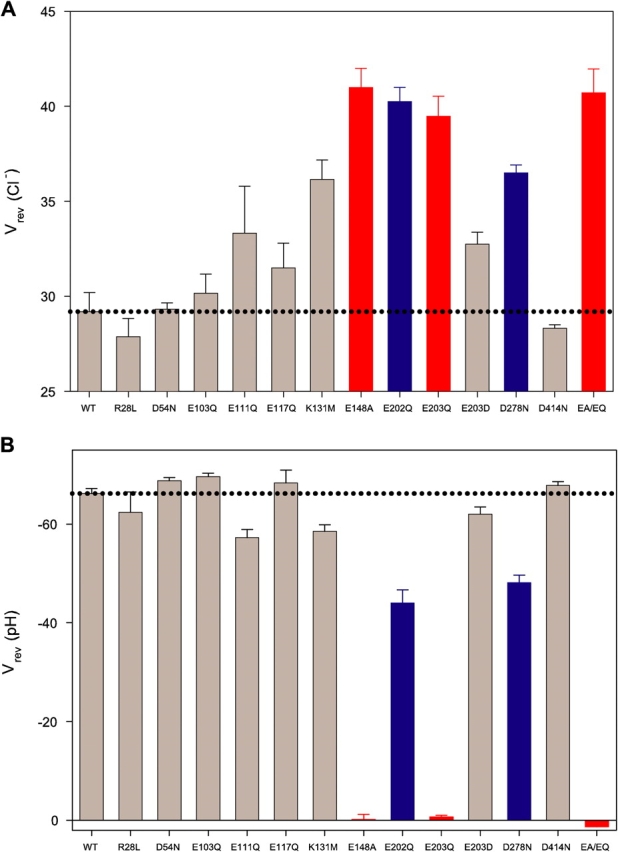
H+/Cl− coupling for CLC-ec-1 mutants. Reversal potentials of indicated mutants in presence of (A) a Cl− gradient (300 mM/45 mM) at symmetrical pH 3.0 or (B) a pH gradient (3/7) at symmetrical 300 mM Cl−. Dotted lines represent wild-type values under the same conditions. Data for wild type and E148A taken from Accardi et al. (2004); EA/EQ denotes the double mutant E148A/E203Q. Colored bars indicate mutants with H+ transport inhibited completely (red) or partially (blue).
pH Dependence of E203Q
Since neutralization of either E148 or E203 abolishes H+ transport, these two residues appear to have identical functions. However, this is not strictly true. While mutation of E148 leads to complete loss of the pH dependence of Cl− transport rates (Accardi et al., 2004), neutralization of E203 merely weakens it, as shown by both fluxes and currents (Fig. 2 B and Fig. 3 B). For example, while wild type and E203Q have similar influx time courses at pH 4.5, the mutant influx is well over 10-fold faster than wild type at pH 7. Thus, if E148 is neutralized, Cl− flux rates become insensitive to the protonation state of E203; in contrast, if E203 is neutralized, Cl− flux rates still respond to pH, presumably through protonation of E148, which appears to be an absolute requirement for Cl− transport. To verify that this residual pH dependence arises from E148 in the E203Q background, we examined the double mutant E148A/E203Q, and found that it displays pH-independent 36Cl− uptake and no active H+ transport (Figs. 3 and 4), as expected.
E203 Is Unique among Internally Facing Carboxylates
To assess whether the loss of H+ transport upon mutation is specific to E203, we performed similar experiments at the seven other carboxylates indicated in Fig. 1 A. For each mutant (except for E113Q, which failed to express protein), proton coupling was gauged electrically, using reversal potential shifts induced by pH or Cl− gradients. These mutants all display H+-coupled Cl− currents (Fig. 4). Cl−–H+ coupling is similar to wild type except with E202Q and D278N, which display distinctly lower coupling efficiency, as indicated by the smaller shifts in reversal potential induced by a pH gradient and the greater shift in a Cl− gradient. We also neutralized two basic residues, K131 and R28. The former, which is strictly conserved, has been proposed to electrostatically influence the two Cl− ions bound to wild-type CLC-ec1 (Faraldo-Gomez and Roux, 2004), while the latter forms an intersubunit salt bridge with E203. However, R28L shows fully wild-type transport behavior, while K131M still mediates robust proton transport, though with slightly altered characteristics. Thus, E203 is unique among all residues tested in leading to complete loss of proton transport.
