

Abstract
The idiopathic inflammatory bowel diseases, Crohn’s disease (CD) and ulcerative colitis (UC), are chronic, frequently disabling diseases of the intestines. Segregation analyses, twin concordance, and ethnic differences in familial risks have established that CD and UC are complex, non-Mendelian, related genetic disorders. We performed a genome-wide screen using 377 autosomal markers, on 297 CD, UC, or mixed relative pairs from 174 families, 37% Ashkenazim. We observed evidence for linkage at 3q for all families (multipoint logarithm of the odds score (MLod) = 2.29, P = 5.7 × 10−4), with greatest significance for non-Ashkenazim Caucasians (MLod = 3.39, P = 3.92 × 10−5), and at chromosome 1p (MLod = 2.65, P = 2.4 × 10−4) for all families. In a limited subset of mixed families (containing one member with CD and another with UC), evidence for linkage was observed on chromosome 4q (MLod = 2.76, P = 1.9 × 10−4), especially among Ashkenazim. There was confirmatory evidence for a CD locus, overlapping IBD1, in the pericentromeric region of chromosome 16 (MLod = 1.69, P = 2.6 × 10−3), particularly among Ashkenazim (MLod = 1.51, P = 7.8 × 10−3); however, positive MLod scores were observed over a very broad region of chromosome 16. Furthermore, evidence for epistasis between IBD1 and chromosome 1p was observed. Thirteen additional loci demonstrated nominal (MLod > 1.0, P < 0.016) evidence for linkage. This screen provides strong evidence that there are several major susceptibility loci contributing to the genetic risk for CD and UC.
Keywords: Crohn’s disease, ulcerative colitis, Ashkenazim, linkage analysis, chromosome 16
The inflammatory bowel diseases, Crohn’s disease (CD) and ulcerative colitis (UC), are chronic, inflammatory diseases of the intestines with a combined prevalence of 200–300/100,000 in the United States (1–3). CD may involve any part of the gastrointestinal tract (most frequently the terminal ileum and colon) (4). Bowel inflammation is transmural, discontinuous, and may contain granulomas or be associated with intestinal or perianal fistulas. In contrast, in UC, the inflammation is continuous, limited to rectal and colonic mucosal layers, and fistulas and granuloma are not observed (4). In approximately 10% of cases confined to the rectum and colon, definitive classification as CD or UC cannot be made and are designated “indeterminate colitis” (5). Both diseases include extraintestinal inflammation of the skin, eyes, or joints. The etiology of inflammatory bowel disease is unknown. CD and UC are commonly classified as autoimmune diseases. The prevalence of inflammatory bowel disease is increased in individuals with other autoimmune diseases, particularly ankylosing spondylitis, psoriasis, sclerosing cholangitis, and multiple sclerosis (6).
There is strong evidence from twin studies, familial risk data, and segregation analyses that inflammatory bowel disease, especially CD, is genetic (6–11). CD and UC are considered complex genetic traits as inheritance does not follow any simple Mendelian models (6, 10–12). The degree of genetic clustering in siblings, λs, (prevalence in siblings divided by population prevalence) has been estimated at 36.5 for CD, 16.6 for UC, and 24.7 for inflammatory bowel disease (UC, CD, and indeterminate colitis) (13). The λs values reflect the feasibility of identifying disease genes through genome-wide searches and compares favorably with other complex genetic disorders, such as insulin-dependent diabetes mellitus (λs = 15) (12). The cross-disease relative risks are 3.85 for UC given a CD proband, and 1.72 conversely (8), suggesting the presence of shared susceptibility genes between the two diseases. Ashkenazi Jews have 2–8 times greater prevalence of inflammatory bowel disease compared with non-Ashkenazim Caucasians in all geographic areas studied and a 2-fold familial risk (14, 15).