Structure and Halide Binding of E203Q
We crystallized E203Q and determined its structure at 3.3 Å resolution by molecular replacement (Fig. 5; Table I). The mutant crystallized isomorphically to wild type with a virtually identical structure (backbone rmsd = 0.4 Å) except close to the site of mutation. In wild-type protein, E203 forms a salt bridge with R28 from the other subunit of the homodimer and an H bond with E113 in the same subunit. In the mutant structure, the Q203 side chain occupies the same position as E203 in the wild type, but R28 lacks side chain density and so is most likely disordered. This result raises the intriguing possibility that crosstalk between the two subunits is involved in the proton-coupling mechanism; however, the wild-type behavior shown by R28L (Fig. 4) eliminates this cross-subunit salt bridge as a necessary element of the transport cycle.
Figure 5.

Crystal structure and halide binding. (A) Wild type; (B) E203Q. Top panel, 2FO-FC electron density maps (blue) contoured at 1.2 σ, with protein model shown in stick representation. Wild-type map was calculated to 3.3 Å resolution from deposited structure factors (accession no. 1OTS). Bottom panel, anomalous difference density maps (green) of crystals grown in NaBr, determined from data collected at the Br− absorption edge, contoured at 5 σ, with the same wild-type protein model (1OTS) shown for both maps, with subunit A in red and subunit B in blue.
TABLE I.
Crystallographic Statistics
| E203Q | E203Q | Wild-type CLC-ec1 | |
|---|---|---|---|
| Anion | Cl− | Br− | Br− |
| Resolution | 3.3 Å | 3.90 Å | 3.2 Å |
| Number of reflections | 36,343 | 23,320 | 43,955 |
| Completeness | 88.3% (85.5%) | 98.5% (95.6%) | 99.0% (98.4%) |
| R-sym | 9.6 (47.1) | 8.5 (49.4) | 7.1 (52.3) |
| <I>/σI | 13.4 (2.1) | 13.3 (2.9) | 19.1 (2.3) |
| R-factor | 24.8 | 26.9 | 25.1 |
| R-free | 28.7 | 32.5 | 29.3 |
| Bond length RMSD | 0.007 | 0.009 | 0.013 |
| Bond angle RMSD | 1.082 | 1.068 | 1.466 |
Crystals of CLC-ec1-FAB complex were formed in 3-μl sitting drops, in the presence of 100–250 mM NaCl or NaBr. All crystals were of spacegroup C2 with cell dimensions similar to those of the wild-type complex (1OTS).
Crystals grown in Br−, a functionally faithful Cl− substitute (Accardi et al., 2004), are particularly advantageous for studying the anion-binding properties of CLC-ec1, since this halide can be visualized by anomalous diffraction, as has been done for wild-type protein and several mutants (Dutzler et al., 2003; Lobet and Dutzler, 2005). When crystallized from NaBr solutions, wild-type protein shows two such sites, both prominently occupied, and Br− density shows up identically in the E203Q mutant (Fig. 5). Thus, the crystal structures indicate that this mutant with profoundly altered function adopts an unperturbed conformation with normal anion-binding characteristics.
DISCUSSION
All exchange transport mechanisms involve protein conformational changes linked to sided binding and release of substrates (Maloney, 1994). For CLC Cl−/H+ exchangers, only the sketchiest picture of the exchange mechanism has yet emerged. X-ray structures of CLC-ec1 tell us where the Cl− ions bind (Fig. 1). In combination with functional analysis, the structures also suggest a plausible mechanism for concerted movement of Cl− and H+ linked to a small conformational change in one part of the transport cycle: simple rotation of the E148 side chain that alters access of both H+ and Cl− to the protein interior from the extracellular solution (Dutzler et al., 2003; Accardi and Miller, 2004). For these reasons, E148 is well established as serving a dual role at the extracellular face of the protein: handing off protons to, and allowing concomitant entry of Cl− ions from, the external solution. (This coupled reaction is of course reversible and must operate in both directions.)
Since E148 faces the extracellular solution, we supposed that an analogous proton transfer site might exist near the intracellular surface of the protein. This is by no means required in coupled transport mechanisms. Indeed, in a conventional exchange mechanism, a single protonatable locus alternates its exposure to the two sides of the membrane through a cycle of conformational changes, as proposed in detail for the lactose permease (Kaback, 2005). But the structure of CLC-ec1, in contrast to that of lactose permease, does not suggest any obvious conformational changes that would alternate exposure of E148. For this reason, we conjectured that protons may somehow move through the protein during transport. Such a picture envisions a proton pathway connecting distant residues on each side of the membrane, each mediating proton transfer between the protein and its corresponding aqueous solution, as in the well-characterized proton transfer residues in the bacterial reaction center, for example (Ädelroth and Brzezinski, 2004). These speculative considerations provided the motivation for the carboxylate scan described here.
E203 reveals itself as the internal proton transfer site anticipated by this “pathway” hypothesis. Our results do not nail down this conclusion rigorously, but they do suggest it strongly via several lines of argument. First, neutralization of E203 completely and specifically abolishes all proton transport, as expected if the carboxyl group were required for shuttling protons to and from the protein interior, a prerequisite for participation in the coupled-transport cycle. Second, E203 is partially buried near the intracellular solution, as is required for this proposed function. Third, E203 is unique among all groups tested in being absolutely necessary for proton coupling; while mutations at E202 and at D278 lower coupling, mutation at E203 alone fully ablates it. Fourth, the only other residue that behaves this way is the externally facing E148, for which a proton transfer function is strongly supported by previous work. Finally, the loss of H+ transport in E203Q cannot be attributed to mutagenic disruption, since the structure of this mutant is similar to wild type and specific Cl− transport is maintained. These considerations support the idea that E203 directly catalyzes proton transfer between protein and intracellular solution as a required step in the coupled transport cycle.
This conclusion leads to two further inferences. It argues that the protein contains a pathway along which protons move between E148 on the extracellular side and E203 on the intracellular. Proton movement along this pathway must be somehow coupled to occupancy of the Cl− binding sites, which themselves imply a pathway for anions. Discovering the still-unknown rules of this coupling is required for understanding the transport mechanism (Jencks, 1980). We do not yet know how protonation of E203 influences the Cl− sites, but it is intriguing that computational studies (Faraldo-Gomez and Roux, 2004) suggest a strong electrostatic influence of the protonation state of E203 (as well as of other nearby residues) on the affinity of Cl− binding to the protein. A second inference is that this ion transport pathway is bifurcated, as illustrated in Fig. 6. The structure of E148Q, which mimics the “external open” form with E148 protonated, shows an additional Cl− ion occupying the location vacated by the Q148 side chain (Dutzler et al., 2003). Thus, on the external side of the protein, the Cl− and H+ pathways overlap; but they diverge on the intracellular side, since E203 is distant from the internal Cl− site. A future challenge will be to explore the terra incognita between the two aqueous transfer sites, where H+ must bind as it traverses the membrane, where the central Cl− ion is coordinated by hydroxyls of S107 and Y445, and where the two pathways appear to separate. This proposed bifurcated pathway is functionally mirrored in the characteristics of E203 and E148. Protonation of E203 is a necessary step only for H+ transport, while protonation of E148 is required for both Cl− and H+ movements.
Figure 6.

Bifurcated pathway. CLC dimer, with the following side chains highlighted: E148, E203 (red); S107, Y445 (blue). Cl− ions shown as green spheres. Proposed Cl− and H+ pathways are indicated in green and red, respectively. The region between E148 and E203, which the H+ must traverse, is indicated by the gray box, with side chains of S107 and Y445 indicated in blue.