A genome-wide screen in 41 equivalent European CD sibling pairs testing 270 markers with replication in a second panel of 71 pairs demonstrated evidence for linkage over a broad, pericentromeric region on chromosome 16, IBD1 (16). This region subsequently has been confirmed for CD but not UC (17–19). A separate genome-wide screen (89 sibling pairs undergoing the initial, genome-wide screen with 260 autosomal markers and 97 pairs used for replication) was performed on families from the United Kingdom with all inflammatory bowel disease phenotypes (UC, CD, and mixed families) (20). Genome-wide evidence for linkage was identified on chromosome 12, and two other inflammatory bowel disease loci with suggestive evidence for linkage were mapped to chromosomes 7 and 3p. The number of affected relative pairs undergoing the initial, genome-wide screen in these studies suggests that additional loci possibly might be identified through genome-wide screening with a significantly larger sample size and more dense marker set. Furthermore, in a detailed replication study of 148 affected relative pairs with CD, one-third Ashkenazim, in the four above inflammatory bowel disease candidate loci, we found significant evidence for only the IBD1 locus (S.R.B. and J.H.C., unpublished observations). We report here the results of a genome-wide screen with 377 markers on 297 CD, UC, or mixed relative pairs from 174 American families, 37% Ashkenazim. We identified two novel regions on chromosomes 1p and 3q with suggestive evidence for linkage, and we report nominal evidence for linkage in 15 additional chromosomal regions.
MATERIALS AND METHOD
Ascertainment and Phenotypic Characterization of Families.
We recruited families containing at least one informative affected relative pair primarily through the inflammatory bowel disease clinical practices at the University of Chicago (S.B.H. and B.S.K.) and the Meyerhoff Inflammatory Bowel Disease Center at the Johns Hopkins Hospital (T.M.B.) as well as additional inflammatory bowel disease referral centers. In all cases, informed consent for a molecular genetic study was obtained, and the study protocol was approved by the institutional review boards of the University of Chicago and the Johns Hopkins University. Confirmation of diagnoses for idiopathic, chronic, inflammatory bowel disease, i.e., CD, UC, or indeterminate colitis, was obtained from primary review of endoscopic, radiologic, and pathologic data (4, 21). Families with members having indeterminate colitis (5) (meet criteria for idiopathic inflammatory bowel disease, but cannot be distinguished from CD or UC) were included in the “all families” category (Fig. 1). Patients were classified as being Ashkenazi Jewish if at least two grandparents were self-defined as Jewish and their families immigrated from countries in Central or Eastern Europe. Table 1 summarizes the families by diagnosis, race, ethnicity, and affected relative pair type.
Figure 1.

Multipoint nonparametric linkage analysis of all families, CD-only families, mixed (families containing at least one or more member with a diagnosis of CD and one or more member with a diagnosis of UC, or any member with indeterminate idiopathic colitis) and UC-only families. The all-families curves reflect the combined contributions of the CD-only families, UC-only families, and mixed families. Map distances were taken from the Weber screening set 8.0. Multipoint analysis was performed by using a modified version of genehunter (25). Information content was 0.48, SD 0.07. ∗, regions implicated in Hugot et al.’s genome-wide screen (16). +, regions implicated in Satsangi et al.’s genome-wide screen (20). MHC, major histocompatibility complex.
Table 1.
Summary of families studied by diagnosis and ethnicity
| CD-only families | UC-only families | Mixed families | All families | |
|---|---|---|---|---|
| Number of families | 99 | 13 | 62 | 174 |
| Affecteds genotyped (n) | ||||
| Non-Ashenazim Caucasians | 156 | 33 | 83 | 272 (62%) |
| Ashkenazim | 100 | 6 | 56 | 162 (37%) |
| Other | 2 | 0 | 3 | 5 (1%) |
| Total | 258 | 39 | 142 | 439 |
| Affected relative pairs (n) | ||||
| Sibling pair | 97 | 18 | 36 | 151 |
| Uncle-niece | 29 | 2 | 25 | 56 |
| Grandparent-grandchild | 7 | 1 | 9 | 17 |
| More distant | 42 | 5 | 26 | 73 |
| Total | 175 | 26 | 96 | 297 |
Genotyping and Map Distances.
Genotyping was performed primarily by the Mammalian Genotyping Service (National Heart, Lung, and Blood Institute) at the Marshfield Center for Medical Genetics by using semiautomated fluorescent genotyping methods (22). The markers used were 371 autosomal, primarily tri- and tetranucleotide repeat microsatellite markers, predominately from the Weber screening set 8.0 (http://www.marshmed.org/genetics/) (23). Two loci, D1S234 and D1S255, were used to narrow relatively larger mapping intervals on chromosome 1p (see Table 3), and these were genotyped by using semiautomated fluorescent methods by the Johns Hopkins Genetic Resources Core Facility Genotyping Service. Map distances were taken from the Weber screening set 8.0 map and the comprehensive Marshfield genetic map, available on their web site. Genotyping error rate was estimated by comparing blinded duplicate genotyping results of eight separate patients.
Table 3.
Two-point and multipoint nonparametric lod scores in peak regions on chromosomes 1p, 3q, 4q, and 16 stratified by ethnicity
| Chr | Position | Locus | All families
|
Ashkenazim
|
White non-Jewish
|
|||
|---|---|---|---|---|---|---|---|---|
| 2-pt lod | MLod | 2-pt lod | MLod | 2-pt lod | MLod | |||
| 1 | 28 | D1S3369 | 0.30 | 0.99 | 0.42 | 0.077 | 0.05 | 0.67 |
| 37 | D1S552 | 2.03 | 2.54 | 0.00 | 0.063 | 3.14 | 2.59 | |
| 39 | — | — | 2.65 | — | 0.075 | — | 2.64 | |
| 46.3 | D1S234 | 0.52 | 2.01 | 0.11 | 0.098 | 0.25 | 1.77 | |
| 49 | D1S1622 | 0.95 | 1.95 | 0.16 | 0.042 | 0.41 | 1.78 | |
| 59 | D1S255 | 0.00 | 1.67 | 0.00 | 0.066 | 0.00 | 1.17 | |
| 68 | D1S3721 | 0.11 | 0.58 | 0.28 | 0.00 | 0.00 | 0.26 | |
| 3 | 186 | D3S1763 | 0.70 | 0.97 | 0.033 | 0.00 | 1.06 | 1.68 |
| 195 | D3S3053 | 0.73 | 2.06 | 0.026 | 0.00 | 1.31 | 3.39 | |
| 200 | — | — | 2.29 | — | 0.013 | — | 3.23 | |
| 207 | D3S2427 | 1.25 | 2.07 | 0.0013 | 0.080 | 2.16 | 2.22 | |
| 217 | D3S1262 | 1.39 | 1.90 | 2.86 | 1.96 | 0.014 | 0.16 | |
| 226 | D3S2398 | 0.27 | 1.32 | 0.96 | 1.59 | 0.00 | 0.15 | |
| 232 | D3S2418 | 0.30 | 0.83 | 1.08 | 1.13 | 0.00 | 0.070 | |
| 4* | 88 | D4S2361 | 0.00 | 0.05 | 0.20 | 0.36 | 0.00 | 0.00 |
| 101 | D4S1647 | 0.61 | 1.15 | 1.11 | 2.11 | 0.00 | 0.031 | |
| 110 | D4S2623 | 3.00 | 2.76 | 2.23 | 2.76 | 0.36 | 0.29 | |
| 126 | D4S2394 | 0.37 | 0.64 | 1.35 | 1.58 | 0.00 | 0.00 | |
| 16† | 11 | D16S748 | 2.53 | 1.76 | 0.57 | 0.83 | 1.67 | 0.85 |
| 20 | D16S764 | 0.36 | 1.28 | 0.63 | 1.37 | 0.0029 | 0.27 | |
| 33 | D16S403 | 0.76 | 1.02 | 0.075 | 0.41 | 0.59 | 0.66 | |
| 39 | D16S769 | 1.03 | 1.69 | 0.97 | 1.51 | 0.17 | 0.61 | |
| 46 | D16S753 | 0.49 | 0.66 | 0.79 | 0.41 | 0.028 | 0.31 | |
| 50 | D16S3396 | 0.12 | 0.51 | 0.11 | 0.11 | 0.0021 | 0.35 | |
| 92 | D16S516 | 1.76 | 1.31 | 1.05 | 0.57 | 1.15 | 1.04 | |
Mixed families.
CD-only families.
Data Analysis.