As a final point, we should note an odd feature of the Cl−/H+ exchange mechanism: that abolition of H+ transport does not impede Cl− permeation. Extensively studied exchange transporters, MFS exchangers (Ruan et al., 1992; Auer et al., 2001), Cl−/HCO3 − exchangers (Knauf et al., 2002), Na+/Ca2+ exchangers (Dong et al., 2002; Kang and Hilgemann, 2004), mitochondrial nucleotide exchangers (Pebay-Peyroula et al., 2003), display obligatory exchange, whereby removal of either substrate completely inhibits net movement of the other. But this is not the case for CLC-ec1. Mutation of either proton transfer residue, E148 or E203, abolishes H+ transport but leaves Cl− transport largely unaffected. Our assays are not sufficiently quantitative to tell us precisely how much the unitary rate of Cl− flux is affected in these mutants, but the overall transport behavior suggests that any effect on Cl− turnover cannot be very large. For this reason, we consider that the mechanism of CLC transporters will turn out to be novel, unconventional, and eerily reminiscent of ion channel permeation, and fundamentally unlike the alternating-site schemes that have so productively guided our understanding of long-studied and more familiar classes of transport proteins.
Acknowledgments
We are grateful to Prof. Dr. Raimund Dutzler for hosting an extended research visit to his laboratory at the University of Zürich and to the John Simon Guggenheim Foundation for a fellowship supporting this visit. We thank J. Cohen and Dr. C. Nimigean for helpful discussions, and acknowledge the National Cell Culture Center (Minneapolis, MN) for efficient production of the 10EC3/G4 antibody. We thank the staff at the Swiss Light Source, Advanced Photon Source and at the Advanced Light Source. We wish to thank Prof. B.V.V. Prasad for help with data collection.
This work was supported in part by National Institutes of Health grant GM-31768. W. Nguitragool is a Howard Hughes Medical Institute Predoctoral Fellow.
Olaf S. Andersen served as editor.
References
- Accardi, A., L. Kolmakova-Partensky, C. Williams, and C. Miller. 2004. Ionic currents mediated by a prokaryotic homologue of CLC Cl− channels. J. Gen. Physiol. 123:109–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Accardi, A., and C. Miller. 2004. Secondary active transport mediated by a prokaryotic homologue of ClC Cl− channels. Nature. 427:803–807. [DOI] [PubMed] [Google Scholar]
- Ädelroth, P., and P. Brzezinski. 2004. Surface-mediated proton-transfer reactions in membrane-bound proteins. Biochim. Biophys. Acta. 1655:102–115. [DOI] [PubMed] [Google Scholar]
- Auer, M., M.J. Kim, M.J. Lemieux, A. Villa, J. Song, X.D. Li, and D.N. Wang. 2001. High-yield expression and functional analysis of Escherichia coli glycerol-3-phosphate transporter. Biochemistry. 40:6628–6635. [DOI] [PubMed] [Google Scholar]
- Chen, M.F., and T.Y. Chen. 2001. Different fast-gate regulation by external Cl− and H+ of the muscle-type ClC chloride channels. J. Gen. Physiol. 118:23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong, H., J. Dunn, and J. Lytton. 2002. Stoichiometry of the cardiac Na+/Ca2+ exchanger NCX1.1 measured in transfected HEK cells. Biophys. J. 82:1943–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutzler, R., E.B. Campbell, M. Cadene, B.T. Chait, and R. MacKinnon. 2002. X-ray structure of a ClC chloride channel at 3.0 Å reveals the molecular basis of anion selectivity. Nature. 415:287–294. [DOI] [PubMed] [Google Scholar]
- Dutzler, R., E.B. Campbell, and R. MacKinnon. 2003. Gating the selectivity filter in ClC chloride channels. Science. 300:108–112. [DOI] [PubMed] [Google Scholar]
- Estevez, R., B.C. Schroeder, A. Accardi, T.J. Jentsch, and M. Pusch. 2003. Conservation of chloride channel structure revealed by an inhibitor binding site in ClC-1. Neuron. 38:47–59. [DOI] [PubMed] [Google Scholar]