Data were analyzed by nonparametric linkage analysis with a modified version of genehunter (24) using the Haldane map function. genehunter uses an inheritance vector to characterize meiosis events and estimates identity by descent through a multipoint, likelihood-based approach (25). This modified version of genehunter uses exact likelihood calculations rather than the perfect data approximation, the latter being overly conservative in datasets with missing information (24). The Spairs scoring function was used, as it follows a more normal distribution than the Sall scoring function (26). The linear model was used except for the stratified analysis of mixed, Jewish families, where the exponential model (24) also was performed. Allele frequencies were estimated from unaffected founders. For subanalysis by ethnicity, allele frequencies specific to the ethnic group (Ashkenazim, non-Ashkenazim Caucasians) were applied. The P-values for subanalyses by ethnicity or diagnosis are uncorrected for multiple comparisons. To determine the robustness of the analysis with respect to incorrect assumptions about marker allele frequencies in regions of maximal linkage, we perturbed the allele frequencies, by simulating from a multinomial distribution, for the markers in the regions giving the peaks at the 1p and 3q loci. In general, the simulated allele frequencies correspond to a similar heterozygosity of the marker, but the values of the frequencies can vary up to 50% from the initial values (the variation is larger for rare alleles and smaller for common alleles). Pairwise comparisons and identity by descent estimates of all genotyped individuals were performed to identify sample duplications and misspecified half-siblings.
RESULTS
We performed a genome-wide screen on 439 affected individuals and 198 connecting relatives from 174 families. Approximately 230,000 genotypes were analyzed and a blinded estimate of the genotype error rate was 0.73%. Fig. 1 demonstrates the multipoint, nonparametric logarithm of the odds (MLod) curves for all, CD-only, mixed, and UC-only families. Seventeen regions showed nominal evidence for linkage (MLod ≥ 1.0, P ≤ 0.016) in either all families or a diagnostic subset (Table 2, Fig. 1). Highly suggestive evidence for linkage (27) was observed on chromosome 1p near D1S552 (MLod = 2.65, P = 2.4 × 10−4) in all families. Most of the evidence for linkage was from the non-Ashkenazim families (MLod = 2.59, P = 2.5 × 10−4), compared with the Ashkenazim (MLod = 0.098, P = 0.24) (Table 3).
Table 2.
Regions showing nominal multipoint evidence of linkage in a genome-wide screen for inflammatory bowel disease
| Region | MLod score | Zmax score | P-value |
|---|---|---|---|
| D1S552 | 2.65 | 3.49 | 0.00024 |
| D1S1609* | 1.74 | 2.83 | 0.0023 |
| D2S2952-D2S1400† | 1.24 | 2.39 | 0.0084 |
| D3S2432† | 1.06 | 2.21 | 0.014 |
| D3S3053-D3S2427 | 2.29 | 3.25 | 0.00057 |
| D4S1647 | 1.71 | 2.81 | 0.0025 |
| D5S1462 | 1.19 | 2.34 | 0.0096 |
| D7S820* | 1.18 | 2.32 | 0.010 |
| D8S256† | 1.49 | 2.62 | 0.0044 |
| D9S2157† | 1.41 | 2.55 | 0.0054 |
| D11S1999† | 1.66 | 2.76 | 0.0028 |
| D12S2070† | 1.15 | 2.31 | 0.011 |
| D14S608* | 1.53 | 2.65 | 0.0040 |
| D16S748-D16S764† | 1.81 | 2.89 | 0.0019 |
| D16S769† | 1.69 | 2.79 | 0.0026 |
| D16S516† | 1.31 | 2.46 | 0.0071 |
| D19S1034-D19S586† | 1.38 | 2.52 | 0.0059 |
Zmax, 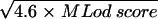 .
.
Linkage evidence in mixed families.
Linkage evidence in CD families.
A broad region of linkage was observed on chromosome 3q for all families (MLod = 2.29, P = 5.7 × 10−4), which represents contributions from both CD (MLod = 1.39, P = 5.7 × 10−3), mixed (MLod = 1.23, P = 8.7 × 10−3), and UC families (MLod = 0.50, P = 0.065) (Fig. 1). We stratified by ethnicity (Table 3) and demonstrated contributions from both non-Ashkenazim Caucasian (MLod = 3.39, P = 3.92 × 10−5) and Ashkenazim families (MLod = 1.96, P = 1.35 × 10−3) (Table 3). To determine the robustness of the results with respect to incorrect assumptions about marker allele frequencies, we perturbed the allele frequencies. The peak lod scores after perturbation were between 2.61 and 2.74 (initially 2.65) on chromosome 1p and between 2.12 and 2.31 (initially 2.29) on chromosome 3q.
For the total data set, the third strongest evidence of linkage was on chromosome 4q (MLod 1.71, P = 2.5 × 10−3). Evidence for linkage in this region was much greater for the limited subset of mixed families (96 affected relative pairs from 62 families); the uncorrected MLod was 2.76 (P = 1.9 × 10−4). Furthermore, in 16 mixed, Ashkenazim families comprising 40 typed affecteds and 29 affected relative pairs, the MLod was 2.80 (P = 1.7 × 10−4) using a linear model and 3.70 (P = 1.81 × 10−5) using the exponential model more applicable to smaller data sets such as these (24).
Of the 14 remaining peaks, three CD peaks (Fig. 1) were observed on chromosome 16, one of which, D16S769 (MLod = 1.69, P = 2.6 × 10−3), lies within the pericentromeric region of IBD1 (16) (Table 1, Fig. 1) and provides additional confirmatory evidence. Throughout chromosome 16, the evidence for linkage is greater in the CD families compared with all families, with no evidence observed in UC families. We observed greater evidence for linkage in Ashkenazim families than non-Ashkenazim Caucasian families (Table 3) for the IBD1 locus. This difference is particularly significant given the smaller number of families in the Ashkenazim group. In addition, a broad region proximal to D16S769 not overlapping the IBD1 locus (16) demonstrates lod scores greater than 1.1 in CD families (Fig. 1).
To separate heterogeneous effects that may account for the broad linkage observed on chromosome 16, we reanalyzed that chromosome. We tested only those CD families demonstrating positive or negative nonparametric linkage (NPL) scores (as determined by the original version of genehunter) (25) on chromosome 1p, the region demonstrating the greatest evidence for linkage in all families (Fig. 2) (Nancy J. Cox, personal communication). The MLod curve in those families with positive NPL scores (25) on chromosome 1p (Fig. 2B) demonstrated a more tightly localized peak at 50 centimorgans (cM) centered in the middle of the previously defined IBD1 region (16). Furthermore, the evidence for linkage on chromosome 16 disappeared throughout the IBD1 region in the subset of those families with negative NPL scores (25) on chromosome 1p with peaks observed in the p and q telomeric regions of the chromosome (Fig. 2C).
Figure 2.

Analysis of CD-only families on chromosome 16, conditioned on results from the region of maximal MLod on chromosome 1p. MLod curves of all CD families (A), CD families with positive NPL scores (25) (B), and negative (25) scores on chromosome 1p (C). IBD1, interpolated region of linkage described by Hugot et al. (16).
We observed no confirmatory evidence supporting linkage to the previously reported chromosomes 12, 7, or 3p loci (Fig. 1) in all families (20). Nominal evidence for linkage was observed in the region just proximal to the chromosome 3p locus in CD families and on the chromosome 7 locus in mixed families (Table 1). We found no significant evidence supporting linkage in the major histocompatibility complex (MHC) region, supporting the concept of a minor role for MHC-linked genes in inflammatory bowel disease susceptibility (Fig. 1) (28, 29).
DISCUSSION
A present hypothesis of inflammatory bowel disease is that several genes are involved, with some genes being common to CD and UC. Two previous genome-wide screens have identified susceptibility loci on chromosome 16 (16) in CD families (subsequently confirmed) (17–19) and loci on chromosomes 12, 7, and 3p in all inflammatory bowel disease families (20). The CD locus on chromosome 16, IBD1 (16), and the inflammatory bowel disease locus on chromosome 12 (20) were reported to have estimated locus-specific λs of 1.3 and 2.0, respectively. Therefore, these loci alone could not possibly account for the total genetic risks observed for inflammatory bowel disease and particularly for CD, and thus additional susceptibility loci are predicted.
Our study differed from the previous screens (16, 20) by the use of a much larger number of affected relative pairs in the screening phase, the study of nonsibling relative pairs and by the inclusion of a significant number of Ashkenazi Jewish pedigrees, who have a greater population and familial risk (14, 15). Furthermore, the use of a denser screen, containing primarily tri- and tetranucleotide repeat markers (30), allowed us to more thoroughly test the genome for linkage and possibly examine regions not well covered in the previous screens. All of these factors likely contributed to our identifying loci not seen in the previous studies.