- Faraldo-Gomez, J.D., and B. Roux. 2004. Electrostatics of ion stabilization in a ClC chloride channel homologue from Escherichia coli. J. Mol. Biol. 339:981–1000. [DOI] [PubMed] [Google Scholar]
- Foster, J.W. 2004. Escherichia coli acid resistance: tales of an amateur acidophile. Nat. Rev. Microbiol. 2:898–907. [DOI] [PubMed] [Google Scholar]
- Iyer, R., T.M. Iverson, A. Accardi, and C. Miller. 2002. A biological role for prokaryotic ClC chloride channels. Nature. 419:715–718. [DOI] [PubMed] [Google Scholar]
- Jencks, W.P. 1980. The utilization of binding energy in coupled vectorial processes. Adv. Enzymol. Relat. Areas Mol. Biol. 51:75–106. [DOI] [PubMed] [Google Scholar]
- Jentsch, T.J., I. Neagoe, and O. Scheel. 2005. CLC chloride channels and transporters. Curr. Opin. Neurobiol. 15:319–325. [DOI] [PubMed] [Google Scholar]
- Kaback, H.R. 2005. Structure and mechanism of the lactose permease. C. R. Biol. 328:557–567. [DOI] [PubMed] [Google Scholar]
- Kang, T.M., and D.W. Hilgemann. 2004. Multiple transport modes of the cardiac Na+/Ca2+ exchanger. Nature. 427:544–548. [DOI] [PubMed] [Google Scholar]
- Kida, Y., S. Uchida, H. Miyazaki, S. Sasaki, and F. Marumo. 2001. Localization of mouse CLC-6 and CLC-7 mRNA and their functional complementation of yeast CLC gene mutant. Histochem. Cell Biol. 115:189–194. [DOI] [PubMed] [Google Scholar]
- Knauf, P.A., F.Y. Law, T.W. Leung, A.U. Gehret, and M.L. Perez. 2002. Substrate-dependent reversal of anion transport site orientation in the human red blood cell anion-exchange protein, AE1. Proc. Natl. Acad. Sci. USA. 99:10861–10864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornak, U., D. Kasper, M.R. Bosl, E. Kaiser, M. Schweizer, A. Schulz, W. Friedrich, G. Delling, and T.J. Jentsch. 2001. Loss of the ClC-7 chloride channel leads to osteopetrosis in mice and man. Cell. 104:205–215. [DOI] [PubMed] [Google Scholar]
- Lobet, S., and R. Dutzler. 2005. Ion binding properties of the CLC chloride selectivity filter. EMBO J. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloney, P.C. 1994. Bacterial transporters. Curr. Opin. Cell Biol. 6:571–582. [DOI] [PubMed] [Google Scholar]
- Niemeyer, M.I., L.P. Cid, L. Zuniga, M. Catalan, and F.V. Sepulveda. 2003. A conserved pore-lining glutamate as a voltage- and chloride-dependent gate in the ClC-2 chloride channel. J. Physiol. 553:873–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pebay-Peyroula, E., C. Dahout-Gonzalez, R. Kahn, V. Trezeguet, G.J. Lauquin, and G. Brandolin. 2003. Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature. 426:39–44. [DOI] [PubMed] [Google Scholar]
- Picollo, A., and M. Pusch. 2005. Chloride/proton antiporter activity of mammalian CLC proteins ClC-4 and ClC-5. Nature. 436:420–423. [DOI] [PubMed] [Google Scholar]
- Richard, H., and J.W. Foster. 2004. Escherichia coli glutamate- and arginine-dependent acid resistance systems increase internal pH and reverse transmembrane potential. J. Bacteriol. 186:6032–6041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan, Z.S., V. Anantharam, I.T. Crawford, S.V. Ambudkar, S.Y. Rhee, M.J. Allison, and P.C. Maloney. 1992. Identification, purification, and reconstitution of OxlT, the oxalate: formate antiport protein of Oxalobacter formigenes. J. Biol. Chem. 267:10537–10543. [PubMed] [Google Scholar]
- Scheel, O., A.A. Zdebik, S. Lourdel, and T.J. Jentsch. 2005. Voltage-dependent electrogenic chloride/proton exchange by endosomal CLC proteins. Nature. 436:424–427. [DOI] [PubMed] [Google Scholar]
- Terwilliger, T.C. 2000. Maximum-likelihood density modification. Acta Crystallogr. D Biol. Crystallogr. 56(Pt 8):965–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin, J., Z. Kuang, U. Mahankali, and T.L. Beck. 2004. Ion transit pathways and gating in ClC chloride channels. Proteins. 57:414–421. [DOI] [PubMed] [Google Scholar]