Our genome-wide screen identifies two susceptibility loci with suggestive evidence (27) for linkage on chromosomes 1p and 3q in all families. The evidence for linkage on chromosome 1p represents contributions from CD- and UC-only families, but not from mixed families. When stratified by ethnicity, evidence for linkage on chromosome 1p results almost exclusively from non-Ashkenazim Caucasian families. In contrast, evidence for linkage on chromosome 3q results from the combined contributions of all diagnostic groups (CD, UC, and mixed families) as well as both Ashkenazim and white non-Jewish families.
In the limited subset of mixed families, we observed an MLod of 2.76 on chromosome 4q, which corresponds to an uncorrected P-value of 1.9 × 10−4. In Jewish families, the MLod was 2.80 (1.65 × 10−4). Using the exponential model (24), the MLod score was 3.70 (1.81 × 10−5) in mixed Jewish families on chromosome 4q. This last result must be interpreted conservatively both because of the number of statistical tests applied (stratification by diagnosis and ethnicity) as well as the relatively small sample size from which the result derives. However, as this region represents the third-highest MLod score in all families (1.71), it clearly warrants further study with additional families. There was no significant evidence for association by transmission/disequilibrium testing (31) at any of the markers on chromosomes 1p, 3q, and 4q (data not shown).
We observed additional confirmatory evidence for IBD1 (16) in CD families at D16S769, which overlaps the previously defined region. However, three general peaks can be identified on chromosome 16, and positive lod scores were observed over very broad regions. In the initial report, the IBD1 region encompassed more than 40 cM and two general peaks were observed (16). Together with subsequent studies, peaks in CD families have been reported for D16S407 (20), D16S748-D16S764 (present report), D16S769 (present report), D16S409 (18), D16S409-D16S419 (16), D16S411 (17, 19), D16S503 (16), and D16S516 (present report). These markers span the majority of chromosome 16. Taken together, these results suggest that more than one gene may be contributing to the evidence for linkage observed on chromosome 16.
Analysis of chromosome 16 in subsets of families based on linkage results from the region with greatest linkage in the genome (chromosome 1p) has the potential to separate heterogeneous contributions. The evidence for linkage in the pericentromeric region of IBD1 disappeared (Fig. 2) when analyzing those families with negative (25) scores on chromosome 1p (with appearance of peaks in the p and q telomeric regions) and increased to a more narrow peak at 50 cM centered in the middle of IBD1 (16) in families with positive NPL scores (25). The observed trends provide additional confirmatory evidence for linkage on chromosome 1p because stratifying on a false positive region of linkage would be expected to exert a random effect (for both positive and negative chromosome 1p NPL scores) on chromosome 16. On the contrary, the observed effects suggest an epistatic interaction between the chromosomes 1p and pericentromeric region of chromosome 16 peaks.
The additional loci that show nominal evidence for linkage (MLod ≥ 1.0, P ≤ 0.016) (Table 1) may potentially point to important inflammatory bowel disease susceptibility genes; follow-up studies with additional markers and families have been advocated in those regions identified in the initial genome screen with pointwise P-values ≤ 0.01 (32) or even, P ≤ 0.05 (27).
The absence of confirmatory evidence for the previously identified region on chromosome 12 (20) is not unexpected as the number of families required to replicate linkage may be significantly greater than the number required to initially identify linkage (33). Furthermore, the marker density in our study was not optimized for confirmation. The previous inflammatory bowel disease screen was primarily of non-Jewish Caucasians (20), however, our chromosome 12 results did not change with stratification by ethnicity. Another possibility is that the chromosome 12 locus represents primarily UC susceptibility factors, given the greater preponderance of UC in that report (20).
In conclusion, we observed suggestive evidence for linkage in all inflammatory bowel disease families in two susceptibility regions on chromosomes 1p and 3q. In a third region on chromosome 4q, an MLod score of 2.76 (P = 1.9 × 10−4) was observed in mixed families, especially pronounced in the Ashkenazim. Confirmatory evidence for linkage was observed in the pericentromeric region of chromosome 16, IBD1 (16), as well as evidence for epistasis between IBD1 and chromosome 1p. Analysis in the Ashkenazim may be of special benefit given the greater homogeneity in that group as well as their utility in refining gene localization using linkage disequilibrium (34). These studies suggest that linkage regions on chromosomes 3q and 4q may be illuminated by such approaches if present evidence for linkage is extended in additional families.
Acknowledgments
Foremost, we thank the patients and their families for their participation in the study. We gratefully acknowledge Joseph B. Kirsner, Graeme I. Bell, and Eugene B. Chang (University of Chicago), and Barton Childs, Cindy Berman, Romulo Baltazar, Eliyahu Kutoff, and Geoffrey Ravenhill (Johns Hopkins). We thank Augustine Kong and Michael Frigge for the use of the modified version of genehunter and most especially thank Nancy J. Cox for her original suggestions on subset analysis. We thank referring physicians Jason Bodzin, Daniel Present, Mary Harris, James Reed, Carl D’Angelo, Lester Rosen, Alan Lake, and Carmen Cuffari. This project was partially funded by the Crohn’s and Colitis Foundation of American (Career Development Award-J.H.C., First Award-S.R.B., family resources-T.M.B.), the Gastrointestinal Research Foundation, the Harvey M. and Lyn P. Meyerhoff Inflammatory Bowel Disease Center, the Mazza Foundation, the Edison Foundation, the Reva and David Logan Center for Gastrointestinal Research, Glaxo Institute for Digestive Health (J.H.C.), National Institutes of Health/National Center for Research Resources GCRC RR00055 (J.H.C.) and RR00052 (S.R.B.), and the Mammalian Genotyping Service (National Heart, Lung, and Blood Institute).
ABBREVIATIONS
- CD
Crohn’s disease
- UC
ulcerative colitis
- MLod
multipoint logarithm of the odds score
- NPL
nonparametric linkage
- cM
centimorgan
References
- 1.Calkins B M, Mendeloff A I. Epidemiol Rev. 1986;8:60–91. doi: 10.1093/oxfordjournals.epirev.a036296. [DOI] [PubMed] [Google Scholar]
- 2.Gollop J H, Phillips S F, Melton L J, III, Zinmeister A R. Gut. 1988;29:1943–1982. doi: 10.1136/gut.29.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stonnington C M, Phillips S F, Melton L J, III, Zinmeister A R. Gut. 1987;28:402–409. doi: 10.1136/gut.28.4.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Podolsky D K. N Engl J Med. 1991;325:928–937. doi: 10.1056/NEJM199109263251306. ; 1008–1016. [DOI] [PubMed] [Google Scholar]
- 5.Lee K S, Medline A, Shockey S. Arch Pathol Lab Med. 1979;103:173–176. [PubMed] [Google Scholar]
- 6.Yang H, Rotter J I. In: Inflammatory Bowel Disease: From Bench to Bedside. Targan S R, Shanahan F, editors. Baltimore: Williams & Wilkins; 1994. pp. 32–64. [Google Scholar]
- 7.Tysk C, Lindberg E, Janerot G, Floderus-Myrhed B. Gut. 1988;29:990–996. doi: 10.1136/gut.29.7.990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Orholm M, Munkholm P, Langholz E, Nielsen O H, Sorensen T I A, Binder V. N Engl J Med. 1991;324:84–88. doi: 10.1056/NEJM199101103240203. [DOI] [PubMed] [Google Scholar]
- 9.Probert C S J, Jayanthi V, Hughes A O, Thompson J R, Wicks A C B, Mayberry J F. Gut. 1993;34:1547–1551. doi: 10.1136/gut.34.11.1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuster W, Pascoe L, Purrmann J, Funk S, Majewski F. Am J Med Genet. 1989;32:105–108. doi: 10.1002/ajmg.1320320122. [DOI] [PubMed] [Google Scholar]
- 11.Orholm M, Iselius L, Sorensen T I, Munkholm P, Langholz E, Binder V. Br Med J. 1993;306:20–24. doi: 10.1136/bmj.306.6869.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lander E S, Schork N J. Science. 1994;265:2037–2048. doi: 10.1126/science.8091226. [DOI] [PubMed] [Google Scholar]
- 13.Satsangi J, Jewell D P, Bell J I. Gut. 1997;40:572–574. doi: 10.1136/gut.40.5.572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sandler R S. In: Inflammatory Bowel Disease: From Bench to Bedside. Targan S R, Shanahan F, editors. Baltimore: Williams & Wilkins; 1994. pp. 5–30. [Google Scholar]
- 15.Yang H, McElree C, Roth M P, Shanahan F, Targan S R, Rotter J I. Gut. 1993;34:517–524. doi: 10.1136/gut.34.4.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hugot J P, Laurent-Puig P, Gower-Rousseau C, Olson J M, Lee J C, Beaugerie L, Naom I, Dupas J L, VanGossum A, Orholm M, et al. Nature (London) 1996;379:821–823. doi: 10.1038/379821a0. [DOI] [PubMed] [Google Scholar]
- 17.Ohmen J D, Yang H Y, Yamamoto K K, Zhao H Y, Yuanhong M, Bentley L G, Haung Z, Gerwehr S, Pressman S, McElree C, et al. Hum Mol Genet. 1996;5:1679–1684. doi: 10.1093/hmg/5.10.1679. [DOI] [PubMed] [Google Scholar]
- 18.Cho J H, Fu Y, Kirschner B S, Hanauer S B. Inflammatory Bowel Dis. 1997;3:186–190. [PubMed] [Google Scholar]
- 19.Parkes M, Satsangi J, Lathrop G M, Bell J I, Jewell D P. Lancet. 1996;348:1588. doi: 10.1016/S0140-6736(05)66204-6. [DOI] [PubMed] [Google Scholar]
- 20.Satsangi J, Parkes M, Louis E, Hashimoto L, Kato N, Welsh K, Terwilliger J D, Lathrop G M, Bell J I, Jewell D P. Nat Genet. 1996;14:199–202. doi: 10.1038/ng1096-199. [DOI] [PubMed] [Google Scholar]
- 21.Gower-Rousseau C, Salomez J L, Dupas J L, Marti R, Nuttens M C, Votte A, Lemahieu M, Lemaire B, Colombel J F, Cortot A. Gut. 1994;35:1433–1438. doi: 10.1136/gut.35.10.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schwengel D A, Jedlicka A E, Nanthakumar E J, Weber J L, Levitt R C. Genomics. 1994;22:46–54. doi: 10.1006/geno.1994.1344. [DOI] [PubMed] [Google Scholar]
- 23.Yuan B, Vaske D, Weber J L, Beck J, Sheffield V C. Am J Hum Genet. 1997;60:459–460. [PMC free article] [PubMed] [Google Scholar]
- 24.Kong A, Cox N J. Am J Hum Genet. 1997;61:1179–1188. doi: 10.1086/301592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kruglyak L, Daly M J, Reeve-Daly M P, Lander E S. Am J Hum Genet. 1996;58:1347–1363. [PMC free article] [PubMed] [Google Scholar]
- 26.McPeek, M. S. (1998) Genet. Epidemiol., in press.
- 27.Lander E, Kruglyak L. Nat Genet. 1995;11:241–247. doi: 10.1038/ng1195-241. [DOI] [PubMed] [Google Scholar]
- 28.Naom I, Lee J, Ford D, Bowman S J, Lanchbury J S, Haris I, Hodgson S V, Easton D, Lennard-Jones J, Mathew C G. Am J Hum Genet. 1996;59:226–233. [PMC free article] [PubMed] [Google Scholar]
- 29.Hugot J P, Laurent-Puig P, Gower-Rousseau C, Caillat-Zucman S, Beaugerie L, Dupas J L, Van Gossum A, Bonaiti-Pellie C, Cortot A, Thomas G. Am J Med Genet. 1994;52:207–213. doi: 10.1002/ajmg.1320520216. [DOI] [PubMed] [Google Scholar]
- 30.Dubovsky J, Sheffield V C, Duyk G M, Weber J L. Hum Mol Genet. 1995;4:449–452. doi: 10.1093/hmg/4.3.449. [DOI] [PubMed] [Google Scholar]
- 31.Spielman R S, Ewens W J. Am J Hum Genet. 1996;59:983–989. [PMC free article] [PubMed] [Google Scholar]
- 32.Ghosh S, Schork N J. Diabetes. 1996;45:1–14. doi: 10.2337/diab.45.1.1. [DOI] [PubMed] [Google Scholar]
- 33.Bell J I, Lathrop G M. Nat Genet. 1996;13:377–378. doi: 10.1038/ng0896-377. [DOI] [PubMed] [Google Scholar]
- 34.Motulsky A G. Nat Genet. 1995;9:99–101. doi: 10.1038/ng0295-99. [DOI] [PubMed] [Google Scholar]